

Abstract
Background
Elevated lipoprotein(a) (Lp[a]) and familial hypercholesterolemia (FH) are inherited lipid disorders. Their frequencies, coexistence, and associations with premature coronary artery disease (CAD) in patients admitted to the coronary care unit (CCU) remain to be defined.
Hypothesis
Elevated Lp(a) and FH are commonly encountered among CCU patients and independently associated with increased premature CAD risk.
Methods
Plasma Lp(a) concentrations were measured in consecutive patients admitted to the CCU with an acute coronary syndrome (ACS) or prior history of CAD for 6.5 months. Elevated Lp(a) was defined as concentrations ≥0.5 g/L. Patients with LDL‐C ≥ 5 mmol/L exhibited phenotypic FH. Premature CAD was diagnosed in those age < 60 years, and the relationship between this and elevated Lp(a) and FH was determined by logistic regression.
Results
316 patients were screened; 163 (51.6%) had premature CAD. Overall, elevated Lp(a) and FH were identified in 27.0% and 11.6% of patients, respectively. Both disorders were detected in 4.4% of individuals. Elevated Lp(a) (32.0% vs 22.2%; P = 0.019) and FH phenotype (15.5% vs 8.0%; P = 0.052) were more common with premature vs nonpremature CAD. Elevated Lp(a) alone conferred a 1.9‐fold, FH alone a 3.2‐fold, and the combination a 5.3‐fold increased risk of premature CAD (P = 0.005).
Conclusions
Elevated Lp(a) and phenotypic FH were commonly encountered and more frequent with premature CAD. The combination of both disorders is especially associated with increased CAD risk. Patients admitted to the CCU with ACS or previously documented CAD should be routinely screened for elevated Lp(a) and FH.
1. INTRODUCTION
Elevated lipoprotein(a) [Lp(a)] and familial hypercholesterolemia (FH) are inherited disorders that are associated with increased coronary artery disease (CAD) risk.1, 2 FH, an autosomal dominant disorder affecting approximately 1 in 250 individuals worldwide, is characterized by markedly elevated levels of low‐density lipoprotein cholesterol (LDL‐C), due primarily to mutations in the gene encoding the LDL receptor (LDLR).1 Lp(a) consists of an LDL‐like particle covalently bound to the glycoprotein, apolipoprotein (a) [apo(a)]. Concentrations of Lp(a) vary by up to 1000‐fold between individuals, with levels exhibiting a pronounced and positively skewed distribution. Approximately 20% of the general population are likely to have elevated Lp(a), defined as a concentration of ≥0.5 g/L.2 Lp(a) is a highly heritable trait, with the gene encoding apo(a), LPA, explaining most of the variation in circulating levels. Epidemiological3, 4 and Mendelian randomization studies5, 6 have provided conclusive evidence for a causal association between Lp(a) and increased risk of CAD.
Because FH and elevated Lp(a) are genetic disorders with risk exposure beginning at birth, early detection and intervention with both pharmacotherapy and lifestyle measures is crucial for mitigating the burden of CAD. The coexistence of both disorders may exacerbate the risk of CAD.7, 8, 9, 10, 11 Nevertheless, there is currently a widespread lack of awareness that both FH and elevated Lp(a) accelerate CAD, so that these disorders remain under‐recognized in clinical practice. Universal screening for FH and elevated Lp(a) in patients presenting to coronary care units (CCU) may afford a unique opportunity for detecting previously undiagnosed individuals. Importantly, given the co‐dominant mode of inheritance, this approach would also be useful for triggering the cascade testing of relatives of index cases, allowing earlier intervention and reducing the risk of CAD in newly identified family members.
Although the frequency of FH has been investigated in several studies of patients with acute coronary syndromes (ACS),12, 13, 14, 15, 16, 17, 18 the prevalence of elevated Lp(a) in patients presenting to the CCU has not been well established. Furthermore, the spectrum of additional cardiovascular (CV) risk factors in patients with elevated Lp(a) and established CAD has not been fully described. This is important because the management of secondary CV risk factors in patients with elevated Lp(a) is at present the most practicable approach for reducing the Lp(a)‐associated risk of CAD until the advent of specific Lp(a)‐lowering therapies.19
We examine the frequency and coexistence of phenotypic FH and elevated Lp(a) and their association with premature CAD in patients consecutively admitted to a CCU.
2. METHODS
2.1. Patients and study design
Between January 25 and August 11, 2016, three hundred sixteen patients consecutively admitted to the CCU at Royal Perth Hospital were screened for FH and elevated Lp(a). Screening was undertaken in 247 patients who had been admitted with an ACS (78.2%); of those with ACS, 125 (50.6%) were admitted with ST‐segment elevation myocardial infarction, 101 (40.9%) with non–ST‐segment elevation myocardial infarction, and 21 (8.5%) with unstable angina. The remaining 69 patients (21.8%) had CAD diagnosed at the time of their admission with coronary angiogram, or alternatively had an established history of CAD and were admitted with complications relating to their disease, including arrhythmias, angina, and heart failure. A fasting venous blood sample for lipid profiling was obtained with the patient recumbent. Elevated Lp(a) was defined as a plasma concentration of ≥0.5 g/L. This threshold was based on the recommendations of published clinical guidelines.2, 20, 21 Phenotypic FH was defined as a treatment and Lp(a) corrected LDL‐C ≥ 5.0 mmol/L; this corresponds to a Dutch Lipid Clinic Network Criteria (DLCNC) score of ≥3 and a diagnosis of at least possible FH. Premature CAD was diagnosed in patients age < 60 years at the time of their hospital admission.
The use of medications, blood test results, and the presence of CV risk factors including type 2 diabetes mellitus (T2DM), hypertension (HTN), and smoking status were recorded from patient discharge summaries. Exclusion criteria were potential secondary causes of hypercholesterolemia, including chronic kidney disease. Clinical audit approval was granted by Royal Perth Hospital (GEKO Quality Activity 10431).
2.2. Biochemical analyses
Plasma total cholesterol, high‐density lipoprotein cholesterol (HDL‐C), and triglyceride (TG) concentrations were measured in fresh samples using standard enzymatic and immunoturbidimetric methods at a National Association of Testing Authorities–accredited laboratory. The Friedewald equation was used to calculate LDL‐C, except in those with TG >4.5 mmol/L, when LDL‐C was measured directly.22 In individuals receiving statin therapy at the time of hospital admission, LDL‐C was corrected for the estimated effect of the statin regimen on LDL‐C concentration.23 Because approximately 30% of Lp(a) mass is cholesterol mass,24 LDL‐C was further adjusted for the cholesterol content of Lp(a). Lp(a) was measured by an isoform‐independent automated latex‐enhanced immunoassay (Quantia assay and standard).25
2.3. Statistical analysis
The primary outcome of the study was the frequency of elevated Lp(a) ≥0.5 g/L and phenotypic FH in patients consecutively admitted to the CCU with either an ACS or prior history of CAD. The frequency of these disorders were compared between patients with premature CAD (age < 60 years) and nonpremature CAD (age > 60 years). Clinical characteristics were compared between groups using unpaired t tests, ANOVA, and χ2 analyses where appropriate. Logistic regression analyses were carried out to investigate associations with premature CAD and adjusted for the established predictors of increased CAD risk: sex, T2DM, HTN, creatinine, smoking status, and statin therapy. Lp(a), TG, and creatinine levels exhibited a skewed distribution and were log‐transformed prior to analysis. All analyses were carried out using SPSS version 21 (IBM Corp., Armonk, NY), with P values <0.05 deemed to be statistically significant.
3. RESULTS
Over the period of study, 316 patients were screened for elevated Lp(a) and phenotypic FH. The patient characteristics at admission are shown in Table 1. Of those screened, 69.9% were male and the average age at admission was 61.0 years (range, 23.0–90.8 years). Overall, 38.3% of patients were receiving statin therapy at the time of their hospitalization. A total of 163 patients (51.6%) were admitted with a premature coronary event, the remainder having nonpremature CAD.
Table 1.
Clinical characteristics at the time of hospital admission in patients admitted to the CCU
| Variable | Value |
|---|---|
| Age, y | 61.02 ± 13.56 |
| Male sex | 69.9 |
| HTN | 51.9 |
| T2DM | 30.4 |
| Former or current smoker | 39.6 |
| Statin therapy at admission | 38.3 |
| Cr, μmol/L, median (IQR) | 75.0 (21.75) |
| TC, mmol/L | 4.87 ± 1.39 |
| HDL‐C, mmol/L | 1.06 ± 0.30 |
| TG, mmol/L, median (IQR) | 1.50 (1.15) |
| LDL‐C, mmol/L | 2.97 ± 1.14 |
| Corrected LDL‐C, mmol/La | 3.53 ± 1.58 |
| Non–HDL‐C, mmol/L | 3.81 ± 1.36 |
| Lp(a), g/L, median (IQR) | 0.16 (0.45) |
| Lp(a) category, g/L | |
| ≥0.3 | 34.2 |
| ≥0.5 | 26.3 |
| ≥1.0 | 7.6 |
| Phenotypic FH | 11.4 |
| Premature CAD | 51.6 |
Abbreviations: CAD, coronary artery disease; CCU, coronary care unit; Cr, creatinine; FH, familial hypercholesterolemia; HDL‐C, high‐density lipoprotein cholesterol; HTN, hypertension; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); SD, standard deviation; T2DM, type 2 diabetes mellitus; TC, total cholesterol; TG, triglycerides.
Data are presented as percentages or mean ± SD, unless otherwise noted.
LDL‐C adjusted for statin therapy and the cholesterol content of Lp(a).
3.1. Frequency of elevated Lp(a) and phenotypic FH
The median Lp(a) level in CCU patients was 0.16 g/L (interquartile range [IQR], 0.45 g/L). The frequency of elevated Lp(a) was 34.2%, 26.3%, and 7.6% with reference to the thresholds ≥0.3 g/L, ≥0.5 g/L, and ≥1.0 g/L, respectively. The frequency of elevated Lp(a) ≥0.5 g/L was significantly higher in individuals experiencing a premature coronary event (31.9%) compared with those without a premature event (20.3%, P = 0.019; Figure 1).
Figure 1.
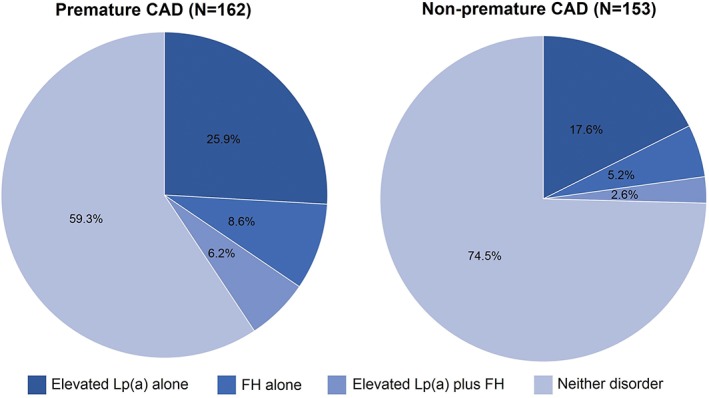
The frequency of elevated Lp(a) and FH phenotype in patients with premature and nonpremature CAD. Abbreviations: CAD, coronary artery disease; FH, familial hypercholesterolemia; Lp(a), lipoprotein(a)
The mean treatment‐ and Lp(a)‐corrected LDL‐C was 3.5 mmol/L (range, 0.4–13.8 mmol/L) and was significantly higher in patients with premature CAD compared with nonpremature CAD (3.9 vs 3.2 mmol/L; P < 0.001). The frequency of phenotypic FH in patients presenting to the CCU was 11.4% (n = 36). In patients with premature CAD, the frequency of phenotypic FH was almost twice as high when compared with nonpremature CAD (14.8% vs 7.8%), but this failed to reach statistical significance (P = 0.052; Figure 1).
The combination of both elevated Lp(a) and phenotypic FH was present in 4.4% (n = 14) of patients; 21.9% (n = 69) had elevated Lp(a) alone, 7.0% (n = 22) had phenotypic FH alone, and 66.7% (n = 210) had neither disorder. As shown in Figure 1, the frequency of having both of these disorders was greater in those with premature CAD (6.2% vs 2.6%; P = 0.033).
3.2. Frequency of CV risk factors
The frequencies of secondary CV risk factors based on the diagnoses of elevated Lp(a) or phenotypic FH are shown in Table 2. There was a significantly higher frequency of females with elevated Lp(a) than without elevated Lp(a) (P = 0.012). The frequencies of other risk factors for CAD did not significantly differ between patients with and without elevated Lp(a). Despite no difference in the proportion of patients on statin therapy at the time of their admission, fewer patients with elevated Lp(a) had achieved a guideline‐recommended LDL‐C target of <1.8 mmol/L (P = 0.035).
Table 2.
Clinical characteristics at the time of hospital admission according to presence or absence of elevated Lp(a) and FH phenotype
| Variable | Lp(a) ≥0.5 g/L | Lp(a) <0.5 g/L | p Value | FH+ | FH– | p Value |
|---|---|---|---|---|---|---|
| Age, y | 58.7 ± 12.5 | 61.8 ± 13.9 | 0.075 | 56.4 ± 10.6 | 61.6 ± 13.9 | 0.028 |
| Male sex | 59.0 | 73.8 | 0.012 | 61.1 | 71.0 | 0.225 |
| HTN | 49.4 | 52.8 | 0.595 | 55.6 | 51.3 | 0.627 |
| T2DM | 28.9 | 30.9 | 0.736 | 44.4 | 28.3 | 0.047 |
| Ever smoked | 36.1 | 40.8 | 0.459 | 41.7 | 39.4 | 0.796 |
| Statin at admission | 42.7 | 36.8 | 0.346 | 80.6 | 32.6 | <0.001 |
| Cr, μmol/L, median (IQR) | 76.6 (72.3–81.0) | 81.7 (78.0–85.5) | 0.128 | 85.1 (75.5–95.9) | 79.7 (76.6–82.9) | 0.266 |
| TC, mmol/L | 5.1 ± 1.4 | 4.8 ± 1.4 | 0.044 | 5.9 ± 1.4 | 4.7 ± 1.2 | <0.001 |
| HDL‐C, mmol/L | 1.1 ± 0.3 | 1.0 ± 0.3 | 0.067 | 1.0 ± 0.2 | 1.1 ± 0.3 | 0.733 |
| TG, mmol/L, median (IQR) | 1.5 (1.4–1.7) | 1.6 (1.5–1.7) | 0.605 | 2.0 (1.6–2.4) | 1.5 (1.4–1.6) | 0.015 |
| LDL‐C, mmol/L | 3.2 ± 1.2 | 2.9 ± 1.1 | 0.011 | 3.9 ± 1.2 | 2.9 ± 1.0 | <0.001 |
| Adjusted LDL‐C, mmol/La | 3.7 ± 2.3 | 3.5 ± 1.2 | 0.149 | 6.7 ± 2.0 | 3.1 ± 0.92 | <0.001 |
| Non–HDL‐C, mmol/L | 4.0 ± 1.4 | 3.7 ± 1.3 | 0.099 | 4.8 ± 1.4 | 3.6 ± 1.2 | <0.001 |
| LDL‐C, mmol/L | ||||||
| >1.8 | 90.4 | 80.2 | 0.035 | 100 | 80.6 | 0.004 |
| >2.5 | 30.1 | 36.6 | 0.285 | 91.7 | 61.6 | <0.001 |
| Lp(a), g/Lb | 0.85 (0.79‐0.92) | 0.13 (0.12‐0.14) | <0.001 | 0.28 (0.19‐0.41) | 0.20 (0.18‐0.22) | 0.062 |
Abbreviations: CI, confidence interval; Cr, creatinine; FH, familial hypercholesterolemia; HDL‐C, high‐density lipoprotein cholesterol; HTN, hypertension; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); SD, standard deviation; T2DM, type 2 diabetes mellitus; TC, total cholesterol; TG, triglycerides.
Data are presented as percentages or mean ± SD, unless otherwise noted.
LDL‐C adjusted for statin therapy and the cholesterol content of Lp(a).
Data presented as the geometric mean and 95% CI.
When comparing patients with and without phenotypic FH, there was no significant difference in the frequency of HTN and a history of smoking (Table 2). Conversely, almost twice as many patients with phenotypic FH had T2DM (44.4% vs 28.3%; P = 0.047), which is consistent with the significantly increased frequency of statin therapy in these patients (80.6% vs 32.6%; P < 0.001). A more adverse lipid profile, including increased TG (P = 0.015), corrected LDL‐C (P < 0.001), and lower attainment of LDL‐C targets, was observed in FH patients (Table 2).
3.3. Associations with premature CAD
At the time of hospitalization, patients without elevated Lp(a) or FH were on average 62.4 years of age. Those with elevated Lp(a) alone were on average 3 years younger (59.4 years), FH alone 5 years younger (57.1 years), and patients with both elevated Lp(a) and FH were 7 years younger (55.3 years) than unaffected individuals (P = 0.062).
Logistic regression analyses were undertaken to investigate the association between elevated Lp(a), FH phenotype, and the combination of both disorders with the risk of experiencing a premature CAD event. When adjusting for additional CAD risk factors, elevated Lp(a) (odds ratio: 1.88, P = 0.024) and FH phenotype (odds ratio: 3.02, P = 0.008) were independently associated with an increased risk of premature CAD (Table 3). When investigating the possible combinations of these disorders, patients with elevated Lp(a) alone had a 1.9‐fold increased risk of premature CAD when compared with individuals with neither disorder (P = 0.031), and a 3.2‐fold increased risk of premature CAD was observed in patients with an FH phenotype alone (P = 0.024). The combination of both elevated Lp(a) and FH phenotype was associated with a 5.3‐fold higher risk of premature CAD when compared with patients with neither disorder (P = 0.011; Table 3 and Figure 2). The associations with premature CAD remained significant when the analysis was restricted to patients admitted to the CCU with an ACS (data not shown). Because a curvilinear association with CAD risk has been observed for Lp(a) levels >0.3 g/L, the association with premature CAD was additionally investigated employing this cutoff. Although Lp(a) ≥0.3 g/L alone was associated with increased risk of premature CAD, in combination with FH phenotype the risk of CAD was not increased beyond that observed for FH alone (data not shown).
Table 3.
Multivariate regression analyses showing the independent association between elevated Lp(a) and FH phenotype and the risk of premature CAD
| OR | 95% CI | p Value | |
|---|---|---|---|
| Model 1a | |||
| Male sex | 1.37 | 0.78‐2.40 | 0.269 |
| T2DM (Y vs N) | 1.52 | 0.88‐2.67 | 0.134 |
| HTN (Y vs N) | 0.98 | 0.60‐1.61 | 0.943 |
| Smoking status (ever vs never) | 1.29 | 0.79‐2.11 | 0.307 |
| Ln Cr | 0.30 | 0.11‐0.78 | 0.014 |
| Statin at admission (Y vs N) | 0.44 | 0.25‐0.77 | 0.004 |
| FH phenotype | 3.02 | 1.33‐6.85 | 0.008 |
| Elevated Lp(a) | 1.88 | 1.09‐3.24 | 0.024 |
| Model 2b | |||
| Male sex | 1.38 | 0.79‐2.41 | 0.266 |
| T2DM (Y vs N) | 1.54 | 0.88‐2.69 | 0.131 |
| HTN (Y vs N) | 0.99 | 0.60‐1.63 | 0.961 |
| Smoking status (ever vs never) | 1.29 | 0.79‐2.11 | 0.305 |
| Ln Cr | 0.30 | 0.11‐0.79 | 0.015 |
| Statin at admission (Y vs N) | 0.44 | 0.25‐0.77 | 0.004 |
| Elevated Lp(a) and FH phenotype composite | — | — | 0.005 |
| Elevated Lp(a) alone vs neither disorder | 1.92 | 1.06‐3.46 | 0.031 |
| FH phenotype alone vs neither disorder | 3.17 | 1.16‐8.63 | 0.024 |
| Elevated Lp(a) + FH phenotype vs neither disorder | 5.27 | 1.47‐18.91 | 0.011 |
Abbreviations: CAD, coronary artery disease; CI, confidence interval; Cr, creatinine; FH, familial hypercholesterolemia; HTN, hypertension; Ln, logarithm; Lp(a), lipoprotein(a); N, no; OR, odds ratio; T2DM, type 2 diabetes mellitus; Y, yes.
Model 1 includes FH phenotype and elevated Lp(a) as separate variables. The significant association for both elevated Lp(a) and FH phenotype demonstrates their independent effects on the risk of developing premature CAD.
Model 2 investigates the interaction between elevated Lp(a) and FH phenotype. Patients with both elevated Lp(a) and phenotypic FH were at the highest risk of developing premature CAD.
Figure 2.
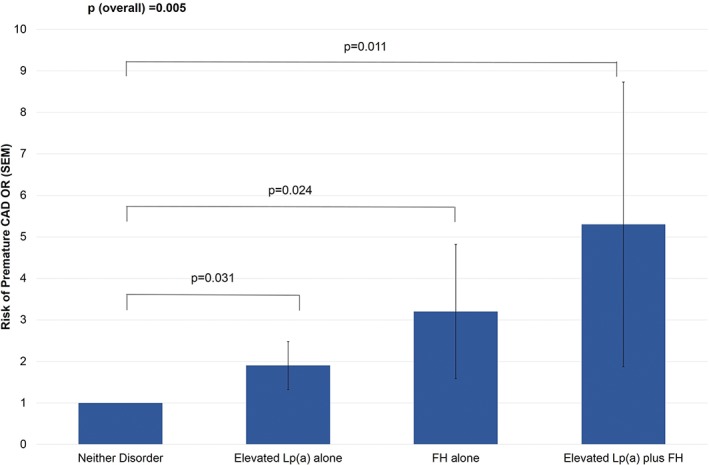
Association between elevated Lp(a) and FH phenotype with the risk of having a premature CAD event. Analyses adjusted for sex, T2DM, HTN, smoking status, Cr, and statin therapy at admission. Abbreviations: CAD, coronary artery disease; Cr, creatinine; FH, familial hypercholesterolemia; HTN, hypertension; Lp(a), lipoprotein(a); OR, odds ratio; SEM, standard error of the mean; T2DM, type 2 diabetes mellitus
4. DISCUSSION
We have demonstrated that elevated Lp(a) and phenotypic FH are frequently detected in patients admitted to the CCU and are independently associated with an increased risk of premature CAD.
Elevated Lp(a) and FH are common inherited disorders of lipid metabolism, affecting 20% and 0.4% of the general population, respectively.1, 2 In our study of patients with CAD, we identified a higher prevalence of elevated Lp(a), with 27% of individuals affected; an even greater frequency of 32% was observed in those with premature CAD. In a recent smaller investigation of high‐risk patients presenting for percutaneous coronary interventions, elevated Lp(a) ≥0.5 g/L was identified in 38% of subjects, and the majority of these patients had premature CAD.26
Consistent with the present study in which phenotypic FH was diagnosed in 11.6% of patients, other recent investigations have also found that FH is relatively common in patients with ACS.12, 13, 14, 15, 16, 17, 18 In the Eastern Danish Heart Registry, a study of 13 174 patients following an acute myocardial infarction, possible FH based on the DLCNC was identified in 9.7% of individuals.14 That study additionally found that FH patients presented with an myocardial infarction 6 years earlier than those with unlikely FH.14 In our study we also detected an earlier age of CAD onset in FH patients and additionally observed that patients with both FH and elevated Lp(a) tended to develop CAD at an even younger age than individuals with FH or elevated Lp(a) alone. Furthermore, patients with FH and elevated Lp(a), approximately 5% of the cohort, were found to be at the greatest risk of premature CAD and were ≥5× more likely to have been admitted for a premature event than those with neither disorder. FH patients with elevated Lp(a) are well recognized to be at exceptionally high CAD risk.7, 8, 9, 10, 11 Furthermore, in FH patients, elevated Lp(a) has been shown to be an independent predictor of CAD risk in a recently described risk equation.27
The markedly elevated CAD risk associated with FH and elevated Lp(a) necessitates early detection. At present, most cases of these dual abnormalities remain undetected, and these disorders are not routinely screened for in patients in the CCU. The relatively high rate of detection of FH and elevated Lp(a) demonstrates the value of screening for these disorders in the CCU. Importantly, the identification of new index cases could also be used to initiate cascade screening of close family members, allowing for targeted preventive measures in newly detected relatives. The effectiveness of cascade screening for FH has been well‐established,28, 29 and although the yield of Lp(a) cascade screening has not been extensively examined, we have preliminary data demonstrating a detection rate of almost 1 in 2 in close relatives of Lp(a) index cases (unpublished data).
The treatment of FH is focused on reducing LDL‐C to target levels with lifestyle modification and pharmacotherapy, usually a statin plus ezetimibe and in some cases additionally a proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibody (mAb). Early initiation of lipid‐lowering therapy reduces the cumulative LDL‐C burden in FH and effectively delays the onset of CAD.30 Conversely, there are currently no therapeutic agents for selectively lowering Lp(a); although newer agents, including PCSK9 mAbs, have been shown to have Lp(a)‐lowering effects, most are not widely available and it has yet to be demonstrated that the achieved reductions in Lp(a) translate to an improvement in CAD risk.31 Hence, in the majority of patients with elevated Lp(a), lifestyle intervention including the management of conventional CV risk factors offers the most practicable approach for mitigating CAD risk. This was recently emphasised in the EPIC‐Norfolk study, in which the American Heart Association metric of ideal CV health was associated with a 75% reduction in the CV risk associated with high Lp(a) levels.19 We found that most patients presenting to the CCU with elevated Lp(a) had ≥1 additional CV risk factor that should be aggressively managed to reduce Lp(a)‐associated CV risk. Novel antisense therapies targeting apo(a) have been shown to have potent Lp(a)‐lowering effects, and it seems likely that in the future these will be the preferred treatment for patients with very elevated Lp(a).32, 33
4.1. Study limitations
There are several limitations to our study, including the relatively small sample size. The adequacy of documentation of both family history of CAD and hypercholesterolemia was lacking in many individuals, and this information could not be considered in the diagnosis of FH. However, in a subset of 119 patients with a recorded family history, 8 (6.7%) individuals met our criteria for phenotypic FH and reported a first‐degree relative with premature CAD. Furthermore, we also used phenotypic as opposed to molecular criteria for diagnosing FH. Although genetic testing affords the most accurate diagnosis of FH, the value of an LDL‐C cutoff of >4.9 mmol/L has been supported by the National Lipid Association.20 An FH phenotype defined using this cutoff has additionally been associated with markedly elevated 30‐year CAD risk,34 substantiating the use of the threshold employed in our study. Nevertheless, it has also recently been demonstrated that individuals with a pathogenic FH‐causing variant are at increased CAD risk when compared with individuals with the same LDL‐C levels without a mutation.35
5. CONCLUSION
The results support the routine screening for FH and elevated Lp(a) in the CCU, particularly among patients with premature CAD. The value of screening for these disorders is for secondary prevention in patients with established CAD, and for primary prevention in cascade‐screened relatives of index cases. Detection enables effective treatment, which involves a statin plus ezetimibe and potentially a PCSK9 mAb in patients with FH. The management of individuals with both FH and elevated Lp(a), who are at exceptionally high CAD risk, should also utilize the above agents; and in patients with markedly elevated Lp(a), apo(a) antisense therapy is likely to be an important therapeutic option in the future. Aggressive management of modifiable CV risk factors with lifestyle and pharamacotherapies, including statins, is important for mitigating Lp(a)‐associated CAD risk,19, 36, 37 pending the imminent availability of therapies for selectively lowering Lp(a).
Conflicts of interest
The authors declare no potential conflicts of interest.
Ellis KL, Pang J, Chieng D, et al. Elevated lipoprotein(a) and familial hypercholesterolemia in the coronary care unit: Between Scylla and Charybdis. Clin Cardiol. 2018;41:378–384. 10.1002/clc.22880
Funding information Royal Perth Hospital Medical Research Foundation, Grant/Award number: N/A; Raine Medical Research Foundation, Grant/Award number: N/A
REFERENCES
- 1. Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel . Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478a–3490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel . Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration . Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kamstrup PR, Benn M, Tybjaerg‐Hansen A, et al. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176–184. [DOI] [PubMed] [Google Scholar]
- 5. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium . Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. [DOI] [PubMed] [Google Scholar]
- 6. Kamstrup PR, Tybjaerg‐Hansen A, Steffensen R, et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. [DOI] [PubMed] [Google Scholar]
- 7. Alonso R, Andres E, Mata N, et al; SAFEHEART Investigators . Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–1989. [DOI] [PubMed] [Google Scholar]
- 8. Alonso R, Mata P, Muñiz O, et al. PCSK9 and lipoprotein(a) levels are two predictors of coronary artery calcification in asymptomatic patients with familial hypercholesterolemia. Atherosclerosis. 2016;254:249–253. [DOI] [PubMed] [Google Scholar]
- 9. Langsted A, Kamstrup PR, Benn M, et al. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–587. [DOI] [PubMed] [Google Scholar]
- 10. Chan DC, Pang J, Hooper AJ, et al. Elevated lipoprotein(a), hypertension and renal insufficiency as predictors of coronary artery disease in patients with genetically confirmed heterozygous familial hypercholesterolemia. Int J Cardiol. 2015;201:633–638. [DOI] [PubMed] [Google Scholar]
- 11. Jansen AC, van Aalst‐Cohen ES, Tanck MW, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med. 2004;256:482–490. [DOI] [PubMed] [Google Scholar]
- 12. Zafrir B, Shapira C, Lavie G, et al. Identification and characterization of severe familial hypercholesterolemia in patients presenting for cardiac catheterization. J Clin Lipidol. 2016;10:1338–1343. [DOI] [PubMed] [Google Scholar]
- 13. Li S, Zhang Y, Zhu CG, et al. Identification of familial hypercholesterolemia in patients with myocardial infarction: a Chinese cohort study. J Clin Lipidol. 2016;10:1344–1352. [DOI] [PubMed] [Google Scholar]
- 14. Rerup SA, Bang LE, Mogensen UM, et al. The prevalence and prognostic importance of possible familial hypercholesterolemia in patients with myocardial infarction. Am Heart J. 2016;181:35–42. [DOI] [PubMed] [Google Scholar]
- 15. Mortensen MB, Kulenovic I, Klausen IC, et al. Familial hypercholesterolemia among unselected contemporary patients presenting with first myocardial infarction: prevalence, risk factor burden, and impact on age at presentation. J Clin Lipidol. 2016;10:1145.e1–1152.e1. [DOI] [PubMed] [Google Scholar]
- 16. Nanchen D, Gencer B, Muller O, et al. Prognosis of patients with familial hypercholesterolemia after acute coronary syndromes. Circulation. 2016;134:698–709. [DOI] [PubMed] [Google Scholar]
- 17. Rallidis LS, Triantafyllis AS, Tsirebolos G, et al. Prevalence of heterozygous familial hypercholesterolaemia and its impact on long‐term prognosis in patients with very early ST‐segment elevation myocardial infarction in the era of statins. Atherosclerosis. 2016;249:17–21. [DOI] [PubMed] [Google Scholar]
- 18. Pang J, Poulter EB, Bell DA, et al. Frequency of familial hypercholesterolemia in patients with early‐onset coronary artery disease admitted to a coronary care unit. J Clin Lipidol. 2015;9:703–708. [DOI] [PubMed] [Google Scholar]
- 19. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: the EPIC‐Norfolk prospective population study. Atherosclerosis. 2017;256:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient‐centered management of dyslipidemia: part 1—full report. J Clin Lipidol. 2015;9:129–169. [DOI] [PubMed] [Google Scholar]
- 21. Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: the Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) . Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis. 2016;253:281–344. [DOI] [PubMed] [Google Scholar]
- 22. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 23. Haralambos K, Whatley SD, Edwards R, et al. Clinical experience of scoring criteria for familial hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis. 2015;240:190–196. [DOI] [PubMed] [Google Scholar]
- 24. Kinpara K, Okada H, Yoneyama A, et al. Lipoprotein(a)‐cholesterol: a significant component of serum cholesterol. Clin Chim Acta. 2011;412:1783–1787. [DOI] [PubMed] [Google Scholar]
- 25. Marcovina SM, Albers JJ, Scanu AM, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000;46:1956–1967. [PubMed] [Google Scholar]
- 26. Weiss MC, Berger JS, Gianos E, et al. Lipoprotein(a) screening in patients with controlled traditional risk factors undergoing percutaneous coronary intervention. J Clin Lipidol. 2017;11:1177–1180. [DOI] [PubMed] [Google Scholar]
- 27. Pérez de Isla L, Alonso R, Mata N, et al. Predicting cardiovascular events in familial hypercholesterolemia: the SAFEHEART Registry (Spanish Familial Hypercholesterolemia Cohort Study). Circulation. 2017;135:2133–2144. [DOI] [PubMed] [Google Scholar]
- 28. Lázaro P, Pérez de Isla L, Watts GF, et al. Cost‐effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J Clin Lipidol. 2017;11:260–271. [DOI] [PubMed] [Google Scholar]
- 29. Ademi Z, Watts GF, Pang J, et al. Cascade screening based on genetic testing is cost‐effective: evidence for the implementation of models of care for familial hypercholesterolemia. J Clin Lipidol. 2014;8:390–400. [DOI] [PubMed] [Google Scholar]
- 30. Ellis KL, Hooper AJ, Burnett JR, et al. Progress in the care of common inherited atherogenic disorders of apolipoprotein B metabolism. Nat Rev Endocrinol. 2016;12:467–484. [DOI] [PubMed] [Google Scholar]
- 31. Ellis KL, Boffa MB, Sahebkar A, et al. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57–82. [DOI] [PubMed] [Google Scholar]
- 32. Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double‐blind, placebo‐controlled phase 1 study. Lancet. 2015;386:1472–1483. [DOI] [PubMed] [Google Scholar]
- 33. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double‐blind, placebo‐controlled, dose‐ranging trials. Lancet. 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 34. Perak AM, Ning H, de Ferranti SD, et al. Long‐term risk of atherosclerotic cardiovascular disease in us adults with the familial hypercholesterolemia phenotype. Circulation. 2016;134:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khera AV, Won HH, Peloso GM, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lloyd‐Jones DM, Hong Y, Labarthe D, et al; American Heart Association Strategic Planning Task Force and Statistics Committee . Defining and setting national goals for cardiovascular health promotion and disease reduction: the American Heart Association's strategic Impact Goal through 2020 and beyond. Circulation. 2010;121:586–613. [DOI] [PubMed] [Google Scholar]