

Summary
Background & Aims
Spinocerebellar ataxia type 3 (SCA3), also known as Machado‐Joseph disease (MJD), is an autosomal dominantly inherited neurodegenerative disorder and the most common form of SCA worldwide. It is caused by the expansion of a polyglutamine (polyQ) tract in the ataxin‐3 protein. Nuclear localization of the affected protein is a key event in the pathology of SCA3 via affecting nuclear organization, transcriptional dysfunction, and seeding aggregations, finally causing neurodegeneration and cell death. So far, there is no effective therapy to prevent or slow the progression of SCA3.
Methods
In this study, we explored the effect of divalproex sodium as an HDACi in SCA3 cell models and explored how divalproex sodium interferes with pathogenetic processes causing SCA3.
Results
We found that divalproex sodium rescues the hypoacetylation levels of histone H3 and attenuates cellular cytotoxicity induced by expanded ataxin‐3 partly via preventing nuclear transport of ataxin‐3 (particularly heat shock‐dependent).
Conclusion
Our study provides novel insights into the mechanisms of action of divalproex sodium as a possible treatment for SCA3, beyond the known regulation of transcription.
Keywords: divalproex sodium, HDAC inhibitors, nuclear localization, SCA3 treatment, transcriptional dysfunction
1. INTRODUCTION
Spinocerebellar ataxia type 3 (SCA3), also known as Machado‐Joseph disease (MJD), is an inherited neurodegenerative disorder and the most common form of autosomal dominant inherited ataxias worldwide.1 Spinocerebellar ataxia type 3 (SCA3) is one of nine polyglutamine (polyQ) expansion diseases which form a group of hereditary neurodegenerative disorders: six types of spinocerebellar ataxias (SCA) such as SCA1, SCA2, SCA6, SCA7, SCA17, and SCA3, Huntington's disease (HD), dentatorubral‐pallidoluysian atrophy (DRPLA), and spinal and bulbar muscular atrophy (SBMA).2, 3, 4, 5, 6, 7, 8, 9
Spinocerebellar ataxia type 3 (SCA3) is caused by the expansion of a CAG repeat in the ATXN3 gene encoding a polyglutamine‐containing protein called ataxin‐3. In general, ataxin‐3 is a primarily cytoplasmic protein, although, depending on the cell type and level of expression, it may also be detected both in the nucleus and in the cytoplasm.10, 11, 12, 13, 14, 15 Beyond doubt, nuclear localization of ataxin‐3 plays an important role in the pathology of SCA3, and ataxin‐3 was shown to be more toxic in the nucleus than in the cytoplasm by disturbing nuclear organization, transcriptional function, and seeding aggregations.11, 16, 17, 18 In a previous in vivo study, we demonstrated that nuclear‐localized expanded ataxin‐3 aggravates the SCA3 phenotype in transgenic mice and increases the presence of aggregations. On the other hand, transgenic mice with an ataxin‐3, which is kept in the cytoplasm, showed much milder symptoms and fewer inclusions.19 This evidence demonstrates that nuclear ataxin‐3 is required for the manifestation of a SCA3 phenotype and proved that strategically preventing nuclear translocation of ataxin‐3 is a promising treatment for SCA3.
Emerging data indicate HDAC inhibitors as potential therapeutic compounds for the treatment of neurodegenerative diseases such as SCA3, Alzheimer's disease, Parkinson's disease, Huntington's disease, and Amyotrophic lateral sclerosis.20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 However, the exact mechanism of action of HDAC inhibitors is still widely unknown. HDACi include aliphatic acids (valproic acid [VPA], sodium butyrate [SB], phenylbutyrate), hydroxamate (trichostatin A [TSA], SAHA, CBHA, LAQ‐824, PXD‐101), cyclic peptide (depsipeptide), benzamide (MS‐275), and others (Tubacin).36 Phenylbutyrate has already entered a phase II clinical trial for the treatment of HD.25 Recent studies show that VPA alleviates neurodegeneration and extends the life span of a Drosophila SCA3 model35 and that VPA rescues hypoacetylated H3 and H4 levels in SCA3 cell models.20 Valproic acid is found to ameliorate dopaminergic neurodegeneration and prolongs the life span in Caenorhabditis elegans.37, 38 Besides, safety and efficacy of VPA has been tested in patients with SCA3/MJD.23 However, the effect of VPA on nuclear transport of ataxin‐3 in a SCA3 cell model has never been tested. Divalproex sodium (or valproate semisodium, a mixture of VPA and sodium valproate) is a member of an aliphatic acids family of HDACi and is FDA‐approved for the treatment of bipolar disorders, epilepsy, and migraine. Therefore, we decided to test whether divalproex sodium modulates the translocation of ataxin‐3 and rescues cytotoxicity induced by the nuclear transport of expanded ataxin‐3 in SCA3 cell models, providing novel clues for the potential mechanisms of HDAC inhibitors as a potential therapy for SCA3.
2. MATERIALS AND METHODS
2.1. Cell culture
Human Embryonic Kidney 293 (HEK293) cells were maintained as previously described.39 Chinese hamster ovary (CHO) cells were obtained from DiscoveRx (San Diego, CA, USA). Cells were cultured in F12 medium (Gibco, Life Technologies, Darmstadt, Germany) containing 10% fetal bovine serum (Gibco, Life Technologies), 1% penicillin/streptomycin (Gibco, Life Technologies), and 300 μg/mL Hygromycin B (Invitrogen, Carlsbad, CA, USA).
2.2. Cell transfection
For cell transfection, human ataxin‐3 was cloned into a pEGFP vector system. The constructs with three different CAG repeats stretches (15, 77, or 148 CAG) named as ataxin‐315CAG, ataxin‐377CAG, and ataxin‐3148CAG encode ataxin‐3 containing 15Q, 77Q, or 148Q, respectively. These constructs were linearized by the ApaI restriction enzyme for stable transfection. CHO cells were stably transfected using FuGENE 6 (Roche, Basel, Switzerland) according to the manufacturer's recommendation. Ataxin‐315CAG, ataxin‐377CAG, and ataxin‐3148CAG were used for transient transfection into HEK293 cells using the Attractene Transfection Reagent (QIAGEN, Hilden, Germany) according to the manufacturer's description.
2.3. Protein extraction
Subcellular fractionation was performed to separate the cytoplasmic and nuclear fractions. The cells were harvested, washed once with 5 mL PBS, and lysed in 100‐200 μL cold cytoplasmic buffer (10 mmol/L HEPES pH 7.5, 10 mmol/L KCl, 0,1 mmol/L EDTA, 0,1 mmol/L EGTA, 1 mmol/L DTT, 0.5% NP40, Complete Protease Inhibitor Cocktail [Roche]). The lysates were placed on ice for 20 minutes, vortexed twice (5 seconds each), and centrifuged at 18 928 g at 4°C for 5 minutes in a tabletop centrifuge. The supernatant was stored as the cytoplasmic fraction. The pellet was resuspended in 200 μL washing buffer (cytoplasmic buffer with final concentration of 1% NP‐40) and recentrifuged as before. The pellet was then again resuspended in 100 μL nuclear buffer (20 mmol/L HEPES pH 7.9, 400 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, Complete Protease Inhibitor Cocktail [Roche]), rotated for 30 minutes at 4°C, and centrifuged at 18 928 g for 20 minutes in a tabletop centrifuge. The supernatant was saved as the nuclear fraction.
For total protein extraction, cells were harvested, washed once with cold PBS, and centrifuged. The pellet was resuspended in 50 μL TNES (50 mmol/L Tris pH 7.5, 400 mmol/L NaCl, 0.5% Tween 20, Complete Protease Inhibitor Cocktail (Roche), incubated for 5 minutes on ice, and then centrifuged for 15 minutes at 4°C at 18 928 g in a tabletop centrifuge. The supernatant contained the total protein lysate.
2.4. SDS PAGE and Western blot
The protein concentration was measured using a Bradford assay40; 30 μg of protein samples and the protein marker (Precision Plus Protein Standards, Bio‐Rad Laboratories, Hercules, CA, USA) were loaded onto the gel (10%) and run as described previously (Schmidt et al11). Separated proteins were transferred onto nitrocellulose (NC) membrane (0.2 μm, GE Healthcare Life Sciences, Chicago, IL, USA). After transfer, the membrane was blocked in 1 × TBST (10 mmol/L Tris pH 7.5; 0.15 mol/L NaCl; 0.1% Tween 20) with 4% low fat dry milk (SlimFast, Allpharm Vertriebs GmbH) for 2 hours and incubated with the primary antibody diluted in 1 × TBST for 2 hours at room temperature or for overnight at 4°C. After washing three times with 1 × TBST, the blot was incubated with the secondary antibody diluted in 1 × TBST for 1 hour, washed again as before, detected with Amersham ECL Western Blotting Detection Kit (GE Healthcare Life Sciences), and processed using an Odyssey Fc system (LI‐COR Biotechnology, Lincoln, NE, USA).
2.5. Filter trap assay
Filter trap assays were used to detect the SDS‐resistant insoluble aggregations of mutant ataxin‐3.41, 42 Seventy‐two hours after transfection, HEK293 cells were harvested, centrifuged at 250 g for 10 minutes, and resuspended in 300 μL PBS with Complete Protease Inhibitor Cocktail (Roche). The lysates were sonicated for 1 minutes (30 seconds twice) for cell lysis and DNA fragmentation. A Bradford assay (see above) was performed to measure the protein concentration. The sample was then supplemented with 2% SDS for the filter trap assay; 100 μg of sample was loaded onto a cellulose acetate membrane (0.2 μm, GE Healthcare Life Sciences) on top of filter paper. The filter trap apparatus was run and washed twice with PBS. The membrane was then prepared for Western blot as described above.
2.6. Cell viability assay
The PrestoBlue Cell Viability Reagent (Life Technologies, Darmstadt, Germany) is a resazurin‐based reagent and was used to detect cell viability. Cells were plated at 5 × 104 cells per well of a 96 well plate. Cell viability was then assessed with the PrestoBlue Cell Viability Reagent after incubating for 10 minutes and measured by the fluorescent intensity using a Synergy HT microplate reader (BioTek, Bad Friedrichshall, Germany) (Excitation: 535 nm [25 nm bandwidth], Emission: 615 nm [10 nm bandwidth]).
2.7. Statistical analyses
Data from multiple experiments were expressed as means ± standard error (SEM). Statistical significance was analyzed by Student's t‐test for data with a normal distribution. The effect of multiple factors was determined by a two‐way analysis of variance (ANOVA) test. Significance levels were described as follows: P < 0.05*; P < 0.01**; P < 0.001***, except where noted.
3. RESULTS
3.1. Divalproex sodium increases the acetylation level of histone H3
A growing number of evidence shows that the acetylation level of histones is reduced in polyQ diseases.35, 43 To determine whether divalproex sodium which is a mixture of sodium valproate and valproic acid indeed leads to an inhibition of histone deacetylation, the effect of divalproex sodium on levels of histone acetylation was investigated. Pronouncedly increased levels of acetylated histone H3 were observed in HEK293 cells expressing ataxin‐315Q or ataxin‐377Q treated with divalproex sodium for 24 hours compared to a control group treated with DMSO (Figure 1), and an increased tendency was observed after treatment of 6 and 12 hours. Thereby, we conclude that divalproex sodium increases levels of acetylated histone H3.
Figure 1.
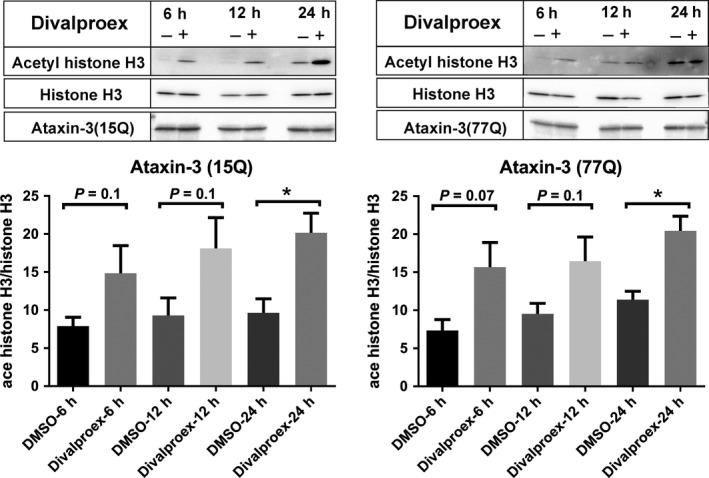
Divalproex sodium increases the level of acetylated histone H3 level in ataxin‐3‐transfected HEK293 cells. Normal (15CAG) or expanded (77CAG) ataxin‐3‐transfected cells were treated with divalproex sodium (Selleck chemical, 0.3 mmol/L in DMSO, (+)) or with DMSO alone (−) 48 h after transfection. The cells were treated with divalproex sodium or DMSO over 6, 12, and 24 h. The Western blot was probed with antiacetylated histone H3 (Abcam, Cambridge, UK), antihistone H3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and antiataxin‐3 (1H9, Merck Millipore, Darmstadt, Germany). Antibodies include antihistone H3 and antiataxin‐3 are used as controls. The x‐axis shows the different treatment group. The y‐axis represents the acetylated histone H3 values normalized to histone H3. Error bars represent the standard error of the mean. Data represent three independent experiments (n = 3). The data with a normal distribution were analyzed with Student's t‐test. *P < 0.05
3.2. Divalproex sodium does not alter soluble mutant ataxin‐3 protein level
After confirming the effect of divalproex sodium on the histone H3 acetylation level, we wanted to know whether divalproex sodium also affects the protein level of ataxin‐3. HEK293 cells expressing ataxin‐315Q or ataxin‐377Q were treated with divalproex sodium for 6, 12, and 24 hours and analyzed. Western blot analysis demonstrated that divalproex sodium does not change soluble mutant ataxin‐3 (ataxin‐377Q) and normal ataxin‐3 (ataxin‐315Q) (Figure 2).
Figure 2.
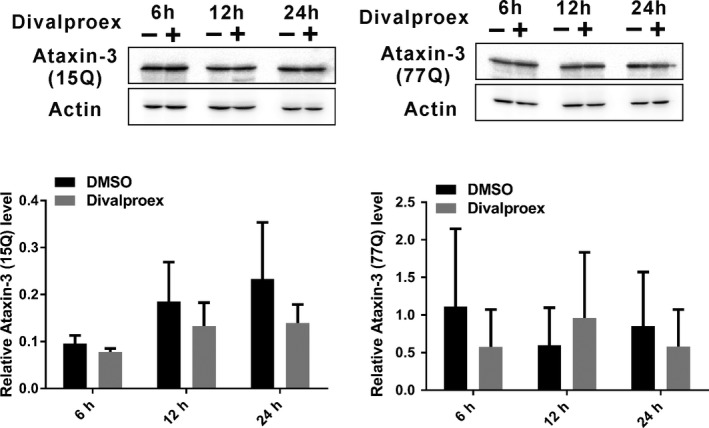
Divalproex sodium does not change the protein levels of ataxin‐3 in HEK293 cells. HEK293 cells were transfected with ataxin‐315 CAG or ataxin‐377 CAG. 48 h after transfection, cells were treated with divalproex sodium (at 0.3 mmol/L in DMSO for 6, 12, or 24 h; +) or DMSO alone as control (−). Western blots were probed with antiataxin‐3 (1H9, Merck Millipore) and antiactin (Sigma Aldrich, St. Louis, MO, USA) as a loading control. Error bars represent the standard error of the mean. Data represent three independent experiments (n = 3). The data in each time point with a normal distribution were analyzed with Student's t‐test
3.3. Divalproex sodium does not significantly affect aggregate formation of expanded ataxin‐3
As the level of expanded ataxin‐3 is not altered upon treatment with divalproex sodium, we evaluated whether protein aggregates formed by expanded ataxin‐3 is reduced after treatment with divalproex sodium. We therefore used HEK293 cells expressing ataxin‐3148Q and treated them with divalproex sodium and DMSO as a control. We applied a filter trap assay to quantify the level of protein aggregates (Figure 3). The filter trap assay revealed that divalproex sodium exhibits a negative effect on the aggregates in comparison with the DMSO‐treated group and with the untreated group.
Figure 3.
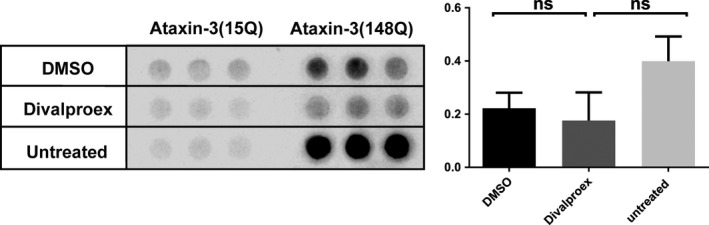
Divalproex sodium does not affect ataxin‐3148Q aggregation as measured by filter trap assay. Three days after transfection, HEK293 cells expressing ataxin‐3148Q or ataxin‐315Q were treated with DMSO alone as a control and divalproex sodium at 10 μmol/L in DMSO for 24 h. The untreated cells were used as blank control. Error bars represent standard error of the mean over four independent experiments (n = 4). Values with a normal distribution were assessed by Student's t‐test
3.4. Divalproex sodium alleviates heat‐shock‐induced nuclear localization of ataxin‐3
It is known that protein aggregates are easily formed when ataxin‐3 is localized in the nucleus.10, 11, 16, 17, 18, 19 Although divalproex sodium had no influence on the aggregation, we next explore whether it affects the nuclear localization of ataxin‐3 as the early pathological stage before aggregation forms. It has been shown that certain cellular stress such as heat shock promotes ataxin‐3′s translocation into the nucleus.44 Therefore, we asked whether this effect of cellular stress‐induced nuclear presence of ataxin‐3 can be directly alleviated using divalproex sodium. To do this, we stressed the cells by heat shock and investigated whether treatment with divalproex sodium would alleviate the nuclear localization of ataxin‐3. CHO cells expressing ataxin‐377Q were pretreated with divalproex sodium for 24 hours and then heat‐shocked for 1.5 hours. Fractionation data indicated that heat shock increased the nuclear abundance of ataxin‐3, whereas divalproex sodium alleviated the heat‐shock‐induced nuclear translocation of ataxin‐3 (Figure 4).
Figure 4.
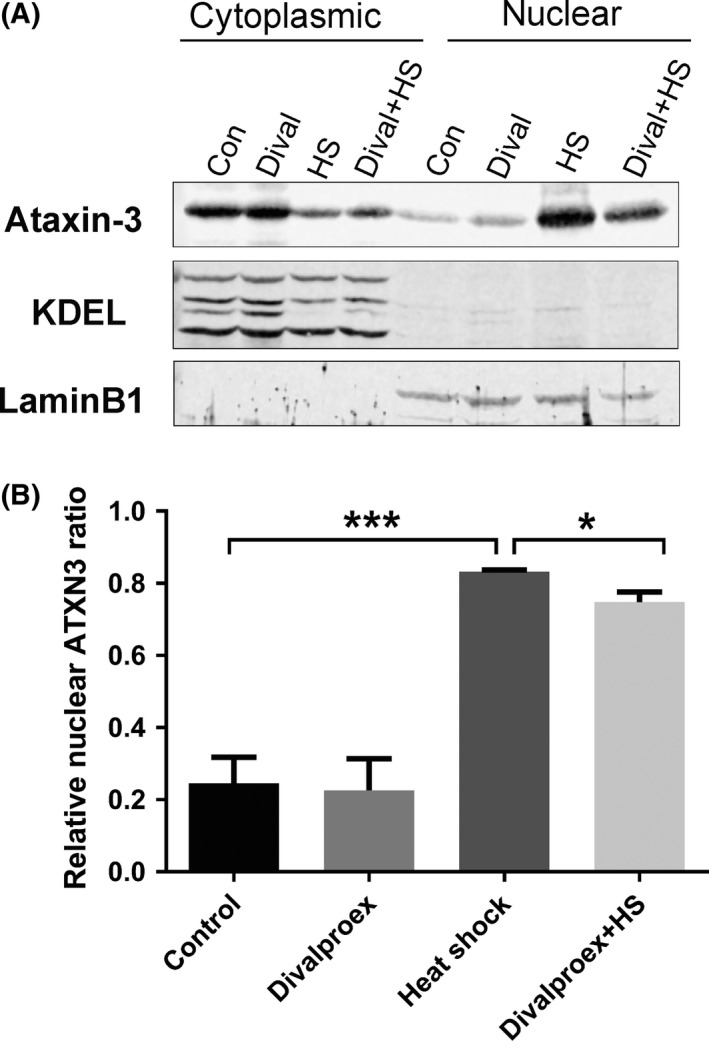
Divalproex sodium alleviates heat‐shock‐induced nuclear uptake of ataxin‐3. A, Western blot of the subcellular fraction of CHO cells expressing ataxin‐377Q was probed with antiataxin‐3 (1H9, Merck Millipore), anti‐KDEL (cytoplasmic marker, Enzo Life Sciences, Farmingdale, NY, USA), and antilamin B1 (nuclear marker, Santa Cruz Biotechnology). Four groups of cells with different treatments are shown. Those without heat shock were treated with either divalproex sodium (0.3 mmol/L, 24 h; Dival) or DMSO as a control (Con). The heat‐shocked cells were pretreated with either divalproex sodium (0.3 mmol/L, 24 h; Dival + HS) or DMSO as control before heat shock at 43°C for 1.5 h (HS). B, Quantification of Western blot data showed that heat shock induces a strong shift of ataxin‐3 from the cytoplasm to the nucleus and significantly reduced levels of nuclear ataxin‐3 in cells pretreated with divalproex sodium before heat shock compared with the control. Error bars represent standard error of the mean in four independent experiments (n = 4). Values with a normal distribution were assessed by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001
3.5. Divalproex sodium suppresses the cytotoxicity of expanded ataxin‐3
PolyQ proteins commonly show stronger toxicity in the nucleus than in the cytoplasm.19, 28, 45 If divalproex sodium prevents the stress‐induced nuclear entry of expanded ataxin‐3, we would expect an alleviated cellular viability of CHO cells expressing expanded ataxin‐3 (77Q) upon divalproex sodium treatment. To assess a rescuing effect of divalproex sodium on the cytotoxicity caused by expanded ataxin‐3, cell viability tests using PrestoBlue were performed. We observed that divalproex sodium successfully rescued the cytotoxicity induced by expanded ataxin‐3 (77Q) at concentrations ranging from 100 to 1000 μmol/L; divalproex sodium on its own at 1000 μmol/L showed weak toxicity to the cells (Figure 5). However, the extent of toxicity was still weaker than the effect of DMSO, suggesting that the toxicity was induced by high concentration of DMSO rather than divalproex sodium. In addition, the rescuing effect was observed in cells expressing ataxin‐377Q either at 2 hours or at 24 hours of treatment (Figure 5).
Figure 5.
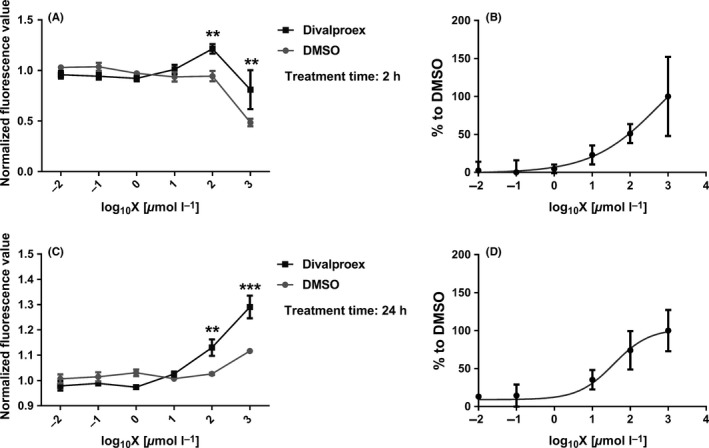
Divalproex sodium alleviates the cytotoxicity induced by expanded ataxin‐3 as assessed by cell viability assay using the PrestoBlue cell viability reagent (Life Technologies). CHO cells expressing ataxin‐377Q were seeded in a 96 well plate and treated with various concentrations of divalproex sodium for 2 h (A,B) and 24 h (C,D). The x‐axis shows the log10 of concentration of divalproex sodium. The y‐axes of the left diagrams (A,C) represent fluorescence values normalized to untreated cells. The diagrams on the right (B,D) show on the y‐axis values normalized to the DMSO‐treated group as sigmoidal curves. Error bars represent standard error of the mean in three independent experiments (n = 3). The data with a normal distribution were assessed by two‐way ANOVA. **P < 0.01, ***P < 0.001
4. DISCUSSION
Transcriptional disturbance has been suggested as a potential mechanism for the pathology of polyQ neurodegenerative diseases. Pathological polyQ protein or inclusions formed by expanded polyQ protein bind transcription factors, and these aberrant interactions cause transcriptional dysregulation, resulting in neuronal toxicity.46, 47, 48, 49 Moreover, histone acetylation and deacetylation, regulated by histone acetyltransferases and histone deacetylases, are responsible for the transcriptional regulation of eukaryotic cells. Histone acetylation activates transcription by facilitating more relaxed chromatin structure, and histone deacetylases suppress transcription by facilitating chromatin condensation.50, 51, 52, 53 A reduction of acetylated H3 and H4 induced by mutant Htt was observed in vitro and in vivo, reducing the histone acetyltransferases activity of the CREB‐binding protein (CBP).29, 31, 32 Therefore, HDAC inhibitors rescuing transcriptional dysfunction and rescuing the damage to histone acetylation homeostasis are proposed as a potential therapeutic strategy. In this study, we found that divalproex sodium increased the acetylation levels of histone H3. These results are consistent with previous studies and indicate that divalproex sodium prevents the cytotoxicity partly via rescuing hypoacetylation of histones in the polyQ diseases in combination with cell viability results.29, 35
Previous studies indicated that none of the HDAC inhibitors could prevent aggregations of mutant Htt.30, 31, 32 Similar observations were described for VPA tested in transgenic mouse models of SCA3.35 In agreement with this, our study also found that divalproex sodium at the concentration of 10 μmol/L and 0.3 mmol/L (data not shown) both did not inhibit aggregation formation of mutant ataxin‐3. Aggregations caused by the mutant ataxin‐3 are a hallmark of SCA3/MJD disease; however, the effect of aggregation has been reported to be dichotomous.54 Our results suggested they might mediate neuroprotective effects.
In addition, we evaluated the effect of divalproex sodium on the translocation of ataxin‐3 and observed that divalproex sodium decreases heat‐shock‐induced nuclear accumulation of ataxin‐3 as the early stage before aggregates forms. However, how does it achieve this effect? Proteins are transported between the cytoplasm and nucleus through the nuclear pore complex (NPC) which bridges the nuclear envelope, connecting the nucleus to the cytoplasm.55, 56 Only proteins less than around 40‐60 kDa in size can be shuttled into nucleus via passive diffusion.57, 58 Other large proteins must enter the nucleus through the NPC via an active transport system regulated by a signaling pathway.59 It is generally hypothesized that normal ataxin‐3, which is around 42 kDa, can pass through NPC by passive diffusion, whereas expanded ataxin‐3 is actively transported into the nucleus required for transport proteins. Up to this study, no import protein was reported to shuttle ataxin‐3 and only one export protein was indentified to modulate polyQ protein transport.60
Importins are a class of Karyopherins which modulate the nucleocytoplasmic transporting in eukaryotic cells through the NPC.57, 61, 62 Acetylases can acetylate histones and also target other proteins. CBP, one acetylase, acetylates the nuclear import proteins (importins) such as importin‐α2.63 HDACi, like TSA, could increase this acetylation process and also regulate the localization of importin‐α2.63, 64 These results indicate that nuclear import is likely to be regulated by acetylation and HDACi could modulate this process.
In our primary experiments (data not shown), over‐expressing an importin isoform was found to increase the nuclear amount of ataxin‐3, suggesting that it may be involved in the nuclear transport of ataxin‐3. In addition, over‐expressing the importin isoform also augmented the aggregation formation of mutant ataxin‐3. The divalproex sodium‐treated importin‐expressing cells showed less tendency to form aggregations in comparison with the importin isoform coexpressing cells and had the same level of aggregations as DMSO‐treated cells. These data indicate that this importin isoform may play a role in nuclear import of ataxin‐3 and divalproex sodium prevents it to perform this function likely via arresting the localization of the importin subunit and subsequently alters the transport of ataxin‐3. This suggests that divalproex sodium affects localization of ataxin‐3, as mediated by this importin isoform.
More importantly, nuclear polyQ proteins are more toxic than those in the cytoplasm. Cell viability data demonstrated that divalproex sodium increased cell viability, suggesting it could prevent the cytotoxicity caused by nuclear expanded ataxin‐3. Our finding supports that HDAC inhibitors reduce the polyQ toxicity28, 65, 66 and also demonstrates that this beneficial effect may result from the inhibition of nuclear accumulation of mutant polyQ, subsequently reduction of nuclear‐localized polyQ‐induced toxicity. In addition to mutant polyQ‐induced toxicity, another key mechanism of cell death in the polyQ diseases may be the cause of decreasing histone acetylation and HDACi can reduce this cytotoxicity by rescuing histone acetylation.28, 65
5. CONCLUSIONS
These results suggest divalproex sodium, as an HDAC inhibitor, not only rescues transcriptional dysfunction, but also affects nuclear transport. Therefore, our data provide evidence for the mechanism of divalproex sodium (a HDAC inhibitor) as a promising therapeutic strategy for SCA3 treatment via regulating the translocation of ataxin‐3 and preventing the cytotoxicity induced by expanded ataxin‐3.
CONFLICT OF INTEREST
The authors declares no conflict of interest.
ACKNOWLEDGMENT
This study was supported by the Chinese Scholarship Council (CSC) to Z.W. The Natural Science Basic Research Plan in Shaanxi Province of China (2017JQ8029), Scientific Technology Program & Innovation Fund of Xi'an City Special Project (2016CXWL04) and Social Development & Scientific Technology Project of Shaanxi Province (2016S‐079) supported this work. The authors also thank Ryan Price language edits.
Wang Z‐J, Hanet A, Weishäupl D, et al. Divalproex sodium modulates nuclear localization of ataxin‐3 and prevents cellular toxicity caused by expanded ataxin‐3. CNS Neurosci Ther. 2018;24:404–411. 10.1111/cns.12795
REFERENCES
- 1. Takiyama Y, Nishizawa M, Tanaka H, et al. The gene for Machado‐Joseph disease maps to human chromosome 14q. Nat Genet. 1993;4:300‐304. [DOI] [PubMed] [Google Scholar]
- 2. Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221‐226. [DOI] [PubMed] [Google Scholar]
- 3. Gispert S, Twells R, Orozco G, et al. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23‐24.1. Nat Genet. 1993;4:295‐299. [DOI] [PubMed] [Google Scholar]
- 4. Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A‐voltage‐dependent calcium channel. Nat Genet. 1997;15:62‐69. [DOI] [PubMed] [Google Scholar]
- 5. Michalík A, Del‐Favero J, Mauger C, Löfgren A, Van Broeckhoven C. Genomic organisation of the spinocerebellar ataxia type 7 (SCA7) gene responsible for autosomal dominant cerebellar ataxia with retinal degeneration. Hum Genet. 1999;105:410‐417. [DOI] [PubMed] [Google Scholar]
- 6. Koide R, Kobayashi S, Shimohata T, et al. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA‐binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047‐2053. [DOI] [PubMed] [Google Scholar]
- 7. Walker FO. Huntington's disease. Lancet. 2007;369:218‐228. [DOI] [PubMed] [Google Scholar]
- 8. Kanazawa I. Molecular pathology of dentatorubral‐pallidoluysian atrophy. Philos Trans R Soc Lond B Biol Sci. 1999;354:1069‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X‐linked spinal and bulbar muscular atrophy. Nature. 1991;352:77‐79. [DOI] [PubMed] [Google Scholar]
- 10. Paulson HL, Das SS, Crino PB, et al. Machado‐Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann Neurol. 1997;41:453‐462. [DOI] [PubMed] [Google Scholar]
- 11. Schmidt T, Landwehrmeyer GB, Schmitt I, et al. An isoform of ataxin‐3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998;8:669‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tait D, Riccio M, Sittler A, et al. Ataxin‐3 is transported into the nucleus and associates with the nuclear matrix. Hum Mol Genet. 1998;7:991‐997. [DOI] [PubMed] [Google Scholar]
- 13. Trottier Y, Cancel G, An‐Gourfinkel I, et al. Heterogeneous intracellular localization and expression of ataxin‐3. Neurobiol Dis. 1998;5:335‐347. [DOI] [PubMed] [Google Scholar]
- 14. Macedo‐Ribeiro S, Cortes L, Maciel P, Carvalho AL. Nucleocytoplasmic shuttling activity of ataxin‐3. PLoS ONE. 2009;4:e5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pozzi C, Valtorta M, Tedeschi G, et al. Study of subcellular localization and proteolysis of ataxin‐3. Neurobiol Dis. 2008;30:190‐200. [DOI] [PubMed] [Google Scholar]
- 16. Paulson HL, Perez MK, Trottier Y, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333‐344. [DOI] [PubMed] [Google Scholar]
- 17. Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291‐304. [DOI] [PubMed] [Google Scholar]
- 18. Sun J, Xu H, Negi S, Subramony SH, Hebert MD. Differential effects of polyglutamine proteins on nuclear organization and artificial reporter splicing. J Neurosci Res. 2007;85:2306‐2317. [DOI] [PubMed] [Google Scholar]
- 19. Bichelmeier U, Schmidt T, Hübener J, et al. Nuclear localization of ataxin‐3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J Neurosci. 2007;27:7418‐7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin XP, Feng L, Xie CG, Chen DB, Pei Z, Liang XL, et al. Valproic acid attenuates the suppression of acetyl histone H3 and CREB activity in an inducible cell model of Machado‐Joseph disease. Int J Dev Neurosci. 2014;38:17‐22. [DOI] [PubMed] [Google Scholar]
- 21. Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis. 2011;41:481‐488. [DOI] [PubMed] [Google Scholar]
- 22. Chou AH, Chen YL, Hu SH, Chang YM, Wang HL. Polyglutamine‐expanded ataxin‐3 impairs long‐term depression in Purkinje neurons of SCA3 transgenic mouse by inhibiting HAT and impairing histone acetylation. Brain Res. 2014;1583:220‐229. [DOI] [PubMed] [Google Scholar]
- 23. Lei LF, Yang GP, Wang JL, Chuang DM, Song WH, Tang BS, et al. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat Disord. 2016;26:55‐61. [DOI] [PubMed] [Google Scholar]
- 24. Esteves S, Duarte‐Silva S, Naia L, Neves‐Carvalho A, Teixeira‐Castro A, Rego AC, et al. Limited effect of chronic valproic acid treatment in a mouse model of machado‐joseph disease. PLoS ONE. 2015;10:e0141610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784‐796. [DOI] [PubMed] [Google Scholar]
- 26. Hahnen E, Hauke J, Tränkle C, Eyüpoglu IY, Wirth B, Blümcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs. 2008;17:169‐184. [DOI] [PubMed] [Google Scholar]
- 27. Ying M, Xu R, Wu X, et al. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. 2006;281:12580‐12586. [DOI] [PubMed] [Google Scholar]
- 28. McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci U S A. 2001;98:15179‐15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine‐dependent neurodegeneration in Drosophila. Nature. 2001;413:739‐743. [DOI] [PubMed] [Google Scholar]
- 30. Hockly E, Richon VM, Woodman B, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041‐2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418‐9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171‐82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556‐563. [DOI] [PubMed] [Google Scholar]
- 33. Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830‐2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mielcarek M, Benn CL, Franklin SA, et al. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease. PLoS ONE. 2011;6:e27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi J, Zhang L, Tang B, et al. Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS ONE. 2013;8:e54792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kautu BB, Carrasquilla A, Hicks ML, Caldwell KA, Caldwell GA. Valproic acid ameliorates C. elegans dopaminergic neurodegeneration with implications for ERK‐MAPK signaling. Neurosci Lett. 2013;541:116‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evason K, Collins JJ, Huang C, Hughes S, Kornfeld K. Valproic acid extends Caenorhabditis elegans lifespan. Aging Cell. 2008;7:305‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Antony PM, Mäntele S, Mollenkopf P, et al. Identification and functional dissection of localization signals within ataxin‐3. Neurobiol Dis. 2009;36:280‐292. [DOI] [PubMed] [Google Scholar]
- 40. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem. 1976;72:248‐254. [DOI] [PubMed] [Google Scholar]
- 41. Scherzinger E, Lurz R, Turmaine M, et al. Huntingtin‐encoded polyglutamine expansions form amyloid‐like protein aggregates in vitro and in vivo. Cell. 1997;90:549‐558. [DOI] [PubMed] [Google Scholar]
- 42. Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid‐like polyglutamine‐containing protein aggregates. Methods Enzymol. 1999;309:375‐386. [DOI] [PubMed] [Google Scholar]
- 43. Minamiyama M, Katsuno M, Adachi H, et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183‐1192. [DOI] [PubMed] [Google Scholar]
- 44. Reina CP, Zhong X, Pittman RN. Proteotoxic stress increases nuclear localization of ataxin‐3. Hum Mol Genet. 2010;19:235‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klement IA, Skinner PJ, Kaytor MD, et al. Ataxin‐1 nuclear localization and aggregation: role in polyglutamine‐induced disease in SCA1 transgenic mice. Cell. 1998;95:41‐53. [DOI] [PubMed] [Google Scholar]
- 46. Steffan JS, Kazantsev A, Spasic‐Boskovic O, et al. The Huntington's disease protein interacts with p53 and CREB‐binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763‐6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wood JD, Nucifora FC Jr, Duan K, et al. Atrophin‐1, the dentato‐rubral and pallido‐luysian atrophy gene product, interacts with ETO/MTG8 in the nuclear matrix and represses transcription. J Cell Biol. 2000;150:939‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li F, Macfarlan T, Pittman RN, Chakravarti D. Ataxin‐3 is a histone‐binding protein with two independent transcriptional corepressor activities. J Biol Chem. 2002;277:45004‐45012. [DOI] [PubMed] [Google Scholar]
- 49. Evert BO, Araujo J, Vieira‐Saecker AM, et al. Ataxin‐3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J Neurosci. 2006;26:11474‐11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Norton VG, Marvin KW, Yau P, Bradbury EM. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J Biol Chem. 1990;265:19848‐19852. [PubMed] [Google Scholar]
- 51. Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73‐84. [DOI] [PubMed] [Google Scholar]
- 52. Hebbes TR, Thorne AW, Crane‐Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988;7:1395‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A. 1996;93:14503‐14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Teixeira‐Castro A, Jalles A, Esteves S, et al. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado‐Joseph disease. Brain. 2015;138:3221‐3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fabre E, Hurt E. Yeast genetics to dissect the nuclear pore complex and nucleocytoplasmic trafficking. Annu Rev Genet. 1997;31:277‐313. [DOI] [PubMed] [Google Scholar]
- 56. Feldherr CM, Akin D. EM visualization of nucleocytoplasmic transport processes. Electron Microsc Rev. 1990;3:73‐86. [DOI] [PubMed] [Google Scholar]
- 57. Davis LI. The nuclear pore complex. Annu Rev Biochem. 1995;64:865‐896. [DOI] [PubMed] [Google Scholar]
- 58. Panté N, Aebi U. Molecular dissection of the nuclear pore complex. Crit Rev Biochem Mol Biol. 1996;31:153‐199. [DOI] [PubMed] [Google Scholar]
- 59. Schwamborn K, Albig W, Doenecke D. The histone H1(0) contains multiple sequence elements for nuclear targeting. Exp Cell Res. 1998;244:206‐217. [DOI] [PubMed] [Google Scholar]
- 60. Chan WM, Tsoi H, Wu CC, et al. Expanded polyglutamine domain possesses nuclear export activity which modulates subcellular localization and toxicity of polyQ disease protein via exportin‐1. Hum Mol Genet. 2011;20:1738‐1750. [DOI] [PubMed] [Google Scholar]
- 61. Stoffler D, Fahrenkrog B, Aebi U. The nuclear pore complex: from molecular architecture to functional dynamics. Curr Opin Cell Biol. 1999;11:391‐401. [DOI] [PubMed] [Google Scholar]
- 62. Poon IK, Jans DA. Regulation of nuclear transport: central role in development and transformation? Traffic. 2005;6:173‐186. [DOI] [PubMed] [Google Scholar]
- 63. Bannister AJ, Miska EA, Görlich D, Kouzarides T. Acetylation of importin‐alpha nuclear import factors by CBP/p300. Curr Biol. 2000;10:467‐470. [DOI] [PubMed] [Google Scholar]
- 64. Ryan CM, Harries JC, Kindle KB, Collins HM, Heery DM. Functional interaction of CREB binding protein (CBP) with nuclear transport proteins and modulation by HDAC inhibitors. Cell Cycle. 2006;5:2146‐2152. [DOI] [PubMed] [Google Scholar]
- 65. Ghosh S, Feany MB. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum Mol Genet. 2004;13:2011‐2018. [DOI] [PubMed] [Google Scholar]
- 66. Marinova Z, Ren M, Wendland JR, et al. Valproic acid induces functional heat‐shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111:976‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]