

Summary
Huntington's disease (HD) is a fatal neurodegenerative condition, due to a mutation in the IT15 gene encoding for huntingtin. Currently, disease‐modifying therapy is not available for HD, and only symptomatic drugs are administered for the management of symptoms. In the last few years, preclinical and clinical studies have indicated that pharmacological strategies aimed at inhibiting cyclic nucleotide phosphodiesterase (PDEs) may develop into a novel therapeutic approach in neurodegenerative disorders. PDEs are a family of enzymes that hydrolyze cyclic nucleotides into monophosphate isoforms. Cyclic nucleotides are second messengers that transduce the signal of hormones and neurotransmitters in many physiological processes, such as protein kinase cascades and synaptic transmission. An alteration in their balance results in the dysregulation of different biological mechanisms (transcriptional dysregulation, immune cell activation, inflammatory mechanisms, and regeneration) that are involved in neurological diseases. In this review, we discuss the action of phosphodiesterase inhibitors and their role as therapeutic agents in HD.
Keywords: cyclic nucleotides, Huntington's disease, neuroprotection, PDE inhibitors, phosphodiesterase (PDEs)
1. INTRODUCTION
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder and is characterized by motor impairment and cognitive and psychiatric symptoms.1, 2 Currently, only symptomatic drugs are available to cure HD. Tetrabenazine was approved by FDA in 2008 for the treatment of choreic movements in HD. Other symptoms, such as hypokinesia and rigidity and/or depressive and behavioral defects, are usually treated with antiparkinson, antidepressant, and antipsycotic drugs.3, 4, 5, 6, 7
The neuropathological hallmark of HD is the degeneration of striatum. However, several studies have shown repercussions on other parts of the brain, such as cerebral cortex, globus pallidus, thalamus, substantia nigra, and cerebellum. Moreover, not all the striatal neurons are affected in the same way: While aspiny neurons (cholinergic interneurons marked with somatostatin, neuropeptide y) are spared, medium spiny neurons (MSNs) undergo a massive degeneration that begins at the early stages of the disease. MSNs are divided into two different subtypes: enkephalin‐containing neurons project to the external segment of globus pallidus (GPe) (indirect pathway) and substance P neurons projecting to internal segment of globus pallidus (GPi) (direct pathway). Neurons of the indirect pathway are more vulnerable to HD degeneration.8, 9, 10, 11, 12
Many mechanisms are involved in HD neurodegeneration and include both loss and gain of function mutated huntingtin–polyglutamine aggregates formation of mutant protein, alteration of axonal transport and energy metabolism, oxidative stress, excitotoxicity, impairment of synaptic signals, and transcriptional dysregulation.13 In particular, it is known that transcriptional dysregulation plays a major role in HD, since mutated huntingtin alters the cAMP response element‐binding protein (CREB) and its related transcriptional action.14, 15, 16, 17, 18 Brain‐derived neurotrophic factor (BDNF) gene transcription is regulated by CREB,19 and its expression is stimulated by huntingtin that lost this function when mutated.20, 21 Several studies have shown a downregulation of BDNF levels in cellular and animal models,19, 21, 22, 23 as well as in postmortem brain of HD patients.21, 24
Thus, strategies aimed at increasing CREB transcription—and, consequently, the expression of BDNF—have neuroprotective effects in HD animals.25, 26, 27 CREB activation is mediated by a cAMP‐dependent protein kinase (PKA). The balance of intracellular cAMP/cGMP levels, thus, represents an important aspect in the regulation of neuronal processes.
In light of this, PDEs and their inhibitors can play an important role in a therapeutic scenario for HD. In fact, PDEs are enzymes that catabolize cAMP and/or cGMP in the cell. Thus, their inhibition could be beneficial in the neuronal degeneration in the central nervous system (CNS).
In this review, we will discuss the inhibition of PDEs and its neuroprotective action in HD by upregulation of the cyclic nucleotide signaling.
2. MOLECULAR MECHANISM OF PDES FUNCTION
Phosphodiesterases (PDEs) are fundamental enzymes that belong to intracellular signal transduction cascade, hydrolyzing cyclic nucleotides cAMP, and/or cGMP. PDEs are divided into 11 families and variants, encoded by 21 genes. Moreover, they are classified as cAMP (PDEs 4,7,8), cGMP (PDEs 5,6,9) or double specific PDEs (PDEs 1,2,3,10,11).28, 29
Cyclic nucleotides are second messengers in the signal transduction process and synaptic transmission in dopaminergic, noradrenergic and glutamatergic systems in neurons.30, 31, 32, 33 cAMP and cGMP derives, respectively, from ATP and GTP by the reaction of adenylyl (AC) and guanylyl cyclase (GC). In particular, the cellular mechanism involved is the following: the binding of hormones, neurotransmitters, chemokines, autocrine, and paracrine receptor factors to GPCR receptors (G protein‐coupled receptors) activates the heterotrimeric G proteins, consisting of the three subunits (alpha, beta and gamma).34, 35 In fact, the trans‐membrane adenylated cyclic (tAC), activated by the stimulatory G protein (Gs) and inhibited by the inhibitory G protein (Gi), produces 3′,5′‐cyclic adenosine monophosphate (cAMP) from ATP; however, it is also synthesized in the brain by soluble adenylate cyclase (AC), whose production is stimulated by bicarbonate (HCO3) and calcium in neuronal cells and in glia. At this point, cyclic AMP stimulates the protein kinase A, the exchange factors directly activated by cAMP 1 (EPAC1 and EPAC2), and/or the cyclic nucleotides gated channels (CNGs). In the case of the production of 3′,5′‐cyclic guanosine monophosphate (cGMP), however, nitrogen monoxide (NO) activates soluble GC (sGC), whereas particulate guanylyl cyclase (pGC) is activated through binding of natriuretic peptides (NPs) to coupled receptors. The cGMP, produced from the GTP, will in turn activate the protein kinase G and CNGs. Formation of both cAMP and cGMP involves activation of CREB by phosphorylation, generating the transcription of several genes. As previously mentioned, both cAMP and cGMP are neutralized through the hydrolysis performed by phosphodiesterases.36, 37, 38, 39, 40, 41, 42
3. PDES IN NEURODEGENERATIVE DISORDERS
Many cellular processes are impaired in neurodegenerative disorders, as an imbalance of cyclic nucleotides with consequent alteration of PDEs functions and neuronal survival.43 Cyclic nucleotides can be considered a central player in many cellular processes, such as long‐term potentiation, synaptic plasticity, memory, neurogenesis, neurotransmission, in which they regulate—by PKA (protein kinase A) and ERK (extracellular regulated kinase) signaling pathway—many proteins and their transcription and translation.44, 45 As mentioned above, CREB is an important target of cAMP and cGMP.46, 47 CREB has a pivotal role in neuronal survival and plasticity, and one of its target gene is BDNF, a key growth factor for the striatum. BDNF is a neurotrophin involved in synaptic plasticity, neuronal survival, and differentiation. It is known that BDNF expression is altered in neurodegenerative diseases, and a reduction in its levels occurs in people affected by Alzheimer's disease, Parkinson's disease, and Huntington's disease. Particularly in HD, BDNF shows a dysregulation in cellular transport mechanisms, and in transcription and postsynaptic signaling. Moreover, only in HD, BDNF levels are linked to huntingtin mutation. In fact, wild‐type huntingtin has a stimulating effect on BDNF expression. Consequently, a reduction in BDNF protein level occurs in the striatum.21, 22, 48, 49 PDEs are largely diffused in neuronal cells, and thanks to modulating cAMP/cGMP content, they have attracted the attention of neuroscientists as possible key molecules in the battle against neurodegenerative diseases.50, 51, 52, 53 Furthermore, a bulk of data point out the role of PDEs in dopaminergic pathways. Thus, their inhibition seems to be a useful strategy to fight diseases such as Alzheimer's disease, Parkinson's disease, HD, depressive and cognition disorders.54
4. PHOSPHODIESTERASES IN DOPAMINERGIC PATHWAYS
As mentioned previously, the dopaminergic system is altered in neurodegenerative and neuropsychiatric diseases, and among these also in Huntington's disease. These diseases have in common the cortico‐striato‐thalamic systems, which work in a synergistic and complex way,55, 56, 57, 58, 59 and in which the alteration of the balance involves the appearance of motor, cognitive, and behavioral dysfunctions typical of these disorders. Dopamine appears to be the modulator of these circuits both in the frontal cortex and in the striatum, regions in which dopamine receptors are strongly distributed.57, 60, 61 In fact, as it is well known, some of these diseases, such as PD, use dopaminergic therapy to fight its symptoms, although it subsequently involves various side effects. Dopamine, originating from the pars compacta of the substantia nigra or from the VTA, is bound to the D1 receptors on the MSNs neurons of the direct pathway activating the stimulatory (Golf, Gs) and stimulating the production of cAMP, or to the D2 receptors of the indirect pathway that bind to the inhibitory G protein (Gi) that inhibits the production of cAMP.57, 62, 63, 64, 65, 66 This information therefore establishes the central role played by dopamine in the regulation of psychomotor mechanisms. As mentioned above, the action of dopamine occurs through the signal transduction mechanism operated by the cAMP/PKA system and is also controlled by phosphodiesterases.67 In the striatum, the regulation of cAMP and cGMP turnover plays a fundamental role because of their inhibitory and excitatory effects of the nigro‐striated‐pallid circuits, mentioned before, in neurons. In the striatum are expressed many phosphodiesterase isoforms, which have various localizations and functions influencing synaptic plasticity and excitability of the membrane.68 Furthermore, the activation of the signal transduction mechanism of cAMP and cGMP, as mentioned, involves the activation of a series of downstream proteins, such as PKA and PKG, respectively, which in turn activates other modulators of neuronal excitability as ion channels and phosphatases. For example, in the striatum, their activation involves the phosphorylation of DARPP‐32, which plays a central role in the regulation of dopamine and its action on the GABAergic and glutamminergic pathway. The phosphorylation of DARPP‐32 involves the activation of its inhibitory phosphatase activity of PP1, which is inactivated and makes PKA unable to stimulate other substrates such as CREB.30, 69, 70, 71 All this information highlights the importance of the PKA/PKG pathway in the execution of complex motor mechanisms through the regulation of striatal synaptic transmission.72, 73 In fact, MSNs, striatal projection neurons, have high levels of ACs and GCs, whose activation is regulated by various neurotransmitters (eg, dopamine, 5HT, neuropeptide Y, adenosine, glutamate) that act through GPCRs and NO synthesis stimulation, respectively. The different isoforms of PDEs are highly expressed in the striatum.30, 74, 75 The inhibition of phosphodiesterases involves a regulation of the cAMP/PKA signal transduction mechanism, resulting in the stimulation of dopamine synthesis at the dopaminergic terminal level, the inhibition of dopamine signal in the D2 receptors in striato‐pallidal neurons, and stimulation of dopamine D1 receptors in striatonigral neurons.67 In fact, numerous studies have shown that, under physiological conditions, compounds that activate the production of cAMP and cGMP in the MSNs (such as the aforementioned PDEs inhibitors or cyclase activators) positively regulate cortico‐striatal transmission. In the opposite case, when drugs, such as cyclase inhibitors, decrease the levels of these species in neuronal cells, there is a reduction in synaptic activity.76, 77, 78, 79 All these considerations lead to the hypothesis of the central role played by AC‐cAMP‐PKA mechanism in MSNs, enhancing the activation of AMPA and NMDA receptors during cortico‐striatal transmission. As mentioned previously, the excitatory effect of NMDA receptors is due to dopamine binding to D1 receptors, with activation of the AC‐cAMP‐PKA signal transduction system. On the contrary, binding to D2 receptors inhibits the cascades of the AC‐cAMP‐PKA signal.80, 81, 82, 83, 84, 85 As mentioned above, these mechanisms, widely described so far, are altered in neurodegenerative and neuropsychiatric diseases, and therefore also in Huntington's disease. In HD, the pathological mechanism of the disease is the neuronal death of striatal MSNs neurons, and in particular of the striatal cells of the indirect pathway. The death of these neurons appears at least in part due to the overexpression of mutant huntingtin, although the underlying mechanism is not yet clear.86 Various studies, conducted in vitro and in vivo (using the different HD animal models available) but also in patients with HD, have highlighted the reduced level of expression of cAMP, CREB, and nNOS mRNA.47, 87, 88, 89 These data underline the alteration of the cAMP/cGMP mechanism and of the PDE signals. These reports confirm what was said previously: the activity of phosphodiesterase inhibitors which increase the levels of cyclic nucleotides in striatal neurons seems to regulate the striatal cortical transmission in a positive way.68 Moreover, it is known that dopamine undergoes changes in the levels of expression and release in the course of the HD. Dopamine has a biphasic modulation in the striatum during the course of HD disease. In particular, it seems that in the initial stages of the disease there is an increase in dopamine neurotransmission which involves hyperkinesia in the movement. On the contrary, in the late stages of the pathology, there is a reduction in dopamine neurotransmission with the consequent appearance of hypokinesia. Various studies have also observed the loss of dopaminergic innervation and a reduction in TH + neurons in the striatum of postmortem brains of HD patients.90 These observations were confirmed in PET studies conducted both on patients with overt disease and on patients with the modified gene but who still had not shown the typical symptoms of the disease.91, 92 These data have been confirmed in animal models.93, 94, 95 Furthermore, a reduction in dopamine D1 and D2 receptor levels was also evidenced.96
5. PDES IN HUNTINGTON'S DISEASE
In the following section, we examine the different PDEs that play a role in HD pathology, with regard to their tissue distribution, and to the inhibitors that have been tested so far in clinical and preclinical trials in HD (Table 1).
Table 1.
The principal phosphodiesterases in HD and their relative inhibitors
| Phosphodiesterase | Substrate | Isoforms | Distribution | Modulation | Inhibitors |
|---|---|---|---|---|---|
| PDE1 | cAMP/cGMP |
PDE1A PDE1B PDE1C |
Cortex, Hippocampus, Striatum | Ca2+/Calmodulin | Vinpocetine |
| PDE4 | cAMP |
PDE4A PDE4B PDE4D |
Cortex, Hippocampus, Striatum | Phosphorylation | Rolipram |
| PDE5 | cGMP | PDE5A | Spinal cord, Cerebellum | cAMP/Phosphorylation | Sildenafil, Vardenafil, Tadalafil, |
| PDE10 | cAMP/cGMP | PDE10A | Striatum | cAMP/Phosphorylation |
TP 10 TAK‐063 PF‐0254920 |
PDE, Phosphodiesterase.
As mentioned above, PDEs and their relative inhibitors could be considered a new therapeutic strategy in HD. In fact, various studies showed beneficial, neuroprotective effects of PDEs inhibitors in animal models of HD improving motor and cognitive problems but also increasing the expression of species such as pCREB and BDNF, altered, as mentioned, in Huntington's disease 97, 98, 99, 100 (Figure 1).
Figure 1.
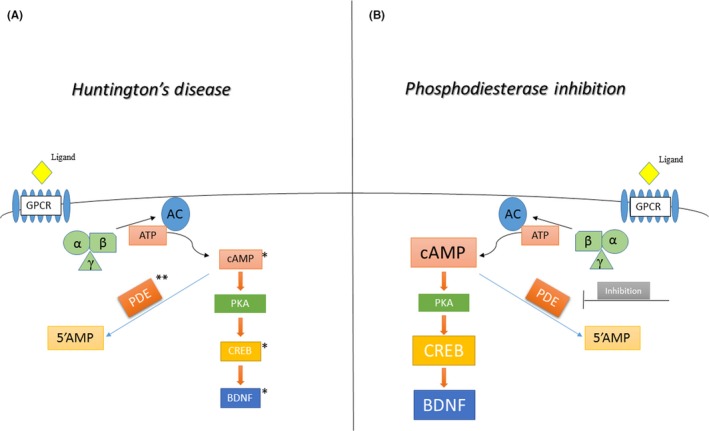
Figure shows the signaling cascade of cAMP (cGMP not shown in figure) in Huntington's disease, where there is a reduction in cAMP levels and cAMP response element‐binding protein (CREB) activity of transcription gene. Inhibition of phopshodiesterases seems to improve motor and cognitive deficits and restore cAMP and CREB normal expression. *Reduced levels of cAMP, CREB, and brain‐derived neurotrophic factor (BDNF) levels. **Phosphodiesterase (PDEs) levels changes according to the different isoforms analyzed (please see main text)
5.1. PDE 1
Phosphodiesterase 1 (PDE1) hydrolyzes both cAMP and cGMP. It has three isoforms (PDE1A, PDE1B, PDE1C), two of which (PDE1A and PDE1B) are expressed in striatum, as well as in cortex and hippocampus as PDE1C. Particularly, PDE1B is ubiquitously distributed in spiny projection neurons and colocalizes with D1 receptors,101 suggesting a possible involvement of this isoform in striatal neurodegeneration and dopaminergic signaling.102, 103 PDE1 is not localized only in cytoplasm, but, for example, PDE1A is expressed in nucleus, which has a regulation activity of gene transcription.104 For its high expression in striatum and frontal cortex and its colocalization with D1 receptor, PDE1B could be considered a good target for phosphodiesterase inhibitors in disorders characterized by cognitive complications (schizophrenia) and motor dysfunction. Therefore, PDE1 inhibition was studied in Parkinson's disease. In fact, vinpocetine is a PDE1 inhibitor characterized by the capacity to reduce neuronal inflammation and the expression of TNF‐α and IL‐1β.105 In many studies, this compound has shown neuroprotective properties: regulate oxidative stress and enhance cognition in behavioral test and memory both in animal models and patients.106, 107, 108
Interestingly, PDE1A2 isoform was found to be preferentially distributed in cholinergic interneurons,103 suggesting a role in the survival of striatal interneurons in HD.
With the aim of shedding light on the role of PDEs in HD, vinpocetine treatment was shown to ameliorate impaired cognition and motor coordination in a 3‐nitroproprionic acid‐induced HD rat model, by reducing oxidative species, inflammation, and mitochondrial dysfunction.109
Recently, a new molecule capable of inhibiting PDE1 has been discovered. It has been tested in PHASE 1 of a clinical trial and has shown positive effects not only in Alzheimer's disease and schizophrenia, but also in movement disorders.110
Thus, inhibition of PDE1 could be seen as a potential drug target in HD treatment, although more precise studies are necessary in both clinical and preclinical research in HD.
5.2. PDE 4
The best described PDEs are the PDE4 family, composed by four different enzymes (PDE4A, PDE4B, PDE4C, and PDE4D). With the exception of PDE4C, the other isoforms are ubiquitous in the central nervous systems, and their distribution is particularly high in the striatum, cortex, and hippocampus.111
PDE4 has distinct functions in the dopaminergic system because of its different distribution to the various striatal neuronal subtypes. Moreover, indirect pathway presents a higher expression of PDE4B than direct pathway. Thus inhibition of PDE4 regulates and ameliorates cAMP/PKA signaling in the indirect pathway neurons, and at the same time, upregulating TH activation and dopamine formation.112
In fact, regulating cAMP balance PDE4 is essential in the PKA/CREB/BDNF pathway, as demonstrated by the CREB‐upregulating effects of PDE inhibition in depressive behavior.113, 114
As mentioned earlier, CREB represents an important transcription factor, as it is needed for adult neuronal survival and for mediating nuclear calcium‐regulated gene transcription. CREB is activated by cAMP‐dependent protein kinase (PKA) by the phosphorylation of its Ser133. Consequently, the activated form pCREB binds to CREs elements (cyclic AMP response elements) on the promoter region of DNA and promotes the transcription of various genes involved in memory and neuronal plasticity, such as BDNF.115, 116, 117 Our group previously confirmed the abnormalities in CREB transcription in the quinolinic‐induced rat model of HD, describing a differential modulation of pCREB in the striatal neuronal population.46 Particularly, a decreased expression in the neurons most vulnerable to HD (medium spiny neurons, parvalbumin and carletinin positive interneuron) was observed. On the other hand, cholinergic interneurons conserve adequate pCREB expression, and probably this event confers their neuroprotection.46
Interestingly, however, PDE4 in HD was described to be decreased in R6/2 mice, suggesting a compensatory mechanism due to a concurrent decrease in CREB activation, as seen above.118
The first generation of PDE4 inhibitor is rolipram. Previous studies of our group showed that this PDE4 inhibitor is able to increase the levels of pCREB (the activated form of CREB) in the medium spiny neurons, with neuroprotective effects both in HD rat model induced by quinolinic acid, and in R6/2 transgenic HD mice. These effects were demonstrated by the reduction of intranuclear formation of mutant huntingtin inclusions, sparing of striatal neurons, decrease in microglial activation, the delay of onset, and decrease in severity of neurological impairment and movement defects.119, 120, 121
In spite of the promising results obtained in animal models, these inhibitors have not proved to be successful in human clinical trials: rolipram have various side effects, such as nausea and emesis, 122 and in multiple sclerosis MRI showed an increase in brain inflammatory processes measured by brain lesions.123
Thus, recently, the new purpose of the research will be to study new inhibitors that do not have the aforementioned side effects.124, 125 The last clinical study in HD is represented by the PDE4 inhibitor GSK356278 (GlaxoSmithKline) that has shown good tolerability in patients although the improvement in motor and cognitive symptoms is not clear.66
5.3. PDE 5
PDE 5 is specific to cGMP and is abundant in striatum, cortex, and hippocampus.126
Inhibition of these phosphodiesterases showed positive effects in rats as far as it concerns memory amelioration,127, 128 synaptic plasticity,129 and depressive symptoms.130
Sildenafil, known primarily for its use in the erectile dysfunction and pulmonary arterial hypertension due to its vasodilator effects, also showed to exert a neuroprotective effect in various animal models of disease.126, 131 Indeed, Puerta et al132 demonstrated that sildenafil and vardenafil ameliorate neurological impairment, decrease death of medium spiny neurons, and upregulate pCREB and BDNF expression in a rat model of HD induced by 3 nitroproprionic acid. This confirms the importance of cGMP pathway in HD pathology, suggesting PDE5 inhibitors as a possible therapeutic strategy in HD.
5.4. PDE 10
Phosphodiesterase 10 is an enzyme with a double specificity for cAMP and cGMP, characterized by its high level of expression in striatum, nucleus accumbens, and olfactory tubercle. PDE10A is also distributed in hippocampus, thalamus, cerebellum, and spinal cord.133, 134, 135, 136 PDE10 expression in the caudate portion of the basal ganglia suggests a role of this enzyme in striatonigral and striato‐pallidal pathways.137 Xie et al137 described PDE10A localization only in medium spiny neurons, whereas it was not expressed in interneurons. Because of such peculiar expression in medium spiny neurons (MSNs), PDE10A involvement was studied in dopamine signaling. Many studies showed that inhibition of PDE10A involves the activation of D1‐direct and D2‐indirect pathway.138, 139, 140, 141 In 2008, Nishi et al142 showed the effects of papaverine, a PDE10A inhibitor, in D1‐DARPP‐32‐FLAG/D2 DARPP‐32‐Myc mice and demonstrated that PDE10A controls cAMP/PKA pathway as a dopamine D2 antagonist, activating the indirect pathway.
However, our group also found PDE10A protein expressed in interneurons, with a nuclear distribution, suggesting a specific role of the PDEs determined by their distribution.143, 144, 145 Studies described a decrease in PDE10A mRNA expression in striatum of R6/2 mice and in brain samples of HD affected people118; also, low cAMP expression was recorded in the striatum of HD patients and in STHdh Q111 cell HD model.88, 146 It is possible that the latter mechanism determines the decrease in PDE10A expression as a compensatory mechanism.147 Interestingly, in contrast with the findings by Hebb et al 2004,118 our group found a dramatic increase of PDE10A in MSN of R6/2 mice compared to WT mice. This pattern is repeated in all types of interneurons (parvalbuminergic, somatostatinergic, calretininergic), except in cholinergic ones where PDE10A showed low expression levels during the disease progression.143 On the basis of these considerations, our group has previously tested the PDE10A inhibitor TP10 (Pfizer) in rats and R62 mice, obtaining a reduction in striatal neuronal loss and an increase in life span. This was associated with the upregulation of pCREB and BDNF protein expression.148, 149 Those results were later confirmed by the study by Beaumont et al.150 obtained by using PDE10 inhibitors in the Q175 mice that show activation of CREB pathway and of MAP kinase signaling cascades.
Recently, a novel PDE10A inhibitor 1‐[2‐fluoro‐4‐(1H‐pyrazol‐1yl)phenyl]‐5‐methoxy‐3‐(1‐phenyl‐1H‐pyrazol‐5‐yl)pyridazin‐4(1H)‐one (TAK‐063) has been tested in R6/2 mouse HD model. This new compound produces an activation of both indirect and direct pathway MSNs. In this study, TAK‐063 showed beneficial effects in mice with amelioration of behavioral and neurological impairment, reduction in neurons loss in striatum, and upregulation of BDNF expression.151
The effects of PDE10A inhibition are still under investigation. A Phase II clinical trial has just been concluded: PF‐0254920 drug was used to taste safety and tolerability in HD patients. Unfortunately, with the data available so far, the drug does not ameliorate movement and/or behavioral problems.152
6. CONCLUSIONS
This review highlighted the important role that phosphodiesterases plays in many cellular processes under physiological and/or pathological conditions. Therefore, we have described in more detail the phosphodiesterases expressed in the striatum and related brain regions, the main target of the Huntington pathology, and more involved in the regulation of the dopaminergic system, universally recognized as altered in the aforesaid pathology. All the studies presented here suggest the use of PDE inhibitors in HD, in order to potentiate cAMP signaling in the striatum.
As mentioned elsewhere, in recent years the therapeutic role of phosphodiesterase regulation has emerged through their inhibition. Without a doubt, the most known drug among antiphosphodiesterase drugs is viagra, which inhibits phosphodiestrase 5 and improves erectile dysfunction. Few other drugs capable of inhibition phosphodiestrase are used in clinical settings: for example, in the dysregulation of various diseases such as pulmonary hypertension, acute refractory cardiac failure, intermittent claudication, and chronic obstructive pulmonary disease. Moreover, the importance of PDEs inhibition is demonstrated by the NIH clinical trials Web site.153 In fact, some of FDA‐approved PDE drugs in Phase Trials are being tested in AD, HD, and/or other neurodegenerative disease patients.
Therefore, in the last few years, clinical neuroscientists have shifted their attention to the development of new candidates for PDE inhibition, developing isoform selective inhibitors.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Cardinale A, Fusco FR. Inhibition of phosphodiesterases as a strategy to achieve neuroprotection in Huntington's disease. CNS Neurosci Ther. 2018;24:319–328. 10.1111/cns.12834
REFERENCES
- 1. Albin RL, Tagle DA. Genetics and molecular biology of Huntington's disease. Trends Neurosci. 1995;18:11‐14. [DOI] [PubMed] [Google Scholar]
- 2. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971‐983. [DOI] [PubMed] [Google Scholar]
- 3. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369‐384. [DOI] [PubMed] [Google Scholar]
- 4. Levesque M, Bedard A, Cossette M, Parent A. Novel aspects of the chemical anatomy of the striatum and its efferents projections. J Chem Neuroanat. 2003;26:271‐281. [DOI] [PubMed] [Google Scholar]
- 5. Rubinsztein DC, Carmichael J. Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5:1‐21. [DOI] [PubMed] [Google Scholar]
- 6. Conneally PM. Huntington disease: genetics and epidemiology. Am J Hum Genet. 1984;36:506‐526. [PMC free article] [PubMed] [Google Scholar]
- 7. Walker FO. Huntington's disease. Lancet. 2007;369:218‐228. [DOI] [PubMed] [Google Scholar]
- 8. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559‐577. [DOI] [PubMed] [Google Scholar]
- 9. Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733‐5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sapp E, Ge P, Aizawa H, et al. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington's disease using high resolution image analysis. Neuroscience. 1995;64:397‐404. [DOI] [PubMed] [Google Scholar]
- 12. Galvan L, Andre VM, Wang EA, Cepeda C, Levine MS. Functional differences between direct and indirect striatal output pathways in Huntington's disease. J Huntingtons Dis. 2012;1:17‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27:2803‐2820. [DOI] [PubMed] [Google Scholar]
- 14. Steffan JS, Kazantsev A, Spasic‐Boskovic O, et al. The Huntington's disease protein interacts with p53 and CREB‐binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763‐6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine‐dependent neurodegeneration in Drosophila. Nature. 2001;413:739‐743. [DOI] [PubMed] [Google Scholar]
- 16. Nucifora FC Jr, Sasaki M, Peters MF, et al. Interference by huntingtin and atrophin‐1 with cbp‐mediated transcription leading to cellular toxicity. Science. 2001;291:2423‐2428. [DOI] [PubMed] [Google Scholar]
- 17. Choi YS, Lee B, Cho HY, et al. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's disease. Neurobiol Dis. 2009;36:259‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang H, Nucifora FC Jr, Ross CA, DeFranco DB. Cell death triggered by polyglutamine‐expanded huntingtin in a neuronal cell line is associated with degradation of CREB‐binding protein. Hum Mol Genet. 2003;12:1‐12. [DOI] [PubMed] [Google Scholar]
- 19. Zuccato C, Cattaneo E. Role of brain‐derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294‐330. [DOI] [PubMed] [Google Scholar]
- 20. Zuccato C, Belyaev N, Conforti P, et al. Widespread disruption of repressor element‐1 silencing transcription factor/neuron‐restrictive silencer factor occupancy at its target genes in Huntington's disease. J Neurosci. 2007;27:6972‐6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin‐mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493‐498. [DOI] [PubMed] [Google Scholar]
- 22. Fusco FR, Zuccato C, Tartari M, et al. Co‐localization of brain‐derived neurotrophic factor (BDNF) and wild‐type huntingtin in normal and quinolinic acid‐lesioned rat brain. Eur J Neurosci. 2003;18:1093‐1102. [DOI] [PubMed] [Google Scholar]
- 23. Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919‐930. [DOI] [PubMed] [Google Scholar]
- 24. Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain‐derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257‐261. [DOI] [PubMed] [Google Scholar]
- 25. Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain‐derived neurotrophic factor over‐expression in the forebrain ameliorates Huntington's disease phenotypes in mice. J Neurochem. 2008;105:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708‐14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giampa C, Montagna E, Dato C, Melone MA, Bernardi G, Fusco FR. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington's disease. PLoS ONE. 2013;8:e64037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488‐520. [DOI] [PubMed] [Google Scholar]
- 29. Sharma S, Kumar K, Deshmukh R, Sharma PL. Phosphodiesterases: regulators of cyclic nucleotide signals and novel molecular target for movement disorders. Eur J Pharmacol. 2013;714:486‐497. [DOI] [PubMed] [Google Scholar]
- 30. Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024‐1030. [DOI] [PubMed] [Google Scholar]
- 31. Lesch KP, Lerer B. The 5‐HT receptor–G‐protein–effector system complex in depression. I. Effect of glucocorticoids. J Neural Transm Gen Sect. 1991;84:3‐18. [DOI] [PubMed] [Google Scholar]
- 32. Majewski HK, Musgrave IF. Second messenger pathways in the modulation of neurotransmitter release. Aust N Z J Med. 1995;25:817‐821. [DOI] [PubMed] [Google Scholar]
- 33. Neve KA, Seamans JK, Trantham‐Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165‐205. [DOI] [PubMed] [Google Scholar]
- 34. Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven‐transmembrane receptors. Trends Pharmacol Sci. 2004;25:413‐422. [DOI] [PubMed] [Google Scholar]
- 35. Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636‐1639. [DOI] [PubMed] [Google Scholar]
- 36. Das A, Xi L, Kukreja RC. Phosphodiesterase‐5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280:12944‐12955. [DOI] [PubMed] [Google Scholar]
- 37. Corbin JD, Turko IV, Beasley A, Francis SH. Phosphorylation of phosphodiesterase‐5 by cyclic nucleotide‐dependent protein kinase alters its catalytic and allosteric cGMP‐binding activities. Eur J Biochem. 2000;267:2760‐2767. [DOI] [PubMed] [Google Scholar]
- 38. Cheung WY. Cyclic 3′,5′‐nucleotide phosphodiesterase. Evidence for and properties of a protein activator. J Biol Chem. 1971;246:2859‐2869. [PubMed] [Google Scholar]
- 39. Goraya TA, Cooper DM. Ca2 + ‐calmodulin‐dependent phosphodiesterase (PDE1): current perspectives. Cell Signal. 2005;17:789‐797. [DOI] [PubMed] [Google Scholar]
- 40. Chen J, Martinez J, Milner TA, Buck J, Levin LR. Neuronal expression of soluble adenylyl cyclase in the mammalian brain. Brain Res. 2013;1518:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kobialka M, Witwicka H, Siednienko J, Gorczyca WA. Metabolism of cyclic GMP in peritoneal macrophages of rat and guinea pig. Acta Biochim Pol. 2003;50:837‐848. [PubMed] [Google Scholar]
- 42. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91:651‐690. [DOI] [PubMed] [Google Scholar]
- 43. Cui Q, So KF. Involvement of cAMP in neuronal survival and axonal regeneration. Anat Sci Int. 2004;79:209‐212. [DOI] [PubMed] [Google Scholar]
- 44. Kleppisch T. Phosphodiesterases in the central nervous system. Handb Exp Pharmacol. 2009;191:71‐92. [DOI] [PubMed] [Google Scholar]
- 45. Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011;79:1277‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giampa C, DeMarch Z, D'Angelo V, et al. Striatal modulation of cAMP‐response‐element‐binding protein (CREB) after excitotoxic lesions: implications with neuronal vulnerability in Huntington's disease. Eur J Neurosci. 2006;23:11‐20. [DOI] [PubMed] [Google Scholar]
- 47. Gines S, Seong IS, Fossale E, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock‐in mice. Hum Mol Genet. 2003;12:497‐508. [DOI] [PubMed] [Google Scholar]
- 48. Jancic D, Lopez de Armentia M, Valor LM, Olivares R, Barco A. Inhibition of cAMP response element‐binding protein reduces neuronal excitability and plasticity, and triggers neurodegeneration. Cereb Cortex. 2009;19:2535‐2547. [DOI] [PubMed] [Google Scholar]
- 49. Zuccato C, Cattaneo E. Brain‐derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311‐322. [DOI] [PubMed] [Google Scholar]
- 50. Bollen E, Prickaerts J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life. 2012;64:965‐970. [DOI] [PubMed] [Google Scholar]
- 51. Perez‐Torres S, Cortes R, Tolnay M, Probst A, Palacios JM, Mengod G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer's disease brains examined by in situ hybridization. Exp Neurol. 2003;182:322‐334. [DOI] [PubMed] [Google Scholar]
- 52. Garcia‐Osta A, Cuadrado‐Tejedor M, Garcia‐Barroso C, Oyarzabal J, Franco R. Phosphodiesterases as therapeutic targets for Alzheimer's disease. ACS Chem Neurosci. 2012;3:832‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang HT, Huang Y, Masood A, et al. Anxiogenic‐like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology. 2008;33:1611‐1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramirez AD, Smith SM. Regulation of dopamine signaling in the striatum by phosphodiesterase inhibitors: novel therapeutics to treat neurological and psychiatric disorders. Cent Nerv Syst Agents Med Chem. 2014;14:72‐82. [DOI] [PubMed] [Google Scholar]
- 55. Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine‐receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228‐235. [DOI] [PubMed] [Google Scholar]
- 56. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Surmeier DJ, Carrillo‐Reid L, Bargas J. Dopaminergic modulation of striatal neurons, circuits, and assemblies. Neuroscience. 2011;198:3‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 60. Nishi A, Kuroiwa M, Shuto T. Mechanisms for the modulation of dopamine d(1) receptor signaling in striatal neurons. Front Neuroanat. 2011;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuroiwa M, Snyder GL, Shuto T, et al. Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP‐32 signaling cascade in frontal cortex. Psychopharmacology. 2012;219:1065‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor‐regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429‐1432. [DOI] [PubMed] [Google Scholar]
- 63. Ferre S, Quiroz C, Orru M, et al. Adenosine A(2A) receptors and A(2A) receptor heteromers as key players in striatal function. Front Neuroanat. 2011;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sibley DR, Monsma FJ Jr, Shen Y. Molecular neurobiology of dopaminergic receptors. Int Rev Neurobiol. 1993;35:391‐415. [DOI] [PubMed] [Google Scholar]
- 65. Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182‐217. [DOI] [PubMed] [Google Scholar]
- 66. Heckman PR, van Duinen MA, Bollen EP, et al. Phosphodiesterase inhibition and regulation of dopaminergic frontal and striatal functioning: clinical implications. Int J Neuropsychopharmacol. 2016;19:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nishi A, Snyder GL. Advanced research on dopamine signaling to develop drugs for the treatment of mental disorders: biochemical and behavioral profiles of phosphodiesterase inhibition in dopaminergic neurotransmission. J Pharmacol Sci. 2010;114:6‐16. [DOI] [PubMed] [Google Scholar]
- 68. Threlfell S, West AR. Review: modulation of striatal neuron activity by cyclic nucleotide signaling and phosphodiesterase inhibition. Basal Ganglia. 2013;3:137‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89‐101. [DOI] [PubMed] [Google Scholar]
- 70. Deisseroth K, Tsien RW. Dynamic multiphosphorylation passwords for activity‐dependent gene expression. Neuron. 2002;34:179‐182. [DOI] [PubMed] [Google Scholar]
- 71. Frank DA, Greenberg ME. CREB: a mediator of long‐term memory from mollusks to mammals. Cell. 1994;79:5‐8. [DOI] [PubMed] [Google Scholar]
- 72. Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine‐mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211‐219. [DOI] [PubMed] [Google Scholar]
- 73. Del Bel EA, Guimaraes FS, Bermudez‐Echeverry M, et al. Role of nitric oxide on motor behavior. Cell Mol Neurobiol. 2005;25:371‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366‐398. [DOI] [PubMed] [Google Scholar]
- 75. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309‐327. [DOI] [PubMed] [Google Scholar]
- 76. Colwell CS, Levine MS. Excitatory synaptic transmission in neostriatal neurons: regulation by cyclic AMP‐dependent mechanisms. J Neurosci. 1995;15(3 Pt 1):1704‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Threlfell S, Sammut S, Menniti FS, Schmidt CJ, West AR. Inhibition of Phosphodiesterase 10A Increases the Responsiveness of Striatal Projection Neurons to Cortical Stimulation. J Pharmacol Exp Ther. 2009;328:785‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. West AR, Grace AA. The nitric oxide‐guanylyl cyclase signaling pathway modulates membrane activity States and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J Neurosci. 2004;24:1924‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sammut S, Threlfell S, West AR. Nitric oxide‐soluble guanylyl cyclase signaling regulates corticostriatal transmission and short‐term synaptic plasticity of striatal projection neurons recorded in vivo. Neuropharmacology. 2010;58:624‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Brady AM, O'Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci. 2004;24:1040‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576‐9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chergui K, Lacey MG. Modulation by dopamine D1‐like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32‐0432. Neuropharmacology. 1999;38:223‐231. [DOI] [PubMed] [Google Scholar]
- 83. Levine MS, Li Z, Cepeda C, Cromwell HC, Altemus KL. Neuromodulatory actions of dopamine on synaptically‐evoked neostriatal responses in slices. Synapse. 1996;24:65‐78. [DOI] [PubMed] [Google Scholar]
- 84. Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine‐ and cAMP‐regulated phosphoprotein (Mr 32 kDa)/protein phosphatase‐1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297‐10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jocoy EL, Andre VM, Cummings DM, et al. Dissecting the contribution of individual receptor subunits to the enhancement of N‐methyl‐d‐aspartate currents by dopamine D1 receptor activation in striatum. Front Syst Neurosci. 2011;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gusella JF, MacDonald ME, Ambrose CM, Duyao MP. Molecular genetics of Huntington's disease. Arch Neurol. 1993;50:1157‐1163. [DOI] [PubMed] [Google Scholar]
- 87. Cramer H, Kohler J, Oepen G, Schomburg G, Schroter E. Huntington's chorea– measurements of somatostatin, substance P and cyclic nucleotides in the cerebrospinal fluid. J Neurol. 1981;225:183‐187. [DOI] [PubMed] [Google Scholar]
- 88. Cramer H, Warter JM, Renaud B. Analysis of neurotransmitter metabolites and adenosine 3′,5′‐monophosphate in the CSF of patients with extrapyramidal motor disorders. Adv Neurol. 1984;40:431‐435. [PubMed] [Google Scholar]
- 89. Norris PJ, Waldvogel HJ, Faull RL, Love DR, Emson PC. Decreased neuronal nitric oxide synthase messenger RNA and somatostatin messenger RNA in the striatum of Huntington's disease. Neuroscience. 1996;72:1037‐1047. [DOI] [PubMed] [Google Scholar]
- 90. Bedard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson's disease and Huntington's chorea. Parkinsonism Relat Disord. 2011;17:593‐598. [DOI] [PubMed] [Google Scholar]
- 91. Richfield EK, O'Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington's disease. Neurosci Lett. 1991;132:121‐126. [DOI] [PubMed] [Google Scholar]
- 92. van Oostrom JC, Dekker M, Willemsen AT, de Jong BM, Roos RA, Leenders KL. Changes in striatal dopamine D2 receptor binding in pre‐clinical Huntington's disease. Eur J Neurol. 2009;16:226‐231. [DOI] [PubMed] [Google Scholar]
- 93. Cha JH, Kosinski CM, Kerner JA, et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. 1998;95:6480‐6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bibb JA, Yan Z, Svenningsson P, et al. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci U S A. 2000;97:6809‐6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ariano MA, Aronin N, Difiglia M, et al. Striatal neurochemical changes in transgenic models of Huntington's disease. J Neurosci Res. 2002;68:716‐729. [DOI] [PubMed] [Google Scholar]
- 96. Cepeda C, Murphy KP, Parent M, Levine MS. The role of dopamine in Huntington's disease. Prog Brain Res. 2014;211:235‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci. 2017;18:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fusco FR, Paldino E. Role of phosphodiesterases in Huntington's disease. Adv Neurobiol. 2017;17:285‐304. [DOI] [PubMed] [Google Scholar]
- 99. Fusco FR, Giampa C. Phosphodiesterases as therapeutic targets for Huntington's disease. Curr Pharm Des. 2015;21:365‐377. [DOI] [PubMed] [Google Scholar]
- 100. Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660‐670. [DOI] [PubMed] [Google Scholar]
- 101. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology. 2010;59:367‐374. [DOI] [PubMed] [Google Scholar]
- 102. Polli JW, Kincaid RL. Expression of a calmodulin‐dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J Neurosci. 1994;14(3 Pt 1):1251‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kakkar R, Raju RV, Sharma RK. Calmodulin‐dependent cyclic nucleotide phosphodiesterase (PDE1). Cell Mol Life Sci. 1999;55:1164‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nagel DJ, Aizawa T, Jeon KI, et al. Role of nuclear Ca2 + /calmodulin‐stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res. 2006;98:777‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gomez CD, Buijs RM, Sitges M. The anti‐seizure drugs vinpocetine and carbamazepine, but not valproic acid, reduce inflammatory IL‐1beta and TNF‐alpha expression in rat hippocampus. J Neurochem. 2014;130:770‐779. [DOI] [PubMed] [Google Scholar]
- 106. DeNoble VJ. Vinpocetine enhances retrieval of a step‐through passive avoidance response in rats. Pharmacol Biochem Behav. 1987;26:183‐186. [DOI] [PubMed] [Google Scholar]
- 107. Deshmukh R, Sharma V, Mehan S, Sharma N, Bedi KL. Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine – a PDE1 inhibitor. Eur J Pharmacol. 2009;620:49‐56. [DOI] [PubMed] [Google Scholar]
- 108. Hindmarch I, Fuchs HH, Erzigkeit H. Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int Clin Psychopharmacol. 1991;6:31‐43. [DOI] [PubMed] [Google Scholar]
- 109. Gupta S, Sharma B. Protective effects of phosphodiesterase‐1 (PDE1) and ATP sensitive potassium (KATP) channel modulators against 3‐nitropropionic acid induced behavioral and biochemical toxicities in experimental Huntingtons disease. Eur J Pharmacol. 2014;732:111‐122. [DOI] [PubMed] [Google Scholar]
- 110. Li P, Zheng H, Zhao J, et al. Discovery of potent and selective inhibitors of phosphodiesterase 1 for the treatment of cognitive impairment associated with neurodegenerative and neuropsychiatric diseases. J Med Chem. 2016;59:1149‐1164. [DOI] [PubMed] [Google Scholar]
- 111. Cherry JA, Davis RL. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol. 1999;407:287‐301. [PubMed] [Google Scholar]
- 112. Yamashita N, Hayashi A, Baba J, Sawa A. Rolipram, a phosphodiesterase‐4‐selective inhibitor, promotes the survival of cultured rat dopaminergic neurons. Jpn J Pharmacol. 1997;75:155‐159. [DOI] [PubMed] [Google Scholar]
- 113. Krebs EG, Beavo JA. Phosphorylation‐dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923‐959. [DOI] [PubMed] [Google Scholar]
- 114. Itoh T, Tokumura M, Abe K. Effects of rolipram, a phosphodiesterase 4 inhibitor, in combination with imipramine on depressive behavior, CRE‐binding activity and BDNF level in learned helplessness rats. Eur J Pharmacol. 2004;498:135‐142. [DOI] [PubMed] [Google Scholar]
- 115. Vallejo M. Transcriptional control of gene expression by cAMP‐response element binding proteins. J Neuroendocrinol. 1994;6:587‐596. [DOI] [PubMed] [Google Scholar]
- 116. Kobierski LA, Wong AE, Srivastava S, Borsook D, Hyman SE. Cyclic AMP‐dependent activation of the proenkephalin gene requires phosphorylation of CREB at serine‐133 and a Src‐related kinase. J Neurochem. 1999;73:129‐138. [DOI] [PubMed] [Google Scholar]
- 117. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605‐623. [DOI] [PubMed] [Google Scholar]
- 118. Hebb AL, Robertson HA, Denovan‐Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington's disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123:967‐981. [DOI] [PubMed] [Google Scholar]
- 119. DeMarch Z, Giampa C, Patassini S, Martorana A, Bernardi G, Fusco FR. Beneficial effects of rolipram in a quinolinic acid model of striatal excitotoxicity. Neurobiol Dis. 2007;25:266‐273. [DOI] [PubMed] [Google Scholar]
- 120. DeMarch Z, Giampa C, Patassini S, Bernardi G, Fusco FR. Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2008;30:375‐387. [DOI] [PubMed] [Google Scholar]
- 121. Giampa C, Middei S, Patassini S, et al. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902‐910. [DOI] [PubMed] [Google Scholar]
- 122. Fleischhacker WW, Hinterhuber H, Bauer H, et al. A multicenter double‐blind study of three different doses of the new cAMP‐phosphodiesterase inhibitor rolipram in patients with major depressive disorder. Neuropsychobiology. 1992;26:59‐64. [DOI] [PubMed] [Google Scholar]
- 123. Bielekova B, Richert N, Howard T, et al. Treatment with the phosphodiesterase type‐4 inhibitor rolipram fails to inhibit blood–brain barrier disruption in multiple sclerosis. Mult Scler. 2009;15:1206‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Man HW, Schafer P, Wong LM, et al. Discovery of (S)‐N‐[2‐[1‐(3‐ethoxy‐4‐methoxyphenyl)‐2‐methanesulfonylethyl]‐1,3‐dioxo‐2,3‐dihy dro‐1H‐isoindol‐4‐yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor‐alpha inhibitor. J Med Chem. 2009;52:1522‐1524. [DOI] [PubMed] [Google Scholar]
- 125. Dart‐Neuroscience . Clinical Trials. Available online: http://www.dartneuroscience.com/ClinicalTrials.php. Accessed February 23, 2018.
- 126. Puerta E, Hervias I, Goni‐Allo B, Lasheras B, Jordan J, Aguirre N. Phosphodiesterase 5 inhibitors prevent 3,4‐methylenedioxymethamphetamine‐induced 5‐HT deficits in the rat. J Neurochem. 2009;108:755‐766. [DOI] [PubMed] [Google Scholar]
- 127. Prickaerts J, Steinbusch HW, Smits JF, de Vente J. Possible role of nitric oxide‐cyclic GMP pathway in object recognition memory: effects of 7‐nitroindazole and zaprinast. Eur J Pharmacol. 1997;337:125‐136. [DOI] [PubMed] [Google Scholar]
- 128. Prickaerts J, van Staveren WC, Sik A, et al. Effects of two selective phosphodiesterase type 5 inhibitors, sildenafil and vardenafil, on object recognition memory and hippocampal cyclic GMP levels in the rat. Neuroscience. 2002;113:351‐361. [DOI] [PubMed] [Google Scholar]
- 129. Picconi B, Bagetta V, Ghiglieri V, et al. Inhibition of phosphodiesterases rescues striatal long‐term depression and reduces levodopa‐induced dyskinesia. Brain. 2011;134(Pt 2):375‐387. [DOI] [PubMed] [Google Scholar]
- 130. Liebenberg N, Harvey BH, Brand L, Brink CB. Antidepressant‐like properties of phosphodiesterase type 5 inhibitors and cholinergic dependency in a genetic rat model of depression. Behav Pharmacol. 2010;21:540‐547. [DOI] [PubMed] [Google Scholar]
- 131. Puzzo D, Staniszewski A, Deng SX, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid‐beta load in an Alzheimer's disease mouse model. J Neurosci. 2009;29:8075‐8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Puerta E, Hervias I, Barros‐Minones L, et al. Sildenafil protects against 3‐nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol Dis. 2010;38:237‐245. [DOI] [PubMed] [Google Scholar]
- 133. Fujishige K, Kotera J, Michibata H, et al. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem. 1999;274:18438‐18445. [DOI] [PubMed] [Google Scholar]
- 134. Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA. Isolation and characterization of PDE10A, a novel human 3′, 5′‐cyclic nucleotide phosphodiesterase. Gene. 1999;234:109‐117. [DOI] [PubMed] [Google Scholar]
- 135. Seeger TF, Bartlett B, Coskran TM, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113‐126. [DOI] [PubMed] [Google Scholar]
- 136. Coskran TM, Morton D, Menniti FS, et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54:1205‐1213. [DOI] [PubMed] [Google Scholar]
- 137. Xie Z, Adamowicz WO, Eldred WD, et al. Cellular and subcellular localization of PDE10A, a striatum‐enriched phosphodiesterase. Neuroscience. 2006;139:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Smith SM, Uslaner JM, Cox CD, et al. The novel phosphodiesterase 10A inhibitor THPP‐1 has antipsychotic‐like effects in rat and improves cognition in rat and rhesus monkey. Neuropharmacology. 2013;64:215‐223. [DOI] [PubMed] [Google Scholar]
- 139. Grauer SM, Pulito VL, Navarra RL, et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331:574‐590. [DOI] [PubMed] [Google Scholar]
- 140. Schmidt CJ, Chapin DS, Cianfrogna J, et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325:681‐690. [DOI] [PubMed] [Google Scholar]
- 141. Strick CA, James LC, Fox CB, Seeger TF, Menniti FS, Schmidt CJ. Alterations in gene regulation following inhibition of the striatum‐enriched phosphodiesterase, PDE10A. Neuropharmacology. 2010;58:444‐451. [DOI] [PubMed] [Google Scholar]
- 142. Nishi A, Kuroiwa M, Miller DB, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460‐10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Leuti A, Laurenti D, Giampa C, et al. Phosphodiesterase 10A (PDE10A) localization in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2013;52:104‐116. [DOI] [PubMed] [Google Scholar]
- 144. Houslay MD. Compartmentalization of cyclic AMP phosphodiesterases, signalling ‘crosstalk’, desensitization and the phosphorylation of Gi‐2 add cell specific personalization to the control of the levels of the second messenger cyclic AMP. Adv Enzyme Regul. 1995;35:303‐338. [DOI] [PubMed] [Google Scholar]
- 145. Houslay MD, Milligan G. Tailoring cAMP‐signalling responses through isoform multiplicity. Trends Biochem Sci. 1997;22:217‐224. [DOI] [PubMed] [Google Scholar]
- 146. Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME. Enhanced Akt signaling is an early pro‐survival response that reflects N‐methyl‐D‐aspartate receptor activation in Huntington's disease knock‐in striatal cells. J Biol Chem. 2003;278:50514‐50522. [DOI] [PubMed] [Google Scholar]
- 147. Kleiman RJ, Kimmel LH, Bove SE, et al. Chronic suppression of phosphodiesterase 10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission, and signaling pathways implicated in Huntington's disease. J Pharmacol Exp Ther. 2011;336:64‐76. [DOI] [PubMed] [Google Scholar]
- 148. Giampa C, Patassini S, Borreca A, et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington's disease. Neurobiol Dis. 2009;34:450‐456. [DOI] [PubMed] [Google Scholar]
- 149. Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS ONE. 2010;5:e13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Beaumont V, Zhong S, Lin H. et al. Phosphodiesterase 10A Inhibition Improves Cortico‐Basal Ganglia Function in Huntington's Disease Models. Neuron. 2016;92:1220‐1237. [DOI] [PubMed] [Google Scholar]
- 151. Harada A, Suzuki K, Kimura H. TAK‐063, a Novel Phosphodiesterase 10A Inhibitor, Protects from Striatal Neurodegeneration and Ameliorates Behavioral Deficits in the R6/2 Mouse Model of Huntington's Disease. J Pharmacol Exp Ther. 2017;360:75‐83. [DOI] [PubMed] [Google Scholar]
- 152. Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington's disease: what's in the pipeline? Mov Disord. 2014;29:1434‐1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. NIH‐USA . https://clinicaltrials.gov. Accessed February 23, 2018.