

Summary
Aims
Depression is currently the most common mood disorder. Regulation of intracellular cyclic adenosine monophosphate (cAMP) and/or cyclic guanosine monophosphate (cGMP) signaling by phosphodiesterase (PDE) inhibition has been paid much attention for treatment of depression. This study aimed to investigate the neuroprotective effects of Hcyb1, a novel PDE2 inhibitor, in HT‐22 cells and antidepressant‐like effects in mouse models of depression.
Methods
Hcyb1 was synthesized and its selectivity upon PDE2 was tested. Moreover, HT‐22 hippocampal cells were used to determine the effects of Hcyb1 on cell viability, cyclic nucleotide levels, and the downstream molecules related to cAMP/cGMP signaling by neurochemical, enzyme‐linked immunosorbent, and immunoblot assays in vitro. The antidepressant‐like effects of Hcyb1 were also determined in the forced swimming and tail suspension tests in mice.
Results
Hcyb1 had a highly selective inhibition of PDE2A (IC 50 = 0.57 ± 0.03 μmol/L) and over 250‐fold selectivity against other recombinant PDE family members. Hcyb1 at concentrations of 10−10 and 10−9 mol/L significantly increased cell viability after treatment for 24 hours. At concentrations of 10−9~10−7 mol/L, Hcyb1 also increased cGMP levels by 1.7~2.3 folds after 10‐minute treatment. Furthermore, Hcyb1 at the concentrations of 10−9 mol/L increased both cGMP and cAMP levels 24 hours after treatment. The levels of phosphorylation of CREB and BDNF were also increased by Hcyb1 treatment in HT‐22 cells for 24 hours. Finally, in the in vivo tests, Hcyb1 (0.5, 1, and 2 mg/kg, i.g.) decreased the immobility time in both forced swimming and tail suspension tests, without altering locomotor activity.
Conclusion
These results suggest that the novel PDE2 inhibitor Hcyb1 produced neuroprotective and antidepressant‐like effects most likely mediated by cAMP/cGMP‐CREB‐BDNF signaling.
Keywords: antidepressant, cell viability, cyclic nucleotide, forced swim test, Hcyb1, phosphodiesterase 2(PDE2) inhibitor, tail suspension test
1. INTRODUCTION
Depression is currently the most common mood disorder with the incidence of 18%‐20% in the population. It is ranked as the second leading cause of disability worldwide and the eleventh leading cause of global disease burden.1 The currently available treatments for depression are primarily drugs reducing noradrenergic and serotonergic reuptake, inhibiting monoamine oxidase, and ameliorating inflammation.2, 3 However, despite the variety of therapeutics, approximately one‐third of the patients respond to the first treatment, and remaining up to one‐third never respond to antidepressants, which are considered as treatment resistant.4, 5 In addition, many of the current antidepressants frequently produce undesirable side effects, while the action mechanism has not been fully elucidated. Therefore, it is important and necessary to develop novel antidepressants for achieving desired therapeutic efficacy.
Phosphodiesterases (PDEs), a class of enzymes that mainly catalyze the hydrolysis of the secondary messengers, cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate (cGMP), have gained attention as potential pharmacological targets for neuropsychiatric diseases.6, 7 Currently, mammalian PDEs comprise at least 11 gene families. Among them, PDE2 is a dual substrate enzyme which is highly expressed in various brain regions, including the cerebral cortex, hippocampus, amygdale, and striatum, implicating that PDE2 may play a critical role in emotion, perception, concentration, and cognition.8, 9, 10 Inhibition of PDE2 activity in the brain can lead to increased intracellular cAMP and cGMP and their downstream protein kinases, which activate transcription factors, ultimately regulate the expression of specific genes that are involved in the expression of depressive symptoms or symptom relief.11, 12 The main evidence has been generated from studies using Bay 60‐7550, the most widely characterized selective PDE2A inhibitor, which not only increases cGMP in neuronal cultures, but also produces antidepressant‐like effects in the mouse model of depression.13, 14, 15 However, Bay 60‐7550 also potently inhibits PDE1 and has poor penetration into the brain, leading to the limit use of this drug simply in basic research, rather than clinical studies.16, 17 During the last several years, many other potent PDE2A inhibitors with various degrees of selectivity have been developed as indicated by patent applications published from January 2010 to February 2016.9 However, the early clinical trials of these PDE2 inhibitors were terminated, and the reasons were unclear.9 Therefore, further studies are urgent to develop novel PDE2 inhibitors to meet clinical needs.
In this study, a novel and highly selective PDE2 inhibitor Hcyb1 was first synthesized and chosen to determine its neuroprotective profile and the related mechanism. Moreover, the antidepressant‐like effects of Hcyb1 were investigated in mouse model of despair tests, that is, forced swimming and tail suspension tests.
2. MATERIALS AND METHODS
2.1. Synthesis of Hcyb1
2.1.1. Ethyl ‐N‐(2‐amino‐1, 2‐dicyanovinyl) formimidate (3)
2‐3‐diamino‐2‐butanedinitrile (3.0 g, 27.8 mmol) and triethyl orthoformate (4.5 g, 30.5 mmol) were refluxed in 1, 4‐dioxane (22 mL) for 0.5 hour. The mixture is poured into water. After cooling to ambient temperature, the solid was filtered off and dried to afford ethyl ‐N‐(2‐amino‐1, 2‐dicyanovinyl) formimidate (3.8 g, 83%) [Mp: 134.3‐135.4°C, ESI MS: 165 [M + H] + 1, H NMR (400 MHz, DMSO) δ 7.95 (s, 1H), 6.99 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H)].
2.1.2. 5‐Amino‐1‐(1‐(naphthalen‐1‐yl) methyl)‐1H‐imidazole‐4‐carbonitrile (5)
Ethyl‐N‐(2‐amino‐1, 2‐dicyanovinyl) formimidate (2.0 g, 12 mmol) and 1‐(1‐naphthyl) ethylamine (2.18 g. 15 mmol) were suspended in EtOH (20 mL). The mixture was stirred at 30°C overnight. Next morning, the 1 mol/L KOH (30 mL) solution was added and stirred for 1 hour. The mixture is poured into water, and the solid was filtered off and dried to afford 5‐amino‐1‐(1‐(naphthalen‐1‐yl) methyl)‐1H‐imidazole‐4‐carbonitrile (2.98 g, 93%) [Mp: 193‐194°C, ESI MS: 263 [M + H] + 1, 1H NMR (400 MHz, DMSO) δ 7.96 (dd, J = 17.7, 7.6 Hz, 3H), 7.57 (dd, J = 15.8, 6.4 Hz, 3H), 7.36 (d, J = 7.1 Hz, 1H), 7.05 (s, 1H), 6.35 (s, 2H), 6.14 (d, J = 6.7 Hz, 1H), 1.83 (d, J = 6.7 Hz, 3H).].
2.1.3. 5‐Amino‐1‐(1‐(naphthalen‐1‐yl) ethyl)‐1H‐imidazole‐4‐carboxamide (6)
5‐amino‐1‐(1‐(naphthalen‐1‐yl) methyl)‐1H‐imidazole‐4‐carbonitrile (2.87 g, 11.9 mmol) was suspended in a mixture of EtOH (19 mL) and NH4OH (17 mL). Slowly drop the 50% H2O2 (6 mL) at 30°C for 6 hours. After washing with water, the solid was dried to afford 5‐amino‐1‐(1‐(naphthalen‐1‐yl) ethyl)‐1H‐imidazole‐4‐carboxamide (1.6 g, 57%) [ESI MS: 281 [M + H] + 1, 1H NMR (400 MHz, DMSO) δ 8.05 (d, J = 8.1 Hz, 1H), 7.96 (dd, J = 21.7, 7.8 Hz, 2H), 7.56 (dt, J = 21.1, 7.2 Hz, 4H), 7.35 (d, J = 7.2 Hz, 1H), 6.99 (s, 1H), 6.72 (d, J = 57.0 Hz, 2H), 6.13 (d, J = 6.8 Hz, 1H), 5.89 (s, 2H), 1.85 (d, J = 6.7 Hz, 3H).].
2.1.4. 2‐Benzyl‐9‐(1‐(naphthalen‐1‐yl) ethyl)‐1H‐purin‐6(9H)‐one (Hcyb1)
Of 55% NaH (3.29 g, 75.5 mmol) was added to the water‐removed 1,4‐dioxane (20 mL). After vigorously stirring at 30°C, 5‐amino‐1‐(1‐(naphthalen‐1‐yl)ethyl)‐1H‐imidazole‐4‐carboxamide (1.3 g, 5.04 mmol) and phenyl ethyl acetate (4.13 g, 25.2 mmol) were added overnight. Next morning, slowly drop ethanol in the mixture until no bubbles are produced. The crude mixture was concentrated in vacuo. The oil was stirred vigorously in a biphasic mixture of water (200 mL) and EtOAc (300 mL) at 60°C for 0.5 hour. After cooling to ambient temperature, the organic phase was concentrated in vacuo after drying over MgSO4. The residual solid was recrystallized from EtOAc to afford the target molecule as a white solid (0.46 g, 27%) [Mp: 239‐241°C, ESI MS: 381 [M + H] + 1. 1H NMR (300 MHz, DMSO) δ 12.43 (s, 1H), 8.23 (d, J = 8.3 Hz, 1H), 8.18 (s, 1H), 7.94 (dd, J = 14.6, 7.8 Hz, 2H), 7.60 (d, J = 6.3 Hz, 1H), 7.56‐7.39 (m, 3H), 7.35‐7.19 (m, 5H), 6.59 (q, J = 7.1 Hz, 1H), 5.76 (s, 1H), 3.91 (d, J = 14.3 Hz, 2H), 2.00 (d, J = 7.1 Hz, 3H).].
2.2. PDE2 selectivity assay
The enzymatic activities of PDEs were assayed using 3H‐cAMP and 3H‐cGMP as substrates. PDEs were firstly incubated with the reaction mixture of 50 mmol/L Tris‐HCl, pH 7.8, 10 mmol/L MgCl2, and 3H‐cAMP or 3H‐cGMP (40 000 cpm/assay) at 24°C for 30 minutes. Then, the reaction was terminated by addition of ZnSO4 and Ba(OH)2. The reaction product 3H‐AMP or 3H‐GMP was precipitated by BaSO4, while unreacted 3H‐cAMP or 3H‐cGMP remained in the supernatant. Radioactivity in the supernatant was measured by liquid scintillation. For the potent inhibitors, a double or triple measurement was performed, while a single measurement was carried out for the poor inhibitors.
2.3. Drugs preparation and treatments
For experiments on cell cultures, Hcyb1 and Bay 60‐7550 were dissolved in 100% dimethyl sulfoxide (DMSO) at a concentration of 10−2 mol/L and then diluted into culture media. The final concentration of DMSO did not exceed 0.1% of the total volume.
For animal behavioral tests, Hcyb1 and Bay 60‐7550 were dissolved in saline containing 5% DMSO and 5% solutol; the positive control drug diazepam was dissolved simply in saline. Mice were given via gavage (for Hcyb1, i.g.) or intraperitoneal (i.p.) injections of Bay 60‐7550 and/or diazepam, in a volume of 10 mL/kg body weight.
2.4. Cell viability assay
HT‐22 cells, purchased from the American Type Culture Collection (ATCC), were routinely cultured in a mixture of Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) under a humidified atmosphere of 5% CO2‐95% atmosphere at 37°C. Cells were plated onto 96‐well plates at 1 × 105 cells/well density and treated with Hcyb1 at different concentrations and incubation time. Following the indicated treatments, cell viability was assessed using the MTS method according to a previous description.18 MTS solution (120 μL, MTS: DMEM = 1:5) was added into each well, and cells were incubated at 37°C for additional 2 hours. Absorbance at 490 nm was measured using a microplate reader (BioTek, USA).
2.5. Cyclic AMP and GMP assays
Cells were rinsed after treatment with various concentrations of Hcyb1 or Bay 60‐7550 for 24 hours and then incubated with 0.1 mol/L HCl containing 0.5% Triton X‐100 for 10 minutes at room temperature to lyse the cells. Samples were then scraped and centrifuged at 800 g for 15 minutes to remove cellular debris. The supernatants were then collected for immediate assay or stored frozen for assay later using cAMP or cGMP ELISA kits (Enzo Life Sciences, Farmingdale, NY). Absorbance at 405 nm was measured using a microplate reader (BioTek, USA).
A separate set of HT‐22 cells was plated onto 6‐well plates at 1 × 105 cells/well density. On the day of treatment, the cells were rinsed and then incubated for 10 minutes at 37°C with phosphate‐buffered saline (PBS) supplemented with various concentrations of Hcyb1 or Bay 60‐7550 in the presence of 20 μmol/L NMDA for another 15 minutes. After incubation, PBS containing the drugs was removed roughly, and 200 μL HCl (0.1 mol/L) was added to each well to lyse the cells. Cell lysates were collected and centrifuged at 800 g for 15 minutes to remove cellular debris. Supernatants were then collected for immediate assay or stored frozen for assay later using the cAMP or cGMP complete ELISA kit (Enzo Life Sciences, Farmingdale, NY). Absorbance at 405 nm was measured using a microplate reader (BioTek, USA).
2.6. Immunoblot analysis
HT‐22 cells were lysed with RIPA lysis buffer (Upstate Chemicon, USA) containing protease and phosphatase inhibitors (Pierce Biotechnology, USA) and centrifuged at 4000 g for 30 minutes at 4°C. The supernatant was assayed for total protein concentrations using the BCA assay kit (Thermo Scientific, USA). Samples were separated using 10% SDS‐PAGE, and the separated proteins were transferred onto polyvinylidene difluoride membranes. Blots were then incubated in blocking buffer (phosphate‐buffered saline containing 3% BSA and 0.1% sodium azide) for 2 hours at room temperature, washed in tris‐buffered saline with 0.1% Tween‐20 (TBST), and incubated at overnight 4°C with the appropriate primary antibodies, including antiphosphorylated cAMP response element‐binding protein (pCREB) at Ser 133 (1:1000), anti‐CREB (1:200), anti‐brain‐derived neurotrophic factor (BDNF) (1:200) (Santa Cruz, CA), and anti‐β‐actin (1:1000; Abcam, USA) antibodies. Following three washes with TBST, the blots were incubated with the secondary antibodies (goat anti‐mouse IgG or goat anti‐rabbit IgG, 1:10 000) for 1 hours at room temperature. The blots were washed again for three times by TBST buffer, and the immunoreactive bands were detected using the enhanced chemiluminescence (ECL) method. Densitometer readings were used to quantify the amount of protein in each treatment situation.
2.7. Animals and behavioral tests
Male imprinting control region (ICR) mice, weighing between 20 and 25 g, were obtained from the Animal Center at Changzhou University. Mice were housed in a temperature‐controlled room under standard laboratory conditions, with a 12 hours light/12 hours dark cycle. All animals were allowed at least 1 w habituation before experiment. Water and food were freely available in their home cages. The animal studies were carried out according to the National Institute of Health Guide for Care and Use of Laboratory Animals (1996) and were approved by the Institutional Animal Care and Use Committee of Changzhou University.
Tail suspension test (TST): Mice were individually suspended from a bar 50 cm above the floor by means of an adhesive tape for 6 minutes, placed approximately 1 cm from the tip of the tail, as described previously.19 The duration the mouse remained immobile during the last 4‐minute period of the test was recorded. Mice were only considered immobile when they hung passively and completely motionless.
Forced swim test (FST): Mice were individually placed in glass cylinders (25 cm height, 10 cm diameter) containing 10 cm depth of water at 24 ± 1°C for 6 minutes. A mouse was determined to be immobile when there were only small movements necessary to keep its head above water. The immobility duration was recorded during the last 4‐minute period of the test.20
Locomotor activity (LA) test: LA test was assessed using an open‐field chamber.21 The floor of the chamber was divided into sixteen identical squares. Mice were individually placed in the center of the chamber and allowed to acclimatize for 15 minutes. The locomotor counts of mice were measured by counting the number of line crossings (with all four paws crossed the line) in the following 10 minutes.
2.8. Statistical analysis
All data are expressed as the mean ± SEM and were analyzed statistically using one‐way analysis of variance (ANOVA), followed by post hoc Dunnett's tests. Statistical significance was set at P < 0.05.
3. RESULTS
3.1. Hcyb1 synthesis
The lead compound is synthesized according to Figure 1. Diaminomaleonitrile 1 was refluxed with triethyl orthoformate, in dioxane, to generate compound 2. This compound was combined with one equivalent of amine in ethanol, followed by treatment with an aqueous solution of potassium hydroxide to generate compound of structure 4. The remaining cyano group could be oxidized with 30% peroxide in aqueous ammonium hydroxide. Cyclization with esters proceeded in dioxane with sodium hydride as the base followed and heating to provide the desired pyrazolopyrimidines. The structure of compounds was confirmed by MS and NMR.
Figure 1.
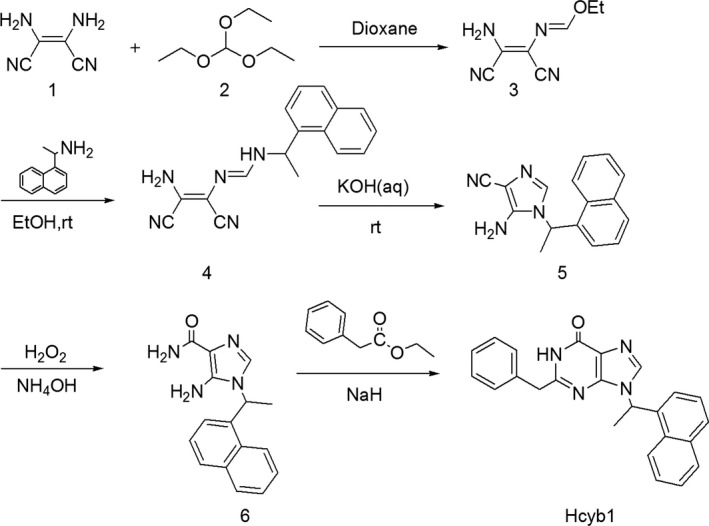
Synthesis and chemical structure of Hcyb1
3.2. PDE2 selectivity
Hcyb1 showed a high PDE2 selectivity with a IC50 value of 0.57 ± 0.03 μmol/L against full‐length recombinant hPDE2A and also displayed >250‐fold over PDE1B, PDE4D, PDE5A, and PDE10A, respectively, revealing that it was inactive against other PDE family members (Table 1).
Table 1.
IC50 values (μmol/L) of Hcyb1 on various PDEs
| Enzyme | IC50(μmol/L) |
|---|---|
| PDE2A | 0.57 ± 0.03 |
| PDE1B | >500 |
| PDE4D | >500 |
| PDE5A | >500 |
| PDE10A | >500 |
3.3. The concentration‐ and time‐dependent effects of Hcyb1 on cell viability in HT‐22 cells
To determine whether Hcyb1 could promote neurons proliferation, the protective effects of Hcyb1 at different concentrations and time points were determined. As shown in Figure 2A, the cell viability was significantly increased when treatment HT‐22 cells with Hcyb1 at concentrations of 10−10 mol/L and 10−9 mol/L for 24 hours (P < 0.05). The time‐dependent effects showed that the cell viability was significantly increased from 12 to 24 hours when treatment of Hcyb1 at concentration of 10−9 mol/L (Figure 2B, P < 0.05; P < 0.01). The maximal effects peaked at 24 hours after treatment.
Figure 2.
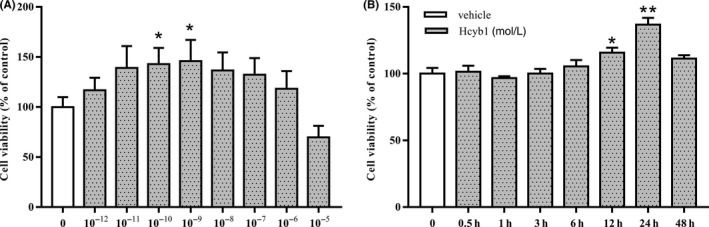
The concentration‐ and time‐dependent effects of Hcyb1 on cell viability in HT‐22 cells. (A) HT‐22 cells were treated with various concentrations of Hcyb1 for 24 hours. (B) HT‐22 cells were treated with 10−9 mol/L Hcyb1 for the indicated periods. Results are expressed as mean ± SEM (n = 6). *P < 0.05 and **P < 0.01 vs vehicle‐treated control group
3.4. The effects of Hcyb1 on cGMP and cAMP levels in HT‐22 cells
The effects of Hcyb1 on accumulation of cGMP and cAMP in HT‐22 cells are shown in Figure 3A,B. cGMP levels were increased significantly after treatment with Hcyb1 at concentrations of 10−9, 10−8, and 10−7 mol/L for 10 minutes (P's < 0.05; P < 0.01). However, cAMP levels did not show any significant changes even though there was a trend toward increase. These effects were similar to those of positive drug Bay 60‐7550 at concentration of 10−6 mol/L. Further study revealed the significant increases in both cGMP and cAMP levels when treating cells with 10−9 mol/L Hcyb1 for 24 hours (P's < 0.05) (Figure 4A,B), which suggest that PDE2 inhibitor prevented hydrolysis of both cGMP and cAMP in HT‐22 cells.
Figure 3.
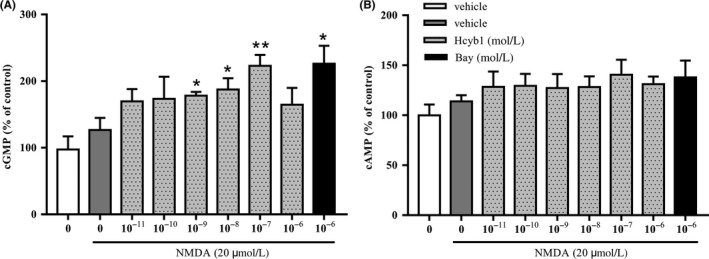
The effects of Hcyb1 on cGMP (A) and cAMP (B) levels in HT‐22 cells after treatment for 10 minutes. The HT‐22 cells were treated with various concentrations of Hcyb1 for 10 minutes and stimulated with NMDA (20 μmol/L) for measurable cGMP and cAMP signal. Results are expressed as mean ± SEM (n = 6). *P < 0.05 and **P < 0.01 vs vehicle‐treated NMDA‐only group
Figure 4.
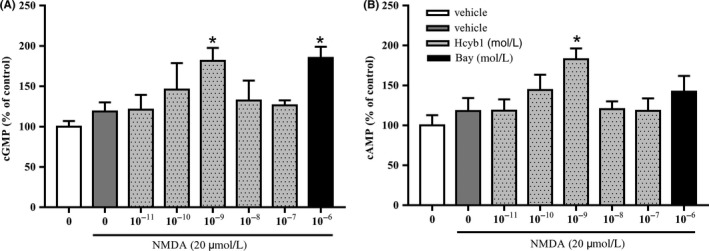
The effects of Hcyb1 on cGMP (A) and cAMP (B) levels in HT‐22 cells after treatment for 24 hours. The HT‐22 cells were treated with various concentrations of Hcyb1 for 24 hours and stimulated with NMDA (20 μmol/L) for measurable cGMP and cAMP signal. Results are expressed as mean ± SEM (n = 6). *P < 0.05 vs vehicle‐treated NMDA‐only group
3.5. The effects of Hcyb1 on pCREB/CREB and BDNF expression in HT‐22 cells
To determine whether PDE2 inhibition by Hcyb1 upregulated downstream CREB phosphorylation and BDNF expression, we analyzed the ratio of pCREB/CREB and BDNF levels 24 hours after treatment with Hcyb1. As shown in Figure 5, exposure of HT‐22 cells to 10−9 mol/L Hcyb1 for 24 hours induced a significant increase in the phosphorylation of CREB (P < 0.05). BDNF expression was also significantly upregulated at the same concentration (P < 0.01). Bay 60‐7550 at 10−6 mol/L also increased both the ratio of pCREB/CREB and BDNF levels.
Figure 5.
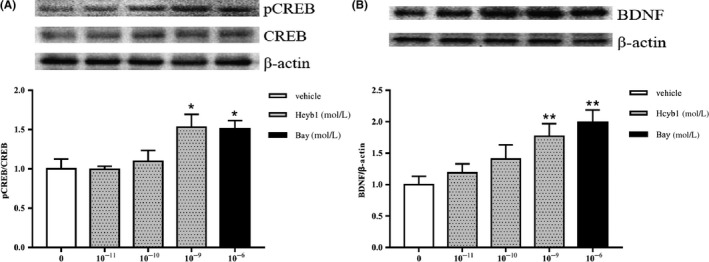
The effects of Hcyb1 on the CREB phosphorylation (A) and BDNF expression (B) in HT‐22 cells after treatment for 24 hours. The HT‐22 cells were treated with Hcyb1 and Bay 60‐7550 for 24 hours before sample collection. Results are expressed as mean ± SEM (n = 6). *P < 0.05 and **P < 0.01 vs vehicle‐treated control group
3.6. The antidepressant‐like effects of Hcyb1 in forced swimming and tail suspension tests (FST and TST)
As shown in Figure 6A,B, the antidepressant‐like effects of Hcyb1 were evaluated in the FST and TST 30 minutes after treatment with Hcyb1. Hcyb1 exhibited dose‐dependent reduction in immobility time when mice were treated with Hcyb1 at doses of 0.5, 1, 2 mg/kg (i.g.) in both FTS and TST [F (3, 35) = 3.578, P < 0.05; F (3, 35) = 3.459, P < 0.05]. The classical antidepressant desipramine and the PDE2 inhibitor Bay 60‐7550 exhibited similar anti‐immobility effects at doses of 10 mg/kg and 3 mg/kg via i.p.
Figure 6.
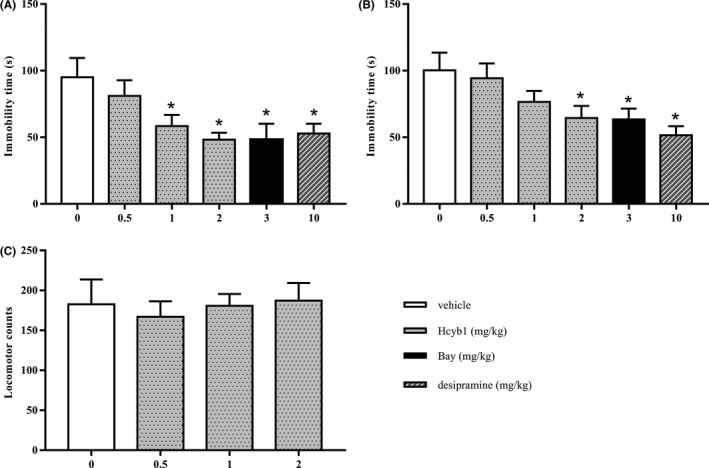
The antidepressant‐like effects of Hcyb1 in mice. Mice were treated with vehicle, Hcyb1, Bay 60‐7550, and desipramine 30 minutes before being subjected to the FST (A) and TST (B). C, Mice were treated with various doses of Hcyb1 30 minutes before the locomotor activity test. Results are expressed as mean ± SEM (n = 9‐10). *P < 0.05 vs vehicle‐treated control group
To rule out whether Hcyb1 had any central stimulating or inhibiting effects on mice, we measured their locomotion counts 10 minutes after a single treatment. As shown in Figure 6C, none of doses of Hcyb1 that were used to induce reduction in immobility time affected locomotor activity, indicating the antidepressant‐like effects of Hcyb1 were not due to central stimulation or inhibition.
4. DISCUSSION
Phosphodiesterase 2 is a novel and valuable pharmacological target in the central nervous system (CNS). Its characteristics of hydrolyzation of cyclic nucleotides and selective distribution in forebrain structures make it critical in controlling several CNS disorders including depression, anxiety, and learning and memory disabilities.8 As most current PDE2 inhibitors contain a pharmacophore of pyrazolopyrimidinone, we designed and synthesized a novel selective PDE2 inhibitor Hcyb1 by introducing more lipophilic groups with polar functionality to the scaffold pyrazolopyrimidinone to improve the cell membrane penetration and then evaluated its protective effects in cultured HT‐22 cells and the antidepressant‐like effects in mouse models of depression. In the present study, Hcyb1 increased the cell viability, cGMP, and cAMP levels and the phosphorylation of CREB and BDNF expression in HT‐22 cells. Moreover, behavioral tests suggested that Hcyb1 produced antidepressant‐like effects that were parallel to the commercial PDE2 inhibitor Bay 60‐7550 and classical antidepressant desipramine.
HT‐22 cells are a subline derived from parent HT4 cells originally immortalized from primary mouse hippocampal neuronal culture. They provide a model in vitro system to study the characteristic of many neurodegenerative disorders and also been widely used in understanding of signal pathways involved in different cellular processes such as proliferation, survival, and apoptosis.22 In this study, the selective PDE2 inhibitor Hcyb1 significantly increased the cell viability, which was considered to be associated with the PDE2 inhibition. PDE2 is a dual substrate PDE, which catalyzes the hydrolysis of both cAMP and cGMP. However, only cGMP level was significantly improved after Hcyb1 treatment for 10 minutes in the present study. A possible explanation is that PDE2 has slightly higher affinity for cGMP than cAMP in the hippocampus and exerts its action mainly via the control of intracellular second messengers, cGMP and, to a lesser extent, cAMP.9, 23 Further study suggested that Hcyb1 increased both cAMP and cGMP levels after treatment for 24 hours, which was consistent with cell viability results. The possible reason is that increasing penetration of Hcyb1 to cells as time goes on, which induces increases in both cAMP and cGMP. The commercial PDE2 inhibitor Bay 60‐7550 did not increase cAMP level even if it was incubated with HT‐22 cells for 24 hours. This finding was consistent with our previous study that the neuroprotective effects of Bay 60‐7550 could be blocked by pretreatment with protein kinase G (PKG) inhibitor KT5823, but not protein kinase A (PKA) inhibitor H89.24
In the signaling pathways involving cAMP and cGMP, respective downstream protein kinases, PKA and PKG, regulate the expression of key genes in central nervous system by activating some transcription factors.25, 26 Cyclic AMP response element‐binding protein (CREB) is one of the transcription factors mainly activated by phosphorylation on the Serine 133 residue.27 And the major transcriptional outcome of CREB phosphorylation/activation is thought to be the production of brain‐derived neurotrophic factor (BDNF).28, 29 In neurons, cAMP and cGMP‐dependent CREB/BDNF signaling cascades lead to diverse cellular responses, including neurotransmitter release, synaptic plasticity, gene transcription, and morphological changes, mediating a wide range of physiological functions and pathological disorders in CNS.30 As the main enzyme to catalyze both cAMP and cGMP, pharmacological inhibition of PDE2 can result in boosted cAMP and cGMP‐dependent signaling pathway which ultimately provoke downstream CREB phosphorylation and BDNF expression.31, 32, 33 As could be predicted by the role of PDE2, previous study has demonstrated that the antidepressant‐like effects of the selective PDE2 inhibitor Bay 60‐7550 were mediated through the cGMP/PKG pathway.34 Bay 60‐7550 was also found to increase the phosphorylation of CREB and BDNF expression in the brain of chronic stress‐induced mice.24 Our study indicated the critical role of cAMP and cGMP signaling regulated by PDE2 in HT‐22 cells. Moreover, both the ratio of pCREB/CREB and BDNF expression in HT‐22 cells were highly increased after Hcyb1 treatment, which was associated with PDE2 inhibition‐dependent increase in cGMP and cAMP. These findings further confirmed that PDE2 inhibitor Hcyb1‐induced neuroprotective effects were probably mediated by cAMP and cGMP‐dependent pCREB/CREB and BDNF expression.
Behavioral studies have previously been shown to play an important role in the evaluation of drugs in CNS. Previous studies showed that inhibitors of PDEs, such as PDE4 and PDE5, could ameliorate depression‐like behaviors through regulating the cAMP and cGMP signaling, respectively.35, 36 Hcyb1 has been confirmed to upregulate both the cAMP and cGMP signaling by PDE2 inhibition. Our results demonstrated that Hcyb1 was effective in both tail suspension and forced swimming tests. Both tests present nonescapable stressful situation to the animals and are sensitive to antidepressant treatment thus providing validation for drug efficacy. As the observed “antidepressant‐like” effects of drugs can sometimes be interpreted as a general stimulation of the central nervous system of an individual, the mice were also tested in a locomotor activity chamber. The results showed that Hcyb1 did not affect the locomotor activity at doses that produced antidepressant‐like effects, indicating the specific antidepressant‐like effects of Hcyb1.
In summary, the present study showed the synthesis of a novel selective PDE2 inhibitor Hcyb1 and its selectivity upon PDE2. The in vitro cell‐based assays indicated that Hcyb1 had significant neuroprotective effects by increasing the viability of HT‐22 cells and enhancing the cAMP and cGMP‐dependent signaling, such as increased the ratio of pCREB/CREB and BDNF expression. The animal study suggested that Hcyb1 exhibited anti‐immobility effects, which further support that Hcyb1 had potential for treatment of depression.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 81603336).
Liu L, Zheng J, Huang X‐F, et al. The neuroprotective and antidepressant‐like effects of Hcyb1, a novel selective PDE2 inhibitor. CNS Neurosci Ther. 2018;24:652–660. 10.1111/cns.12863
The first two authors contributed equally to this work
Contributor Information
Han‐Ting Zhang, Email: hzhang@hsc.wvu.edu.
Guo‐Qiang Song, Email: sgq@cczu.edu.cn.
Ying Xu, Email: yxu9@buffalo.edu.
REFERENCES
- 1. Marcus M, Yasamy MT, Ommeren MV, Chisholm D, Saxena S. Depression, a Global Public Health Concern. Geneva, Switzerland: WHO; 2012. [Google Scholar]
- 2. Aan het Rot M, Mathew SJ, Charney DS. Neurobiological mechanisms in major depressive disorder. CMAJ. 2009;180:305‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Covington HE, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58:683‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gelenberg AJ. A review of the current guidelines for depression treatment. J Clin Psychiatry. 2010;71:e15. [DOI] [PubMed] [Google Scholar]
- 5. Kennedy SH. A review of antidepressant therapy in primary care: current practices and future directions. Prim Care Companion CNS Disord. 2013;15: PCC.12r01420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481‐511. [DOI] [PubMed] [Google Scholar]
- 7. Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci. 2017;18:E696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang C, Yu Y, Ruan L, et al. The roles of phosphodiesterase 2 in the central nervous and peripheral systems. Curr Pharm Des. 2015;21:274‐290. [DOI] [PubMed] [Google Scholar]
- 9. Trabanco AA, Buijnsters P, Rombouts FJ. Towards selective phosphodiesterase 2A (PDE2A) inhibitors: a patent review (2010 ‐ present). Expert Opin Ther Pat. 2016;26:933‐946. [DOI] [PubMed] [Google Scholar]
- 10. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology. 2010;59:367‐374. [DOI] [PubMed] [Google Scholar]
- 11. Reierson GW, Guo S, Mastronardi C, Licinio J, Wong ML. cGMP signaling, phosphodiesterases and major depressive disorder. Curr Neuropharmacol. 2011;9:715‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Esposito K, Reierson GW, Luo HR, et al. Phosphodiesterase genes and antidepressant treatment response: a review. Ann Med. 2009;41:177‐185. [DOI] [PubMed] [Google Scholar]
- 13. Xu Y, Pan J, Chen L, et al. Phosphodiesterase‐2 inhibitor reverses corticosterone‐induced neurotoxicity and related behavioural changes via cGMP/PKG dependent pathway. Int J Neuropsychopharmacol. 2013;16:835‐847. [DOI] [PubMed] [Google Scholar]
- 14. Ding L, Zhang C, Masood A, et al. Protective effects of phosphodiesterase 2 inhibitor on depression ‐ and anxiety ‐ like behaviors: involvement of antioxidant and anti‐apoptotic mechanisms. Behav Brain Res. 2014;268:150‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masood A, Huang Y, Hajjhussein H, et al. Anxiolytic effects of phosphodiesterase‐2 inhibitors associated with increased cGMP signaling. J Pharmacol Exp Ther. 2009;331:690‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomez L, Breitenbucher JG. PDE2 inhibition: potential for the treatment of cognitive disorders. Bioorg Med Chem Lett. 2013;23:6522‐6527. [DOI] [PubMed] [Google Scholar]
- 17. Reneerkens OA, Rutten K, Bollen E, et al. Inhibition of phosphodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK‐801. Behav Brain Res. 2013;236:16‐22. [DOI] [PubMed] [Google Scholar]
- 18. Xu Y, Zhang C, Wang R, et al. Corticosterone induced morphological changes of hippocampal and amygdaloid cell lines are dependent on 5‐HT7 receptor related signal pathway. Neuroscience. 2011;182:71‐81. [DOI] [PubMed] [Google Scholar]
- 19. Xu Y, Ku BS, Yao HY, et al. The effects of curcumin on depressive‐like behaviors in mice. Eur J Pharmacol. 2005;518:40‐46. [DOI] [PubMed] [Google Scholar]
- 20. Porsolt RD, Bertin A, Jalfre M. “Behavioural despair” in rats and mice: strain differences and the effects of imipramine. Eur J Pharmacol. 1978;51:291‐294. [DOI] [PubMed] [Google Scholar]
- 21. Ma X, Wang R, Zhao X, et al. Antidepressant‐like effect of flaxseed secoisolariciresinol diglycoside in ovariectomized mice subjected to unpredictable chronic stress. Metab Brain Dis. 2013;28:77‐84. [DOI] [PubMed] [Google Scholar]
- 22. Liu J, Li L, Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267‐271. [DOI] [PubMed] [Google Scholar]
- 23. García‐Osta A, Cuadrado‐Tejedor M, García‐Barroso C, Oyarzábal J, Franco R. Phosphodiesterases as therapeutic targets for Alzheimer's disease. ACS Chem Neurosci. 2012;3:832‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y, Pan J, Sun J, et al. Inhibition of phosphodiesterase 2 reverses impaired cognition and neuronal remodeling caused by chronic stress. Neurobiol Aging. 2015;36:955‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleppisch T. Phosphodiesterases in the central nervous system. Handb Exp Pharmacol. 2009;191:71‐92. [DOI] [PubMed] [Google Scholar]
- 26. Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660‐670. [DOI] [PubMed] [Google Scholar]
- 27. Shaywitz Adam J, Greenberg Michael E. CREB: a stimulus‐induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821‐861. [DOI] [PubMed] [Google Scholar]
- 28. Shieh PB, Ghosh A. Molecular mechanisms underlying activity‐dependent regulation of BDNF expression. J Neurobiol. 1999;41:127‐134. [PubMed] [Google Scholar]
- 29. Bollen E, Puzzo D, Rutten K, et al. Improved long‐term memory via enhancing cGMP‐PKG signaling requires cAMP‐PKA signaling. Neuropsychopharmacol. 2014;39:2497‐2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang ZZ, Zhang Y, Zhang HT, Li YF. Phosphodiesterase: an interface connecting cognitive deficits to neuropsychiatric and neurodegenerative diseases. Curr Pharm Des. 2015;21:303‐316. [DOI] [PubMed] [Google Scholar]
- 31. Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology. 2009;202:419‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masood A, Nadeem A, Mustafa SJ, O'Donnell JM. Reversal of oxidative stress‐induced anxiety by inhibition of phosphodiesterase‐2 in mice. J Pharmacol Exp Ther. 2008;326:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu Y, Chen Z, Pan J, Zhang HT, O'Donnell JM. Memory enhancement induced by PDE2 knockdown in an Alzheimer's disease model of mice. FASEB J. 2016;30(Supplement707):6. [Google Scholar]
- 34. Liu X, Liu TT, Bai WW, et al. Encoding of rat working memory by power of multi‐channel local field potentials via sparse non‐negative matrix factorization. Neurosci Bull. 2013;29:279‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang C, Xu Y, Zhang HT, Gurney ME, O'Donnell JM. Comparison of the pharmacological profiles of selective PDE4B and PDE4D inhibitors in the central nervous system. Sci Rep. 2017;7:40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang M, Urenjak J, Fedele E, Obrenovitch TP. Effects of phosphodiesterase inhibition on cortical spreading depression and associated changes in extracellular cyclic GMP. Biochem Pharmacol. 2004;67:1619‐1627. [DOI] [PubMed] [Google Scholar]