

Summary
Aims
West syndrome (WS) is a classic form of early infantile epileptic encephalopathy (EIEE) characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on electroencephalography. Genetic defects play a critical role in the pathology of WS, and 54 EIEE genes have been identified till date. This study was designed to uncover new candidate genes for West syndrome.
Methods
In this study, we recruited 56 Chinese families with WS of unknown etiology. Whole exome sequencing (WES) was performed to identify Mendelian inheritance rare or novel variants. The association between candidate genes and WS was analyzed from many aspects, including recurrent genes in patients, predicted variant effect on genes, human tolerance to deficient genes, gene expression in the nervous system, coexpression with EIEE genes, mutual interaction with known EIEE proteins, genes related to ion channel or fragile X mental retardation protein function, and mouse models with manifestation of seizures. Genes with supporting evidence from those aspects were defined as highlight candidate genes.
Results
Whole exome sequencing identified 112 candidate variants in 89 genes. Among the candidate genes, 33 were autosomal dominant, 22 were autosomal recessive, and 34 were X‐linked. Complex bioinformatic analysis revealed 17 highlight candidate genes: ATP2A2, CD99L2, CLCN6, CYFIP1, CYFIP2, GNB1, GPT2, HUWE1, KMT2D, MYO18A, NOS3, RYR1, RYR2, RYR3, TAF1, TECTA, and UBA1. The majority of highlight candidate genes are calcium‐signaling pathway and mental retardation genes.
Conclusions
This is the first WES study of Chinese WS patients with unknown etiology. This combination of phenotypic and genomic data will enable further testing to elucidate mechanisms underlying the pathogenesis of WS.
Keywords: calcium‐signaling pathway genes, mental retardation genes, west syndrome, whole exome sequencing
1. INTRODUCTION
Infantile spasms syndrome (ISs) is a classic form of early infantile epileptic encephalopathy (EIEE, OMIM 308350, http://www.omim.org/). ISs is an epileptic syndrome occurring in children aged <1 year (rarely >2 years), with clinical spasms usually occurring in clusters whose most characteristic electroencephalography (EEG) finding is hypsarrhythmia.1 West syndrome (WS) refers to a subset of ISs and is characterized by a combination of clustered spasms, hypsarrhythmia on EEG, and delayed brain development or regression.2, 3 In WS patients, 70%‐90% have developmental delay or mental retardation, 70% have intractable epilepsy, and 45% present with an autism spectrum phenotype.1, 3 Until now, 54 genes have been identified as causal genes of EIEE. Previous whole exome sequencing (WES) studies in patients with infantile spasms and Lennox‐Gastaut syndrome revealed hundreds of genes associated with classical epileptic encephalopathies.4, 5 In this study, WES was performed on a cohort of Chinese WS patients and their parents (trios) to identify novel de novo and inherited variants.
2. SUBJECTS AND METHODS
2.1. Subjects
This study was approved by an ethics committee at Xiangya Hospital of Central South University. This study has been performed in accordance with the ethical standards laid down in an appropriate version of the Declaration of Helsinki. All persons gave their informed consent prior to their inclusion in the study. The study included 72 Chinese WS patients with no family history and with negative findings in the following: (i) metabolic, structural, immunological, and infectious etiologies (International League Against Epilepsy recommendations); (ii) chromosomal disorders (ie, Down's syndrome), copy number variation (CNV) diseases, and monogenic disorders (ie, Angelman syndrome or tuberous sclerosis complex); and (iii) Genetic‐structural modification of the brain (ie, tuberous sclerosis or DCX mutations leading to lissencephaly).6 Peripheral blood samples were collected from patients and their parents via venipuncture.7
2.2. Whole exome sequencing
DNA samples were collected using the SureSelect Human All Exon V5 Kit following the manufacturer's protocol (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq X Ten (Illumina, San Diego, CA, USA) with 150‐bp paired‐end reads.8
2.3. Data analysis
Sequence reads were aligned to the human genome reference (UCSC hg 19 version) with the Burrows‐Wheeler aligner and recalibrated with the genome analysis toolkit (GATK) and Picard 1.14. Reads around insertion/deletion sites were realigned with the GATK IndelRealigner. Single nucleotide variants and small insertions and deletions (InDels) were identified with the GATK Unified Genotyper in parallel with SAMtools and were annotated with ANNOVAR.9, 10, 11
Candidate variants in known epileptic encephalopathy genes were first analyzed according to the American College of Medical Genetics and Genomics (ACMG) guidelines.12
Fifty‐six patients (38 males, 18 females) negative for pathogenic or likely pathogenic variants were referred to the next step (see Table S1). WES was performed for unaffected parents of these 56 patients as described above and combined with patient data (trios). Only loss‐of‐function (LoF) or protein‐altering variants cosegregated within each family were analyzed. Variant types included nonsense, missense, frameshift, in‐frame InDel, splice site, start‐loss, and stop‐loss. Depth of variants alleles should be 10 at least. Variants recorded in 1000 g (http://browser.1000genomes.org) and ESP6500 (http://evs.gs.washington.edu/EVS) were ruled out. Variants segregated within a family in an autosomal recessive or X‐linked manner were ruled out if the frequency in ExAC (http://exac.broadinstitute.org/) was >0.001. De novo variants in trios were ruled out if the frequency in ExAC was higher than 0.0001. The Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), issued on February 27, 2017, includes data from 123 136 exomes and 15 496 genomes, including 8624 exomes and 811 genomes from the East Asian ethnic group. Filtered variants were reevaluated with gnomAD to compare the coincidence between variation frequency and prevalence of WS. Variants located on the X chromosome with more than 1 incidence of hemizygosity in gnomAD were excluded. Sanger sequencing was used to confirm candidate variants in these families.
2.4. Molecular diagnosis
Variants verified by Sanger sequencing were assessed with the HGMD pro database (http://www.hgmd.org). Candidate genes were searched in the OMIM database, and genes listed as causal genes for Mendelian diseases with genotypes fitting the recorded inheritance pattern were assessed for pathogenicity using the ACMG guidelines.13, 14
2.5. Bioinformatics
Fifty‐four known EIEE genes (OMIM, July 2017 update) were listed as positive genes. Gene ontology (GO) and KEGG analysis were performed for enrichment analyses (http://www.webgestalt.org/).
mRNA expression data of candidate genes in 350 normal brains of fetuses and children aged <3 years generated by RNA‐seq (Poly A library) were provided by the authors of Brain Gene EXPression database (BrainEXP, www.brainexp.org, the data used in this article are not fully uploaded to the database server yet). For age and sex information of samples, see Table S2. Expression data included 16 brain regions (see Table S3), and candidate genes expressed in more than 2 brain areas were defined as positive expression. Furthermore, the database was used to study coexpression patterns between EIEE genes and brain‐expressed candidate genes. Based on the same RNA‐seq database, EIEE genes and brain‐expressed candidate genes were deemed to have a coexpression pattern if weight >0.3 combined with a value of >0.5 or <−0.5.
Protein‐protein interaction analysis of candidate and EIEE genes was performed by geneMANIA (http://genemania.org). Candidate genes were compared with reported ion channel genes,15 Fragile X protein‐inhibited genes,16 and genes known to cause mouse models with seizures from Mouse Genome Informatics database (http://www.informatics.jax.org/).
2.6. Highlight candidate genes
The association between candidate genes and WS was analyzed from many aspects, including recurrent genes in patients, predicted variant effect on genes (Combined Annotation Dependent Depletion [CADD]; http://cadd.gs.washington.edu/), human tolerance to deficient genes (Residual Variation Intolerance Score [RVIS]; http://genic-intolerance.org/), gene expression in the nervous system, coexpression with EIEE genes, mutual interaction with known EIEE proteins, genes related to ion channel15 or fragile X mental retardation protein (FMRP) function,16 and mouse models with manifestation of seizures. Genes with supporting evidence from those aspects were defined as highlight candidate genes.
3. RESULTS
3.1. Pathogenic and likely pathogenic variants in known causal genes
Pathogenic or likely pathogenic variants of known genes such as SCN8A, STXBP1, and CDKL5 confirmed by Sanger sequencing have been detected among 16 of 72 patients, who were excluded from this trial. The remaining 56 patients were referred for followed study (Jing Peng, 2018, unpublished data).
3.2. NGS data statistics for 56 trios
We performed WES for 56 trios, average sequencing depth on target was 152.6 (116.8‐219.6), and fraction of target covered with at least 20× was 98.4% (96.9%‐99.3%) on average (see Table S1).
3.3. Candidate variants
Sanger sequencing identified 112 candidate variants in 89 genes, including 3 nonsense, 98 missense, 3 frameshift deletions, 4 splice site variants, 3 in‐frame InDels, and 1 start‐loss variant. No sign of somatic variants was observed. Nonsense variants, frameshift deletions, start‐loss, and splice site variants were classified as LoF, which accounts for 9.8% of all candidates. A start‐loss variant in mitochondrial ribosomal protein L17 (MRPL17) was identified in patient WS36‐P. This patient had a de novo heterozygous variant that mutated the start codon ATG to CTG. Among the 112 variants, 62 were not reported in 1000 g, ESP6500, ExAC, or gnomAD.
Among 89 genes, 33 presented an autosomal dominant pattern, including 1 de novo variant for 32 genes and 1 de novo plus an additional novel paternal variant in MYCBP2, which might have a modifier effect. Additionally, 22 genes presented with an autosomal recessive pattern, with 2 compound heterozygous variants for 21 genes and 1 homozygous variant in C9orf163 of patient WS37‐P. X‐linked recessive patterns were seen in 33 genes with 1 hemizygote variant from carrier mothers for each gene. One de novo variant in HUWE1 at Xp11 was detected in a male patient WS40‐P. Patients WS2‐P and WS25‐P shared the same nonframeshift deletion in CD99L2. Patients WS15‐P and WS51‐P shared different missense variants in TAF1. No genes presented conflicting inherence patterns. Among the 89 genes, 10 are located on Xp11, 10 on Xp22, and 6 on Xq13 (see Table S4 and Figure S1).
3.4. Molecular diagnosis for OMIM genes
Among 89 candidate genes, 18 have been recorded as causal genes by OMIM. Five genes had pathogenic or likely pathogenic variants cosegregated within families including GNB1 (mental retardation, autosomal dominant 42, OMIM 616973), HUWE1 (mental retardation, X‐linked syndromic, Turner type, OMIM 300706), and KMT2D (Kabuki syndrome 1, OMIM 147920), which are all causal genes for mental retardation‐related syndromes.17, 18, 19 However, HGMD has not recorded these variants. The GNB1 variant has been classified as pathogenic, and variants in HUWE1 and KMT2D are both likely pathogenic. Classic facial features due to KMT2D defects were not present in the patients in this study.19 It is worth noting that TAF1 is known to cause mental retardation, X‐linked, syndromic 33 (OMIM 300966), 2 patients each have a variant classified as VUS.20 And GPT2 is known to cause mental retardation, autosomal recessive 49 (OMIM 616281), and the patient WS31‐P only has a heterozygous de novo variant.21 Table S5 presents the main clinical manifestations of our patients compared with reported phenotypes of corresponding gene defects.
Two variants of CAT have been demonstrated to be pathogenic: Maternal c.480 + 5G > A has impact on gene splicing,22 and paternal c.136delC can result in a premature termination codon (PTC). LoF variants can lead to autosomal recessive acatalasemia (OMIM 614097)22; however, patient WS32‐P did not present this manifestation. According to the ACMG guide, ATP2A2 p. G860S has been defined as likely pathogenic, but patient WS16‐P did not present with acrokeratosis verruciformis (OMIM 101900) or Darier's disease (OMIM 124200).23
3.5. Bioinformatic analysis
Gene ontology analysis of candidate genes and EIEE genes has revealed high similarity in all 3 slim classifications: biological process, molecular function, and cellular component (Figure 1). KEGG analysis revealed enrichment of 6 genes in the calcium‐signaling pathway: ATP2A2, NOS3, PHKA1, RYR1, RYR2, and RYR3 (Figure 2).
Figure 1.
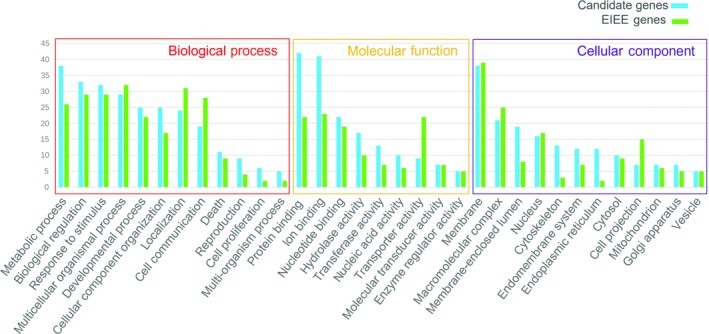
Gene ontology (GO) analysis of the 89 candidate genes and 54 early infantile epileptic encephalopathy (EIEE) genes. Previously identified EIEE genes are marked in green, and candidate genes are marked in blue. The height of the bar represents the number of genes observed in the category. Only categories with 5 or more candidate genes are shown
Figure 2.
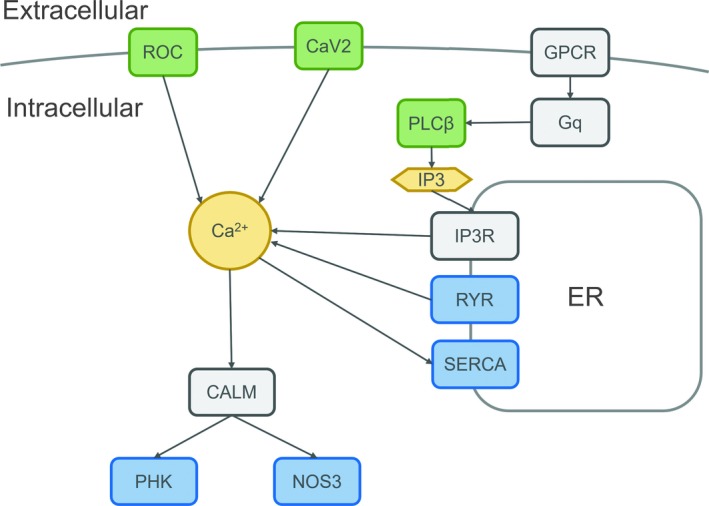
Causal and candidate genes in the calcium‐signaling pathway. Previously identified early infantile epileptic encephalopathy (EIEE) genes are marked in green, and candidate genes are marked in blue. Dark color represents other genes within the calcium‐signaling pathway. ATP2A2 encodes the sarco(endo)plasmic reticulum Ca2+ ATPase2 (SERCA2), which catalyzes the hydrolysis of ATP coupled with the translocation of calcium from the cytosol into the endoplasmic reticulum lumen. RYR1, RYR2, and RYR3 are the only 3 genes in the ryanodine receptor family, which serve as a calcium release channel in the endoplasmic reticulum. Calmodulin (CALM) is the most important calcium‐binding protein, triggering almost all downstream progress of the calcium‐signaling pathway. NOS3 and PHKA1 are downstream genes of CALM. NOS3 generates NO by catalyzing arginine in brain tissue. PHKA1 codes for the alpha subunit of phosphorylase kinase. Both genes regulate a wide variety of downstream cell actions. CaV2: voltage‐gated calcium channel encoded by CACNA1A; PLCβ: Phospholipase C‐beta encoded by PLCB1; ROC: receptor‐operated channel, associated genes include GRIN2D and GRIN1; ER: endoplasmic reticulum; GPCR: G protein‐coupled receptor; Gq: guanine nucleotide‐binding protein; IP3: inositol 1,4,5‐triphosphate; IP3R: inositol 1,4,5‐triphosphate receptor
According to the BrainEXP database, 63 genes are expressed in fetuses or children under 3 years (Table S3). Among those 63 genes, 21 have coexpression with 15 EIEE genes (weight >0.3, value >0.5). However, the coexpression does not have a negative correlation (Figure 3A). Analysis with geneMANIA indicated that 16 proteins encoded by candidate genes interact physically with 13 proteins encoded by EIEE genes (Figure 3B). Among the 89 candidate genes, 5 are ion channel genes, 4 of which are calcium channels (ATP2A2, RYR1, RYR2, and RYR3), and CLCN6 is a chloride channel. Fifteen genes are FMRP‐regulated genes (see Table S4). Three genes are involved in mouse models with seizures: Nos3 (MGI: 2150145), Ryr2 (MGI: 3802940), and Tecta (MGI: 5527094, 5527095, 5527096). Taken together, 17 genes can be considered highlight candidate genes for WS (table 1).
Figure 3.
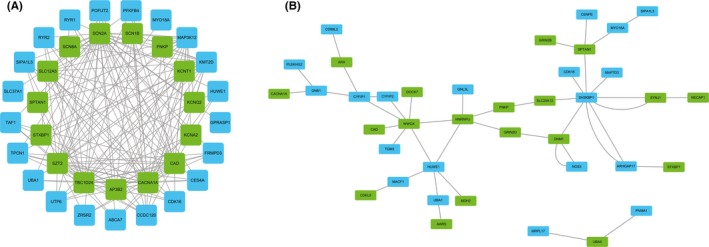
Coexpression and Protein‐protein interactions of candidate genes and early infantile epileptic encephalopathy (EIEE) genes. A, Coexpression of candidate genes and EIEE genes; B, Protein‐protein interactions of candidate genes and EIEE genes. EIEE genes are marked in green, and candidate genes are marked in blue
Table 1.
Detail of highlight candidate genes
| Genes | Family ID | Inheritance patterns | Inherited from | variant types | Cytoband | POS | REF | ALT | variant descriptions | Frequency in gnomAD East Asian | CADD | RVIS | Epression in brain | Coexpression with EIEE genes | Physical interactions with EIEE protein | Additional commands |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATP2A2 | WS16 | AD | de novo | Missense SNV | 12q24.11 | 110782747 | G | A |
NM_001681:exon17 c.G2578A p. Gly860Ser |
22.4 | −1.9026 (3.4897%) | + | − | − | Calcium‐signaling pathway, FMRP | |
| CD99L2 | WS25 | XL | M | In‐frame deletion | Xq28 | 149937525 | GGGC | G |
NM_001184808:exon8 c.549_551del p. Pro184del |
0.000 959 693 | 7.491 | 0.8208 (85.4232%) | + | − | + | Recurrent variant |
| CD99L2 | WS2 | XL | M | In‐frame deletion | Xq28 | 149937525 | GGGC | G |
NM_001184808:exon8 c.549_551del p. Pro184del |
7.491 | + | − | + | Recurrent variant | ||
| CLCN6 | WS22 | AD | de novo | Missense SNV | 1p36.22 | 11884561 | A | C |
NM_001256959:exon7 c.A533C p. Glu178Ala |
17.32 | −1.5930 (5.3763%) | + | − | − | Chloride channel | |
| CYFIP1 | WS46 | AD | de novo | In‐frame deletion | 15q11.2 | 22939182 | AGGT | A |
NM_014608:exon10 c.909_911del p. 303_304del |
−1.8425 (3.7830%) | + | − | + | FMRP | ||
| CYFIP2 | WS1 | AD | de novo | Missense SNV | 5q33.3 | 156721843 | C | T |
NM_001037333:exon4 c.C259T p. Arg87Cys |
23.6 | −1.5154 (5.9238%) | + | − | + | FMRP | |
| GNB1 | WS43 | AD | de novo | Missense SNV | 1p36.33 | 1720572 | G | A |
NM_001282538:exon8 c.C536T p. Ser179Phe |
34 | −0.4777 (28.3969%) | + | − | + | Mental retardation gene, FMRP | |
| GPT2 | WS31 | AD | de novo | Frameshift deletion | 16q11.2 | 46950598 | GCT | G |
NM_001142466:exon7 c.582_583del p. Phe195Serfs*4 |
−0.1023 (44.6041%) | + | − | − | Mental retardation gene | ||
| HUWE1 | WS40 | XL | de novo | Missense SNV | Xp11.22 | 53561638 | C | A |
NM_031407:exon82 c.G12670T p. Ala4224Ser |
0.000 155 848 | 12.68 | −7.0236 (0.1567%) | + | + | + | Mental retardation gene, FMRP |
| KMT2D | WS1 | AD | de novo | Missense SNV | 12q13.12 | 49427485 | G | A |
NM_003482:exon39 c.C11003T p. Pro3668Leu |
0 | 10.77 | −6.4882 (0.0880%) | + | + | − | Mental retardation gene, FMRP, recurrent in Epi4K study |
| MYO18A | WS5 | AR | M | Missense SNV | 17q11.2 | 27493915 | C | T |
NM_078471:exon2 c.G44A p. Arg15Gln |
0.000 194 818 | 21.8 | −0.5903 (24.3597%) | + | + | + | FMRP |
| MYO18A | WS5 | AR | F | Missense SNV | 17q11.2 | 27437031 | C | T |
NM_078471:exon19 c.G3176A p. Arg1059His |
0 | 19.11 | + | + | + | FMRP | |
| NOS3 | WS13 | AD | de novo | Missense SNV | 7q36.1 | 150696101 | C | T |
NM_001160109:exon7 c.C884T p. Ala295Val |
17.61 | 0.1800 (58.1623%) | + | − | + | Calcium‐signaling pathway, mouse model | |
| RYR1 | WS21 | AR | F | Missense SNV | 19q13.2 | 38946118 | A | G |
NM_000540:exon15 c.A1604G p. Asn535Ser |
0.000 173 933 | 15.5 | −5.4856 (0.1271%) | + | + | − | Calcium‐signaling pathway |
| RYR1 | WS21 | AR | M | Missense SNV | 19q13.2 | 38935236 | G | A |
NM_000540:exon7 c.G550A p. Ala184Thr |
0.000 406 457 | 12.97 | + | + | − | Calcium‐signaling pathway | |
| RYR2 | WS29 | AR | M | Missense SNV | 1q43 | 237729900 | A | G |
NM_001035:exon28 c.A3248G p. Glu1083Gly |
0.000 106 078 | 17.67 | −6.5324 (0.0782%) | + | + | − | Calcium‐signaling pathway, mouse model, FMRP, recurrent in Epi4K study |
| RYR2 | WS29 | AR | F | Missense SNV | 1q43 | 237798279 | C | T |
NM_001035:exon44 c.C6779T p. Arg2260Leu |
0.000 616 523 | 22.6 | + | + | − | Calcium‐signaling pathway, mouse model, FMRP, recurrent in Epi4K study | |
| RYR3 | WS56 | AR | F | Missense SNV | 15q14 | 33936671 | A | G |
NM_001036:exon28 c.A3716G p. Lys1239Arg |
0.0 001 161 | 24 | −4.2466 (0.3421%) | + | − | − | Calcium‐signaling pathway |
| RYR3 | WS56 | AR | M | Missense SNV | 15q14 | 33941340 | C | T |
NM_001036:exon31 c.C4046T p. Thr1349Ile |
0.000 116 | 27.7 | + | − | − | Calcium‐signaling pathway | |
| TAF1 | WS51 | XL | M | Missense SNV | Xq13.1 | 70587404 | C | A |
NM_001286074:exon2 c.C236A p. Thr79Asn |
0 | 18.13 | −2.7937 (0.9404%) | + | + | − | Mental retardation gene, recurrent in this and Epi4K study |
| TAF1 | WS15 | XL | M | Missense SNV | Xq13.1 | 70644046 | G | A |
NM_001286074:exon32 c.G4771A p. Asp1591Asn |
12.31 | + | + | − | Mental retardation gene, recurrent in this and Epi4K study | ||
| TECTA | WS28 | AR | M | Missense SNV | 11q23.3 | 121039502 | G | A |
NM_005422:exon19 c.G5867A p. Arg1956Gln |
0.000 106 | 36 | −1.0022 (13.1672%) | − | − | − | Mouse model |
| TECTA | WS28 | AR | F | Missense SNV | 11q23.3 | 120996138 | A | G |
NM_005422:exon7 c.A1331G p. Tyr444Cys |
5.79845E‐05 | 19.55 | − | − | − | Mouse model | |
| UBA1 | WS45 | XL | M | Missense SNV | Xp11.23 | 47071859 | C | T |
NM_003334:exon21 c.C2501T p. Pro834Leu |
14.42 | −1.3428 (6.7398%) | + | + | + | FMRP |
CADD, Combined Annotation Dependent Depletion; FMRP, fragile X mental retardation protein; RVIS, Residual Variation Intolerance Score.
Inherited from: F‐father, M‐mother, de novo‐neither from father or mother; Frequency in gnomAD East Asian: 0‐ not recorded but different variant recorded at the same loci, NA‐none variant recorded in this loci.
4. DISCUSSION
Previous WS and LGS studies have reported that the average number of de novo functional variants in each family is 0.99 and 0.95, respectively.4, 5 In this study, the first for Chinese WS patients, the average number of de novo variants in each family is 0.41, which may result from the use of different bioinformatics pipelines or different ethnic group. Among these 3 studies, only TAF1, RYR2, and KMT2D were overlapped reported, indicated high heterogeneity in WS. Of the 54 EIEE genes, 23 are autosomal dominant, 24 are autosomal recessive, and 7 are X‐linked. While previous studies focused on the dominant hereditary pattern, our study investigated all variants based on Mendelian inheritance, identifying 22 autosomal recessive and 34 X‐linked genes (male patients accounted for 67.9% of the cohort, and each male patient had 0.95 chromosome X candidate variants). Generally, patients with clinical manifestations of Kabuki syndrome receive a KMT2D gene test. However, patients with KMT2D de novo mutations did not present with Kabuki face or skeletal abnormalities, which imply KMT2D could only cause neurodevelopmental symptoms. Additionally, we found 4 families with autosomal recessive variants in TTN (see Table S6). Titin encoded by TTN, the largest protein in the human body, forms powerful elastic filaments along the sarcomere of cardiomyocytes, and TTN is known to cause several Mendelian cardiomyopathy and myopathy diseases.24 We compared our data and parallel data from another Chinese WES study (data not shown) and concluded that the TTN variants we observed were due to the size and mutation rates of the gene rather than a direct association with WS.
We detected nonsense variants in PARS2, ADGRG4, and GOLGA5 and frameshift deletions in GPT2, CAT, and MAP3K15, which all caused a PTC and likely induces nonsense‐mediated decay (NMD); however, because the entire ORF of PARS2 is located on exon 2, NMD would be triggered by an extended 3′‐untranslated region rather than exon junction complex.25 Splice junction variants detected in MYO1H and ME3 were located in the +1 position, which likely causes splicing errors. Splice junction variants in CAT (described before) and VWA3A are located in the +3 and +5 positions, respectively. CADD scores of these 2 splice variants are 14.89 and 13.49, respectively. The VWA3A variant (inherited from the patient's father) is in trans with a novel missense variant (inherited from the patient's mother). Based on these data, we consider these splice junction variants as candidates.
We identified a de novo variant in patient WS36‐P converting the start codon to CTG in MRPL17. The Drosophila homologue of FMR1 (the causative gene for fragile X syndrome, FXS), dFMR1, is known to express 2 main isoforms; the slowly migrating isoform is generated through a “CTG” translational start site in the dfmr1 5′UTR. This isoform is individually sufficient for proper dFMR1 localization in the nervous system. Functional studies demonstrated that both isoforms can function independently to rescue dfmr1 null defects in synaptogenesis and axon guidance.26 Further experimentation is needed to determine whether MRPL17 start‐loss allele is expressed, which ORF is working, and whether the polypeptide has an unknown gain of function leading to cytotoxicity. However, we believe that the original MRPL17 function was disrupted, and this gene may be linked to WS onset.
Among 89 genes, we found 5 genes reported in the OMIM database with mental retardation and related diseases. Additionally, HGMD database revealed that 11% of the candidate genes have been reported to be associated with mental retardation, including CDK16, CXorf36, PLEKHG2, PRICKLE3, REPS2, TDRD6, ULK4, UTP6, ZCCHC16, and ZRSR2. ASD is associated with 15% of the candidate genes, including ABCA7 (which is also known as susceptible gene of Alzheimer's disease 9, OMIM 608907), ARSF, CCDC14, CLCN6, CYFIP1, FAT3, MACF1, MAP3K15, MAP7D3, MYCBP2, TECTA, UTP6, and ZCCHC16. Additionally, 7% of candidate genes are associated with schizophrenia including CES4A, EGR3, FBXO39, MYO18A, TDRD6, and ULK4. In neurodevelopmental diseases, overlap of causal genes is very common. Many genes related to mental retardation (and ASD, Schizophrenia etc.) were indicated in WS in our study. Expansion of genotype‐phenotype association in these genes would aid in elucidating the mechanism and key roles in neural development.
FMR1 encodes FMRP, which regulates many synaptic functions and autism‐related genes.16 These genes also have a strong relation to epilepsy.27 Fifteen genes in our study were reported to be regulated by FMRP, including some calcium‐signaling genes and mental retardation genes discussed above. We also focused on 2 other genes of interest: CYFIP1 and CYFIP2. CYFIP (cytoplasmic FMRP‐interacting protein) family, including CYFIP1 and CYFIP2, which have 88% homology, is a highly conserved protein family expressed highly in synapse of neurons.28 CYFIP proteins play an important role in FMRP function, but CYFIP1 and CYFIP2 have different expression pattern and regulation mechanism in the brain development process: CYFIP1 and FMRP are directly bonded, form complex with translation initiation factor eIF4E, and modulate the affinity of FMRP and mRNAs; CYFIP2 not only has an effort similar with CYFIP1, but also regulated by FMRP through CYFIP2 mRNA translation.29, 30, 31 FXR1P and FXR2P are highly homologous to FMRP and have a similar function.30 CYFIP2 interacts with both FXR1P and FXR2P.29 Downregulation of CYFIP2 leads to hippocampal neuron and synaptic abnormalities, interrupting spatial memory.29 Cyfip1 null is lethal during murine embryonic development, Cyfip1+/− and fmr1 (tm1Cgr) mouse both have seizures that are alleviated by the mGluR5 antagonist MPEP;32 however, the mechanism is not well understood. Furthermore, Cyfip1 heterozygous mutant mice and Fmr1 KO mice have similar behavior.33 Cyfip2 homozygous mutations in mice are lethal.34 Additionally, abnormal maturation of dendritic spines has been observed in FXS patients and Cyfip1 and Cyfip2 mutant mouse models, which are similar to FXS mice.28, 33
CYFIP1 plays an important role in classic CNV diseases: Prader‐Willi syndrome (OMIM 176270) and Angelman syndrome (OMIM 105830). The key region of both syndromes is 15q11‐13, containing 3 critical breakpoints: BP1 (15q11.2), BP2 (15q11.2), and BP3 (15q13.1).35 Deletion from BP1 to BP3 called Type I deletion, deletion from BP2 to BP3 called Type II deletion, and deletion from BP1 to BP2 causes Burnside‐Butler syndrome (CYFIP1 located in this region).36 Patients with a Type I deletion usually have more severe behavioral and psychological problems compared with those with a Type II deletion.37, 38 Deletion from BP1 to BP2 causes more serious global developmental delay, mental behavioral problems, and seizures.37, 38, 39 Above all, genes located in the BP1‐BP2 region play an important role in neural development. CYFIP1 mRNA is upregulated in the peripheral blood of ASD patients.40 A 15q11.2‐q13.1 duplication has also been found in infantile spasm patients.41 According to the Decipher database (https://decipher.sanger.ac.uk/), CNVs involving CYFIP2 were reported in several patients with neurodevelopmental symptoms such as absence seizures, EEG abnormality, attention deficit hyperactivity disorder, autistic behavior, microcephaly, and global developmental delay. However, these CNVs are relatively large (3.84‐20.25 Mb); hence, contribution of single gene is not clear. This is the first report of de novo variants in these 2 genes associated with neurodevelopmental diseases; therefore, further functional studies are needed to clarify the underlying molecular pathological mechanisms.
Several causal genes of EIEE and related diseases are involved in the calcium‐signaling pathway, including PLCB1 (EIEE12, OMIM 613722), CACNA1 (EIEE42, OMIM 617106), GRIN2D (EIEE46, OMIM 617162), GRIN2A (epilepsy, focal, with speech disorder and with or without mental retardation, OMIM 245570), and GRIN1 (mental retardation, autosomal dominant 8, OMIM 614254).42, 43, 44, 45, 46, 47 CACNA1A encodes the P/Q‐type calcium channel alpha subunit (CaV2.1), and a nonsense mutation at CACNA1A p.R1820* results in Ca2+ channel function impairment with a dominant negative effect.48 The functional consequences of an E147K mutation in Xenopus oocytes showed that mutant CaV2.1α1 was functional but caused a decrease in current amplitude and prolongation of the slow inactivation process.49 Dysfunction of CaV2.1 increases excitability of neurons, leading to epileptiform discharge.49 Pharmacological evidence suggests that the antiepileptic effects of lamotrigine and levetiracetam are due to inhibition of N‐ or P/Q‐type calcium channels.50 Mutation of GRIN2D in the M3 transmembrane domain, which forms the ion channel pore, increases receptor responsiveness to glutamate and glycine agonists, decreases sensitivity to negative allosteric modulators, prolongs deactivation time, and increases channel‐opening probability, all consistent with a gain‐of‐function effect in the NMDA receptor.44 Transfection of the mutation into rat cortical neurons resulted in increased neuronal excitotoxicity that could be blocked by the NMDAR antagonist memantine.44
Here, we report 6 calcium‐signaling genes: ATP2A2, NOS3, PHKA1, RYR1, RYR2, and RYR3. ATP2A2 encodes SERCA2, which catalyzes the hydrolysis of ATP coupled with the translocation of calcium from the cytosol into the endoplasmic reticulum lumen.51 RYR family has 3 different ryanodine receptor genes, RYR1, RYR2 and RYR3, which conduct 3 isoforms of RyRs with 70% sequence homology. RyRs serve as a calcium release channel in the endoplasmic reticulum and are broadly expressed across all organs in different patterns, including the central nervous system.52 Many neuromuscular diseases are caused by mutations in RYR1 and RYR2.53 Mice with RYR2 mutations presented with seizures. ATP2A2, RYR1, RYR2, and RYR3 are all directly involved in the regulation of calcium homeostasis. Three patients with variants in ATP2A2, RYR2, or RYR3 are seizure‐free by administration of calcium blocker drugs such as sodium valproate, topiramate, and lamotrigine, suggesting that seizures in these 3 patients may be related to calcium channel defects (see Table S7).
Mutations in NOS3 are associated with late‐onset Alzheimer's disease (OMIM 104300), and cardiovascular diseases.54, 55 NO is a pivotal signaling molecule in several processes including neurotransmission, oxidative stress, activation of immunization, regulation of blood pressure, and calcium homeostasis.56 NOS3 deficiency in mice caused seizures. The regulatory alpha subunit of phosphorylase kinase, a key regulatory enzyme of glycogen metabolism, is encoded by PHKA1.57 PHKA1 is a causal gene for muscle glycogenosis (OMIM 300559) characterized by variable exercise‐induced muscle weakness or stiffness.57Mutations in genes upstream of Calmodulin (CALM) directly affect electrical activity of neurons, leading to the onset of seizures. Mutations in genes downstream of CALM show an association with neural symptoms. Whether disrupted CALM signaling is related to WS onset is worth further study.
This report highlights 17 genes based on genetic supporting evidence and molecular epileptic pathology. In addition to the aforementioned genes, CD99L2 is the only gene hit by recurrent variants and is reported to play a role in the pathogenesis of autism.58 CD99L2 is expressed in the human brain and directly interacts with the protein encoded by a classic EIEE gene, ARX. CLCN6, a member of the mammalian CLCN family of voltage‐gated chloride channels, is expressed in the human brain and has an RVIS score higher than that of 42 EIEE genes. Furthermore, the de novo variant we report in this gene has a high CADD score. Variants of MYO18A and UBA1 have high RVIS and CADD scores,and both genes are regulated by FMRP, coexpressed with EIEE genes, and physically interact with proteins encoded by EIEE genes. TECTA has high RVIS and CADD scores and Tecta mutant mice present with seizures. Multiple lines of evidence indicate association of the above genes with WS, which is worth further functional studies.
This is the first WES study in a Chinese WS cohort. We found 112 candidate variants in 89 candidate genes. Bioinformatic analysis identified 17 highlight candidates, the majority of which are calcium‐signaling pathway and mental retardation genes. A larger cohort, advanced bioinformatic analysis methods, and functional studies would help to identify novel WS genes. Furthering the field will improve our understanding of epileptic encephalopathy and neural development and lead to improvements in genetic diagnosis, treatment, and development of new drugs for patients with neuropsychiatric diseases.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the patients and relatives for their participation in this study. We thank Dr. Yongyi Zou and Chuan Jiao for providing help for GOLGA5‐related experiments and bioinformatics analysis. This work was supported by grants from the National Natural Science Foundation of China (NO. 81771408, NO. 81771409, NO. 81701541, NO. 81671297, NO. 81370771, NO. 81371434, and NO. 81301031), The National Key Research and Development Program of China (NO. 2016YFC1306202; NO. 2016YFC0904400).
Peng J, Wang Y, He F, et al. Novel West syndrome candidate genes in a Chinese cohort. CNS Neurosci Ther. 2018;24:1196–1206. 10.1111/cns.12860
The last two authors contributed equally to this work.
Contributor Information
Fei Yin, Email: yinfei@csu.edu.cn.
Nan Pang, Email: nanpang@csu.edu.cn.
REFERENCES
- 1. Zupanc ML. Infantile spasms. Expert Opin Pharmacol. 2005;4:2039‐2048. [DOI] [PubMed] [Google Scholar]
- 2. Wilmshurst JM, Gaillard WD, Vinayan KP, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia. 2015;56:1185‐1197. [DOI] [PubMed] [Google Scholar]
- 3. Carmant L. Infantile spasms: West syndrome. Arch Neurol. 2002;59:317‐318. [DOI] [PubMed] [Google Scholar]
- 4. Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Consortium E , Project EP , Consortium E . De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95:360‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma Y, Chen C, Wang Y, et al. Analysis copy number variation of Chinese children in early‐onset epileptic encephalopathies with unknown cause. Clin Genet. 2016;90:428‐436. [DOI] [PubMed] [Google Scholar]
- 7. Arafat A, Jing P, Ma Y, et al. Unexplained early infantile epileptic encephalopathy in Han Chinese children: next‐generation sequencing and phenotype enriching. Sci Rep. 2017;7:46227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gai N, Jiang C, Zou YY, Zheng Y, Liang DS, Wu LQ. Novel SIL1 nonstop mutation in a Chinese consanguineous family with Marinesco‐Sjogren syndrome and Dandy‐Walker syndrome. Clin Chim Acta. 2016;458:1‐4. [DOI] [PubMed] [Google Scholar]
- 9. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang C, Gai N, Zou Y, et al. WDR73 missense mutation causes infantile onset intellectual disability and cerebellar hypoplasia in a consanguineous family. Clin Chim Acta. 2017;464:24‐29. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, He F, Yin F, et al. The use of targeted genomic capture and massively parallel sequencing in diagnosis of Chinese Leukoencephalopathies. Sci Rep. 2016;6:35936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harmar AJ, Hills RA, Rosser EM, et al. IUPHAR‐DB: the IUPHAR database of G protein‐coupled receptors and ion channels. Nucleic Acids Res. 2009;37:D680‐D685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrovski S, Kury S, Myers CT, et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am J Hum Genet. 2016;98:1001‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Froyen G, Corbett M, Vandewalle J, et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet. 2008;82:432‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O'Rawe JA, Wu Y, Dörfel MJ, et al. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. 2015;97:922‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ouyang Q, Nakayama T, Baytas O, et al. Mutations in mitochondrial enzyme GPT2 cause metabolic dysfunction and neurological disease with developmental and progressive features. Proc Natl Acad Sci U S A. 2016;113:E5598‐E5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wen J, Osumi T, Hashimoto T, Ogata M. Molecular analysis of human acatalasemia. J Mol Biol. 1990;211:383‐393. [DOI] [PubMed] [Google Scholar]
- 23. Dhitavat J, Macfarlane S, Dode L, et al. Acrokeratosis verruciformis of Hopf is caused by mutation in ATP2A2: evidence that it is allelic to Darier's disease. J Invest Dermatol. 2003;120:229‐232. [DOI] [PubMed] [Google Scholar]
- 24. Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat. 2014;35:1046‐1059. [DOI] [PubMed] [Google Scholar]
- 25. Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19:594‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beerman RW, Jongens TA. A non‐canonical start codon in the Drosophila fragile X gene yields two functional isoforms. Neuroscience. 2011;181:48‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kidd SA, Lachiewicz A, Barbouth D, et al. Fragile X syndrome: a review of associated medical problems. Pediatrics. 2014;134:995‐1005. [DOI] [PubMed] [Google Scholar]
- 28. Oguro‐Ando A, Rosensweig C, Herman E, et al. Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol Psychiatry. 2015;20:1069‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP‐related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A. 2001;98:8844‐8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siomi MC, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among t. Mol Cell Biol. 1996;16:3825‐3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra‐1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887‐898. [DOI] [PubMed] [Google Scholar]
- 32. Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053‐1066. [DOI] [PubMed] [Google Scholar]
- 33. Bozdagi O, Sakurai T, Dorr N, Pilorge M, Takahashi N. Haploinsufficiency of Cyfip1 produces fragile X‐like phenotypes in mice. PLoS One. 2012;7:e42422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han K, Chen H, Gennarino VA, Richman R, Lu HC. Fragile X‐like behaviors and abnormal cortical dendritic spines in Cytoplasmic FMR1‐interacting protein 2‐mutant mice. Hum Mol Genet. 2015;24:1813‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sahoo T. Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype‐phenotype correlations. J Med Genet. 2006;43:512‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Butler MG. Clinical and genetic aspects of the 15q11.2 BP1‐BP2 microdeletion disorder. J Intellect Disabil Res. 2017;61:568‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buiting K. Prader‐Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365‐376. [DOI] [PubMed] [Google Scholar]
- 38. Peters SU, Horowitz L, Barbieri‐Welge R, Taylor JL, Hundley RJ. Longitudinal follow‐up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class. J Child Psychol Psychiatry. 2012;53:152‐159. [DOI] [PubMed] [Google Scholar]
- 39. Abekhoukh S, Bardoni B. CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome. Front Cell Neurosci. 2014;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ingason A, Kirov G, Giegling I, et al. Maternally derived microduplications at 15q11‐q13: implication of imprinted genes in psychotic illness. Am J Psychiatry. 2011;168:408‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riikonen RS, Wallden T, Kokkonen H. Infantile spasms and 15q11.2q13.1 chromosome duplication in two successive generations. Eur J Paediatr Neurol. 2016;20:164‐167. [DOI] [PubMed] [Google Scholar]
- 42. Kurian MA, Meyer E, Vassallo G, et al. Phospholipase C beta 1 deficiency is associated with early‐onset epileptic encephalopathy. Brain. 2010;133:2964‐2970. [DOI] [PubMed] [Google Scholar]
- 43. Epi4K Consortium . De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. 2016;99:287‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li D, Yuan H, Ortiz‐Gonzalez XR, et al. GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet. 2016;99:802‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy‐aphasia spectrum disorders. Nat Genet. 2013;45:1073‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;45:1061‐1066. [DOI] [PubMed] [Google Scholar]
- 47. Chen W, Shieh C, Swanger SA, et al. GRIN1 mutation associated with intellectual disability alters NMDA receptor trafficking and function. J Hum Genet. 2017;62:589‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jouvenceau A, Eunson LH, Spauschus A, et al. Human epilepsy associated with dysfunction of the brain P/Q‐type calcium channel. Lancet. 2001;358:801‐807. [DOI] [PubMed] [Google Scholar]
- 49. Imbrici P, Jaffe SL, Eunson LH, et al. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain. 2004;127:2682‐2692. [DOI] [PubMed] [Google Scholar]
- 50. Pisani A, Bonsi P, Martella G, et al. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45:719‐728. [DOI] [PubMed] [Google Scholar]
- 51. Chernorudskiy AL, Zito E. Regulation of calcium homeostasis by ER redox: a close‐up of the ER/mitochondria connection. J Mol Biol. 2017;429:620‐632. [DOI] [PubMed] [Google Scholar]
- 52. Abu‐Omar N, Das J, Szeto V, Feng ZP. Neuronal ryanodine receptors in development and aging. Mol Neurobiol. 2018;55:1183‐1192. [DOI] [PubMed] [Google Scholar]
- 53. Reddish FN, Miller CL, Gorkhali R, Yang JJ. Calcium dynamics mediated by the endoplasmic/sarcoplasmic reticulum and related diseases. Int J Mol Sci. 2017;18:pii: E1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dahiyat M, Cumming A, Harrington C, et al. Association between Alzheimer's disease and the NOS3 gene. Ann Neurol. 1999;46:664‐667. [DOI] [PubMed] [Google Scholar]
- 55. Oliveira‐Paula GH, Lacchini R, Tanus‐Santos JE. Endothelial nitric oxide synthase: from biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene. 2016;575:584‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cossenza M, Socodato R, Portugal CC, et al. Nitric oxide in the nervous system: biochemical, developmental, and neurobiological aspects. Vitam Horm. 2014;96:79‐125. [DOI] [PubMed] [Google Scholar]
- 57. Burwinkel B, Hu B, Schroers A, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003;11:516‐526. [DOI] [PubMed] [Google Scholar]
- 58. Ramos PS, Sajuthi S, Langefeld CD, Walker SJ. Immune function genes CD99L2, JARID2 and TPO show association with autism spectrum disorder. Mol Autism. 2012;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials