

Summary
Background
Environmental enrichment (EE) has been shown to enhance cognitive function in mouse models of Alzheimer's disease (AD). Magnesium‐L‐threonate (MgT) is a compound with a newly discovered effect to rescue learning and memory function in aging and AD mice.
Aim
To study the additive therapeutic effect of EE combined with MgT (EM) and the potential mechanism underlying the effects.
Materials and Methods
APP/PS1 mice were treated with EE, MgT, or combination of EE and MgT (EM) and compared for restored memory function.
Results
EM was more effective in improving cognition and spatial memory than either treatment alone in either long‐term (12 months, started at 3 months old, which was before disease manifestation) or short‐term (3 months, started at 6 months old, which was after disease manifestation) treatment. The behavioral improvement has coincided with rescue of synaptic contacts in the hippocampal region of the AD mouse brain. Immunoblots also showed that EM but neither single treatment rescued the activity reduction in CaMKII and CREB, two important downstream molecules in the N‐methyl‐D‐aspartate receptor (NMDAR) pathway.
Conclusion
Environmental enrichment and MgT may synergistically improve recognition and spatial memory by reducing synaptic loss and restoring the NMDAR signaling pathway in AD mice, which suggests that combination of EE and MgT may be a novel therapeutic strategy for AD.
Keywords: Alzheimer's disease, APP/PS1 mouse, environmental enrichment, magnesium, N‐methyl‐D‐aspartate receptor signaling
1. INTRODUCTION
Alzheimer's disease (AD) is the most common form of dementia and currently lacks an effective cure. The most prominent symptom of AD is progressive memory loss. Hypotheses of pathology assume that AD is caused by deposition of extracellular amyloid‐β (Aβ) plaques or intracellular tangles composed of hyperphosphorylated microtubule‐associated protein, tau.1, 2 Hippocampal neurons are primarily affected due to the vulnerability of pyramidal cell synapses at the early stage of AD, which eventually leads to functional impairment in cognition and memory.3
Alzheimer's disease can induce dysfunction of various signaling pathways of the nervous system. Of these pathways, N‐methyl‐D‐aspartate receptor (NMDAR) signaling has been extensively studied.4 The NMDAR is an important calcium channel and a glutamate receptor located in the membrane of both somas and dendrites.5 NMDAR‐mediated signaling pathways are closely related to the pathology of AD. Several downstream signaling molecules in this pathway, such as CREB and CaMKII, are essential for synaptic plasticity and memory formation.6, 7 Disturbance of intracellular calcium homeostasis and CaMKII/CREB activation are associated with AD. Reduction in phosphorylated CREB has been observed in postmortem brains of patients with AD8 and AD mouse models.9 In addition, dysregulation of CaMKII has been found in dendritic arbors in AD.6 On the other hand, spatial training increases phosphorylation of CaMKII in the hippocampus and rescues deficits in contextual memory formation of Tg2576 mice.10 Bilateral infusion of CREB into dorsal hippocampus CA1 region of TgCRND8, one kind of the AD model mice, was sufficient to restore the hippocampal‐dependent spatial memory of these animals.11
Magnesium cation (Mg2+) is vital for many physiological processes. Mental health depends on normal Mg2+ intake, absorption, and metabolism. Hypomagnesium was found in many brain regions of patients with AD when compared with age‐matched controls.12 Compared with other Mg2+ compounds (eg, Mg2+ chloride, Mg2+ citrate, and Mg2+ gluconate), dietary intake magnesium‐L‐threonate (MgT) could significantly increase Mg2+ levels in the brain and improves cognitive abilities of aged animals.13 MgT has also been shown to exert beneficial cognitive effects in genetically targeted mouse models of AD.14, 15 Moreover, elevation of Mg2+ in the hippocampus can enhance phosphorylation of CaMKII and CREB and activate their downstream pathways.13
Although intensifies surrounding physical and social stimulation of the nervous system,environmental enrichment (EE) can improve physical and psychological well‐being, refine physiological responses, and induce synaptic growth in laboratory animals.16, 17 EE could increase expression of both presynaptic synaptophysin and postsynaptic PSD‐95 in rodent brains.18, 19 In the hippocampus of adult mouse, EE is also demonstrated to facilitate cell proliferation and neurogenesis of the dentate gyrus.20 Moreover, studies using various behavioral tests have revealed EE‐induced spatial memory improvement in mouse models of AD.21, 22, 23 In the hippocampus and cortex of rat brains, EE stimulation can lead to increased levels of brain‐derived neurotrophic factor (BDNF), which is an essential protein for central nervous system development and functions.24, 25 Expression of NMDAR and the downstream signaling proteins, such as CaMKII, CREB, and ARC, are induced by EE.26, 27, 28
There are no effective cures to AD currently. New insight and treatment strategies are in urgent need. Therefore, we sought to see whether combining EE with MgT, both of which have significant therapeutic effects on AD, would yield greater benefit than either used alone.
2. MATERIALS AND METHODS
2.1. Experimental animals
Transgenic heterozygous female mice (APPswe/PSEN1dE9)29 and wild‐type (WT) female mice were obtained from the Model Animal Research Center of Nanjing University. Mice were housed under 2 types of environmental conditions: EE or non‐EE conditions. Under EE conditions, mice were housed at 5‐6 animals per large cage (70 cm × 60 cm × 30 cm) with several running wheels, toys, and tunnels. Under non‐EE conditions, mice were housed individually in standard cages (30 cm × 12 cm × 12 cm) without enrichment objects. Animals were housed in a controlled environment (temperature = 21 ± 1°C, humidity = 50 ± 10%) under an inverted light cycle (light off 9:00 am to 9:00 pm). Behavior experiments were performed under dim red light. All animal experiments were approved by the Committees on Animal Care of Tsinghua University.
2.2. MgT treatment
Magnesium‐L‐threonate (Neurocentria, Inc, CA, USA) was administered via drinking water at a dose of 910 mg/kg/d for APP/PS1 mice (about 75 mg/kg/d elemental Mg2+). This is consistent with the effective dose described previously.14 For MgT‐treated mice, drinking water was switched to MgT‐containing water at 3 months of age (to evaluate long‐term therapeutic effects) or 6 months of age (to evaluate short‐term therapeutic effects) until the animals were killed. All mice were provided with standard food containing Mg2+ (0.15% elemental Mg2+).
2.3. Short‐term environmental stimulus
As shown in the previous study,14 for short‐term environmental stimuli, 18‐month‐old mice were exposed to a relatively new context before being killed. Briefly, all animals were transferred to new EE cages (5‐6 animals per cage) with new companion mice 24 hours before being killed. After 21 hours, toys and running wheels were placed in the cages. Three hours later, animals were killed. It is important to note that for each mouse raised under EE conditions, the cages and EE stimuli were novel, and the companion animals were introduced from other cages (either housed individually or housed under EE conditions in another cage).
2.4. Novel object recognition test
The apparatus consisted of a square box (60 cm × 60 cm × 30 cm) made of polyvinyl chloride with white walls and floor. Mice were handled by the experimenter for 5 minutes once a day for 1 week. One day before the experiment, each mouse received 2 sessions of habituation to the arena without any object (10 min/session). Four kinds of objects that were adopted for mice did not show natural preference among them. Each object has 4 identical copies. In the sample phase, mice were placed in the center of the box containing 2 identical objects. Each mouse was allowed to explore the box for 5 minutes and then returned to its home cage. After a short‐term (1 hours) or long‐term (24 hours) retention interval, the mouse was placed in the box for 5 minutes and exposed to a copy of former object in the sample phase and a novel object that the mouse had never seen before. During the trials, exploration of an object was defined as directing the nose to the object at a distance of less than 2 cm and/or touching it with the nose or forepaws. Data will not be adopted when a mouse spent less than 10 seconds on exploring objects in 1 test. Recognition ratio was calculated as a percentage ratio of time of each object over total time. The box and objects were cleaned with 20% ethanol between trials to avoid olfactory cues.
2.5. Barnes maze task
The Barnes maze consisted of a white circular table (92 cm in diameter) with 20 circular holes (5 cm in diameter) around its circumference. The circular platform was 150 cm in height from the floor. The table surface was brightly lit by overhead lighting, and air flow was provided by a set of fans that forced the mice to find shelter. Under one of the holes was an escape box that could be reached through the corresponding hole on the table. The maze was rotated daily, and the spatial location of the target remained unchanged with respect to distal visual cues of the room.
At the beginning of a trial, a mouse was placed at the center of the table in a black plastic can. The can was then raised out of sight of the mouse. The mouse was then free to explore the maze and find the hidden escape box. After each trial, the platform and escape box were cleaned with 20% ethanol. If a mouse did not find the escape box within 5 minutes, it was gently placed in the escape box by the experimenter. The test was performed 2 times per day for 6 consecutive days. The latencies for finding the escape box were recorded.30
2.6. Immunofluorescence
Synaptophysin immunoreactive presynaptic terminals density was quantified as described previously.14 In brief, 5 mice in each group were anesthetized with chloral hydrate through intraperitoneal injection (ip, 250 mg/kg) and perfused with phosphate‐buffered saline (PBS), followed by 4% paraformaldehyde. The dissected brains were postfixed in 4% paraformaldehyde for 24 hours and then transferred to 30% sucrose in PBS for another 48 hours. Each right hemisphere brain was cut into coronal sections (6 μm thick). After 10‐12 hours blocking (5% normal serum in PBS with 0.1% Triton X‐100, 4°C), tissue sections were incubated overnight at with the synaptophysin primary antibody (1:500, Millipore, Billerica, MA, USA. 4°C) in PBS with 3% goat serum. Then, the brain slices were washed in PBS and incubated overnight with CF488‐conjugated secondary antibody (CF TM488‐conjugated goat anti‐mouse IgG (H+L), 1:200, Biotium Inc., CA, USA. 4°C). Finally, sections were mounted with antifade mounting medium (Vectashield, Vector Laboratories). Stained brain sections were scanned by IX‐70, Olympus with a 60 × water immersion lens (N/A = 1.2, 3 × zoom), and generated an image with dimensions 78.6 μm × 78.6 μm. Serial z‐sectioning was performed (thickness of 0.6 μm), and the best 3 z‐sections (with highest number of puncta) were collected and merged into a single image. Therefore, the volume of brain tissue per image was ~78.6 × 78.6 × 1.8 × μm3. Three parallel sections of each mouse were performed scanning. The synapses number in the outer molecular layer of dentate gyrus (DG‐OML) was estimated using Image‐Pro Plus version 6.0 software (Media‐Cybernetics). Special filters to separate fluorescent puncta were applied and background levels were equalized.
2.7. Western blotting analysis
Bilateral hippocampal tissues from 18‐month‐old mice which began EE and/or MgT treatment at 6 months of age were homogenized in lysis buffer containing 50 mM Tris‐HCl (pH 8.0), 250 mM NaCl, 1% NP‐40, 0.5% deoxycholate, 0.1% SDS, protease inhibitors and phosphatase inhibitors (Roche Diagnostics). Equal amounts of protein were loaded on polyacrylamide gels and then transferred to PVDF membranes (Millipore). Membranes were blocked and then probed with primary antibodies against CREB (Ser133, 1:1000), pCREB (1:50), CaMKII (1:1000), pCaMKII (Thr286, 1:1000), and GAPDH (1:6000) (all from CST, rabbit host, Danvers, MA, USA) overnight at 4°C. The membranes were then incubated with HRP‐conjugated secondary antibody (ZSGB‐BIO, Beijing, China or CST, 1:10 000) at room temperature for 1 hours. Proteins on the PVDF membranes were visualized by ECL detection reagent and captured on autoradiography film in a dark room. The integrated OD was determined using Image‐Pro Plus software. GAPDH signals on the same membrane served as loading controls. Quantitative analysis was performed by experimenters who were blinded to the treatment and genotype of each group.
2.8. Statistics
Paired Student's t test was used to recognitive index of novel and familiar objects in NORT. Learning curves in the Barnes maze task were analyzed using two‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. Unpaired Student's t test was used to analyze whether STE could enhance CaMKII and CREB phosphorylation ratio in each group. One‐way ANOVA followed by LSD post hoc test for individual comparisons was used to analyze synaptophysin immunofluorescence result. P < 0.05 was considered statistically significant. All data were presented as mean ± SD.
3. RESULT
3.1. Long‐term combination therapy with EE and MgT (EM) ameliorates memory deficit of AD mice
To investigate whether MgT could enhance effects of EE, APP/PS1 mice were exposed to EE (AE) or combination with EE and MgT (EM) treatment at 3 months of age. The NORT and Barnes maze were assessed at 15 and 17 months of age, respectively (Figure 1A).
Figure 1.
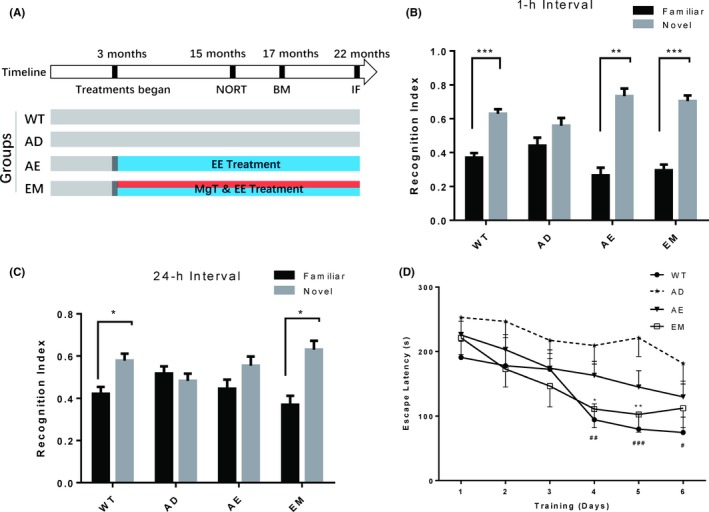
MgT‐enhanced therapeutic effects of EE in preventing memory impairment of AD mice. A, Schematic diagram of the experimental design. Four groups of mice were used: WT (n = 10), AD (n = 11), AE (n = 10), and EM (n = 9). EE and/or MgT treatment started at the age of 3 mos. B, Short‐term (1 h) and C, long‐term (24 h) NORT performed when the mice were 15 mos old. Recognition index was calculated as the percentage of time spent exploring each object. Black bars and gray bars indicate familiar and novel objects, respectively. Paired t tests analysis of percentage of time spent on novel and familiar object, * P < 0.05, ** P < 0.01, *** P < 0.001. Data are expressed as means ± SEM. D, Escape latencies during training in the Barnes maze performed on the same groups at 17 mos of age for 6 consecutive days. Two‐way ANOVA analysis of effects of various treatments, F(3, 216) = 13.4, P < 0.0001. Multiple comparisons: * denotes significant differences between the EM and AD groups (*P < 0.05, **P < 0.01, ***P < 0.001), and # denotes significant differences between the WT and AD groups (#P < 0.05, ##P < 0.01). Data are expressed as means ± SEM
In both short‐term and long‐term NORT, EM mice exhibited a significant preference for the novel object, and this preference was comparable to that of WT mice (paired t test P values: WT, P = 0.0008, EM, P = 0.0003 in 1‐hours interval test; WT, P = 0.0405, EM, P = 0.0153 in 24‐hours interval test, n = 9‐10, novel vs familiar object; Figure 1B,C). In contrast, APP/PS1 transgenic mice (AD) did not spend more time exploring the new object in either short‐ or long‐term NORT experiments (paired t test P values: P = 0.2327 in 1‐hours interval test, P = 0.6224 in 24‐hours interval test, n = 10, novel vs familiar object; Figure 1B, C). EE alone could rescue recognition memory in the short‐term NORT of AD mice, but it had no effect on the long‐term paradigm (paired t test P values: P = 0.0012 in 1‐hours interval test, P = 0.2496 in 24‐hours interval test, n = 10, novel vs familiar object; Figure 1B,C). This result indicates that EM is more potent in ameliorating AD‐related recognition memory impairment of AD mice.
Next, we tested the mice in the Barnes maze to investigate effects of the above treatments on spatial memory deficits. Overall, WT, AE, and EM mice found the hidden escape box faster than AD mice (treatment interaction: F(3, 216) = 13.4, P < 0.0001, Figure 1D), especially on training days 4‐6 of WT and EM groups (Multiple comparisons with AD: WT, P = 0.0057, AE, P = 0.5017, EM, P = 0.0269, in day 4; WT, P = 0.0005, AE, P = 0.1004, EM, P = 0.0055, in day 5, WT, P = 0.0115, AE, P = 0.3622, EM, P = 0.1639, in day 6). Notably, EM and WT groups showed better spatial memory than the AE group. These results suggest that spatial memory impairment in AD mice could be restored by EM treatment and that the preventive effect is more efficient than EE treatment alone.
3.2. Long‐term combination therapy with EE and MgT (EM) prevents synaptic loss in AD mice
A main characteristic of AD is progressive synaptic transmission dysfunction that eventually leads to synaptic loss.3 Thus, we investigated whether changes in synapse number were associated with different treatments. Synaptophysin is a presynaptic marker that is commonly used to determine the number of synapses. In DG region of AD mouse hippocampi, synaptophysin‐positive puncta were remarkably reduced. EE treatment could completely prevent this loss, with puncta at normal levels (ANOVA effect of treatment, F(3, 16) = 11.74, P = 0.0003; Multiple comparisons with AD: WT, P = 0.0057, AE, P = 0.01, EM, P = 0.0001). Interestingly, compared to WT or AE mice, EM mice have shown higher synaptic densities in the outer molecular layer of dentate gyrus (DG‐OML), which indicates that MgT has an additive effect on synaptic restoration in AD mice when coupled with EE(compared with EM, WT, P = 0.0471, AE, P = 0.0279 Figure 2A, B).
Figure 2.
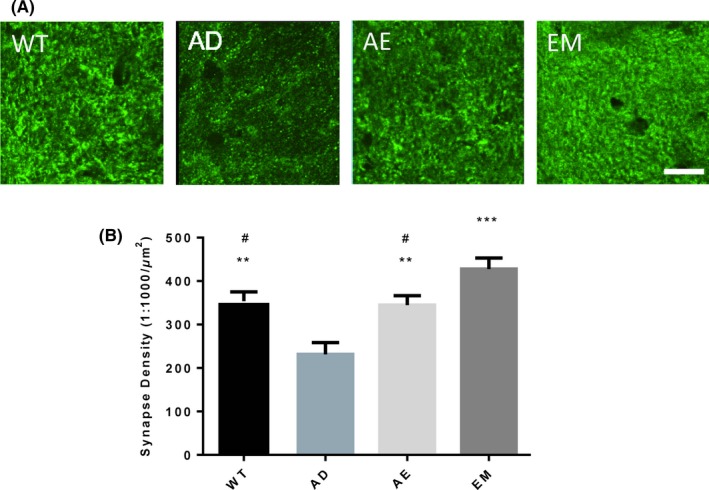
EE‐MgT (EM) prevented synaptic loss in the hippocampi of AD mice. Mice were sacrificed for immunofluorescence when they were 22 mos old. A, Representative fluorescence images of synaptophysin‐positive puncta in the outer molecular layer of the dentate gyrus (DG). B, Quantitative analysis of synaptophysin puncta, WT, AD, AE, and EM, n = 5/group. One‐way ANOVA analysis of effects of various treatments: F = 11.74, P = 0.003; Multiple comparisons: **P < 0.01, ***P < 0.0001, compared to AD; # P < 0.05, compared to EM. Data are expressed as means ± SEM. The scale bar represents 10 μm
3.3. Short‐term EE‐MgT (EM) treatment reverses learning and memory deficits in AD mice
Having demonstrated that long‐term (over 12 months) EM treatment which began at 3 months of age was the most effective therapy for AD model mice, we assessed the therapeutic effects of this combination after the onset of AD for a relatively short term (3 months) in another group of mice. In this experiment, AD mice received treatments at 6 months of age when memory deficits had already occurred as reported previously.31 To clarify the effects of each treatment and their potential synergistic effects, in addition to the AD, WT, AE, and EM groups defined in the last experiment, we added a group of mice receiving MgT alone (AM) (Figure 3A).
Figure 3.
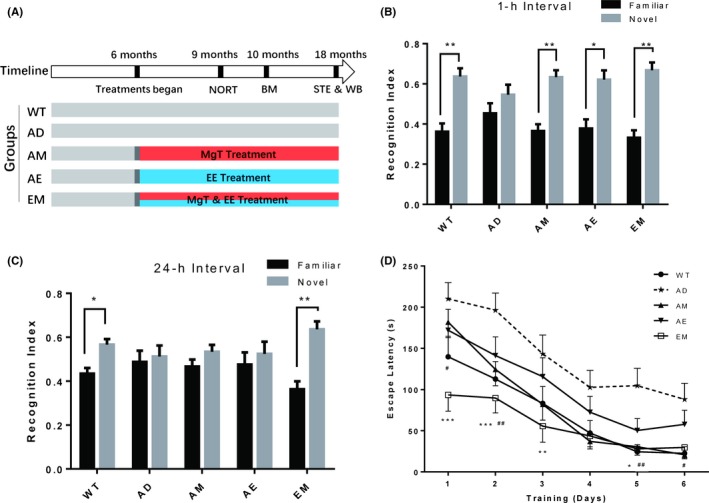
Short‐term EE‐MgT (EM) treatment reversed recognition and spatial memory impairment in AD mice. A, Schematic diagram of the experimental design. EE and/or MgT treatment started at 6 mos of age. Five groups of mice were used: WT (n = 11), AD (n = 16), AM (n = 14), AE (n = 13), and EM (n = 12). Behavioral testing was performed when the mice were 9 to 10 mos old. B, Short‐term (1 h) NORT was performed when the mice were 9 mos old. C, Same as B, and from the same groups of mice but representing long‐term (24 h) NORT performance. Black bars and gray bars indicate familiar and novel objects, respectively. Recognition index was calculated as percentage of time spent exploring each object. Paired t tests analysis of percentage of time spent on novel and familiar object, * P < 0.05, ** P < 0.01. D, Latency to find the hidden escape box during Barnes maze training trials. Two‐way ANOVA analysis of the effects of various treatments, F(4, 366) = 18.14, P < 0.0001,* denotes significant differences between the WT and AD (*P < 0.05, **P < 0.01, ***P < 0.001), and # denotes significant differences between the EM and AD (#P < 0.05, ##P < 0.01). Data are expressed as means ± SEM
After 3 months of short‐term treatment (compared with the previous 12‐month long‐term treatment), we performed the behavioral experiments. In the short‐term interval (1 hours) NORT experiment, AD mice exhibited clear recognition deficits (novel vs familiar object: P = 0.3825, n = 16 Figure 3B). In contrast, mice from the AM, AE, and EM groups performed quite similar to WT mice in which they spent more time exploring the novel object (novel vs familiar object: WT, P = 0.008, n = 11; AM, P = 0.003, n = 14; AE, P = 0.024, n = 13; EM, P = 0.0012, n = 12; Figure 3B). We then increased the difficulty of the test by extending the forgetting interval to 24 hours. As expected, the AM and AE mice did not show a preference for the novel object (novel vs familiar object: AD, P = 0.818, n = 16; AM, P = 0.318, n = 14; AE, P = 0.6675, n = 13; Figure 3C), while the AEM mice still spent more time on the new object as WT mice did (novel vs familiar object: EM, P = 0.0037, n = 12; WT, P = 0.0294, n = 11 Figure 3C).
In the Barnes maze test, our results showed that AM, AE, and EM mice spent less time finding the escape box compared with AD mice throughout the 6 test days (treatment interaction: F(4, 366) = 18.41, P < 0.0001). It is interesting that EM mice exhibited a shorter latency to locate the escape box from the first day of the test, and this latency decreased more rapidly than AE, AM (Multiple comparisons: AE, P = 0.0139, AM, P = 0.0036, in day 1 compared to EM), which indicates that EM mice may have a greater ability in learning process. In the last 3 days, the latency of AM and EM mice became comparable to that of WT mice (Multiple comparisons: AM, P = 0.9876, EM, P = 0.9998, in day 4; AM, P = 0.9981, EM, P = 0.9998, in day 5; AE, P = 0.9999, EM, P = 0.9970, in day 6; compared to WT. Figure 3D).
Thus, the combined treatment produced significant advantages over MgT or EE monotherapy to rescue cognitive memory and spatial memory in APP/PS1 mice, even when the treatment started when memory deficits had already occurred and a relatively shorter treatment period was used.
3.4. The combination therapy (EM) activates the NMDAR pathway
Environmental enrichment and drug treatment can change the response of the central nervous system to external stimuli by adjusting protein expression levels and modifications.32 The NMDAR signaling pathway has been shown to have a crucial role in cognition. Environmental stimulation and MgT improved recognition of WT animals through activation of the NMDAR signaling pathway.14
To explore further the underlying mechanism of behavioral improvement achieved by EM treatment, we compared activities of key proteins in the NMDAR pathway. To this end, each group of mice was randomly divided into 2 subgroups at the age of 18 months (Figure 3A). One subgroup was used to collect baseline phosphorylation levels, while the other subgroup was exposed to short‐term environmental stimulation (STE) and then analyzed for phosphorylation levels.14
In WT mice, STE significantly elevated phosphorylation levels of CREB, but it had no effect on CaMKII phosphorylation (baseline vs STE, CaMKII, P = 0.4286; CREB, P = 0.0173; Figure 4A). Similar results were obtained in the AM group (baseline vs STE, CaMKII, P = 0.556; CREB, P = 0.0159; Figure 4C). In AD mice, STE did not enhance phosphorylation of either CaMKII or CREB (baseline vs STE, CaMKII, P = 0.5368; CREB, P = 0.7922; Figure 4B). It is interesting that although EE could reverse memory impairment of AD mice, STE did not stimulate phosphorylation of either CaMKII or CREB (baseline vs STE, CaMKII, P = 0.329; CREB, P = 0.4286; Figure 4D), which indicates that other pathways may be involved. It is noteworthy that phosphorylation levels of CREB and CaMKII in mice treated with EM were significantly increased by STE (baseline vs STE, CaMKII, P = 0.0079; CREB, 0.0079; Figure 4E). The above results suggest that although both MgT and EE can partially improve recognition and memory of AD mice, the internal mechanism could be diverse. Nonetheless, synergetic effects of MgT and EE, especially in CaMKII activation, were observed in mice treated with both MgT and EE.
Figure 4.
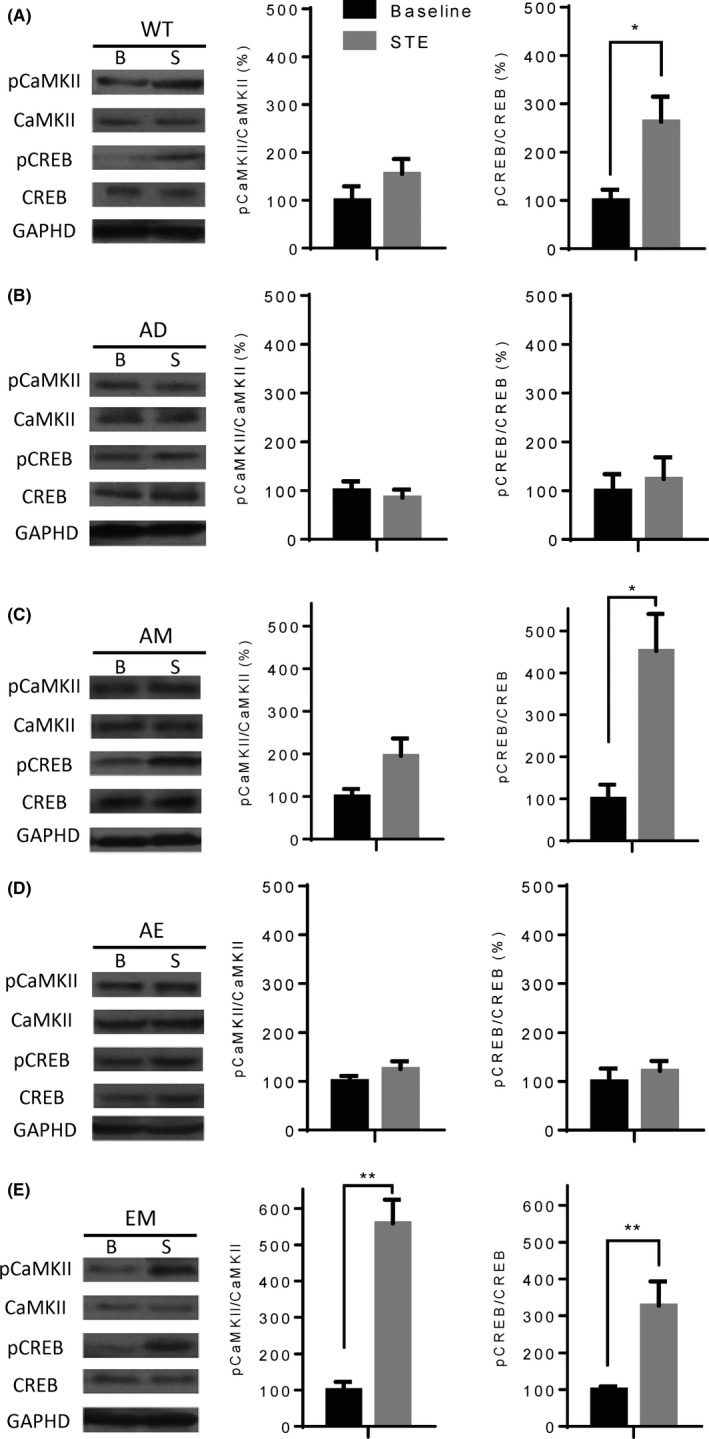
EE‐MgT (EM)‐enhanced STE‐induced CaMKII and CREB activation in the hippocampus of AD mice. A, Left: Representative Western blots showing phosphorylation and expression levels of CaMKII and CREB in the hippocampi of WT mice either after home cage basal conditions (B) or after 24 h of STE (S). GAPDH expression levels served as loading controls. Right: Quantitative analysis of pCaMKII/CaMKII and pCREB/CREB ratios. B, C, D, E, Same A, but representing AD, AM, AE, and EM mice, respectively. Two‐tailed unpaired t tests, *P < 0.05, **P < 0.01, baseline vs STE. Mice used under basal conditions: WT (n = 5), AD (n = 5), AM (n = 6), AE (n = 5), and EM (n = 5); Mice used in STE: WT (n = 6), AD (n = 6), AM (n = 5), AE (n = 6), and EM (n = 5). Data are expressed as means ± SEM
4. DISCUSSION
Environmental enrichment has been demonstrated to slow memory loss associated with AD.33 Human APP and PS1 transgenic mice are often studied as high‐frequency mutations have been found in these 2 genes in early‐stage or familial patients with AD.34 Using various behavioral tests, such as a water maze, Barnes maze, and NORT, EE was proven effective in rescuing brain functions of AD model animals.21, 23, 35 In addition, EE‐treated AD11 mice, a mouse model of sporadic AD, could perform comparably to WT mice in behavioral tests.36 In our study, without MgT, EE along could rescue memory deficit of APP/PS1 mice in 1‐hours interval test of NORT, whereas did not show obvious effect when the interval extended to 24 hours. The results indicate that magnesium can further enhance the beneficial effects of EE.
Magnesium plays a critical role in the nervous system and the level was significantly lower in the brains of patients with AD than in age‐matched controls.37 It was found that elevating Mg2+ levels in the brain can improve the memory of both young and aged rats.13 Shortly thereafter, it was found that MgT significantly improved spatial memory and recognition in 16‐month‐old APPswe/PS1 mice.14 Consistent with former results, we also found that mice fed MgT demonstrated better performance in both NORT and Barnes maze tests than untreated AD mice.
Paradoxically, magnesium is an NMDAR channel blocker, which means high Mg2+ level may downregulate NMDAR‐dependent signaling, and therefore, induce learning and memory impairment. Interestingly, it was observed that in physiological condition, slight and transient elevation of Mg2+ concentration (from 0.8 mM as basal to 1.2 mM within 1 hours) in the culture medium of hippocampal neurons would result in significantly decreased of the amplitudes of NMDA currents of resting membrane potential. However, when the elevation of Mg2+ lasted enough long time (neurons were grown under 1.2 mM), the recorded NMDA currents of resting membrane potential almost identical to the basal condition. Meanwhile, the amount of NMDA receptors located in the membrane increased, and the positive membrane potential displayed with much higher amplitude. As a result, this chronical elevation of Mg2+ enhanced the NMDAR‐dependent signaling.38
Memantine is in a novel class of AD medications that act by blocking NMDA receptors.39 Combination of memantine and EE exhibited higher efficacy than either single treatment in improving the spatial memory of SAMP8 mice and simultaneously in reducing expression of APP and formation of NTF in the hippocampus.40 However, memantine is limited in its recommendation for patients who fail other therapeutic options due to severe side effects, especially before the onset of this disease.41 In contrast, appropriate elevation of Mg2+ in the brain has not shown any adverse effects,13, 14 this advantage may possibly due to both Mg2+ and threonate are normally found in the brain.42
Synaptic loss is the pathological hallmark of cognitive dysfunction in AD. Among all cortical areas analyzed, the hippocampus appears to be the most severely affected region due to loss of synaptic proteins.43 Synaptophysin is a calcium‐binding protein located in vesicle membranes of presynaptic neurons.44 Previous studies have shown a positive correlation between the number of synaptophysin puncta and synaptic density.45 Thus, this protein is often used as a marker for the number of synapses. In our experiment, synaptophysin immunoreactivity in AD mice decreased about 35% in the DG region of the hippocampus compared with WT controls. Consistent with previous studies, EE treatment alone could prevent synaptic loss in the DG region.46 It is even more remarkable that the number of synaptophysin‐positive puncta of mice treated with EM was greater than that of WT or AE mice. This result indicates that in addition to alleviating AD‐induced synaptic loss, treatment combining EE and MgT may contribute to a delay in brain aging.
CaMKII and CREB are important downstream molecules in the NMDAR signaling pathway which involved in memory formation and long‐term potentiation (LTP).47, 48 Both of these 2 molecules are directly linked to AD.49, 50 Therefore, we examined whether activation of CaMKII and CREB could be affected by STE, which is a paradigm to evoke physiological neuronal input.14 STE could induce a much higher level of CaMKII phosphorylation than basal controls in mice treated with EM. In contrast, even though mice from the WT and AM groups showed superior performance in recognition and memory than AD mice, the ratio of p‐CaMKII/CaMKII did not change. This suggests that CaMKII activity may not be involved in the mechanism of MgT‐induced memory restoration.
CREB phosphorylation is a crucial step in initiation of learning and memory‐related gene transcription. Phosphorylation of CREB increased significantly in the hippocampi of WT mice exposed to EE but not in APPswe/PS1 mice.50 Unsurprisingly, our WT mice showed greater CREB activation after STE stimulation, whereas AD mice remain less sensitive. In our experiment, STE significantly induced activation of CREB but not CaMKII in the hippocampi of WT‐ and EE‐treated mice. This result may explain that compared with CREB, activation of CaMKII is more vulnerable to brain changes associated with aging, and exposure to EE alone could not rescue its activity in aged AD mice. However, EM significantly increased the activity of CaMKII, which suggests that EE and MgT function synergistically in NMDAR signaling and neuronal circuits.
Our results have shown that 3 months of EE treatment may restore performance of AD mice in the NORT and Barnes maze test without affecting CaMKII or CREB phosphorylation levels. In fact, a series of studies using multiple methods have suggested that rescuing memory function is related to modulation of expression or activity of CaMKII and/or CREB.27, 50, 51, 52 However, in addition to NMDAR signaling, several other pathways and molecules participate in EE‐induced biochemical changes. Hence, it is possible that memory improved by EE may exploit other pathways such as enhancing BDNF‐TrkB signaling,53 reducing expression and penetration of proinflammatory molecules,54 or enhancing mitochondrial functions for better energy supply.55 Therefore, more research is required to elucidate the precise mechanism underlying EE‐dependent cognitive improvement.
In conclusion, the present study suggests that MgT can amplify therapeutic effects of EE treatment in preventing and reversing neuronal deficits in AD mice. In addition, compared with EE or MgT alone, EM can facilitate phosphorylation of CaMKII and CREB after STE. Thus, our data suggest that combination of EE and MgT may be a novel therapeutic strategy for AD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENT
We express our sincere thanks to Guosong Liu for providing MgT and offering critical ideas. We gratefully acknowledge Wei Guo, David Grace for comments and discussions on the manuscript. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. This work was supported by the National Basic Research Program of China (973 program, Grant 2009CB941303), and National High Technology Research and Development Program (863 program, Grant 2007AA02Z443).
Huang Y, Huang X, Zhang L, et al. Magnesium boosts the memory restorative effect of environmental enrichment in Alzheimer's disease mice. CNS Neurosci Ther. 2018;24:70–79. 10.1111/cns.12775
The first two authors contributed equally to this work.
REFERENCES
- 1. Sinha S, Anderson JP, Barbour R, et al. Purification and cloning of amyloid precursor protein beta‐secretase from human brain. Nature. 1999;402:537‐540. [DOI] [PubMed] [Google Scholar]
- 2. Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell‐surface receptor. Nature. 1987;325:733‐736. [DOI] [PubMed] [Google Scholar]
- 3. Lu B, Nagappan G, Nathan PJ, Blin O. Synaptic function as a preclinical and experimental medicine readout for disease‐modifying therapy in Alzheimer's Disease. Drug Discov Today Ther Strateg. 2013;10:e99‐e104. [Google Scholar]
- 4. Danysz W, Parsons CG. Alzheimer's disease, beta‐amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br J Pharmacol. 2012;167:324‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer's disease. Neurol Sci. 2016;37:1039‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ly PT, Song W. Loss of activated CaMKII at the synapse underlies Alzheimer's disease memory loss. J Neurochem. 2011;119:673‐675. [DOI] [PubMed] [Google Scholar]
- 7. Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges. Curr Drug Targets CNS Neurol Disord. 2005;4:481‐497. [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto‐Sasaki M, Ozawa H, Saito T, Rosler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300‐303. [DOI] [PubMed] [Google Scholar]
- 9. Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta‐amyloid activates the mitogen‐activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125‐4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang X, Chai GS, Wang ZH, et al. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci Rep. 2015;5:9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yiu AP, Rashid AJ, Josselyn SA. Increasing CREB function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2011;36:2169‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andrasi E, Pali N, Molnar Z, Kosel S. Brain aluminum, magnesium and phosphorus contents of control and Alzheimer‐diseased patients. J Alzheimers Dis. 2005;7:273‐284. [DOI] [PubMed] [Google Scholar]
- 13. Slutsky I, Abumaria N, Wu LJ, et al. Enhancement of learning and memory by elevating brain magnesium. Neuron. 2010;65:165‐177. [DOI] [PubMed] [Google Scholar]
- 14. Li W, Yu J, Liu Y, et al. Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer's disease mouse model. Mol Brain. 2014;7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu X, Guan PP, Guo JW, et al. By suppressing the expression of anterior pharynx‐defective‐1alpha and ‐1beta and inhibiting the aggregation of beta‐amyloid protein, magnesium ions inhibit the cognitive decline of amyloid precursor protein/presenilin 1 transgenic mice. FASEB J. 2015;29:5044‐5058. [DOI] [PubMed] [Google Scholar]
- 16. Redolat R, Mesa‐Gresa P. Potential benefits and limitations of enriched environments and cognitive activity on age‐related behavioural decline. Curr Top Behav Neurosci. 2012;10:293‐316. [DOI] [PubMed] [Google Scholar]
- 17. Rosenzweig MR, Bennett EL. Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav Brain Res. 1996;78:57‐65. [DOI] [PubMed] [Google Scholar]
- 18. Nithianantharajah J, Levis H, Murphy M. Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD‐95 proteins. Neurobiol Learn Mem. 2004;81:200‐210. [DOI] [PubMed] [Google Scholar]
- 19. Lambert TJ, Fernandez SM, Frick KM. Different types of environmental enrichment have discrepant effects on spatial memory and synaptophysin levels in female mice. Neurobiol Learn Mem. 2005;83:206‐216. [DOI] [PubMed] [Google Scholar]
- 20. van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266‐270. [DOI] [PubMed] [Google Scholar]
- 21. Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H. Environmental enrichment improves cognition in aged Alzheimer's transgenic mice despite stable beta‐amyloid deposition. NeuroReport. 2004;15:1751‐1754. [DOI] [PubMed] [Google Scholar]
- 22. Costa DA, Cracchiolo JR, Bachstetter AD, et al. Enrichment improves cognition in AD mice by amyloid‐related and unrelated mechanisms. Neurobiol Aging. 2007;28:831‐844. [DOI] [PubMed] [Google Scholar]
- 23. Jankowsky JL, Melnikova T, Fadale DJ, et al. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:5217‐5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N. Increased expression of brain‐derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett. 1992;138:153‐156. [DOI] [PubMed] [Google Scholar]
- 25. Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague‐Dawley rats in vivo. Neuroscience. 2004;124:71‐79. [DOI] [PubMed] [Google Scholar]
- 26. Huang FL, Huang KP, Wu J, Boucheron C. Environmental enrichment enhances neurogranin expression and hippocampal learning and memory but fails to rescue the impairments of neurogranin null mutant mice. J Neurosci. 2006;26:6230‐6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pinaud R, Penner MR, Robertson HA, Currie RW. Upregulation of the immediate early gene arc in the brains of rats exposed to environmental enrichment: implications for molecular plasticity. Brain Res Mol Brain Res. 2001;91:50‐56. [DOI] [PubMed] [Google Scholar]
- 28. Tang YP, Wang H, Feng R, Kyin M, Tsien JZ. Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology. 2001;41:779‐790. [DOI] [PubMed] [Google Scholar]
- 29. Borchelt DR, Ratovitski T, Van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939‐945. [DOI] [PubMed] [Google Scholar]
- 30. Harrison FE, Reiserer RS, Tomarken AJ, McDonald MP. Spatial and nonspatial escape strategies in the Barnes maze. Learn Mem. 2006;13:809‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herring A, Lewejohann L, Panzer AL, et al. Preventive and therapeutic types of environmental enrichment counteract beta amyloid pathology by different molecular mechanisms. Neurobiol Dis. 2011;42:530‐538. [DOI] [PubMed] [Google Scholar]
- 33. Fischer A. Environmental enrichment as a method to improve cognitive function. What can we learn from animal models? NeuroImage. 2016;131:42‐47. [DOI] [PubMed] [Google Scholar]
- 34. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pardon MC, Sarmad S, Rattray I, et al. Repeated novel cage exposure‐induced improvement of early Alzheimer's‐like cognitive and amyloid changes in TASTPM mice is unrelated to changes in brain endocannabinoids levels. Neurobiol Aging. 2009;30:1099‐1113. [DOI] [PubMed] [Google Scholar]
- 36. Berardi N, Braschi C, Capsoni S, Cattaneo A, Maffei L. Environmental enrichment delays the onset of memory deficits and reduces neuropathological hallmarks in a mouse model of Alzheimer‐like neurodegeneration. J Alzheimers Dis. 2007;11:359‐370. [DOI] [PubMed] [Google Scholar]
- 37. Andrasi E, Igaz S, Molnar Z, Mako S. Disturbances of magnesium concentrations in various brain areas in Alzheimer's disease. Magnes Res. 2000;13:189‐196. [PubMed] [Google Scholar]
- 38. Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44:835‐849. [DOI] [PubMed] [Google Scholar]
- 39. Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system–too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699‐723. [DOI] [PubMed] [Google Scholar]
- 40. Dong J, Zhou M, Wu X, Du M, Wang X. Memantine combined with environmental enrichment improves spatial memory and alleviates Alzheimer's disease‐like pathology in senescence‐accelerated prone‐8 (SAMP8) mice. J Biomed Res. 2012;26:439‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang J, Jiang H. Efficacy and adverse effects of memantine treatment for Alzheimer's disease from randomized controlled trials. Neurol Sci. 2015;36:1633‐1641. [DOI] [PubMed] [Google Scholar]
- 42. Sun Q, Weinger JG, Mao F, Liu G. Regulation of structural and functional synapse density by L‐threonate through modulation of intraneuronal magnesium concentration. Neuropharmacology. 2016;108:426‐439. [DOI] [PubMed] [Google Scholar]
- 43. Flood DG, Coleman PD. Hippocampal plasticity in normal aging and decreased plasticity in Alzheimer's disease. Prog Brain Res. 1990;83:435‐443. [DOI] [PubMed] [Google Scholar]
- 44. Thiel G. Synapsin I, synapsin II, and synaptophysin: marker proteins of synaptic vesicles. Brain Pathol. 1993;3:87‐95. [DOI] [PubMed] [Google Scholar]
- 45. Mucke L, Masliah E, Yu GQ, et al. High‐level neuronal expression of abeta 1‐42 in wild‐type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050‐4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nithianantharajah J, Hannan AJ. Enriched environments, experience‐dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697‐709. [DOI] [PubMed] [Google Scholar]
- 47. Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium‐calmodulin kinase II in LTP and learning. Science. 1998;279:870‐873. [DOI] [PubMed] [Google Scholar]
- 48. Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long‐term potentiation in alpha‐calcium‐calmodulin kinase II mutant mice. Science. 1992;257:201‐206. [DOI] [PubMed] [Google Scholar]
- 49. Scott Bitner R. Cyclic AMP response element‐binding protein (CREB) phosphorylation: a mechanistic marker in the development of memory enhancing Alzheimer's disease therapeutics. Biochem Pharmacol. 2012;83:705‐714. [DOI] [PubMed] [Google Scholar]
- 50. Hu YS, Long N, Pigino G, Brady ST, Lazarov O. Molecular mechanisms of environmental enrichment: impairments in Akt/GSK3beta, neurotrophin‐3 and CREB signaling. PLoS One. 2013;8:e64460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saura CA, Valero J. The role of CREB signaling in Alzheimer's disease and other cognitive disorders. Rev Neurosci. 2011;22:153‐169. [DOI] [PubMed] [Google Scholar]
- 52. Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81‐89. [DOI] [PubMed] [Google Scholar]
- 53. Rossi C, Angelucci A, Costantin L, et al. Brain‐derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur J Neurosci. 2006;24:1850‐1856. [DOI] [PubMed] [Google Scholar]
- 54. Briones TL, Woods J, Rogozinska M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol Commun. 2013;1:57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Zigmond MJ, Smeyne RJ. Exercise: is it a neuroprotective and if so, how does it work? Parkinsonism Relat Disord. 2014;20(Suppl 1):S123‐S127. [DOI] [PubMed] [Google Scholar]