

Abstract
Patients with atrial fibrillation (AF) are at an approximately 0.5% to 3% increased risk of thromboembolism during and immediately after catheter ablation. Treatment guidelines recommend periprocedural oral anticoagulation plus unfractionated heparin during ablation. Rivaroxaban and dabigatran are the only non–vitamin K oral anticoagulants for which there are randomized controlled trials assessing uninterrupted anticoagulation in patients undergoing catheter ablation of AF. Edoxaban, a direct factor Xa inhibitor, is noninferior vs warfarin for the prevention of stroke or systemic embolism with less major bleeding in patients with nonvalvular AF. The ELIMINATE‐AF (Evaluation of Edoxaban Compared With VKA in Subjects Undergoing Catheter Ablation of Nonvalvular Atrial Fibrillation) trial is a multinational, multicenter, prospective, randomized, open‐label, parallel‐group, blinded‐endpoint evaluation (PROBE) study to assess the safety and efficacy of once‐daily edoxaban 60 mg (30 mg in patients indicated for a dose reduction) vs vitamin K antagonists (VKA) in patients with nonvalvular AF undergoing catheter ablation (http://www.ClinicalTrials.gov: NCT02942576). A total of 560 patients are planned for randomization to edoxaban or VKA (2:1 ratio) to obtain 450 patients fully compliant with the protocol. Patients will complete 21 to 28 days of anticoagulation prior to the ablation and a 90‐day post‐ablation period. The primary efficacy endpoint is the composite of all‐cause death, stroke, and major bleeding. The primary safety endpoint is major bleeding. A magnetic resonance imaging substudy will assess the incidence of silent cerebral lesions post‐ablation. ELIMINATE‐AF will define the efficacy and safety of edoxaban for uninterrupted oral anticoagulation during catheter ablation of AF.
Keywords: Atrial Fibrillation, Catheter Ablation, Direct Oral Anticoagulant, Direct Oral Anticoagulant Edoxaban, Non–Vitamin K Oral Anticoagulants, Periprocedural
1. INTRODUCTION
Catheter ablation is an effective rhythm‐control strategy in patients with symptomatic atrial fibrillation (AF) who are resistant or intolerant to antiarrhythmic medication or prefer not to take such medications.1, 2, 3, 4, 5 However, patients with AF undergoing ablation are at a small (approximately 0.5% to 3%) increased risk of thromboembolism during and after the procedure.4, 6, 7 Uninterrupted warfarin around the ablation procedure is associated with a lower risk of thromboembolic events and significantly less bleeding complications compared with a bridging strategy utilizing low‐molecular‐weight heparin.8 Therefore, contemporary treatment guidelines recommend uninterrupted oral anticoagulation.3, 4
As a class, non–vitamin K oral anticoagulants (NOACs) have similar efficacy and are associated with less major bleeding than warfarin for the prevention of stroke and systemic embolic events (SEE) in patients with AF.9 Until recently, due to lack of clinical data, there was no consensus regarding recommendations for periprocedural management of NOACs during catheter ablation. However, the 2017 Heart Rhythm Society (HRS)/European Heart Rhythm Association (EHRA)/European Cardiac Arrhythmia Society (ECAS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Society of Electrophysiology and Cardiac Stimulation (SOLAECE) expert consensus statement recommends that patients previously therapeutically anticoagulated with dabigatran or rivaroxaban should continue their anticoagulation regimens uninterrupted before and after undergoing AF catheter ablation.10 This recommendation is based on the only 2 published prospective, randomized, open, parallel‐group, blinded endpoint evaluation (PROBE) trials assessing uninterrupted NOACs in patients undergoing catheter ablation for AF.11, 12
In the Randomized Evaluation of Dabigatran Etexilate Compared to Warfarin in Pulmonary Vein Ablation: Assessment of an Uninterrupted Periprocedural Anticoagulation Strategy (RE‐CIRCUIT) study, uninterrupted dabigatran (150 mg twice daily) was associated with fewer bleeding complications than uninterrupted warfarin in patients undergoing ablation for AF during the 8 weeks post‐ablation.11 In the Active‐Controlled Multi‐Center Study With Blind‐Adjudication Designed to Evaluate the Safety of Uninterrupted Rivaroxaban and Uninterrupted Vitamin K Antagonists in Subjects Undergoing Catheter Ablation for Nonvalvular Atrial Fibrillation (VENTURE‐AF) study, the incidence of major bleeding with uninterrupted rivaroxaban (20 mg once daily) was low and similar to the incidence with an uninterrupted vitamin K antagonist (VKA) during the 4 weeks post‐ablation.12
Edoxaban is a direct oral factor Xa inhibitor with linear and predictable pharmacokinetics indicated for the prevention of stroke or SEE in patients with nonvalvular AF and for the treatment of venous thromboembolism.13, 14 In the Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis In Myocardial Infarction study 48 (ENGAGE AF‐TIMI 48) trial, once‐daily edoxaban high‐dose (60 mg) and low‐dose (30 mg) regimens were noninferior vs well‐managed warfarin for the prevention of stroke or SEE with superior safety (major bleeding).15 The edoxaban dose was halved from 60 mg to 30 mg or from 30 mg to 15 mg, respectively, if any of the following characteristics were present at the time of randomization or during the study: estimated creatinine clearance (CrCl) of 30 to 50 mL/min, a body weight of ≤60 kg, or the concomitant use of potent P‐glycoprotein (P‐gp) inhibitors.
In a pilot evaluation of ENGAGE AF‐TIMI 48, edoxaban was associated with a low risk of ischemic and bleeding events during the first 30 days post‐ablation among 193 catheter ablation procedures performed in 169 patients.16 The median interruption of study drug prior to ablation was 20 days. For 81% of ablations, the study drug was interrupted for >3 days (37 ablations with <5 days, 37 with between 5 and 10 days, 78 with >10 days of interruption, and 41 with no interruption). All bleeding events occurred in patients with ≤10 days of study‐drug interruption, and no ischemic events or deaths occurred in these patients.16 However, specifically evaluating the efficacy and safety of uninterrupted edoxaban in patients with AF undergoing catheter ablation requires a properly designed, prospective, randomized clinical trial.
The Evaluation of Edoxaban Compared With VKA in Subjects Undergoing Catheter Ablation of Nonvalvular Atrial Fibrillation (ELIMINATE‐AF) trial is a multinational, multicenter, PROBE design study assessing the safety and efficacy of once‐daily edoxaban 60 mg (once‐daily 30 mg in patients indicated for a dose reduction) vs VKA in patients with nonvalvular AF undergoing catheter ablation (http://www.ClinicalTrials.gov: NCT02942576; EudraCT: 2016–003069‐25). The primary objective is to descriptively compare the efficacy (composite of all‐cause death, stroke, and International Society on Thrombosis and Haemostasis [ISTH]‐defined major bleeding) and safety (ISTH‐defined major bleeding) of uninterrupted edoxaban vs VKA in patients undergoing catheter ablation. ELIMINATE‐AF will test the hypothesis that AF ablation can be safely and effectively performed with uninterrupted edoxaban therapy.
2. METHODS
2.1. Study design
This study will be conducted at approximately 72 study sites in 11 countries (Appendix). Enrollment commenced in March 2017, and 561 patients have been enrolled as of March 29, 2018.
Eligible patients will be randomized (2:1 ratio) to edoxaban or VKA following enrollment and will complete 21 to 28 days of anticoagulation prior to the ablation procedure in accordance with the current treatment guidelines (Figure 1).4, 5 Patients randomized to edoxaban will receive once‐daily edoxaban 60 mg or a dose reduction to once‐daily edoxaban 30 mg if they have CrCl 15 to 50 mL/min, body weight ≤ 60 kg, or are being treated with select P‐gp inhibitors (eg, cyclosporine, dronedarone, erythromycin, or ketoconazole).
Figure 1.
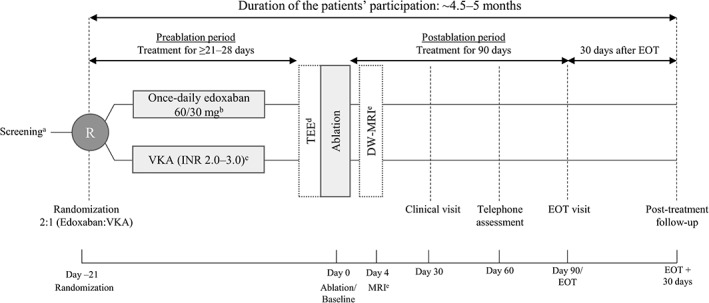
Schematic diagram of study design, including 21 to 28 days of anticoagulation with edoxaban or VKA during the pre‐ablation period, 90 days of post‐ablation treatment, and a 30‐day follow‐up. Abbreviations: CrCl, creatinine clearance; DW‐MRI, diffusion‐weighted magnetic resonance imaging; EOT, end of treatment; INR, international normalized ratio; MRI, magnetic resonance imaging; P‐gp, P‐glycoprotein; TEE, transesophageal echocardiography; VKA, vitamin K antagonist. aScreening and randomization visits may be combined if the inclusion/exclusion criteria can be checked based on health records and actual laboratory results with sufficient accuracy and the patient received informed consent ≥1 day prior to randomization. bPatients will be dose‐reduced to 30 mg edoxaban once daily if they meet any of the following: CrCl 15–50 mL/min, body weight ≤ 60 kg, or being treated with select P‐gp inhibitors. cAll patients randomized to VKA will be required to be in the INR range of 2.0 to 3.0 for the last 10 days prior to the ablation procedure. dTEE in all patients 1 day prior to or on the morning of the ablation procedure. If intracardiac clots are identified, then the ablation procedure will not be performed and the patients will be switched from study medication to standard of care and will enter the 30‐day follow‐up period. eDW‐MRI, diffusion‐weighted magnetic resonance imaging
All patients randomized to VKA will be required to maintain the international normalized ratio (INR) range of 2.0 to 3.0 for the last 10 days prior to ablation. For these patients, INR values will be documented at least once per week during the pre‐ablation period and at least monthly during the post‐ablation treatment period. The ablation procedure may still be performed at the discretion of the investigator, if on the day of or the day prior to ablation, patients in the VKA arm have an INR of 1.5 to 2.0 or 3.0 to 3.5. Otherwise, the patients will not be eligible for catheter ablation and will be switched to standard of care and enter the 30‐day follow‐up period.
Transesophageal echocardiography or intracardiac echocardiography will be performed before catheter ablation to identify potential intracardiac clots. If clots are identified, then the ablation procedure will not be performed and the patients will be switched to standard of care and enter the 30‐day follow‐up period. During catheter ablation, unfractionated heparin will be used in accordance with guidelines to achieve an activated clotting time of 300 to 400 seconds; bridging with low‐molecular‐weight heparin around the ablation procedure is prohibited. Diffusion‐weighted magnetic resonance imaging (DW‐MRI) will be performed at a subset of study sites 4±2 days post ablation to assess the incidence of silent cerebral events or silent cerebral lesions.
Treatment regimens will be continued until end of treatment (EOT) at 90 days post‐ablation, with scheduled clinical visits at 30 and 90 days and a telephone assessment at 60 days post‐ablation (Figure 1). All patients on study medication at 90 days post‐ablation will have a follow‐up at 25 to 35 days after EOT. The overall duration from screening through follow‐up is approximately 4.5 to 5 months. At EOT, patients will either discontinue treatment or will be transitioned to any available oral anticoagulant at the discretion of the investigator.
2.2. Ethics and informed consent
ELIMINATE‐AF will be conducted in accordance with the International Conference on Harmonisation Guidelines on Good Clinical Practice, the Declaration of Helsinki, and local regulations on the conduct of clinical research. An independent Data and Safety Monitoring Board (Appendix) will monitor safety data to protect the rights, safety, and well‐being of patients, and it will make recommendations pertaining to whether to modify the treatment regimens, the study, or the protocol based on prespecified criteria. Strict subject confidentiality will be maintained through subject identification codes. Informed consent will be obtained prior to enrollment.
2.3. Patient population and eligibility
Male and female patients age ≥ 18 years with a documented history of nonvalvular AF (paroxysmal, ≤7 days; persistent, >7 days but ≤12 months; or long‐standing persistent, >12 months) are eligible if they are scheduled for radiofrequency or cryoballoon catheter ablation for AF. Patients undergoing either first or repeated procedures are eligible. Information on the duration of AF will be based upon electrical tracing or medical records.
Patients scheduled for procedures using energy sources other than radiofrequency or cryoballoon are not eligible. In addition, patients with mechanical heart valves, moderate to severe mitral stenosis, and those who have new implantation (≤3 months prior to randomization) of a bioprosthetic heart valve are excluded. Table 1 shows the complete list of exclusion criteria.
Table 1.
Exclusion criteria
| Mechanical heart valves, moderate to severe mitral stenosis, or new implantation (within 3 months prior to randomization) of a bioprosthetic heart valve, with or without AF |
| Transient AF or AF of a reversible nature (eg, myocarditis, postsurgery, ionic disturbances, thyrotoxicosis, pneumonia, or severe anemia) |
| Post‐stroke or a systemic thromboembolic event within the past 6 months prior to randomization |
| Thrombus in the LAA, left atrium, left ventricle, or aorta; an intracardial mass; or history of LAA occlusion/exclusion (either by surgery or by a procedure) |
| MI within 2 months prior to randomization or CABG surgery within 3 months prior to randomization |
| Signs of bleeding, history of clinically relevant ISTH‐defined bleeding, or conditions associated with high risk of bleeding; overt GI bleeding or active ulcer within the previous year |
| Recent severe trauma, major surgery, or deep‐organ biopsy |
| Active infective endocarditis |
| Uncontrolled HTN (BP >170/100 mm Hg) |
| Hemorrhagic disorder including known or suspected hereditary or acquired bleeding or coagulation disorder in the last 12 months prior to randomization |
| Contraindication for edoxaban, VKA, LMWH, or heparin therapy including known allergies, hypersensitivity, or intolerance to any component of these drugs or their excipients |
| Receiving DAPT or planned to receive DAPT during the study |
| Concomitant use of UFH, LMWH, heparin derivatives (eg, fondaparinux), or OACs; bridging with LMWH around the ablation procedure is prohibited |
| Chronic use of medicines affecting hemostasis, including higher doses of ASA (100 mg/d allowed) or chronic oral or parenteral intake of NSAIDs on ≥4 d/wk (use of NSAIDs via other routes is not restricted) |
| Active liver disease or persistent (confirmed by repeat assessments at least a week apart) elevation of liver enzymes/bilirubin: |
| ALT or AST ≥2× ULN |
| TBL ≥1.5× ULN (subjects whose elevated TBL is due to known Gilbert syndrome may be included in the study) |
| Hepatic disease associated with coagulopathy and clinically relevant bleeding risk |
| Kidney failure (calculated CrCl <15 mL/min) |
| Hb <10 g/dL, or platelet count <100 000 cells/μL or WBC count <3000 cells/μL |
| Preplanned invasive diagnostic or therapeutic procedures/interventions (other than endoscopy) during the study period in which bleeding is anticipated; a planned procedure using laser catheter ablation or other forms of catheter ablation different from radiofrequency or cryoballoon |
| Participation in any other interventional trial; previous randomization in this study |
| Active cancer undergoing chemotherapy, radiation, or major surgery within the next 5 months |
| Significant active/uncontrolled concurrent medical illness; life expectancy <6 months |
| Known drug or alcohol dependence within the past 12 months prior to randomization, as judged by the investigators |
| Female patients of childbearing potential not using highly effective contraceptiona; female patients who are pregnant or breastfeeding |
| Patients considered by the investigators to have a condition that would place the patient at increased risk of harm; patients unlikely to comply with the protocol |
Abbreviations: AF, atrial fibrillation; ALT, alanine transaminase; ASA, acetylsalicylic acid (aspirin); AST, aspartate transaminase; BP, blood pressure; CABG, coronary artery bypass grafting; CrCl, creatinine clearance; DAPT, dual antiplatelet therapy; GI, gastrointestinal; Hb, hemoglobin; HTN, hypertension; ISTH, International Society on Thrombosis and Haemostasis; LAA, left atrial appendage; LMWH, low‐molecular‐weight heparin; MI, myocardial infarction; NSAID, nonsteroidal anti‐inflammatory drug; OAC, oral anticoagulant; TBL, total bilirubin; UFH, unfractionated heparin; ULN, upper limit of normal; VKA, vitamin K antagonist; WBC, white blood cell.
Females taking oral contraceptives should have been on therapy for ≥3 months.
2.4. Intervention and randomization
Patients will be randomized (2:1 ratio) to once‐daily edoxaban 60 mg (once‐daily 30 mg in patients meeting the dose‐reduction criteria) or a VKA using a block randomization method. An independent biostatistician will generate the randomization schedule, and randomization will be performed using an interactive voice/web response system. Patients on edoxaban 30 mg due to low body weight will remain on this dose for the duration of the study even if they gain weight; if the patient no longer displays the other dose‐reduction criteria, the dose will be increased to 60 mg. After randomization, patients on edoxaban 60 mg will be dose‐reduced to 30 mg if their weight drops to ≤60 kg, their CrCl decreases to ≤50 mL/min, or if they need concomitant P‐gp inhibitors. Patients randomized to VKAs will have their dose adjusted to maintain an INR within the target range (2.0–3.0). A preferred VKA will be selected for each participating country (see Supporting Information, Table 1, in the online version of this article).
Patients transitioning to edoxaban from a VKA will discontinue the VKA and start edoxaban when INR is ≤2.5; patients transitioning to edoxaban from another NOAC will start edoxaban at the next scheduled NOAC dose. Permanent discontinuation will occur if CrCl falls below 15 mL/min on 2 consecutive occasions or if dialysis is required.
During the study period, including the day before the procedure, once‐daily edoxaban will be taken in the evening. The interval between the last intake of edoxaban and the ablation procedure is not to exceed 18 hours. The rationale for the evening dose is based on the pharmacokinetic (PK) and pharmacodynamic (PD) characteristics of edoxaban. Following once‐daily edoxaban 60‐mg dosing, peak edoxaban plasma concentration (Cmax) occurs at approximately 1.5 hours; the half‐life is 11 to 14 hours.13, 17 In a phase 2 study, after 4 weeks of once‐daily edoxaban 60 mg, the median concentration of edoxaban was approximately 170 ng/mL (interquartile range [IQR], 125–245 ng/mL]) at Cmax and 25 ng/mL (IQR, 10–40 ng/mL) at trough.18 At 12 to 18 hours post–evening dose, during which the ablation procedure should be performed, edoxaban plasma concentrations are expected to be maintained at 17% to 32% of Cmax.17 Consequently, inhibition of intrinsic factor Xa (FXa) activity, which represents the PD effects of edoxaban, is present throughout the ablation period. Based on unpublished PK/PD modeling in the ENGAGE AF‐TIMI 48 trial, once‐daily edoxaban 60 mg would reduce intrinsic FXa activity to approximately 30% at the time of Cmax and 75% at 24 hours post‐dose (Figure 2; data on file).
Figure 2.
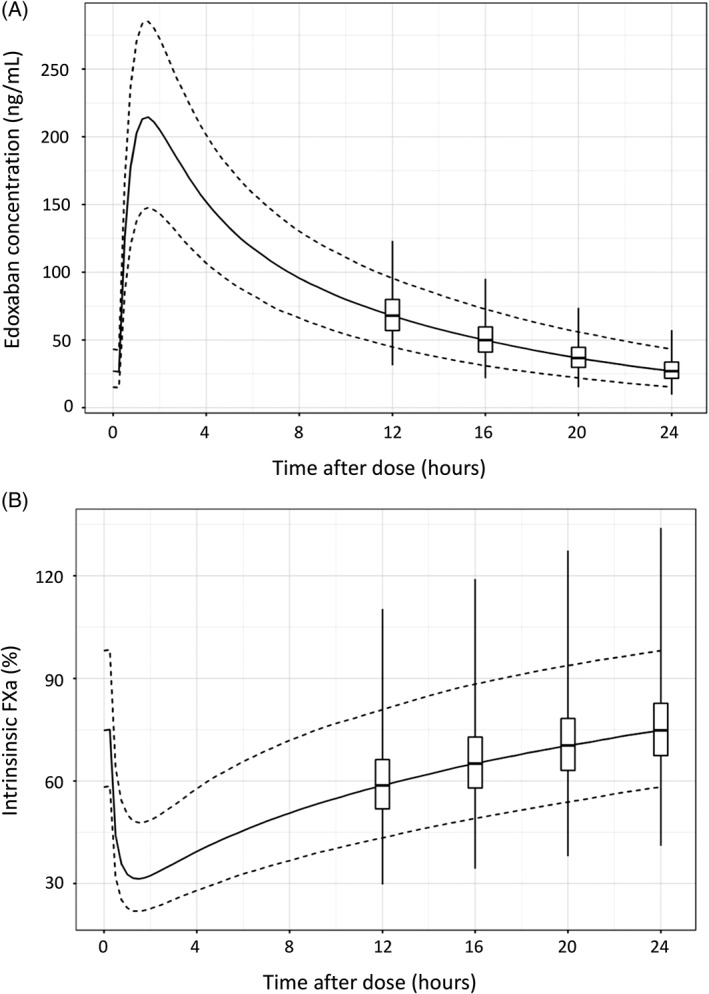
Model predicted (A) steady‐state edoxaban plasma concentration or (B) intrinsic FXa activity over a 24‐hour dosing interval. Prediction was based on PK/PD modeling of edoxaban concentration and intrinsic FXa data collected from 3029 patients in ENGAGE AF‐TIMI 48 trial. Solid line represents the median, and the dashed lines represent the fifth to 95th percentile range of edoxaban plasma concentration or intrinsic FXa activity. Box‐and‐whisker plots shows the distribution of edoxaban plasma concentration or intrinsic FXa activity at specific time points (ie, 12, 16, 20, and 24 hours post‐dose). Boxes represent the first to third quartiles. Whiskers represent the minimum to maximum value range. Abbreviations: ENGAGE AF‐TIMI 48, Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis In Myocardial Infarction study 48; FXa, factor Xa; PK/PD, pharmacokinetic/pharmacodynamic
After the ablation, study medication will be restarted on the day of the procedure. This will occur ≥6 hours post–sheath removal and only after achieving adequate hemostasis.
2.5. Assessments
Baseline demographics and clinical characteristics will include medical history, vital signs, type of AF, HAS‐BLED score, CHA2DS2‐VASc score, and presence of coronary artery disease, heart failure, hypertension, and diabetes mellitus. At each visit, study drug compliance (tablet counts) will be noted.
Clinical endpoints occurring between the end of the catheter ablation procedure and day 90/EOT, including stroke (ischemic, hemorrhagic, or undetermined), SEE, transient ischemic attack (TIA), myocardial infarction, and mortality (all‐cause and cardiovascular) will be assessed. All bleeding events between the date of first intake of study medication to day 90/EOT will also be surveyed.
Stroke is defined as an acute episode of focal or global neurological dysfunction caused by brain, spinal cord, or retinal vascular injury as a result of hemorrhage or infarction. Stroke is distinguished from TIA if the duration of neurological dysfunction is >24 hours, the duration is <24 hours but there is imaging documenting new hemorrhage or infarction, or if the neurological dysfunction results in death. An SEE is defined as an arterial embolism resulting in clinical ischemia, excluding the central nervous system, and coronary and pulmonary arterial circulation. Stroke events (ischemic, hemorrhagic, or undetermined) will be categorized as disabling and nondisabling stroke using the modified Rankin scale. Bleeding events will be classified into major bleeding, clinically relevant nonmajor bleeding, and minor bleeding as defined by the ISTH (see Supporting Information, Table 2, in the online version of this article).19 Bleeding events will also be classified according to Bleeding Academic Research Consortium (BARC) and TIMI classifications for descriptive purposes only (see Supporting Information, Tables 2 and 3, in the online version of this article).20, 21 An independent Clinical Events Committee will review and adjudicate suspected thromboembolic and bleeding events in a blinded manner. All adjudicated endpoints will be analyzed as time to first occurrence.
Table 2.
Study endpoints
| Primary efficacy endpoint |
|---|
| Composite of all‐cause death, strokea (primary definitionb), and major bleeding (ISTH definition) |
| Primary safety endpoint |
| Major bleeding (ISTH definition) |
| Secondary efficacy endpoints |
| Composite of all‐cause death, strokea (alternative definitionc), and major bleeding (ISTH definition) |
| Composite of stroke,a SEE, and CV mortality |
| Composite of stroke,a SEE, and all‐cause mortality |
| Composite of strokea and TIA |
| Strokea |
| Ischemic stroke |
| Hemorrhagic stroke |
| Undetermined stroke |
| SEE |
| TIA |
| Fatal strokea |
| Nonfatal strokea |
| Disabling strokea |
| Nondisabling strokea |
| Secondary safety endpoints |
| Major bleeding (defined by TIMI, BARC [≥2]) |
| Major or CRNM bleeding (defined by ISTH) |
| CRNM bleeding (ISTH definition) |
| Minor bleeding (ISTH definition) |
| Any bleeding |
| ICH |
| Life‐threatening bleeding |
| Fatal major bleeding (ISTH definition) |
| Nonfatal major bleeding (ISTH definition) |
| Fatal major bleeding (defined by TIMI, BARC [≥2]) |
| Nonfatal major bleeding (defined by TIMI, BARC [≥2]) |
| Safety parameters |
| Other endpoints |
| Relevant HEOR parametersd |
| Silent cerebral lesions as defined by DW‐MRIe |
| Cardiac markers (including NT‐proBNP and hs‐cTn) |
Abbreviations: BARC, Bleeding Academic Research Consortium; CRNM, clinically relevant nonmajor; CV, cardiovascular; DW‐MRI, diffusion‐weighted magnetic resonance imaging; HEOR, health economics and outcomes research; hs‐cTn, high‐sensitivity cardiac troponin; ICH, intracranial hemorrhage; ISTH, International Society on Thrombosis and Haemostasis; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide; SEE, systemic embolic events; TIA, transient ischemic event; TIMI, Thrombolysis In Myocardial Infarction.
Includes ischemic, hemorrhagic, and undetermined.
Acute episode of focal or global neurological dysfunction caused by brain, spinal cord, or retinal vascular injury as a result of hemorrhage or infarction. Stroke is distinguished from TIA if the duration of neurological dysfunction is >24 hours, the duration is <24 hours, and there is imaging documenting new hemorrhage or infarction, or if the neurological dysfunction results in death.
Abrupt onset of a focal neurological deficit in the distribution of a single brain artery that is not due to an identifiable nonvascular cause and that either lasts ≥24 hours or results in death within 24 hours of onset.
Includes cancellations of ablation procedure due to inadequate anticoagulation, hospital admissions due to CV causes, length of stay associated with the different types of hospital admissions, and additional outpatient physician or nurse visits that are CV event‐related.
At predetermined select sites.
Table 3.
Comparison between NOAC trials for patients with AF undergoing catheter ablation
| Study design | ELIMINATE‐AF | AXAFA‐AFNET 525 | RE‐CIRCUIT10 | VENTURE‐AF11 |
|---|---|---|---|---|
| PROBE | PROBE | PROBE | PROBE | |
| Study size | 450 planneda | 630 planneda | 635a | 221a |
| Status | Ongoing (NCT02942576) | Ongoing (NCT02227550) | Complete | Complete |
| NOAC | Edoxaban 60/30 mg once dailyb | Apixaban 5/2.5 mg twice dailyc | Dabigatran 150 mg twice daily | Rivaroxaban 20 mg orally once daily with evening meal |
| VKA | Various VKAs (INR 2.0–3.0) | Various VKAs (INR 2.0–3.0) | Warfarin (INR 2.0–3.0) | Various VKAs (INR 2.0–3.0) |
| Anticoagulation prior to ablation | Evening dose prior to day of ablation | Final morning dose on day of ablation | Final morning dose on day of ablation | Preferentially with the evening meal prior to ablation |
| Pre‐ablation TEE | Yes | Yes | Yes | Yes (optional) |
| Pre‐ablation anticoagulation treatment period | 21–28 days | ≥30 daysd | 4–8 weeks | TEE: 1 to 7 days if no thrombuse; no TEE: 3 weeks |
| Post‐ablation anticoagulation treatment period | 90 days | 90 days | 8 weeks | 30 days |
| Primary endpoint | Composite of all‐cause death, stroke, and major bleeding | Composite of all‐cause death, stroke, and major bleeding | Major bleeding | Major bleeding |
| MRI substudy | Yes | Yes | No | No |
Abbreviations: AF, atrial fibrillation; AXAFA‐AFNET 5, Apixaban During Atrial Fibrillation Catheter Ablation: Comparison to Vitamin K Antagonist Therapy; CrCl, creatinine clearance; ELIMINATE‐AF, Evaluation of Edoxaban Compared With VKA in Subjects Undergoing Catheter Ablation of Nonvalvular Atrial Fibrillation; INR, international normalized ratio; MRI, magnetic resonance imaging; NOAC, non–vitamin K antagonist oral anticoagulant; P‐gp, P‐glycoprotein; PROBE, prospective randomized open blinded endpoint; RE‐CIRCUIT, Randomized Evaluation of Dabigatran Etexilate Compared to Warfarin in Pulmonary Vein Ablation: Assessment of an Uninterrupted Periprocedural Anticoagulation Strategy; sCr, serum creatinine; TEE, transesophageal echocardiography; VENTURE‐AF, Active‐Controlled Multi‐Center Study With Blind‐Adjudication Designed to Evaluate the Safety of Uninterrupted Rivaroxaban and Uninterrupted Vitamin K Antagonists in Subjects Undergoing Catheter Ablation for Nonvalvular Atrial Fibrillation; VKA, vitamin K antagonist.
Randomized and having undergone ablation procedure.
Patients will be dose‐reduced to edoxaban 30 mg once daily if they meet any of the following: CrCl 15 to 50 mL/min, body weight ≤ 60 kg, or being treated with select P‐gp inhibitors.
Patients will be dose‐reduced to 2.5 mg apixaban twice daily if they meet 2 of the following: sCr ≥1.5 mg/dL, body weight ≤ 60 kg, or age ≥ 80 years.
Ablation can be performed earlier when atrial thrombi have been excluded by TEE.
Patients with documented anticoagulation for ≥3 weeks prior to randomization only required 1 to 7 days of anticoagulation.
Blood drawn at screening/randomization will be used for analysis of nongenetic cardiac biomarkers, including N‐terminal fragment B‐type natriuretic peptide and high‐sensitivity cardiac troponin. At a predefined subset of study sites, DW‐MRI or diffusion‐weighted imaging fluid‐attenuated inversion recovery will be performed 4 ± 2 days after catheter ablation to assess the incidence of silent cerebral events or silent cerebral lesions. Relevant health economics and outcomes research endpoints will also be assessed, including the cancellation of ablation procedure due to inadequate anticoagulation, hospital admissions due to cardiovascular causes, the length of stay associated with the different types of hospital admissions, and the number of additional outpatient visits that are cardiovascular event–related (see Supporting Information, Table 4, in the online version of this article).
Between randomization and final follow‐up, all adverse events will be surveyed and documented. Adverse events will be coded by organ class and preferred terms using the Medical Dictionary for Regulatory Activities (MedDRA), version 19.1 or newer. Vital signs including heart rate, blood pressure, and body weight will be documented at randomization, at all visits during the study treatment period, and at final follow‐up. Blood samples for laboratory analyses will be collected and CrCl will be calculated using the Cockcroft‐Gault formula at screening/randomization, on the day of catheter ablation, on day 30 post‐ablation, and at the EOT visit.
2.6. Endpoints
The primary efficacy endpoint is the time to first occurrence of all‐cause death, stroke (ischemic, hemorrhagic, or undetermined), or ISTH‐defined major bleeding during the period from the end of the catheter ablation procedure to day 90/EOT. The primary safety endpoint is the time to first occurrence of ISTH‐defined major bleeding from the date of first intake of study medication to day 90/EOT. Table 2 shows the complete list of study endpoints.
2.7. Sample size and statistical analysis
In VENTURE‐AF, the incidence of thromboembolic events with VKA was <1%, as was the incidence of major bleeding events.12 In RE‐CIRCUIT, there were no stroke/SEE events and only 1 TIA in the warfarin group, and the incidence of major bleeding was <7%.11 Based on these data, the incidence of the combined primary endpoint in ELIMINATE‐AF (ie, all‐cause death, stroke, and major bleeding) is estimated to be <3%. Therefore, a sufficiently powered study to test for formal noninferiority or superiority would require a prohibitively large sample size (ie, >8000 patients would be needed to detect a significant difference between the treatment groups with 80% power based on the estimated event rate of 3% in the VKA group and 2% in the edoxaban group); hence, it is not feasible. Enrollment will stop once 450 patients have undergone an ablation procedure without any major protocol violations. To achieve this, approximately 560 patients will need to be enrolled.
An enrollment of 450 patients will provide approximately 81% power to detect differences between the 2 groups based on the assumption that the event rate for major bleeding in the ELIMINATE‐AF study is the same as observed in the RE‐CIRCUIT study (ie, 6.9% in the VKA group and 1.6% in the edoxaban group).11 Moreover, a higher statistical power can be expected due to the longer duration of treatment and follow‐up in ELIMINATE‐AF as compared with RE‐CIRCUIT (in RE‐CIRCUIT, the post‐ablation period was 8 weeks with 1 week of follow‐up; whereas in ELIMINATE‐AF, patients are treated for 3 months after the ablation procedure and followed up for another month).11
All efficacy analyses will be conducted in the per‐protocol population (ie, all randomized patients who received ≥1 dose of the study regimen and do not have any major protocol violations) from the end of catheter ablation until day 90/EOT. The safety analyses will be conducted in all patients who received ≥1 dose of study drug from the time of first dose of study medication to day 90/EOT. All analyses will be based on the randomized treatment regimen, even if a patient inadvertently receives the incorrect drug or dosage or if the edoxaban dose is reduced during the study.
The statistical analysis will be interpreted in a purely descriptive way and no formal confirmatory statistical testing is planned. All analyses will be performed on observed data only and no missing data will be imputed. Data on patients who do not reach a specific endpoint will be censored in the corresponding statistical analyses. All endpoints will be analyzed using a Cox proportional hazard model with treatment regimen as a factor. For time‐to‐first‐event analyses, cumulative event rates over time will be summarized using the Kaplan–Meier method. Adverse events, including treatment emergent adverse events, will be tabulated by event and severity.
3. DISCUSSION
ELIMINATE‐AF is a phase 3, PROBE design study assessing the efficacy and safety of uninterrupted edoxaban vs VKA in patients undergoing catheter ablation for AF. The duration of anticoagulation in the pre‐ablation treatment period (21–28 days) is in agreement with the EHRA guidelines (ie, 3 weeks of systemic anticoagulation at a therapeutic level prior to ablation).4 There will also be an MRI substudy assessing incidence of silent cerebral events or silent cerebral lesions post‐ablation. The MRI substudy will assess the incidence of cerebral ischemia in asymptomatic patients (ie, silent cerebral events or silent cerebral lesions) post‐ablation at a preselected subset of study centers. Following ablation, the incidence of such silent events has been reported to be up to 40% to 50%.22, 23, 24
In a recent meta‐analysis, perioperative NOAC treatment during AF catheter ablation was associated with a lower risk of bleeding vs continuous warfarin (relative risk: 0.78, 95% confidence interval: 0.64–0.95, P = 0.01); however, there were no differences in the risk of thromboembolic events.25 In other meta‐analyses, periprocedural treatment with rivaroxaban or dabigatran during catheter ablation was associated with similar rates of thromboembolic events and major bleeding vs warfarin.26, 27, 28 However, these analyses were limited by a paucity of data. In Ablation Perioperative Dabigatran in Use Envisioning in Japan (ABRIDGE‐J), a brief interruption of dabigatran (1–2 doses) with or without heparin was associated with a lower risk of major bleeding and no increase in thromboembolic events compared with uninterrupted warfarin.29
There have been only 2 PROBE design trials assessing uninterrupted NOAC treatment in AF patients undergoing catheter ablation. In RE‐CIRCUIT, the largest study, uninterrupted dabigatran was associated with significantly lower incidence of major bleeding than warfarin post‐ablation among 704 patients (hazard ratio: 0.22, 95% confidence interval: 0.08–0.59).11 In VENTURE‐AF, the number of adjudicated thromboembolic or bleeding events were similarly low post‐ablation in 248 AF patients randomized to rivaroxaban vs warfarin (26 vs 25).12 There is also an ongoing trial assessing the efficacy and safety of uninterrupted apixaban vs VKA in patients undergoing catheter ablation for AF—Apixaban During Atrial Fibrillation Catheter Ablation: Comparison to Vitamin K Antagonist Therapy (AXAFA‐AFNET 5; http://www.ClinicalTrials.gov: NCT02227550).30
Table 3 shows the differences in the trial designs between ELIMINATE‐AF, RE‐CIRCUIT, VENTURE‐AF, and AXAFA‐AFNET 5.11, 12, 30 Compared with RE‐CIRCUIT, ELIMINATE‐AF has a more systematic pre‐ablation anticoagulation treatment period, which is aligned with current recommendations.4, 11 Unlike RE‐CIRCUIT or VENTURE‐AF, ELIMINATE‐AF includes an MRI substudy assessing silent cerebral lesions.11, 12 ELIMINATE‐AF also allows for the preferred VKA to be selected for each participating country, whereas RE‐CIRCUIT used warfarin.11
Edoxaban will be administered as an evening dose, including the day before the scheduled ablation, to minimize the risk of bleeding and with no interruptions in dosing. In RE‐CIRCUIT, the final morning dose of the dabigatran twice‐daily regimen was taken on the day of the procedure (41.3% received the last dose <4 hours before).11 In the dabigatran group, 3 of 5 major bleeding events occurred in patients receiving the last dose <4 hours before ablation.11 This result suggests the importance of having NOAC levels not at peak during the ablation procedure.
4. CONCLUSION
The results of ELIMINATE‐AF will provide further data to support the growing body of evidence regarding the efficacy and safety of uninterrupted anticoagulation with NOACs during the periprocedural period during catheter ablation for AF.
Supporting information
Supplementary Table 1. VKAs predefined for the various countries
Supplementary Table 2. Definitions of ISTH and TIMI Bleeding
Supplementary Table 3. BARC Bleeding Definition
Supplementary Table 4. Relevant Health Economics Outcome Research Parameters
ACKNOWLEDGMENTS
Assistance in medical writing and editorial support was provided by Stefan Kolata, PhD, of AlphaBioCom, LLC (King of Prussia, PA), and funded by Daiichi‐Sankyo, Inc. (Basking Ridge, NJ).
Conflicts of interest
Stefan H. Hohnloser has received consulting fees from Bayer Healthcare, Boehringer Ingelheim, Bristol‐Myers Squibb, Boston Scientific, Cardiome, Daiichi‐Sankyo, Gilead, Johnson & Johnson, Medtronic, Pfizer, Portola, Sanofi Aventis, Servier, St. Jude Medical, Zoll and lecture fees from Bayer Healthcare, Boehringer Ingelheim, Bristol‐Myers Squibb, Daiichi‐Sankyo, Pfizer, Sanofi Aventis, St. Jude Medical, and Medtronic. John Camm has received institutional research grants from Bayer, Boehringer Ingelheim, Pfizer/BMS, and Daiichi‐Sankyo. Personal fees from Bayer, Boehringer Ingelheim, Pfizer/Bristol‐Myers Squibb, and Daiichi‐Sankyo. Riccardo Cappato has received consulting fees from Zoll and Bayer Healthcare, and has received honoraria for participation in clinical trials or contributions to advisory boards or oral presentations from Medtronic, Boston Scientific, Zoll, Johnson & Johnson, Bayer Healthcare, and Pfizer. He is an owner of stock in Cameron Health. Hans‐Christoph Diener has received honoraria for participation in clinical trials or contributions to advisory boards or oral presentations from Abbott, Achelios, Allergan, AstraZeneca, Bayer Vital, Boehringer Ingelheim, Bristol‐Myers Squibb, CoAxia, Corimmun, Covidien, Daiichi‐Sankyo, D‐Pharm, Fresenius, GlaxoSmithKline, Janssen‐Cilag, Johnson & Johnson, Knoll, Lilly, Merck Sharp and Dohme, Medtronic, MindFrame, Neurobiological Technologies, Novartis, Novo‐Nordisk, Paion, Parke‐Davis, Pfizer, Sanofi‐Aventis, Schering‐Plough, Servier, Solvay, St. Jude, Syngis, Talecris, ThromboGenics, WebMD Global, and Wyeth, and has received financial support for research projects from AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Lundbeck, Novartis, Janssen‐Cilag, Sanofi‐Aventis, Syngis, and Talecris. The Department of Neurology at the University Duisburg‐Essen received research grants from the German Research Council (DFG), German Ministry of Education and Research (BMBF), European Union, US National Institutes of Health, Bertelsmann Foundation, and Heinz‐Nixdorf Foundation. He has no ownership interest and does not own stocks of any pharmaceutical company. Hein Heidbuchel has been member of the scientific advisory boards and/or a lecturer for Boehringer Ingelheim, Bayer, Bristol‐Myers Squibb, Pfizer, Daiichi‐Sankyo, and Cardiome, and has received unconditional research grants through the University of Antwerp from Bracco Imaging Europe and through Hasselt University from Bayer. Hans‐Joachim Lanz, Rüdiger Smolnik, and Ophelia Q.P. Yin are employees of Daiichi‐Sankyo. Lluís Mont has received fees as consultant and for advisory boards and lectures from St. Jude Medical (now Abbott), Medtronic, Boston Scientific, Biotronik, Biosense Webster, and Boehringer Ingelheim. Carlos A. Morillo has received consulting fees from Bayer Healthcare, Biotronik, Boehringer Ingelheim, Boston Scientific, Daiichi‐Sankyo, Medtronic, Pfizer, Sanofi, Servier, and St. Jude Medical (Abbott), and lecture fees from Bayer Healthcare, Biotronik, Daiichi‐Sankyo, Pfizer, St. Jude Medical, and Medtronic. Josef Kautzner has served as a scientific advisor for Bayer, Biosense Webster, Boehringer Ingelheim, Boston Scientific, LivaNova, Medtronic, Merck Sharp and Dohme, and St. Jude Medical (Abbott). He is a co‐Chair of the ELIMINATE‐AF Steering Committee and national coordinator of this study for the Czech Republic. The authors declare no other potential conflicts of interest.
1.
Participating countries for ELIMINATE‐AF: Belgium, Canada, Czech Republic, Germany, Hungary, Italy, Korea, Poland, Spain, Taiwan, and United Kingdom. Executive Steering Committee: Prof. Dr. Stefan H. Hohnloser, MD, Frankfurt, Germany (Chair); Prof. Dr. Josef Kautzner, MD, Prague, Czech Republic (Co‐Chair); Prof. Dr. John Camm, MD, London, United Kingdom; Prof. Dr. Riccardo Cappato, MD, Rozzano (Milan), Italy; Prof. Dr. Hans‐Christoph Diener, MD, PhD, Essen, Germany; Prof. Dr. Hein Heidbüchel, MD, PhD, Antwerp, Belgium; Prof. Dr. Lluís Mont, MD, Barcelona, Spain; Prof. Dr. Carlos A. Morillo, MD, Calgary, Alberta, Canada. Data and Safety Monitoring Board: Prof. Dr. John W.A. Eikelboom, MD, Hamilton, Ontario, Canada; Dr. Jonas Oldgren, MD, PhD, Uppsala, Sweden; Prof. Dr. Robin S. Roberts, PhD, Hamilton, Ontario, Canada. Clinical Events Committee: Prof. Dr. Grethe Andersen, MD, Aarhus, Denmark; Prof. em. Dr. med. Dr. h.c. Günter Breithardt, MD, Münster, Germany; PD Dr. med. Karl Georg Häusler, MD, Berlin, Germany; Prof. Dr. Claude E. Hanet, MD, Yvoir, Belgium; Dr. Eugene P. McFadden, MD, Cork, Ireland; Prof. Dr. med. Ulrich Tebbe, MD, Detmold, Germany. Lead Investigators: Belgium, Prof. Dr. Hein Heidbüchel, MD, PhD, Antwerp; Canada, Prof. Dr. Carlos A. Morillo, MD, Calgary, Alberta; Czech Republic, Prof. Dr. Josef Kautzner, MD, Prague; Germany, Prof. Dr. Stephan Willems, MD, Hamburg; Hungary, Dr. Gabor Duray, MD, PhD, Budapest; Italy, Dr. Massimo Grimaldi, MD, Bari; Korea, Prof. Dr. Moon‐Hyong Lee, MD, Seoul; Poland, Prof. Dr. Piotr Kulakowski, MD, Warsaw; Spain, Prof. Dr. Lluís Mont, MD, Barcelona; Taiwan, Prof. Dr. Shih‐Ann Chen, MD, Taipei; United Kingdom, Prof. Dr. John Camm, MD, London.
Hohnloser SH, Camm J, Cappato R, et al. Uninterrupted administration of edoxaban vs vitamin K antagonists in patients undergoing atrial fibrillation catheter ablation: Rationale and design of the ELIMINATE‐AF study.. Clin Cardiol. 2018;41:440–449. 10.1002/clc.22918
Funding information Daiichi Sankyo Europe GmbH (München, Germany)
REFERENCES
- 1. Calkins H, Reynolds MR, Spector P, et al. Treatment of atrial fibrillation with antiarrhythmic drugs or radiofrequency ablation: two systematic literature reviews and meta‐analyses. Circ Arrhythm Electrophysiol. 2009;2:349–361. [DOI] [PubMed] [Google Scholar]
- 2. Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:32–38. [DOI] [PubMed] [Google Scholar]
- 3. January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society [published correction appears in J Am Coll Cardiol. 2014;64:2305–2307]. J Am Coll Cardiol. 2014;64:e1–e76. [DOI] [PubMed] [Google Scholar]
- 4. Calkins H, Kuck KH, Cappato R, et al. 2012 HRS/EHRA/ECAS Expert Consensus Statement on Catheter and Surgical Ablation of Atrial Fibrillation: recommendations for patient selection, procedural techniques, patient management and follow‐up, definitions, endpoints, and research trial design. Europace. 2012;14:528–606. [DOI] [PubMed] [Google Scholar]
- 5. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the Management of Atrial Fibrillation Developed in Collaboration With EACTS [article in English, Spanish; published correction appears in Rev Esp Cardiol (Engl Ed). 2017;70:1031]. Rev Esp Cardiol (Engl Ed). 2017;70:50.e1–50.e84. [DOI] [PubMed] [Google Scholar]
- 6. Oral H, Chugh A, Ozaydin M, et al. Risk of thromboembolic events after percutaneous left atrial radiofrequency ablation of atrial fibrillation. Circulation. 2006;114:759–765. [DOI] [PubMed] [Google Scholar]
- 7. Vazquez SR, Johnson SA, Rondina MT. Peri‐procedural anticoagulation in patients undergoing ablation for atrial fibrillation. Thromb Res. 2010;126:e69–e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Biase L, Burkhardt JD, Santangeli P, et al. Periprocedural stroke and bleeding complications in patients undergoing catheter ablation of atrial fibrillation with different anticoagulation management: results from the Role of Coumadin in Preventing Thromboembolism in Atrial Fibrillation (AF) Patients Undergoing Catheter Ablation (COMPARE) randomized trial. Circulation. 2014;129:2638–2644. [DOI] [PubMed] [Google Scholar]
- 9. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta‐analysis of randomised trials. Lancet. 2014;383:955–962. [DOI] [PubMed] [Google Scholar]
- 10. Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm. 2017;14:e275–e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calkins H, Willems S, Gerstenfeld EP, et al; RE‐CIRCUIT Investigators . Uninterrupted dabigatran versus warfarin for ablation in atrial fibrillation. N Engl J Med. 2017;376:1627–1636. [DOI] [PubMed] [Google Scholar]
- 12. Cappato R, Marchlinski FE, Hohnloser SH, et al; VENTURE‐AF Investigators . Uninterrupted rivaroxaban vs. uninterrupted vitamin K antagonists for catheter ablation in non‐valvular atrial fibrillation. Eur Heart J. 2015;36:1805–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non‐vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55:641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Savaysa (edoxaban) tablets [full prescribing information] . Parsippany, NJ: Daiichi‐Sankyo Inc., 2015. [Google Scholar]
- 15. Giugliano RP, Ruff CT, Braunwald E, et al; ENGAGE‐AF TIMI 48 Investigators . Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093–2104. [DOI] [PubMed] [Google Scholar]
- 16. Steffel J, Ruff CT, Hamershock RA, et al. First experience with edoxaban and atrial fibrillation ablation: insights from the ENGAGE AF‐TIMI 48 trial. Int J Cardiol. 2017;244:192–195. [DOI] [PubMed] [Google Scholar]
- 17. Lip GY, Agnelli G. Edoxaban: a focused review of its clinical pharmacology. Eur Heart J. 2014;35:1844–1855. [DOI] [PubMed] [Google Scholar]
- 18. Weitz JI, Connolly SJ, Patel I, et al. Randomised, parallel‐group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation. Thromb Haemost. 2010;104:633–641. [DOI] [PubMed] [Google Scholar]
- 19. Rodeghiero F, Tosetto A, Abshire T, et al; ISTH/SSC Joint VWF and Perinatal/Pediatric Hemostasis Subcommittee Working Group . ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–2065. [DOI] [PubMed] [Google Scholar]
- 20. Mehran R, Rao SV, Bhatt DL, et al. Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation. 2011;123:2736–2747. [DOI] [PubMed] [Google Scholar]
- 21. Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation. 1987;76:142–154. [DOI] [PubMed] [Google Scholar]
- 22. Herrera Siklódy C, Deneke T, Hocini M, et al. Incidence of asymptomatic intracranial embolic events after pulmonary vein isolation: comparison of different atrial fibrillation ablation technologies in a multicenter study. J Am Coll Cardiol. 2011;58:681–688. [DOI] [PubMed] [Google Scholar]
- 23. Deneke T, Shin DI, Balta O, et al. Postablation asymptomatic cerebral lesions: long‐term follow‐up using magnetic resonance imaging. Heart Rhythm. 2011;8:1705–1711. [DOI] [PubMed] [Google Scholar]
- 24. Haeusler KG, Koch L, Herm J, et al. 3 Tesla MRI‐detected brain lesions after pulmonary vein isolation for atrial fibrillation: results of the MACPAF study. J Cardiovasc Electrophysiol. 2013;24:142017. [DOI] [PubMed] [Google Scholar]
- 25. Zhao Y, Yang Y, Tang X, et al. New oral anticoagulants compared to warfarin for perioperative anticoagulation in patients undergoing atrial fibrillation catheter ablation: a meta‐analysis of continuous or interrupted new oral anticoagulants during ablation compared to interrupted or continuous warfarin. J Interv Card Electrophysiol. 2017;48:267–282. [DOI] [PubMed] [Google Scholar]
- 26. Aryal MR, Ukaigwe A, Pandit A, et al. Meta‐analysis of efficacy and safety of rivaroxaban compared with warfarin or dabigatran in patients undergoing catheter ablation for atrial fibrillation. Am J Cardiol. 2014;114:577–582. [DOI] [PubMed] [Google Scholar]
- 27. Phan K, Wang N, Pison L, et al. Rivaroxaban versus warfarin or dabigatran in patients undergoing catheter ablation for atrial fibrillation: a meta‐analysis. Int J Cardiol. 2015;185:209–213. [DOI] [PubMed] [Google Scholar]
- 28. Hohnloser SH, Camm AJ. Safety and efficacy of dabigatran etexilate during catheter ablation of atrial fibrillation: a meta‐analysis of the literature. Europace. 2013;15:1407–1411. [DOI] [PubMed] [Google Scholar]
- 29. Nogami A, Machino T, Harada T, et al; ABRIDGE‐J Study Group . Clinical benefit of minimally interrupted dabigatran versus uninterrupted warfarin for catheter ablation of atrial fibrillation: a prospective randomized multicenter trial. Circulation. 2017;136:e448–e467.29229627 [Google Scholar]
- 30. Di Biase L, Callans D, Haeusler KG, et al. Rationale and design of AXAFA‐AFNET 5: an investigator‐initiated, randomized, open, blinded outcome assessment, multi‐centre trial comparing continuous apixaban to vitamin K antagonists in patients undergoing atrial fibrillation catheter ablation. Europace. 2017;19:132–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. VKAs predefined for the various countries
Supplementary Table 2. Definitions of ISTH and TIMI Bleeding
Supplementary Table 3. BARC Bleeding Definition
Supplementary Table 4. Relevant Health Economics Outcome Research Parameters