

Summary
Background
Major depressive disorder (MDD) is a mental disease characterized by depressed mood, lifetime anxiety, and deficits of learning and memory. Inhibition of phosphodiesterase 9 (PDE9) has been reported to improve rodent cognitive and memory function. However, the role of PDE9 in MDD, in particular its manifestations of depression and anxiety, has not been investigated.
Methods
We examined the protective effects of WYQ‐C36D (C36D), a novel PDE9 inhibitor, against corticosterone‐induced cytotoxicity, pCREB/CREB and BDNF expression by cell viability, and immunoblot assays in HT‐22 cells. The potential effects of C36D at doses of 0.1, 0.5, and 1 mg/kg on stress‐induced depression‐ and anxiety‐like behaviors and memory deficits were also examined in mice.
Results
C36D significantly protected HT‐22 cells against corticosterone‐induced cytotoxicity and rescued corticosterone‐induced decreases in cGMP, CREB phosphorylation, and BDNF expression. All these effects were otherwise blocked by the PKG inhibitor Rp‐8‐Br‐PET‐cGMPS (Rp8). In addition, when tested in vivo in stressed mice, C36D produced antidepressant‐like effects on behavior, as shown by decreased immobility time both in the forced swimming and tail suspension tests. C36D also showed anxiolytic‐like and memory‐enhancing effects in the elevated plus‐maze and novel object recognition tests.
Conclusion
Our results show that inhibition of PDE9 by C36D produces antidepressant‐ and anxiolytic‐like behavioral effects and memory enhancement by activating cGMP/PKG signaling pathway. PDE9 inhibitors may have the potential as a novel class of drug to treat MDD.
Keywords: BDNF, C36D, CREB, depression, PDE9 inhibitor
1. INTRODUCTION
Major depressive disorder (MDD) affects large population worldwide including children and adolescents, many of them are suffered from lifetime anxiety and cognitive disabilities besides depressed mood.1, 2 Currently available antidepressant drugs such as tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs), and serotonin and noradrenaline reuptake inhibitor (SNRI) are not sufficient to alleviate all symptoms, such as memory acquisition impairment, difficulty of thinking, and dementia,3 and are particularly ineffective against anxiety and cognitive deficits. Furthermore, some of these drugs have side effects, for example, SSRIs induce long‐term anxiety.4 Thus, discovery of new therapeutic strategies for treatment of MDD is highly desirable. Recent research has suggested that the secondary messenger cyclic guanosine monophosphate (cGMP) signaling is involved in depression‐ and anxiety‐related behaviors.5, 6 cGMP elevation can cause an increase in synaptic transmission and long‐term potentiation, and reverse Aβ induced deficits in long‐term potentiation in hippocampal slices.7, 8 cGMP, formed by the action of the soluble isoform of guanylyl cyclase, is hydrolyzed by enzymes of the PDE superfamily, which has been studied as drug targets for treating various emotional disorders.9, 10, 11, 12, 13 The 21 PDE genes are classified into 11 different families (PDE1−11), among which PDE9A is cGMP‐specific with wide distribution throughout brain regions related to depression, anxiety, and cognitive disorders, such as cortex, hippocampus, and amygdala.14 We hypothesize that inhibiting PDE9 activity may result in an increase in cGMP, which would then counter depression symptoms. Preclinical research has shown that PDE9A inhibition can enhance cognitive function. Indeed, several new PDE9 inhibitors such as PF‐04447943 and BI‐409306 have been developed and evaluated for its treatment of Alzheimer's disease (AD) in Phase II clinical trials.15, 16 However, the antidepressant‐like effects of PDE9 inhibitors have received little attention.
As shown in Figure 1, a novel PDE9 inhibitor WYQ‐C36D (C36D), designed and synthesized by our collaborators Drs. HM Ke and HB Luo, showed higher affinity to PDE9A (IC50 = 21 nmol/L) than the current PDE9 inhibitors and has better PDE9 selectivity over other PDEs.17, 18 This study investigated the protective effects of C36D against corticosterone‐induced neuronal toxicity and the related signaling mechanisms involving the cGMP‐CREB‐BDNF pathway in HT‐22 cells. The in vivo study further supported the in vitro data that suggested C36D reversed acute stress‐induced depression/anxiety‐like behaviors and cognitive impairment in mice.
Figure 1.
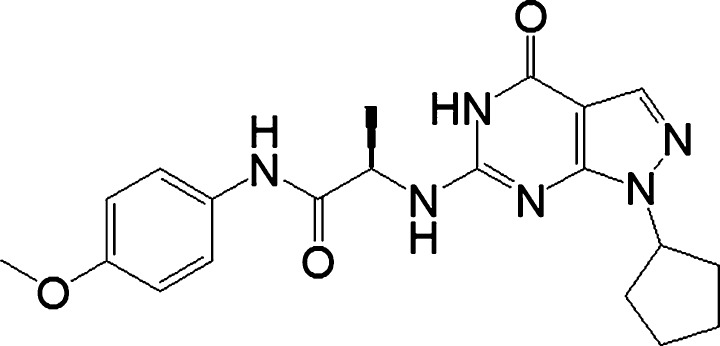
The chemical structure of C36D
2. MATERIALS AND METHODS
2.1. Animals
Male ICR mice, 22‐25 g, were obtained from University at Buffalo. The mice were housed in a temperature‐controlled room under standard laboratory conditions, with controlled ambient temperature (22 ± 1°C), humidity (50 ± 10%), and a 12‐h natural light/dark cycle. Rodent chow and tap water were freely available. Before any treatments, all animals were allowed at least 1 week of habituation. All experiments were carried out according to the National Institute of Health Guide for Care and Use of Laboratory Animals (1996) and were approved by the Institutional Animal Care and Use Committee of University at Buffalo (USA) and Changzhou University (China).
2.2. Drug treatments
Corticosterone, rolipram (Rol), diazepam (DZP), imipramine (IMA), and Rp‐8‐Br‐PET‐cGMPS (Rp8) were purchased from Sigma Chemical Co. (USA). WYQ‐C36D [(R)‐2‐(1‐cyclopentyl‐4‐oxo‐4,5‐dihydro‐1H‐pyrazolo [3,4‐d] pyrimidin‐6‐ylamino)‐N‐(4‐methoxyphenyl) propanamide, C36D] was obtained from Dr. Hengming Ke at University of North Carolina at Chapel Hill. Corticosterone, Rp8, and C36D were dissolved in 0.5% dimethyl sulfoxide (DMSO). Cells were treated with 100 μmol/L corticosterone for 30 minutes before treatment with different doses of C36D. Animals were given bilateral micro‐injections of 2 μL Rp8 (1 nmol, 1 μL/side, i.c.v., dissolved in artificial cerebrospinal fluid) into the CA1 region of the hippocampus. Ten minutes before, C36D, DZP, IMA, or Rol was administered (ip).
2.3. Cell viability assay
Cells were seeded in 96‐well plates at 1 × 105 cells/well and treated with C36D at different concentrations and time point. Following the indicated treatments, cell viability was assessed using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphen‐yl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) method according to a previous description.19 Absorbance at 490 nm was measured using a microplate reader (BioTek, USA).
2.4. Stress procedure
Restraint immobilization (IMO) stress was performed as described previously20 with minor modification. Mice were subjected to a single, 4‐hour episode of acute immobilization stress in ventilated 50‐mL conical tubes (with no access to food or water). The control and stressed mice were treated with vehicle, different doses of C36D, or positive drugs 30 minutes prior to stress procedure. Nonstressed groups (controls) were treated with vehicle, which were kept without food and water for 4 hours. The behavioral tests including forced swimming, tail suspension, elevated plus‐maze, and novel object recognition tests were performed 10 minutes after stress to rule out the false‐negative or false‐positive behaviors.
2.5. Forced swimming test (FST)
The test was performed in mice as described previously.12 Briefly, each mouse is gently introduced into a glass cylinder (20 cm diameter, 45 cm height) filled with water (23 ± 1°C; depth 28 cm) for 6 minutes. Animals were individually placed in the cylinder for 15 minutes and returned to a home cage after being dried quickly with a towel. On the following day, the subject mouse was placed in the cylinder for 6 minutes. The animal was judged to be immobile when it ceased struggling and remained floating motionless in the water, making only small movements necessary to keep its head above water. The immobility time was recorded by video during the last 4‐minute period of the test.
2.6. Tail suspension test (TST)
Mice were placed inside a 3‐sided cubicle and suspended by its tail from a hanger attached to a precision linear load cell that measured activity.21 Mice were acclimated to the testing conditions for at least 1 minute, and this is to account for the fact that mice are uniformly active during this time. The tip of the tail was fixed using adhesive tape. The mouse was suspended by the tail for 6 minutes, and the immobility time was counted for the last 4 minutes.
2.7. Locomotor activity (LA)
The locomotor activity of the mice was measured by an ambulometer with 4 activity chambers according to previous procedure.22 Before testing begins, mice were placed in the chambers. Their paws were contacted or disconnected the active bars which can produce random configurations that were converted into pulses. The pulses were proportional to the locomotor activity of the mice and automatically recorded as the cumulative total counts of motor activity. Fifteen minutes prior to the evaluation for acclimatization, mice were placed in test chambers, and locomotion counts were recorded for a period of 5 minutes.
2.8. Elevated plus‐maze test (EPM)
The elevated plus‐maze test was performed as described previously, and the number of entries into the open arms and the time spent in the open arms are used as indices of open space‐induced anxiety in mice.5 Two open arms (25 × 5 × 0.5 cm) crossed from each other were perpendicular to 2 closed arms (25 × 5 × 16 cm) with a center platform (5 × 5 × 0.5 cm). The maze was elevated 40 cm above the floor. Experiments began by placing a mouse on the central platform facing an open arm. During the first 5 minutes of free exploration, the number of entries (an entry is defined as the center of mass of the mouse enters the arm) into each arm and the time spent in the open arms are recorded. Entries are defined by measuring the center of mass of the mouse as it enters the arm. These measurements serve as an index of anxiety‐like behavior.
2.9. Novel object recognition (NOR)
The NOR test was performed as described previously.23 Briefly, 3 phases, habituation, training (T1), and testing (T2) phase, were included in the procedure. In T1 phase (day 1), each animal was allowed to freely explore the apparatus for 5 minutes. During T2 phase (day 2), a single animal was placed in the center of the open field to explore for 5 minutes. There are 2 identical objects located on the testing area. After a retention interval of 24 hours, the animal was subjected to the T3 phase for 5 minutes. In the T3 retention phase of the NOR test procedure, one familiar object and one novel object were presented. The animals were considered to be exploring the object when directing the nose to the object at a distance of no more than 2 cm and/or touching/sniffing the object. Time spent exploring the objects during T2 and T3 was recorded for each animal and indicated as “a” and “b”, respectively. The discrimination ratio d2 was calculated as d2 = (b − a)/(b + a). If the total of exploration time was less than 20 seconds during T1 or T2, the data are excluded from analysis.
2.10. Measurement of cGMP
The HT‐22 cells were plated at 1 × 105 cells/well onto 6‐well plates and were treated with different drugs for 24 hours before tests.24 Twenty‐four hours after treatment with drugs, cells were treated with 0.1 mol/L HCl containing 0.5% Triton X‐100, and incubated for 10 minutes at room temperature. Samples were then centrifuged to remove cellular debris. cGMP in supernatant was measured by enzyme‐linked immunosorbent assay (Assay Designs, Ann Arbor, MI). Assays were performed according to the manufacturer s protocol.
2.11. Western blot analysis
HT‐22 cells were seeded as a density of 2 × 105 cells/well in 6‐well plates and were washed with phosphate‐buffered saline (PBS) 2 times after PDE9 inhibitor C36D, corticosterone treatment, or Rp8 treatment. They were then lysed with RIPA lysis buffer containing protease and phosphatase inhibitors. The supernatant was assayed for total protein concentrations using BCA assay kit after they were centrifuged at 4000 g for 30 minutes at 4°C. Supernatants containing 20 μg of protein/lane were transferred to polyvinylidene difluoride membranes, then incubated in blocking buffer for 2 hours at room temperature and washed in Tris‐buffered saline (TBS) with 0.1% Tween 20 (TBST), Subsequently, the membranes were incubated with the appropriate primary antibodies over night at 4°C (anti‐CREB: 1:1000, anti‐pCREB: 1:1000, BDNF: 1:1000, 1:1000 and anti‐actin: 1:1000; Abcam, USA). After 3 washes with TBST, the membranes were then incubated with secondary antibody (1:10 000, Abcam, USA) at room temperature for 1 hour and washed for 3 times with TBST. The bands were visualized using the enhanced chemiluminescence method.
3. RESULTS
3.1. The protective effects of C36D against corticosterone‐induced cytotoxicity in HT‐22 cells
To determine the effective dose range of C36D in HT‐22 cells, dose‐dependent and time‐dependent effects were observed after treatment with C36D. The results suggested that C36D at doses of 10−10 to 10−5 mol/L produced inverted U‐shaped effects with the maximal effect at 10−8 mol/L (P < 0.01, Figure 2A). The time‐dependent effects suggested that significant protective effects occur at 12 and 24 hours after C36D treatment. The maximal effect is achieved at 24 hours (P < 0.01, Figure 2B). C36D did not show significant toxicity as the cell viability was increased after treatment with C36D at doses from 10−10 to 10−5 mol/L up to 48 hours. Subsequently, the protective effects of C36D against corticosterone‐induced HT‐22 cell lesion were investigated by assessing cell viability by the MTS assay at 6 concentrations (10−10, 10−9, 10−8, 10−7, 10−6, and 10−5 mol/L), 24 hours after treatment with C36D. The results suggested that C36D protected HT‐22 cells against corticosterone‐induced toxicity at concentrations between 10−8 and 10−7 mol/L, and the best concentration was 10−7 mol/L, at which the cell viability increased to normal level (P < 0.01). C36D had no positive effect at concentrations above 10−6 mol/L or below 10−9 mol/L (Figure 2C). It was noted that the PKG inhibitor Rp8 reversed the protective effects of C36D at a dose of 10−7 mol/L against corticosterone toxicity (P < 0.05). C36D was also shown to significantly rescue HT‐22 cells against corticosterone‐induced cytotoxicity 24 hours after treatment (F(7, 41) = 5.27, P < 0.05; Figure 2D).
Figure 2.
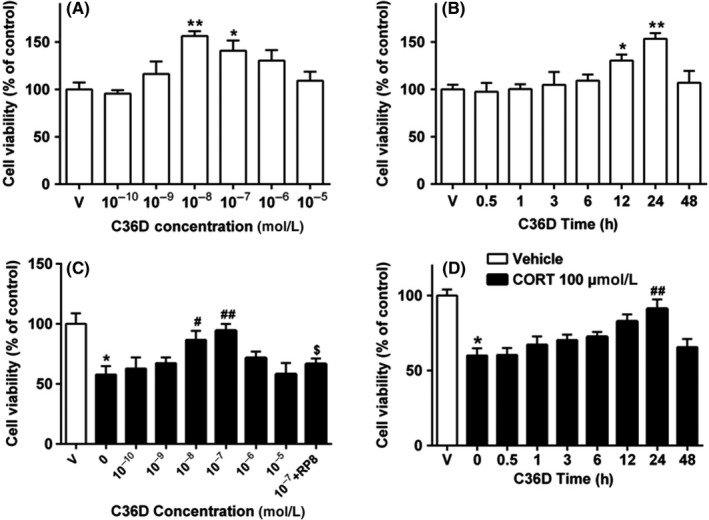
C36D increased HT‐22 cell viability and protected cells against corticosterone (CORT)‐induced cytotoxicity in a concentration‐ and time‐dependent manner. A, HT‐22 cells were treated with various concentrations of C36D for 24 h. B, HT‐22 cells were treated with C36D for the indicated times. C, HT‐22 cells were treated with 100 μmol/L CORT for 30 min, and C36D was added for 24 h. D, HT‐22 cells were treated with 100 μmol/L CORT for 30 min and added C36D for the indicated periods. Cell viability was measured by MTS assay. Results are expressed as the mean ± standard error of the mean (SEM) of 6 independent experiments performed in triplicates. *P < 0.05 and **P < 0.01, compared to control group. # P < 0.05 and ## P < 0.01, compared to vehicle‐treated CORT group. $ P < 0.05, compared to C36D (10−7 mol/L)‐treated CORT group
3.2. The effects of C36D on cGMP levels in corticosterone‐treated HT‐22 cells
The effects of C36D on cGMP level in corticosterone‐treated HT‐22 cells were shown in Figure 3A. The results revealed a significant decrease in cGMP expression when HT‐22 cells were exposed to 100 μmol/L corticosterone (P < 0.01). The decrease in cGMP level was rescued by treatment with C36D at concentrations of 10−8 and 10−7 mol/L, when compared to corticosterone‐treated group (P < 0.05; P < 0.01). Pretreatment with the selective PKG inhibitor Rp8 reversed the protective effect of C36D against corticosterone‐induced cGMP reduction (P < 0.05). C36D was also shown to produce a time‐dependent increase in cGMP (F(6, 24) = 8.23, P < 0.01), from 3 to 24 hours, achieving the maximal effect at 24 hours (Figure 3B).
Figure 3.
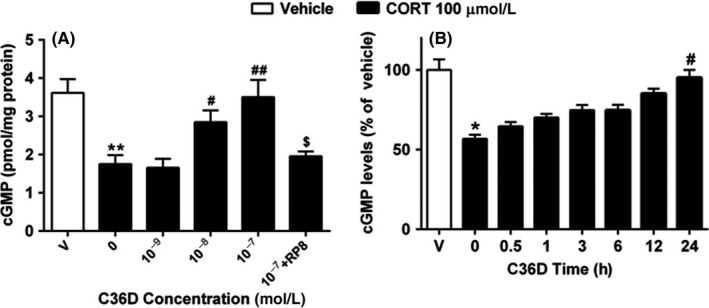
The effects of C36D on CORT‐induced cGMP reduction in concentration‐ and time‐dependent manners in the HT‐22 cells. A, HT‐22 cells were treated with 100 μmol/L CORT for 30 min and added C36D for 24 h. B, HT‐22 cells were treated with 100 μmol/L CORT for 30 min and then with C36D for the indicated periods. The results represent the mean ± SEM, n = 6. *P < 0.05 and **P < 0.01, compared to control group. # P < 0.05 and ## P < 0.01, compared to vehicle‐treated CORT group. $ P < 0.05, compared to C36D (10−7 mol/L)‐treated CORT group
3.3. The effects of C36D on corticosterone‐induced changes in pCREB/CREB and BDNF expression
The ratio of pCREB/CREB was reduced by treatment of HT‐22 cells with corticosterone for 24 hours (P < 0.01). While C36D did not change the reduced ratio of pCREB/CREB below 10−8 mol/L, it significantly increased pCREB/CREB expression ratio at 10−7 mol/L, when compared to vehicle‐treated corticosterone group (P < 0.05; Figure 4A). Similar effects of C36D on BDNF upregulation were shown when treating HT‐22 cells with C36D at 10−7 mol/L (P < 0.05, Figure 4B). However, these effects of C36D on pCREB/CREB and BDNF upregulation were prevented by pretreatment with PKG inhibitor Rp8 (P < 0.05, Figure 4A,B).
Figure 4.
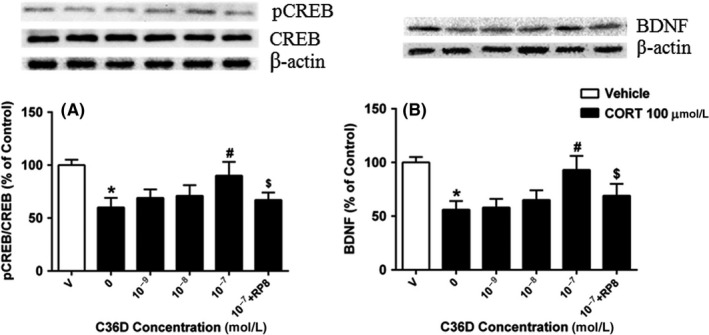
The effects of C36D on CORT‐induced decreased expression of pCREB A, and BDNF B, in the HT‐22 cells. The results represent the mean ± SEM, n = 6. *P < 0.05, compared to control group. # P < 0.05, compared to vehicle‐treated CORT group. $ P < 0.05, compared to C36D (10−7 mol/L)‐treated CORT group
3.4. The effects of C36D on depression, anxiety, and cognitive dysfunction induced by acute stress
To extend the in vitro results, we examined the effects of C36D in vivo and evaluated behavioral changes in acutely stressed mice, which induced depression, anxiety, and cognitive deficits. Figure 5 summarizes the acute stress paradigm; the behaviors were determined 10 minutes after restraint stress for 4 hours to rule out the possible false‐negative or false‐positive results. As shown in Figure 6A,B, acute 4‐hour restraint stress induced a significant increase in immobility time in both tail suspension and forced swimming tests (P < 0.05). Administering C36D at a dose of 1 mg/kg (ip) significantly reduced immobility time in the tail suspension and forced swimming tests, when compared to vehicle‐treated stressed groups (P < 0.001; P < 0.01). Mice treated with PDE4 inhibitor Rolipram (Rol) or classical antidepressant imipramine (IMA)‐treated mice also exhibited significant antidepressant‐like effects (P's < 0.05; P < 0.01). The C36D treatment at the dose that significantly reduced immobility response did not affect the locomotor activity as shown in Figure 6C. In the elevated plus‐maze test, administering C36D produced a significant dose‐dependent increase in the open‐arm entries and of time spent in the open arm compared with vehicle‐treated stressed groups (F(4, 32) = 10.71, P < 0.01; F(4, 32) = 12.35, P < 0.05). The significant increases were observed at a dose of 1.0 mg/kg, respectively (Figure 6D,E). The positive drug diazepam showed similar anxiolytic‐like effects. In the novel object recognition test, the cognitive enhancing effects were significant in acute stress mice with increasing doses of C36D at 0.1, 0.5, and 1 mg/kg, one hour after the training session (F(3, 21) = 3.57, P < 0.05; Figure 6F). The maximal effect of cognitive enhancement induced by C36D was observed at dose of 1 mg/kg (P < 0.05), which was similar to that of rolipram at dose of 0.5 mg/kg. The effects of C36D on behavioral changes in FST, TST, and EPM tests were prevented by pretreatment with the PKG inhibitor Rp8 (P < 0.05) though only tendency to decrease in the discrimination index was observed in the NOR tests.
Figure 5.
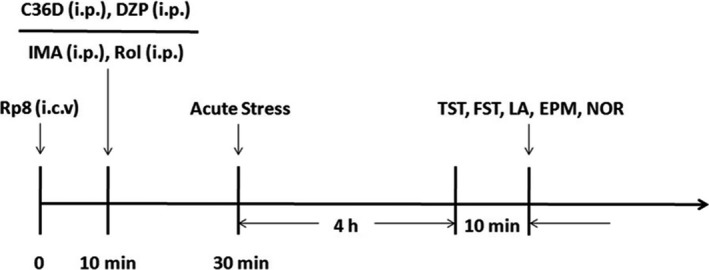
Treatment schedules and test orders. Animals were given bilateral micro‐injections of 2 μL Rp8 (1 nmol, 1 μL/side, i.c.v.), and after 10 min, C36D, DZP, IMA, or Rol was administered (ip). Thirty minutes later, acute stress was performed. Then, the animals were subjected to the behavioral tests including forced swimming (FST), tail suspension (TST), locomotion activity (LA), elevated plus‐maze (EPM), and novel object recognition (NOR) tests 10 min after stress. Rp8 (Rp‐8‐Br‐PET‐cGMPS, 1 nmol, i.c.v.), Rol (rolipram, 0.5 mg/kg, ip). DZP (diazepam, 0.5 mg/kg, ip), IMA (imipramine, 10 mg/kg, ip)
Figure 6.
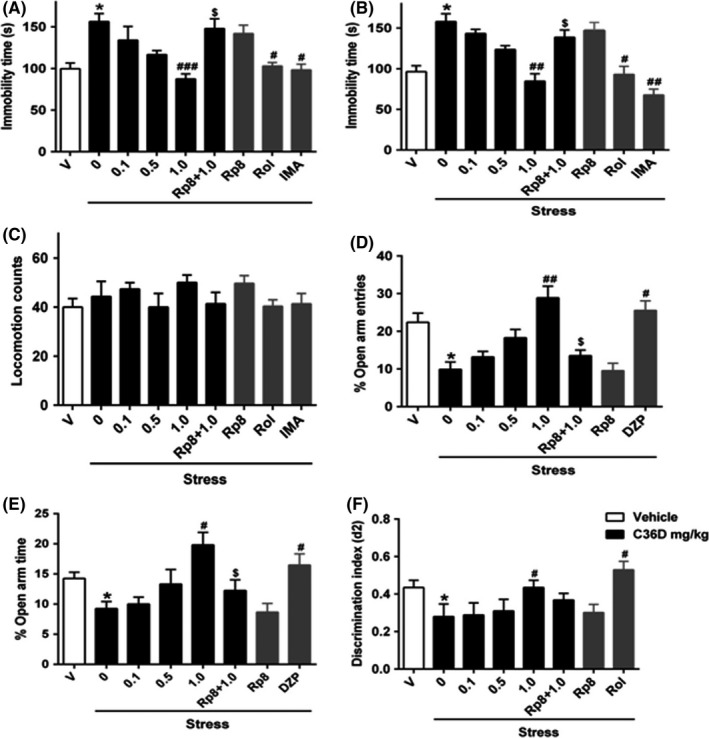
The effects of C36D on acute stress‐induced depression‐/anxiety‐like behaviors and cognitive impairment in TST (A), FST (B), LA (C), EPM (D and E), and NOR (F) tests. The results represent the mean ± SEM, n = 6. *P < 0.05, compared to control group. # P < 0.05, ## P < 0.01, ### P < 0.001, compared to vehicle‐treated stressed group. $ P < 0.05, compared to C36D (1.0 mg/kg)‐treated stressed group
4. DISCUSSION
The distribution of PDE9 protein, a cGMP‐specific phosphodiesterase, has been observed in the cortical areas, including insular, entorhinal, visual cortices, Ammon's horn (CA1, CA2, and CA3) and dentate gyrus of the hippocampus, and amygdala as well. Preclinical research has shown that PDE9A inhibition can enhance cognition and counter the impairment of age‐related memory impairment.25 To date, only a few selective PDE9 inhibitors have been developed, including BAY 73‐6691 and PF‐04447943.15 In a stably transfected PDE9 CHO cell line, Wunder et al have reported, although BAY 73‐6691 is able to dose‐dependently induce intracellular cGMP accumulation, its bioavailability and ability of brain penetration are not ideal. Previous studies have shown that C36D could effectively and selectively inhibit PDE9A as a potential hypoglycemic agent. However, its effects on depression‐ and anxiety‐like behaviors are still unknown. The present study investigated the antidepressant‐ and anxiolytic‐like effects of C36D and assessed the role of cGMP signaling in its action by testing C36D on HT‐22 cells for the first time. The results suggested that C36D could increase cGMP level, upregulated phosphorylation of CREB and BDNF expression in corticosterone‐treated HT‐22 cells. Moreover, the results from behavioral tests supported the in vitro data, which suggested that C36D produced antidepressant‐ and anxiolytic‐like effects and cognitive enhancement that were parallel to positive drugs rolipram, imipramine, or diazepam.
HT‐22 cells are immortalized mouse hippocampal neuronal precursor cells, which served as valuable models to understand cellular and molecular processes relevant to the hippocampus.26 We investigated the effective dose range of C36D to protect HT‐22 cells with or without corticosterone exposure. The results showed dose‐dependent protective effects against corticosterone insults after treatment with C36D for 24 hours. However, a 10‐fold C36D dose increase was required in the presence of corticosterone‐induced cell lesion. It is reasonable that higher doses of C36D would rescue HT‐22 cells against corticosterone‐induced cell lesion. However, these protective effects of C36D against corticosterone‐induced cell lesion were reversed by a PKG inhibitor Rp8, suggesting cGMP signaling involving this protective processes. Cyclic nucleotide (cAMP and cGMP) signaling is fundamentally involved in brain mechanisms that require for the neuronal activity and energy production, metabolic processes, and synaptic physiology.27, 28 The present results indicated that C36D was able to protect HT‐22 cells against corticosterone‐induced cytotoxicity by activation of cGMP/PKG signaling.
Signaling via cGMP is complex and is affected by factors such as cell types and the duration of stress and treatment. Cyclic GMP response element binding protein (CREB) phosphorylation at Ser133 is a key regulatory site by cGMP though there are at least 3 potential phosphorylation sites in human CREB protein related to stress.29 Decreasing cGMP level leads to inactivation of protein kinase (PKG), which in turn reduced the level of phosphorylated CREB. Increasing evidence also suggests that glucocorticoids may interfere phosphorylation of CREB and therefore blocking the expression of CRE‐regulated genes such as BDNF.30, 31 Selective PDE9A inhibitors prevent the breakdown of cGMP, which could have a potential effect on pCREB/CREB and BDNF expression. In accordance with this view, we found that corticosterone induced a significant decrease in pCREB/CREB and BDNF expression in the present study. The corticosterone‐induced decreases in pCREB and BDNF levels were rescued by C36D. Our findings support this chain of events, indicating that C36D is acted mainly by its inhibitory action on PDE9 to protect neuronal cells against corticosterone‐induced cytotoxicity via cGMP‐pCREB‐BDNF pathway.
Behavioral studies play an important role in the evaluation of antidepressant drugs. Van Der Staay et al32 found that PDE9A inhibitor BAY73‐6691 improved performance for several cognitive tasks including social recognition in rats and mice, novel object recognition in rats and scopolamine‐induced impairments in passive avoidance in rats. Considering that cognitive deficits are critical determinant of functional outcome in major depressive disorder, the present study firstly evaluated whether C36D reversed acute stress‐induced depression‐ and anxiety‐like effects including cognitive impairment after treatment with C36D. As expected, C36D produced significant antidepressant‐ and anxiolytic‐like effects, it also ameliorated cognitive deficits induced by acute stress, which further support our in vitro data indicating that C36D exhibits protective effects against corticosterone may be related to its behavioral amelioration induced by acute stress. The detailed molecular cascade involving neuroprotective effects during acute stress and those of C36D treatment is being process to decipher the possible mechanism related to behavioral changes.
In conclusion, the present study firstly demonstrated that C36D protected the HT‐22 cells against corticosterone‐induced cytotoxicity, which might occur by regulating the cGMP/PKG/CREB signaling. The in vivo study extended the in vitro findings that suggested C36D dose‐dependently ameliorated acute stress‐induced depression, anxiety, and cognitive impairment. These results provide novel evidence that targeting PDE9 could be a potential treatment of major depressive disorder.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81603336). The work was also supported by 2015 American Association of Colleges of Pharmacy New Investigator Award and Innovative Micro‐Programs Accelerating Collaboration in Themes: University at Buffalo Internal Funding Program (IMPACT) to Dr. Y. Xu.
Huang X‐F, Jiang W‐T, Liu L, et al. A novel PDE9 inhibitor WYQ‐C36D ameliorates corticosterone‐induced neurotoxicity and depression‐like behaviors by cGMP‐CREB‐related signaling. CNS Neurosci Ther. 2018;24:889–896. 10.1111/cns.12864
Contributor Information
Guo‐Qiang Song, Email: drugs@vip.sina.com.
Ying Xu, Email: yxu9@buffalo.edu.
REFERENCES
- 1. Birmaher B, Ryan ND, Williamson DE, et al. Childhood and adolescent depression: A review of the past 10 years. Part I. J Am Acad Child Adolesc Psychiatry. 1996;35:1427‐1439. [DOI] [PubMed] [Google Scholar]
- 2. Birmaher B, Brent D, Bernet W, et al. Practice parameter for the assessment and treatment of children and adolescents with depressive disorders. J Am Acad Child Adolesc Psychiatry. 2007;46:1503‐1526. [DOI] [PubMed] [Google Scholar]
- 3. Ishihara L, Brayne C. A systematic review of depression and mental illness preceding. Parkinson's disease. Acta Neurol Scand. 2006;113:211‐220. [DOI] [PubMed] [Google Scholar]
- 4. Levinstein MR, Samuels BA. Mechanisms underlying the antidepressant response and treatment resistance. Front Behav Neurosci. 2014;8:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masood A, Nadeem A, Mustafa SJ, et al. Reversal of oxidative stress‐induced anxiety by inhibition of phosphodiesterase‐2 in mice. J Pharmacol Exp Ther. 2008;326:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu Y, Pan J, Chen L, et al. Phosphodiesterase‐2 inhibitor reverses corticosterone‐induced neurotoxicity and related behavioural changes via cGMP/PKG dependent pathway. Int J Neuropsychopharmacol. 2013;16:835‐847. [DOI] [PubMed] [Google Scholar]
- 7. Lu Y, Kandel ER, Hawkins RD. Nitric Oxide signaling contributes to late‐phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250‐10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puzzo D, Vitolo O, Fabrizio T, et al. Amyloid‐beta peptide inhibits activation of the nitric oxide/cGMP/cAMP‐responsive element‐binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887‐6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurice DH, Ke H, Ahmad F, et al. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discovery. 2014;13:290‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481‐511. [DOI] [PubMed] [Google Scholar]
- 11. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309‐327. [DOI] [PubMed] [Google Scholar]
- 12. Rosa JM, Dafre AL, Rodrigues ALS. Antidepressant‐like responses in the forced swimming test elicited by glutathione and redox modulation. Behav Brain Res. 2013;253:165‐172. [DOI] [PubMed] [Google Scholar]
- 13. Ke H, Wang H, Ye M. Structural insight into the substrate specificity of phosphodiesterases. Handb Exp Pharmacol. 2011;204:121‐134. [DOI] [PubMed] [Google Scholar]
- 14. Andreeva SG, Dikkes P, Epstein PM, et al. Expression of cGMP‐Specific phosphodiesterase 9A mRNA in the rat brain. J Neurosci. 2001;15:9068‐9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verhoest PR, Fonseca KR, Hou X, et al. Design and discovery of 6‐[(3S,4S)‐4‐methyl‐1‐(pyrimidin‐2‐ylmethyl)pyrrolidin‐3‐yl]‐1‐(tetrahydro‐2Hpyran‐4‐yl)‐1,5‐dihydro‐4H‐pyrazolo[3,4‐d]pyrimidin‐4‐one(PF‐04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J Med Chem. 2012;55:9045‐9054. [DOI] [PubMed] [Google Scholar]
- 16. Claffey MM, Helal CJ, Verhoest PR, et al. Application of structure‐based drug design and parallel chemistry to identify selective, brain penetrant, in vivo active phosphodiesterase 9A inhibitors. J Med Chem. 2012;55:9055‐9068. [DOI] [PubMed] [Google Scholar]
- 17. Meng F, Hou J, Shao YX, et al. Structure based discovery of highly selective phosphodiesterase‐9A inhibitors and implications for inhibitor design. J Med Chem. 2012;55:8549‐8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao YX, Huang M, Cui WJ, et al. Discovery of a phosphodiesterase 9A inhibitor as a potential hypoglycemic agent. J Med Chem. 2014;57:10304‐10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y, Zhang C, Wang R, et al. Corticosterone induced morphological changes of hippocampal and amygdaloid cell lines are dependent on5‐HT7 receptor related signal pathway. Neuroscience. 2011;182:71‐81. [DOI] [PubMed] [Google Scholar]
- 20. Filipcik P, Novak P, Mravec B, et al. Tau protein phosphorylation in diverse brain areas of normal and CRH deficient mice: up‐regulation by stress. Cell Mol Neurobiol. 2012;32:837‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y, Ku BS, Yao HY, et al. Antidepressant effects of curcumin in the forced swim test and olfactory bulbectomy models of depression in rats. Pharmacol Biochem Behav. 2005;82:200‐206. [DOI] [PubMed] [Google Scholar]
- 22. Li W, Ling S, Yang Y, et al. Systematic hypothesis for post‐stroke depression caused inflammation and neurotransmission and resultant on possible treatments. Neuro Endocrinol Lett. 2014;35:104‐109. [PubMed] [Google Scholar]
- 23. Xu Y, Pan JC, Sun J, et al. Inhibition of phosphodiesterase 2 reverses impaired cognition and neuronal remodeling caused by chronic stress. Neurobiol Aging. 2015;36:955‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y, Lin D, Li S, et al. Curcumin reverses impaired cognition and neuronal plasticity induced by chronic stress. Neuropharmacology. 2009;57:463‐471. [DOI] [PubMed] [Google Scholar]
- 25. Vardigan JD, Converso A, Hutson PH, et al. The selective phosphodiesterase 9 (PDE9) inhibitor PF‐04447943 attenuates a scopolamine‐induced deficit in a novel rodent attention task. J Neurogenet. 2011;25:120‐126. [DOI] [PubMed] [Google Scholar]
- 26. Liu J, Li L, Suo WZ. HT‐22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267‐271. [DOI] [PubMed] [Google Scholar]
- 27. Zhou QG, Zhu LJ, Chen C, et al. Hippocampal neuronal nitric oxide synthase mediates the stress‐related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J Neurosci. 2011;31:7579‐7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen CC, Yang CH, Huang CC, et al. Acute stress impairs hippocampal mossy fiber‐CA3 long‐term potentiation by enhancing cAMP‐specific phosphodiesterase 4 activity. Neuropsychopharmacology. 2010;35:1605‐1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakamoto K, Huang BW, Iwasaki K, et al. Regulation of genotoxic stress response by homeodomain‐interacting protein kinase 2 through phosphorylation of cyclic AMP response element‐binding protein at serine 271. Mol Biol Cell. 2010;21:2966‐2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang C, Cheng Y, Wang H, et al. RNA interference‐mediated knockdown of long‐form phosphodiesterase‐4D (PDE4D) enzyme reverses amyloid‐beta 42‐induced memory deficits in mice. J Alzheimers Dis. 2014;38:269‐280. [DOI] [PubMed] [Google Scholar]
- 31. Maurice T, Duclot F, Meunier J, et al. Altered memory capacities and response to stress inp300/CBP‐associated factor histone acetylase knockout mice. Neuropsychopharmacology. 2008;33:1584‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Der Staay FJ, Rutten K, Barfacker L, et al. The novel selective PDE9 inhibitor BAY73‐6691 improves learning and memory in rodents. Neuropharmacology. 2008;55:908‐918. [DOI] [PubMed] [Google Scholar]