

Summary
Huntington's disease (HD) is a late‐onset fatal neurodegenerative disease, characterized by progressive movement disorders, psychiatric symptoms, and cognitive impairment. The cytosine‐adenine‐guanine (CAG) triplet expansion encoding glutamine present in the protein huntingtin (Htt), produces widespread neuronal and glial pathology. Mutant huntingtin (mHtt) nuclear aggregates are the primary cause of cortical and striatal neuron degeneration, neuronal inflammation, apoptosis and eventual cell loss. The precise mechanisms underlying the pathogenesis of neurodegeneration in HD remain poorly understood and HD patients have no current cure. Potassium channels are widely expressed in most cell types. In neurons, they play a crucial role in setting the resting membrane potential, mediating the rapid repolarization phase of the action potential and controlling sub‐threshold oscillations of membrane potentials. In glial cells, their major contributions are maintaining the resting membrane potential and buffering extracellular K+. Thus, potassium channels have an essential function in both physiological and pathological brain conditions. This review summarizes recent progress on potassium channels involved in the pathology of HD by using different HD mouse models. Exploring the dysfunction of potassium channels in the brain illustrates new approaches for targeting this channel for the treatment of HD.
Keywords: astrocytes, excitotoxicity, Huntington's disease, neurodegeneration, potassium channels
1. INTRODUCTION
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disease, which was first described by George Huntington in 1872. At present, HD is the most common monogenic fatal neurological disorder in western populations, affecting nearly 1 case per 7300 normal individuals based on epidemiological studies.1 HD patients present with progressive movement disorder, psychiatric symptoms, cognitive impairment and usually die from disease complications about 20 years after the onset. In the past few decades there has been a rapid growth in our understanding of the natural history of HD and pathogenesis at both the cellular and macroscopic level. However, few treatments are available and numerous clinical trials have failed to date. In addition, neurodegeneration in HD is also accompanied by signs of systemic neuroinflammation.2, 3 Thus, it is important to explore the precise mechanisms involved in the pathogenesis of HD.
Potassium channels are the most widely distributed type of ion channels and are found in virtually all living organisms.4 In general, potassium channels constitute a ubiquitous family of ion channels with more than 90 genes coding for the principal pore‐forming subunits, which allow K+ ions to cross the plasma membrane when activated.5 They form potassium‐selective pores that span cell membranes and are widely expressed in all cell types in both central and peripheral nervous system. Potassium channels can be categorized into four major classes: voltage‐gated K+ channels (Kv), Ca2+‐activated K+ channels (KCa), two‐pore K+ channels (K2P) and inwardly rectifying K+ channels (Kir).5 All these four classes of potassium channels play important roles in the brain. For instance, in neurons, they are crucial for setting the resting membrane potentials, mediating the rapid repolarization phase of the action potential and controlling sub‐threshold oscillations of membrane potentials.6, 7 In glial cells, the main contributions of potassium channels are to set the resting membrane potential and the buffering of extracellular K+.8, 9, 10, 11 Their unique biophysical properties are also related to neurotransmitter release, hormone secretion, cell proliferation, apoptosis and tumor progression as well.12, 13 The dysfunction of potassium channels can induce numerous CNS disorders, including HD.10, 14, 15, 16 With a tremendous functional relevance in both physiological and pathological brain, potassium channels have become an important drug target in clinical therapies.12, 17, 18, 19, 20 Here we summarize recent progress on understanding the role of potassium channels in the pathology of HD obtained from various mouse models. With a thorough examination of the dysfunction of potassium channels in HD, new insights for the development of therapeutic strategies for the treatment of this devastating neurodegenerative disease could be gained.
2. PATHOGENESIS OF HUNTINGTON'S DISEASE AND HD MURINE MODELS
The prime culprit of HD is the mutant HTT gene, which is located in the short arm of chromosome 4p16.3 and contains a fragment of polyglutamine coding sequences (polyQ).21 In normal populations, the length of polyQ ranges from 6 to 35 repeats, but HD patients carry more than 36 CAG repeats in their HTT gene and exhibit an inverse relationship between CAG repeat length and age of disease onset.22 Mutant Htt proteins tend to form hydrogen bonds with each other and finally develop into complex aggregate inclusion bodies, which can induce cell toxicity.1* Recently, researchers tried to deplete the HTT gene in early mouse embryos. However, those embryos failed to develop healthy brains, indicating that HTT gene is required for normal fetal brain development.23 Thus, the basic function of HTT gene and the pathogenesis of the mutated Htt proteins affecting the brain are not yet fully understood. *[Correction added on 19 February 2018, after first online publication: The word “Mutated” was changed to “Mutant” in the sentence”.]
To explore the mechanisms and pathogenesis of HD, it is necessary to have a general view about the variety of animal models which have been developed and applied to the study of HD.24 In early studies, toxin‐induced models such as the Quinolinic acid (QA) or 3‐nitropropionic (3‐NP) induced HD rat models were most commonly used. These toxins cause striatal neurodegeneration, motor impairments and cognitive deficits.25, 26, 27, 28 Although toxin induced HD models are easy to generate and produce specific symptoms similar to HD, the absence of mutant Htt gene and subsequent Htt aggregate formation prevented these models to precisely study the progressive nature of HD.29 In the mid 1990s, Mangiarini et al30 first created transgenic mouse models (R6 lines) and since then transgenic HD mouse models have become a classical mouse model to study disease progression. The well characterized R6/2 mouse, for example, expresses exon 1 of human Htt with ~144 CAG repeats and displays a rapidly progressive neurological phenotype onset at about 2 months of age, which includes decreased motor skills, tremors, stereotypic grooming and weight loss as well.31 In addition, Hodgson et al and Slow et al32, 33 created and developed a YAC transgenic mouse model. They used a yeast artificial chromosome (YAC) vector system to express a full‐length human Htt allele with 46, 72 or 128 CAG repeats under control of the endogenous Htt promoter. The advantage of this model is that YAC mice live much longer and therefore can be used to study the HD progression over a long period.33 With the development of genetic technology, knock in mouse models (KI models) such as classic chimeric HdhQ line and recent zQ175 mouse line were created and considered to be accurate for mimicking human HD characteristics.34, 35, 36, 37 Besides rodent models, nonhuman primates and sheep models have also been developed and added to the variety of HD animal research tools.38, 39, 40
3. ALTERED K+ CONDUCTANCE AND K+ CHANNEL DYSFUNCTION EXACERBATE STRIATAL NEURONS VULNERABILITY AND INTRINSIC EXCITABILITY
As a neurodegenerative disorder, HD affects the projection neurons mainly in two brain regions, striatum and cerebral cortex. Before distinct cell death and behavioral symptoms in HD, mHtt gene firstly causes neuronal dysfunction and subtle morphological changes.41, 42, 43, 44 The K+ conductances are crucial to maintain the hyperpolarized membrane potential and the slow firing rate of medium‐sized spiny neurons (MSNs) in the striatum.45, 46, 47, 48 Using an aggressive HD mouse model, Klapstein et al42 examined the electrophysiological properties of striatal MSNs in R6/2 mice. They found an increased membrane input resistance and depolarized resting membrane potential (RMP) as the behavioral phenotype developed,41, 42 indicating a possible alteration of K+ conductances in HD mice. Soon after, a study of K+ channel subtypes in HD neurons followed up.49, 50 By combining immunostaining and immunoblotting methods, Ariano et al compared K+ channel subtype expression in MSNs and large aspiny striatal interneurons (LANs) in both R6/2 and R6/1 HD mouse models. They found substantial reduction of Kir2.1, Kir2.3 and Kv2.1 protein expression in MSNs but not in LANs of R6/2 mice.49 These membrane channel protein alterations further enhanced NMDA receptor subunit NR1 expression by upregulation of PKA phosphorylation in MSNs. Consistent with this finding, the Levine group specifically focused on the examination of the inwardly and outwardly rectifying K+ channel currents in striatal MSNs in both R6/2 HD mice and a slowly progressing model, TgCAG100.50 They found that in addition to the decrease in expression of Kir2.1, Kir2.3, and Kv2.1 protein in MSNs, significant reductions in both inwardly and outwardly rectifying K+ channel currents were detected in acutely dissociated MSNs from striatal slices in HD mice50 (Figure 1).
Figure 1.
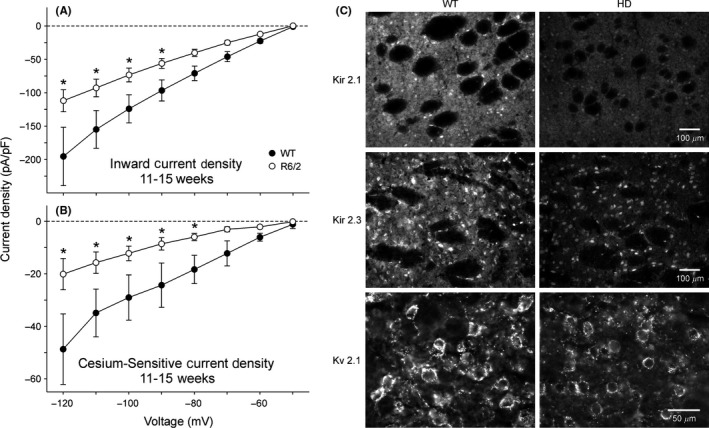
Reduction of inward K+ currents and Kir 2.1, 2.3 and Kv2.1 immuofluorescence intensity in R6/2 transgenic mice. (A‐B). Current‐voltage (I/V) relationships for inward K+ current densities (A) and Cs+‐sensitive K+ current densities (B) obtained from acutely dissociated MSNs from 11‐ to 15‐wk R6/2 transgenics and WTs. In these experiments, a 20 mM K+ concentration in the external solution was used to set the equilibrium potential at approximately −50 mV, corresponding to the holding potential. Obvious reduction of inward K+ current and Cs+‐sensitive K+ current occurred in R6/2 MSNs. (C). Immunofluorescent staining for the K+ channel proteins Kir2.1, Kir2.3 and Kv2.1. Top: Kir2.1 was detected within MSNs and the neuropil but was excluded from the fiber bundles of the internal capsule. The cellular fluorescent signal in R6/2 mice was decreased by 48%. Middle: Kir2.3 was visible within MSNs and the striatal neuropil but not found in fiber bundles. The cellular luminosity was decreased 26% in R6/2 compared with the WT mice. Bottom: Kv2.1 was associated with the membrane rim and initial processes of MSNs. Cellular luminosity decreased 47% in the HD compared with the WT. [Correction added on 19 February 2018, after first online publication: The word “Kv3.1” was changed to “Kv2.1” in the sentence “Reduction of inward K+ currents and Kir 2.1, 2.3 and Kv3.1 immuofluorescence intensity in R6/2 transgenic mice”.]
Interestingly, when the symptoms became more severe to cause apparent locomotor deficits in R6/2 HD model mice at 5‐9 weeks age, Cao et al51 found striatal output neurons (SONs) exhibited higher firing rates and more regular discharge patterns compared with WT SONs, which was caused by an altered activity‐dependent regulation of intrinsic excitability. It is well recognized that a fast activity‐dependent homeostatic control of excitability (fADH) in neurons can modify cells' firing rate, timing of evoked spikes, intrinsic firing patterns and alterations in synaptic transmission.52, 53 With injection of brief trains of bursts in a seconds time scale, fADH can be induced to modify neuron firing rate and convert regular firing patterns to irregular ones through activation of the M current mediated by voltage‐gated potassium channel subfamily 7 or KCNQ channels.54 Cao et al reported a reduction of M currents mediated by KCNQ 2/3 channels caused impairment of fADH in SONs of R6/2 mice. Pharmacological activation of KCNQ channel and chronic treatment with an M‐current activator retigabine, not only restored the hyperactivity network, but also improved motor performance phenotypes of HD mice.51
In summary, the attenuation of K+ conductances and dysfunction of potassium channel subunits in MSNs or SONs not only exacerbates neuronal vulnerability and changes the intrinsic excitability, but also disrupts synaptic integration and both afferent and efferent information processing in HD basal ganglia.
4. KATP CHANNELS ARE IMPAIRED IN HD BASAL GANGLIA NETWORK
The basal ganglia consist of the corpus striatum and related nuclei including, globus pallidus, subthalamic nucleus (STN) and substantia nigra pars reticulata (SNr). It is a network crucial for processing movement, emotion, motivation and cognitive function. Basal ganglia dysfunction is associated with a number of disorders that influence movement including Parkinson's disease, HD and dystonia.55, 56 One of the pathologic mechanisms of HD is involvement of mitochondrial oxidative stress‐activated ATP‐sensitive potassium channel (KATP).57, 58, 59 Marti et al57 first reported the study of a mitochondrial toxin 3‐nitropropionic (3‐NP)‐induced experimental HD model. As a succinate dehydrogenase (SDH) inhibitor, 3‐NP causes a rapid depletion of intracellular ATP in neurons which leads to impairment of cation exchange pumps and progressive membrane depolarization.60 Marti et al found that the acute energy depletion produced by 3‐NP induced two different responses in rat striatal nerve endings. The 3‐NP induced potentiation of GABA release was Ca2+‐dependent and relied on Na+ channel opening. On the other hand, 3‐NP induced inhibition of acetylcholine release, which was tetrodotoxin insensitive but relied on the opening of ATP‐dependent K+ channels to prevent the membrane depolarization caused by mitochondrial impairment, which suggested a mechanism for the selective survival of cholinergic interneurons in an experimental HD model. The protection bestowed by activation of KATP channels could be a therapeutic target for mitigation of HD.57, 59
Although the dysfunction of the striatum and cortex has been extensively studied in HD models, Atherton et al61 recently found a functional impairment of KATP channels in the STN in both BACHD and Q175 knock‐in mouse models. Prior to the onset of major symptoms of HD, STN neurons showed aberrant autonomous firing activity due to KATP channel‐mediated firing disruption in BACHD and Q175 mice. Inhibition of KATP channels with the pharmacological blocker glibenclamide and a membrane permeable form of enzyme catalase to break down H2O2 produced from mitochondrial oxidative phosphorylation,62, 63 STN neurons firing rate and regularity were rapidly rescued, suggesting a direct action on KATP channels in these neurons. Persistent activation of KATP channels in STN neurons in HD eventually led to cell apoptosis and death in this nucleus. In all, the functional switch of KATP channel acting on different types of neurons in HD indicates its important role in keeping the homeostasis of cortico‐striatal‐thalamic circuitry.
5. SK3 CHANNELS CONTRIBUTE TO DOPAMINE REGULATION IN HD
SK3 channels belong to small conductance calcium‐activated potassium channel family (SKCa). They are not voltage dependent and only can be activated by low concentration of intracellular Ca2+. Activation of SK3 channels efficiently limits the AP firing frequency and thus is important for regulation of the afterhyperpolarization (AHP) in most electrically excitable cells.64 Another important characteristic of SK3 channels is this type of channel carries a stretch of polyglutamine residues which is close to its amino terminus. Among the earliest functional changes detected in human HD post‐mortem studies, is a dramatic reduction of D1 and D2 dopamine receptors in the striatum and cortex, suggesting alterations of the dopamine (DA) system.65, 66 Dopamine neurons are characterized electrophysiologically by a long‐duration action potential (AP) and a delayed repolarization following cessation of a hyperpolarizing current pulse.67, 68 The slow firing pattern of DA neurons is mainly regulated by the depolarizing slow oscillatory potential (SOP) and the AHP.69, 70 Dallérac et al used age‐matched transgenic mice 89 and 116 CAG repeats R6/1 line approaching to the characterization of DA dysfunction in models of pre‐manifest and manifest state of HD. Combined with methods of electrophysiology, immunostaining and the dopamine release measurements, Dallérac et al71 found an altered tonic firing rate in HD DA neurons was mediated by SK3 channel.* As the SK3 channel carries a polyglutamine stretch close to its intracellular NH2 terminus, it potentially constitutes a target for the expanded polyglutamine‐containing proteins in HD and prevents the SK3 channel inserting into the membrane of DA neurons from executing its normal function. SK3 staining further illustrated that SK3 channel proteins were redistributed and translocated into the nuclei of TH‐positive DA neurons in HD mice. Thus, the altered sAHP component generated by SK3 channels increased the intrinsic excitability of DA neurons in the substantia nigra pars compacta in R6/1 HD mice.71 *[Correction added on 19 February 2018, after first online publication: The word “medicated” was changed to “mediated” in the sentence.]
6. KIR4.1 CHANNEL DEFICITS IN ASTROCYTES
Inwardly rectifying K+ channel (Kir) has a classical property that allows K+ to flow inwards when the resting membrane potential (RMP) is negative to the equilibrium potential for K+ (EK). This provides the driving force for glial cells to uptake the K+ released during neuronal activity and allow processing functions such as “K+ spatial buffering” or “K+ siphoning”. Glia express multiple Kir channel subtypes, which have distinct functional roles in regulating conductance, sensitivity to intracellular and extracellular factors, including pH, ATP, G‐proteins, neurotransmitters and hormones.72 A key feature of CNS glia is their specific expression of the Kir4.1 subtype, significantly in astrocytes, which maintains the cell membrane potential.8, 9, 73 Kir4.1 plays a fundamental role in the pathogenesis of various brain and neural diseases.10, 11, 74, 75, 76 Since the last decade, it has been reported that mHtt aggregates are distributed in astrocytes both in post mortem specimens from HD patients and transgenic mouse models of HD.10, 11, 77, 78, 79, 80 Tong et al10 first reported that Kir4.1 channel current was robustly decreased in astrocytes with mHtt nuclear inclusions both in R6/2 and Q175 HD mice models (Figure 2). Astrocytic Kir4.1 channel deficits started at the early onset of HD symptoms and impaired spatial K+ buffering, which led to subsequent increased MSN excitability in striatum caused by elevated ambient K+ levels. Through AAV2/5‐mediated delivery of Kir4.1 specifically into astrocytes in vivo, some motor dysfunctions were attenuated and HD mice lifespan prolonged.10 To systematically explore the role of astrocytes in the corticostriatal circuit in HD, Jiang et al combined a genetically encoded Ca2+ indicator, GCaMP3 microinjected into astrocytes and an iGluSnFR measured glutamate uptake. They illustrated alterations in both Ca2+ signaling and GLT1‐mediated glutamate uptake in striatal astrocytes in HD mice. More interesting, those impairments in astrocytes can be rescued by restoring Kir4.1 channels, indicating Kir4.1 channels are a causative key factor for sequential dysfunction in astrocytes, which eventually breaks down the neuronal circuits in HD brains.81 Therefore, as a double‐edged sword, Kir4.1 channels in astrocytes play a crucial role for both K+ and glutamate homeostasis, and their dysfunction leads to excitotoxicity and cell damage.
Figure 2.
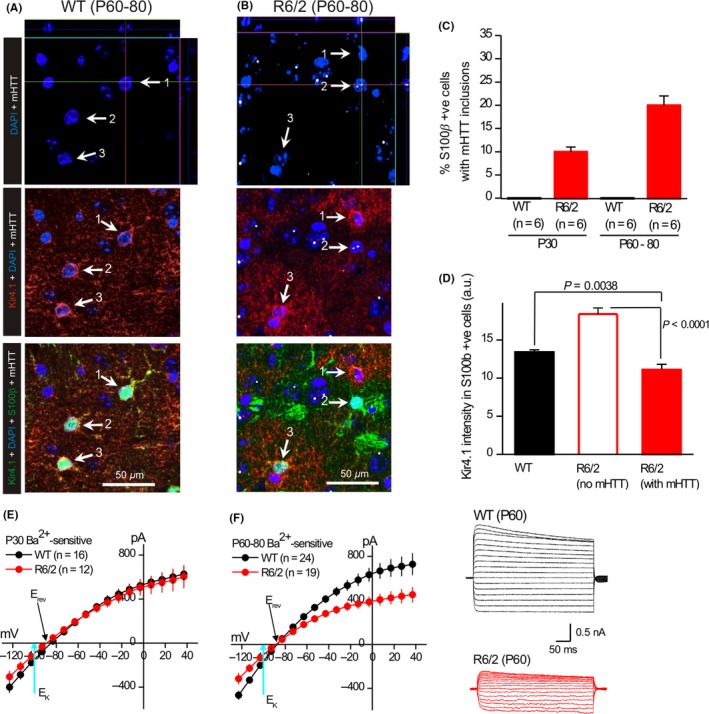
Kir4.1 immunostaining and Ba2+‐sensitive Kir4.1 currents are reduced in striatal astrocytes of R6/2 mice. (A). Representative quadruple color immunofluorescence images of WT striatum at P60‐80, labeled in the indicated colors for DAPI, Kir4.1, S100β and mHTT. In WT controls, no cells expressed mHTT. The white arrows (1‐3) point to cells that were S100β positive: these were also Kir4.1 positive (red). (B). As in (A), but for striatal tissue from R6/2 mice at P60‐80. Here many cells were mHTT and S100β positive but had more reduced Kir4.1 immunostaining (eg, white arrow 2), whereas other cells lacked mHTT and displayed normal Kir4.1 and S100β immunostaining (white arrow 3). (C). Plots the percentage of S100β positive cells that also contained mHTT nuclear inclusions in WT and R6/2 mice at P30 and P60‐80. (D). Plots of Kir4.1 immunostaining intensity for WT mice at P60‐80, as well as for S100β positive astrocytes that contained or did not contain mHTT. (E). I/V plots for Ba2+‐sensitive Kir4.1 currents for WT and R6/2 striatal astrocytes at P30. (F). I/V plots for Ba2+‐sensitive Kir4.1 currents for WT and R6/2 striatal astrocytes at P60, with representative traces to the right. For the I/V plots, in some cases the error bars are smaller than the symbols used (they are thus not always visible)
More recently, Benraiss et al82 supported the causal pathological role of astrocytes in HD by engrafting mHTT‐expressing human glial progenitor cells (hGPCs) in wild type mice. They found that mHTT glia can impart disease phenotype to normal mice and striatal neurons are more hyperexcitable because of the impairment of astrocytic function of K+ buffering. Conversely, replacement of diseased striatal glia with wild‐type CD44+ human glia slowed down the disease progression and restored interstitial K+ homeostasis in R6/2 mice.82 Taken together, the study of astrocytic Kir4.1 channel function and the non‐cell autonomous role of astrocytes provide a completely new research direction for the treatment of HD.
7. K+ CHANNELS IN PERIPHERAL TISSUES
HD is a debilitating and progressive neurodegenerative disorder in the central nervous system, for which currently there is no cure. The symptoms are mostly characterized by severe motor and cognitive defects, which are widely considered as the result of neurodegeneration. However, skeletal muscles have also demonstrated atrophy, metabolic and mitochondrial histological abnormalities in HD.83, 84, 85, 86, 87, 88 Waters et al89 specifically examined the adult skeletal muscles' membrane properties that control motor contraction in R6/2 transgenic mice. They found that the easily triggered and prolonged action potentials in diseased fibers of HD are largely due to decreases of chloride channel CIC‐1 and Kir2.1 potassium channel. The reduction of Clcn1 and Kcnj2 mRNA expression levels could be the cause of muscle hyperexcitability and induced prolonged muscle contractions, which contribute to the chorea, rigidity and dystonia that characterize the disease.89 The K+ channel dysfunction in peripheral organs and tissues broaden our way to explore their roles in the peripheral nervous system (PNS) in brain diseases including HD.
8. CONCLUDING REMARKS
Growing evidence indicates that both neuronal and non‐neuronal mHtt toxicity plays an important role in HD pathology. Although here we summarized the role of K+ channels and their subtypes as a causative factor mainly in striatal neurons and astroglia, it remains largely unknown whether other cell types such as NG2‐glia and/or oligodendrocytes are involved in the pathogenesis of this neurodegenerative disease.90 NG2‐glia and oligodendrocytes constitute two major populations of macroglia in the CNS and they are fundamental to neurons' myelin production and formation.91, 92 A recent study clearly showed K+ channel mRNA and protein functional expression in both cell types.93, 94, 95 It thus becomes necessary and relevant to elucidate a potential interaction of K+ channels with myelin‐related genes. A better understanding of K+ channel function in different kinds of cell populations and the integration of K+ channels into neural networks will provide a promising target to seek out a therapeutic treatment and functional recovery in HD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work is supported by grants from the Natural Science Foundation of China (31571063, 91632104), the program for Professor of Special Appointment (Eastern Scholar for Dr. X.T.) at Shanghai Institutions for Higher Learning (1510000084), Shanghai Pujiang Program (15PJ1404600). Special thanks to Drs Carlos Cepeda and Baljit S. Khakh for providing Figure 1 and Figure 2 image of the review, Dr. Carlos Cepeda and Mr. William R. Bazzarre for comments during manuscript preparation. [Correction added on 19 February 2018, after first online publication: Contribution of Dr. Baljit S. Khakh for Figure 2 was added.]
Zhang X, Wan J‐Q, Tong X‐P. Potassium channel dysfunction in neurons and astrocytes in Huntington's disease. CNS Neurosci Ther. 2018;24:311–318. 10.1111/cns.12804
REFERENCES
- 1. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. [DOI] [PubMed] [Google Scholar]
- 2. Bjorkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869‐1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moller T. Neuroinflammation in Huntington's disease. J Neural Transm (Vienna). 2010;117:1001‐1008. [DOI] [PubMed] [Google Scholar]
- 4. Littleton JT, Ganetzky B. Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron. 2000;26:35‐43. [DOI] [PubMed] [Google Scholar]
- 5. Tian C, Zhu R, Zhu L, et al. Potassium channels: structures, diseases, and modulators. Chem Biol Drug Des. 2014;83:1‐26. [DOI] [PubMed] [Google Scholar]
- 6. Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451‐465. [DOI] [PubMed] [Google Scholar]
- 7. Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage‐dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock‐out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short‐term synaptic potentiation. J Neurosci. 2007;27:11354‐11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tong X, Ao Y, Faas GC, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci. 2014;17:694‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML. The role of glial‐specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016;132:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Humphries ES, Dart C. Neuronal and cardiovascular potassium channels as therapeutic drug targets: promise and pitfalls. J Biomol Screen. 2015;20:1055‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ru Q, Tian X, Wu YX, et al. Voltage‐gated and ATP‐sensitive K+ channels are associated with cell proliferation and tumorigenesis of human glioma. Oncol Rep. 2014;31:842‐848. [DOI] [PubMed] [Google Scholar]
- 14. Hall AM, Throesch BT, Buckingham SC, et al. Tau‐dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease. J Neurosci. 2015;35:6221‐6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kasianowicz JJ. Introduction to ion channels and disease. Chem Rev. 2012;112:6215‐6217. [DOI] [PubMed] [Google Scholar]
- 16. Dragicevic E, Schiemann J, Liss B. Dopamine midbrain neurons in health and Parkinson's disease: emerging roles of voltage‐gated calcium channels and ATP‐sensitive potassium channels. Neuroscience. 2015;284:798‐814. [DOI] [PubMed] [Google Scholar]
- 17. Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389‐397. [DOI] [PubMed] [Google Scholar]
- 18. De Franco E, Flanagan SE, Houghton JA, et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet. 2015;386:957‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. 2016;53:217‐225. [DOI] [PubMed] [Google Scholar]
- 20. Yeh CY, Bulas AM, Moutal A, et al. Targeting a potassium channel/syntaxin interaction ameliorates cell death in ischemic stroke. J Neurosci. 2017;37:5648‐5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Group THsDCR . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971‐983. [DOI] [PubMed] [Google Scholar]
- 22. Rosenblatt A, Kumar BV, Mo A, et al. Age, CAG repeat length, and clinical progression in Huntington's disease. Mov Disord. 2012;27:272‐276. [DOI] [PubMed] [Google Scholar]
- 23. Barnat M, Le Friec J, Benstaali C, Humbert S. Huntingtin‐mediated multipolar‐bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron. 2017;93:99‐114. [DOI] [PubMed] [Google Scholar]
- 24. Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Genetic mouse models of Huntington's and Parkinson's diseases: illuminating but imperfect. Trends Neurosci. 2004;27:691‐697. [DOI] [PubMed] [Google Scholar]
- 25. Bordelon YM, Chesselet MF, Nelson D, Welsh F, Erecinska M. Energetic dysfunction in quinolinic acid‐lesioned rat striatum. J Neurochem. 1997;69:1629‐1639. [DOI] [PubMed] [Google Scholar]
- 26. Borlongan CV, Randall TS, Cahill DW, Sanberg PR. Asymmetrical motor behavior in rats with unilateral striatal excitotoxic lesions as revealed by the elevated body swing test. Brain Res. 1995;676:231‐234. [DOI] [PubMed] [Google Scholar]
- 27. Furtado JC, Mazurek MF. Behavioral characterization of quinolinate‐induced lesions of the medial striatum: relevance for Huntington's disease. Exp Neurol. 1996;138:158‐168. [DOI] [PubMed] [Google Scholar]
- 28. Brouillet E. The 3‐NP Model of Striatal Neurodegeneration. Curr Protoc Neurosci. 2014;67:9‐48‐41‐14. [DOI] [PubMed] [Google Scholar]
- 29. Ramaswamy S, McBride JL, Kordower JH. Animal models of Huntington's disease. ILAR J. 2007;48:356‐373. [DOI] [PubMed] [Google Scholar]
- 30. Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493‐506. [DOI] [PubMed] [Google Scholar]
- 31. Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537‐548. [DOI] [PubMed] [Google Scholar]
- 32. Hodgson JG, Agopyan N, Gutekunst CA, et al. A YAC mouse model for Huntington's disease with full‐length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181‐192. [DOI] [PubMed] [Google Scholar]
- 33. Slow EJ, van Raamsdonk J, Rogers D, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555‐1567. [DOI] [PubMed] [Google Scholar]
- 34. Wheeler VC, White JK, Gutekunst CA, et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock‐in mice. Hum Mol Genet. 2000;9:503‐513. [DOI] [PubMed] [Google Scholar]
- 35. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock‐in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11‐26. [DOI] [PubMed] [Google Scholar]
- 36. Lin CH, Tallaksen‐Greene S, Chien WM, et al. Neurological abnormalities in a knock‐in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137‐144. [DOI] [PubMed] [Google Scholar]
- 37. Menalled LB, Kudwa AE, Miller S, et al. Comprehensive behavioral and molecular characterization of a new knock‐in mouse model of Huntington's disease: zQ175. PLoS ONE. 2012;7:e49838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang SH, Cheng PH, Banta H, et al. Towards a transgenic model of Huntington's disease in a non‐human primate. Nature. 2008;453:921‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skene DJ, Middleton B, Fraser CK, et al. Metabolic profiling of presymptomatic Huntington's disease sheep reveals novel biomarkers. Sci Rep. 2017;7:43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morton AJ, Howland DS. Large genetic animal models of Huntington's Disease. J Huntingtons Dis. 2013;2:3‐19. [DOI] [PubMed] [Google Scholar]
- 41. Levine MS, Klapstein GJ, Koppel A, et al. Enhanced sensitivity to N‐methyl‐D‐aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515‐532. [PubMed] [Google Scholar]
- 42. Klapstein GJ, Fisher RS, Zanjani H, et al. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667‐2677. [DOI] [PubMed] [Google Scholar]
- 43. Cepeda C, Hurst RS, Calvert CR, et al. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laforet GA, Sapp E, Chase K, et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112‐9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bargas J, Galarraga E, Aceves J. Electrotonic properties of neostriatal neurons are modulated by extracellular potassium. Exp Brain Res. 1988;72:390‐398. [DOI] [PubMed] [Google Scholar]
- 46. Bargas J, Galarraga E, Aceves J. An early outward conductance modulates the firing latency and frequency of neostriatal neurons of the rat brain. Exp Brain Res. 1989;75:146‐156. [DOI] [PubMed] [Google Scholar]
- 47. Nisenbaum ES, Wilson CJ. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci. 1995;15:4449‐4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nisenbaum ES, Wilson CJ, Foehring RC, Surmeier DJ. Isolation and characterization of a persistent potassium current in neostriatal neurons. J Neurophysiol. 1996;76:1180‐1194. [DOI] [PubMed] [Google Scholar]
- 49. Ariano MA, Wagle N, Grissell AE. Neuronal vulnerability in mouse models of Huntington's disease: membrane channel protein changes. J Neurosci Res. 2005;80:634‐645. [DOI] [PubMed] [Google Scholar]
- 50. Ariano MA, Cepeda C, Calvert CR, et al. Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol. 2005;93:2565‐2574. [DOI] [PubMed] [Google Scholar]
- 51. Cao Y, Bartolome‐Martin D, Rotem N, et al. Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2015;112:2239‐2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu J, Kang N, Jiang L, Nedergaard M, Kang J. Activity‐dependent long‐term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:1750‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu WW, Chan CS, Surmeier DJ, Disterhoft JF. Coupling of L‐type Ca2 + channels to KV7/KCNQ channels creates a novel, activity‐dependent, homeostatic intrinsic plasticity. J Neurophysiol. 2008;100:1897‐1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Misonou H. Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist. 2010;16:51‐64. [DOI] [PubMed] [Google Scholar]
- 55. Obeso JA, Rodriguez‐Oroz MC, Benitez‐Temino B, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Mov Disord. 2008;23(Suppl 3):S548‐S559. [DOI] [PubMed] [Google Scholar]
- 56. Bunner KD, Rebec GV. Corticostriatal dysfunction in Huntington's disease: the basics. Front Hum Neurosci. 2016;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marti M, Mela F, Ulazzi L, et al. Differential responsiveness of rat striatal nerve endings to the mitochondrial toxin 3‐nitropropionic acid: implications for Huntington's disease. Eur J Neurosci. 2003;18:759‐767. [DOI] [PubMed] [Google Scholar]
- 58. Liot G, Valette J, Pepin J, Flament J, Brouillet E. Energy defects in Huntington's disease: why “in vivo” evidence matters. Biochem Biophys Res Commun. 2017;483:1084‐1095. [DOI] [PubMed] [Google Scholar]
- 59. Gupta S, Sharma B. Protective effects of phosphodiesterase‐1 (PDE1) and ATP sensitive potassium (KATP) channel modulators against 3‐nitropropionic acid induced behavioral and biochemical toxicities in experimental Huntingtons disease. Eur J Pharmacol. 2014;732:111‐122. [DOI] [PubMed] [Google Scholar]
- 60. Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427‐468. [DOI] [PubMed] [Google Scholar]
- 61. Atherton JF, McIver EL, Mullen MR, et al. Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington's disease. Elife. 2016;5:e21616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adam‐Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non‐electron transport chain sources. Antioxid Redox Signal. 2005;7:1140‐1149. [DOI] [PubMed] [Google Scholar]
- 63. Avshalumov MV, Chen BT, Koos T, Tepper JM, Rice ME. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP‐sensitive potassium channels. J Neurosci. 2005;25:4222‐4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Girault A, Haelters JP, Potier‐Cartereau M, et al. Targeting SKCa channels in cancer: potential new therapeutic approaches. Curr Med Chem. 2012;19:697‐713. [DOI] [PubMed] [Google Scholar]
- 65. Ginovart N, Lundin A, Farde L, et al. PET study of the pre‐ and post‐synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain. 1997;120(Pt 3):503‐514. [DOI] [PubMed] [Google Scholar]
- 66. Pavese N, Andrews TC, Brooks DJ, et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington's disease: a PET study. Brain. 2003;126:1127‐1135. [DOI] [PubMed] [Google Scholar]
- 67. Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463‐3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yung WH, Hausser MA, Jack JJ. Electrophysiology of dopaminergic and non‐dopaminergic neurones of the guinea‐pig substantia nigra pars compacta in vitro. J Physiol. 1991;436:643‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ping HX, Shepard PD. Apamin‐sensitive Ca(2 + )‐activated K+ channels regulate pacemaker activity in nigral dopamine neurons. NeuroReport. 1996;7:809‐814. [DOI] [PubMed] [Google Scholar]
- 70. Wolfart J, Roeper J. Selective coupling of T‐type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404‐3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dallerac GM, Levasseur G, Vatsavayai SC, et al. Dysfunctional dopaminergic neurones in mouse models of Huntington's disease: a role for SK3 channels. Neurodegener Dis. 2015;15:93‐108. [DOI] [PubMed] [Google Scholar]
- 72. Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kofuji P, Ceelen P, Zahs KR, et al. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733‐5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429‐5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shin JY, Fang ZH, Yu ZX, et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Song FE, Huang JL, Lin SH, et al. Roles of NG2‐glia in ischemic stroke. CNS Neurosci Ther. 2017;23:547‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bradford J, Shin JY, Roberts M, et al. Expression of mutant huntingtin in mouse brain astrocytes causes age‐dependent neurological symptoms. Proc Natl Acad Sci USA. 2009;106:22480‐22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Faideau M, Kim J, Cormier K, et al. In vivo expression of polyglutamine‐expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet. 2010;19:3053‐3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jansen AH, van Hal M, Op den Kelder IC, et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell‐type‐specific. Glia. 2017;65:50‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Khakh BS, Beaumont V, Cachope R, et al. Unravelling and exploiting astrocyte dysfunction in Huntington's disease. Trends Neurosci. 2017;40:422‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jiang R, Diaz‐Castro B, Looger LL, Khakh BS. dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington's disease model mice. J Neurosci. 2016;36:3453‐3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Benraiss A, Wang S, Herrlinger S, et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun. 2016;7:11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lodi R, Schapira AH, Manners D, et al. Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann Neurol. 2000;48:72‐76. [PubMed] [Google Scholar]
- 84. Ribchester RR, Thomson D, Wood NI, et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci. 2004;20:3092‐3114. [DOI] [PubMed] [Google Scholar]
- 85. Saft C, Zange J, Andrich J, et al. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov Disord. 2005;20:674‐679. [DOI] [PubMed] [Google Scholar]
- 86. Strand AD, Aragaki AK, Shaw D, et al. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863‐1876. [DOI] [PubMed] [Google Scholar]
- 87. Turner C, Cooper JM, Schapira AH. Clinical correlates of mitochondrial function in Huntington's disease muscle. Mov Disord. 2007;22:1715‐1721. [DOI] [PubMed] [Google Scholar]
- 88. She P, Zhang Z, Marchionini D, et al. Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington's disease. Am J Physiol Endocrinol Metab. 2011;301:E49‐E61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Waters CW, Varuzhanyan G, Talmadge RJ, Voss AA. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci USA. 2013;110:9160‐9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Huang B, Wei W, Wang G, et al. Mutant huntingtin downregulates myelin regulatory factor‐mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85:1212‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9‐22. [DOI] [PubMed] [Google Scholar]
- 92. Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2 + CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tang X, Taniguchi K, Kofuji P. Heterogeneity of Kir4.1 channel expression in glia revealed by mouse transgenesis. Glia. 2009;57:1706‐1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929‐11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brasko C, Hawkins V, La D, Rocha IC, Butt AM. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct Funct. 2017;222:41‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]