

Abstract
Acromegalic cardiomyopathy is the leading cause of morbidity and all‐cause mortality in patients with acromegaly. Though acromegaly is a rare condition, the associated derangements are vast and severe. Stemming from an increase in circulating growth hormone (GH) and insulin‐like growth factor‐1 levels (IGF‐1), acromegalic cardiomyopathy results in pathological changes in myocyte growth and structure, cardiac contractility, and vascular function. These molecular changes manifest commonly as biventricular hypertrophy, diastolic and systolic dysfunction, and valvular regurgitation. Early recognition of the condition is paramount, though the insidious progression of the disease commonly results in a late diagnosis. Biochemical testing, based on IGF‐1 measurements, is the gold standard of diagnosis. Management should be centered on normalizing serum levels of both IGF‐1 and GH. Transsphenoidal resection remains the most cost‐effective and permanent treatment for acromegaly, though medical therapy possesses benefit for those who are not surgical candidates. Ultimately, achieving control of hormone levels results in a severe reduction in mortality rate, underscoring the importance of early recognition and treatment.
Keywords: Acromegalic Cardiomyopathy, Acromegaly, Diagnosis, Epidemiology, Clinical Manifestations, Prognosis
1. INTRODUCTION
Cardiac complications associated with acromegaly are collectively known as acromegalic cardiomyopathy and involve nearly all aspects of the cardiovascular system: myocyte and intercellular myocardial composition, systolic and diastolic ventricular function, valvular heart disease, and heart electrical disturbances. The fundamental physiologic derangement in acromegaly is a pathologically elevated serum concentration of circulating growth hormone (GH) and insulin‐like growth factor‐1 (IGF‐1), most commonly produced by a pituitary tumor.1 Because the diagnosis of acromegaly is often delayed by many years due to insidious progression of the disease process, the necessity of early recognition and management is paramount to avert chronic cardiovascular damage.
2. EPIDEMIOLOGY
Acromegaly is a relatively rare condition with a prevalence of approximately 60 cases per million and an incidence of 3 to 4 new cases per million per year in the United States.2 The slow progression of acromegaly leads to a diagnosis 4 to 10 years after onset, at an average age of 40 years in both men and women.3 Though males and females are equally affected, younger patients tend to possess more aggressive tumors and consequently have higher GH concentrations.3
Increases in GH and IGF‐1 result in facial, extremity, cardiac, rheumatologic, respiratory, and metabolic derangements.4 Though acromegaly possesses a wide range of characteristic symptoms, acromegalic cardiomyopathy specifically refers to the associated cardiac conditions of the parent disease. Although only two‐thirds of patients show evidence of cardiomyopathy at the time acromegaly is diagnosed, about 90% of patients will develop cardiomyopathy in their lifetime.5 In a study of 37 patients, the most common cardiac complications were left ventricular hypertrophy (56.8%), diastolic dysfunction (51.4%), and hypertension (87.5%).6
3. PATHOGENESIS
GH production is stimulated by a variety of signals, including growth hormone–releasing hormone (GHRH) and is suppressed by somatostatin signaling. Most acromegalic patients have a growth hormone–secreting pituitary adenoma.7 In rare cases, excess GHRH can originate from a neuroendocrine tumor or hypothalamic tumor, but ectopic GH can also derive from a different tumor, such as a bronchial carcinoid tumor. Elevated GH stimulates the liver to synthesize increased IGF‐1, a mediator of muscle growth, which contributes to the somatic manifestations of the disease.8
The GH/IGF‐1 axis influences 3 major aspects of the cardiovascular system: myocyte growth and structure, cardiac contractility, and vascular function (Figure 1). The major processes of myocyte growth regulation include an increase in amino acid uptake/protein synthesis, cardiomyocyte size, and cardiac muscle gene expression.1 The level of serum GH/IGF‐1 activates cardiac growth in a parallel fashion, resulting in a characteristic, biventricular hypertrophic response.9 Overexpression of IGF‐1 independently increases the transcription of major cardiac muscle–specific genes, including troponin 1, myosin light chain‐2, α actin, and IGF1‐binding protein, leading to fibrosis and sarcomerogenesis.1 IGF‐1 in particular promotes collagen synthesis by fibroblasts, whereas GH increases the rate of the cardiac collagen deposition.1 With this interstitial remodeling, ventricular relaxation is impaired, eventually resulting in initial diastolic dysfunction and ensuing systolic dysfunction.10 Furthermore, the inhibitory influence of IGF‐1 on apoptosis prevents myocyte loss, adding to the development of an acromegalic heart when present in excess.11 In addition to possessing both GH and IGF‐1 receptors, myocardium and endothelium can produce IGF‐1 as a result of a GH‐mediated autocrine and paracrine response, thus exacerbating the effects of a GH‐producing tumor.10
Figure 1.
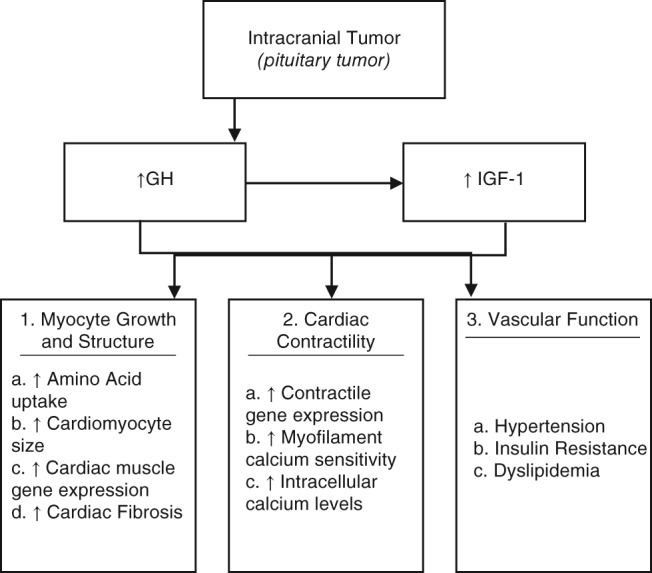
Pathogenesis of acromegalic cardiomyopathy. Abbreviations: GH, growth hormone; IGF‐1, insulin‐like growth factor 1
GH/IGF‐1 affects cardiac contractility in a similar manner to its effect on myocyte growth and structure. IGF‐1 promotes transcription of certain contractile genes, and within myocyte cells, the GH/IGF‐1 axis also increases intracellular calcium levels.12
Vascular‐related comorbidities associated with acromegaly include hypertension, insulin resistance, and dyslipidemia.4 Hypertension is seen in 30% to 45% of cases, primarily due to the antinatriuretic action of GH body fluid expansion, increased arterial stiffness, and endothelial dysfunction, though the exact mechanism of which is unclear.13 Also well documented is the role of GH/IGF‐1 in insulin resistance; the resulting hyperglycemia is reversible with normalization of GH levels. In addition, GH acts as a regulator of lipolysis, leading to dyslipidemia.14
4. CLINICAL MANIFESTATIONS
A clear set of clinical manifestations arise in acromegalic cardiomyopathy, the most common being biventricular hypertrophy, diastolic and systolic dysfunction, and valvular regurgitation (Table 1). Because there is a tendency to diagnose acromegaly late due to its subtle onset, many of these characteristic changes develop over years prior to recognition.
Table 1.
Clinical stages of acromegalic cardiomyopathy1
| Stage | Years of Active Disease | Characteristics |
|---|---|---|
| Early | <5 years | Enhanced contractility, decreased peripheral vascular resistance, increased cardiac output |
| Middle | >5 years | Biventricular hypertrophy, diastolic dysfunction, impaired cardiac performance |
| Late | >15 years | Systolic dysfunction, diastolic dysfunction (congestive heart failure), valvular disease, coronary artery disease, arrhythmias |
The thickening of cardiac walls is a result of chronic growth stimulus. This concentric hypertrophy results from an increase in cardiac myocyte cellular width due to assembly of new sarcomeres in parallel, a commonality between acromegalic and other types of cardiac pathology.15 Though the hypertrophy of both ventricular walls increases significantly, left ventricular hypertrophy and interventricular septal thickening are most frequent, found in over half of acromegalic patients.16 The severity of these derangements is primarily determined by aging and the duration of acromegaly, but it is often exacerbated by long‐standing hypertension.17
Though many of the clinical presentations of acromegalic cardiomyopathy are similar to the abnormalities that are produced by chronic hypertension, multiple analyses have revealed certain effects on cardiac morphology and function specific to acromegaly, including impaired systolic function and increased diastolic filling time.18 The main histological finding unique to acromegalic cardiomyopathy is the widespread accretion of interstitial fibrosis that impairs heart architecture.17 This leads to abnormal ventricular relaxation that causes diastolic dysfunction, though contractility and ejection fraction tend to remain unchanged.19 Isovolumic relaxation time is commonly prolonged, and transvalvular (mitral and tricuspid) flow velocities are markedly decreased.20 Similarly, a 19% increase in the likelihood of developing regurgitant valvular dysfunction has been reported in those with acromegaly, likely due to abnormalities in matrix regulation and subsequent myxoid degeneration.21 In this regard, pathologic aortic valve regurgitation has been found to be more common than mitral regurgitation (20% vs 5%). 21
Abnormalities of cardiac rhythm are not as frequent as the aforementioned cardiac abnormalities. Ectopic beats, paroxysmal atrial fibrillation, and supraventricular tachycardia, sick sinus syndrome, ventricular tachycardia, and bundle branch blocks are all more frequent in acromegaly than in controls.17
A literature review on pertinent clinical case series was conducted to outline the major cardiac‐related outcomes documented in acromegalic patients (Table 2). The majority of these studies found similar differentiating characteristics between those with acromegalic cardiomyopathy and age‐ and sex‐matched controls.
Table 2.
Cardiac‐related outcomes in acromegalic patients 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54
| Author (Year) | PMID | No. of Patients | Statistically Significant Differences (Compared to Controls) |
|---|---|---|---|
| Iida et al (1990) | 2145456 | 8 | ↑: LV mass (n = 8); end‐diastolic diameter (n = 5), concentric LV hypertrophy (n = 3), normal fractional shortening of LV (n = 7) |
| Mercuro et al (2000) | 11022178 | 18 | ↑: LV mass, isovolumetric relaxation time; unchanged EF |
| Twardowski et al (2001) | 11926140 | 28 | ↑: LV mass; ventricular premature complexes (n = 18), ventricular filling dysfunction (n = 25), increased frequency of ectopic beats, normal LV ejection fraction |
| Damjanovic et al (2002) | 12034409 | 102 | ↑: LV mass; heart failure (n = 10) |
| Herrmann et al (2002) | 12030909 | 13 | ↓: Early diastolic annular velocity |
| Vianna et al (2002) | 12040351 | 15 | ↑: LV mass, relative wall thickness; diastolic dysfunction |
| Galderisi et al (2006) | 16584510 | 30 | ↑: LV mass; diastolic dysfunction (impaired Doppler indexes of LV and RV diastolic function without any difference in global systolic function) |
| Colao et al (2007) | 17646725 | 57 | ↑: LV mass, lipid levels, insulin levels |
| van der Klaauw et al (2008) | 18495693 | 37 | ↑: Aortic root diameter |
| Kiris et al (2013) | 23254835 | 30 | ↑: Left ventricular dyssynchrony |
| Unubol et al (2014) | 24901024 | 28 | ↑: Aortic pulse wave velocity, mean platelet volume |
| Guo et al (2015) | 26600803 | 108 | ↑: LV end‐diastolic and end‐systolic diameter, myocardial thickness, great vessel diameter; diastolic dysfunction, unchanged systolic function |
| Orosz et al (2015) | 25915951 | 30 | ↑: LV mass, relative wall thickness; short‐term beat‐to‐beat QT variability |
| Sanchez‐Ortiga et al (2015) | 26122359 | 32 | ↑: LV mass; diastolic dysfunction, sleep apnea‐hypopnea syndrome (n = 26) |
Abbreviations: ↑ or ↓, variable that increased or decreased in measurement; LV, left ventricle; n, number of patients; RV, right ventricle.
5. DIAGNOSIS
Early recognition of acromegalic cardiomyopathy is essential given the frequency with which diagnosis is commonly delayed. Internists most commonly diagnose acromegaly (40%), though ophthalmologists, dental surgeons, gynecologists, rheumatologists, and sleep specialists often identify the condition independently, likely due to recognition of the systemic manifestations of the disease relevant to their respective areas of interest.5
Given that the early symptoms of acromegaly are not particularly sensitive, a focused history and physical examination are the most effective means of confirming suspicion of the condition. Rather than relying on the assessment of comorbidities, which often appear late in the disease, familiarization of the phenotype of the general acromegalic condition may be fruitful in early detection.2 External manifestations indicative of acromegaly include macroglossia, jaw malocclusion, sleep disturbances, ventilatory dysfunction, arthritis, carpal tunnel syndrome, decreased libido, and menstrual abnormalities.2 These developments often arise subtly over time, often unbeknownst to the patient. Inquiries such as those concerning a changing ring or shoe size, pain when writing, or shifting teeth, in addition to a comparison of current facial structure to past photographs, may be valuable in confirming clinical suspicion. A striking example of the clinical consequences resulting from overlooking acromegaly as the etiology of heart failure is provided by the case of a 35‐year‐old man with repetitive admissions for cardiac decompensation over a 15‐year period (Figure 2). When acromegalic heart failure was finally identified, eventual transsphenoidal resection of the microadenoma resulted in normalization of hormone levels and alleviation of the clinical syndrome.4
Figure 2.
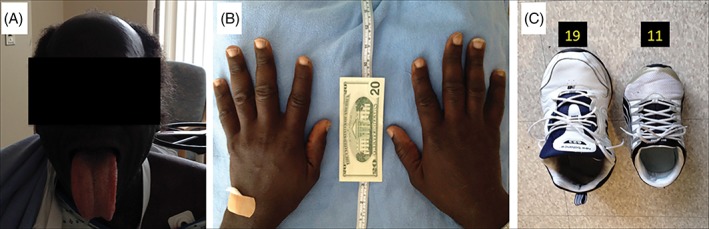
Physical manifestations of acromegaly.4 Patient is a 6 ft 8 in., 280‐pound African American male. (A) The patient had bossing and macroglossia. (B) Enlargement of the hands due to swelling of the soft tissue, and growth of bone, cartilage, and muscle. (C) The patient's shoe, size 19, compared with a size‐11 shoe
Eventually, untreated acromegaly can and will manifest into heart failure. Most patients fail to present until the late stage of acromegalic cardiomyopathy, at which point systolic dysfunction is found in conjunction with a host of the previously noted cardiac abnormalities. Typical symptoms of right heart failure include peripheral edema, abdominal fullness, nausea, and weight gain, whereas left heart failure leads to dyspnea on exertion, rest, and during sleep, orthopnea, and chest pain.5 Cardiac examination may reveal third and fourth heart sounds, pulmonary crackles, and left ventricular heave.5 These signs and symptoms are common to virtually all patients with cardiac failure (congestive heart failure [CHF]), presenting a challenge to clinicians to distinguish late‐stage acromegalic cardiomyopathy from other causes of heart failure. Echocardiography is the most important noninvasive tool for confirming the presence and severity of the aforementioned structural and functional cardiac abnormalities. However, the etiology of these abnormalities relies on specific endocrine evidence.
Biochemical testing is based on measurement of IGF‐1, the single best test for the diagnosis of acromegaly, as elevated IGF‐1 is present in almost all acromegalic patients and has a very high sensitivity for detecting the endocrine abnormality (concentrations are age and sex specific).22 However, IGF‐1 results must be age adjusted, as serum IGF‐1 concentrations peak during puberty and decline thereafter, potentially misleading clinicians in classifying the elevated IGF‐1 serum values of elderly acromegaly patients as normal. In addition, some conditions may lead to falsely low IGF‐1 levels, including liver failure, renal failure, and malnutrition. Random serum GH concentrations are not a useful initial diagnostic tool, because normal GH secretion is pulsatile, diurnal, and stimulated by various factors of daily living (fasting, exercise, stress, sleep).23 Nevertheless, dynamic GH testing can serve as a subsequent and confirmatory test for an elevated or equivocal IGF‐1. The gold standard to confirm acromegaly is a 75‐g oral glucose tolerance test. A GH concentration above 1 to 2 ng/mL (depending on the assay) 2 hours after ingestion of 75 g of glucose is consistent with acromegaly.24 After biochemical diagnosis of GH excess, the source must be determined, usually starting with pituitary magnetic resonance imaging (Figure 3).
Figure 3.
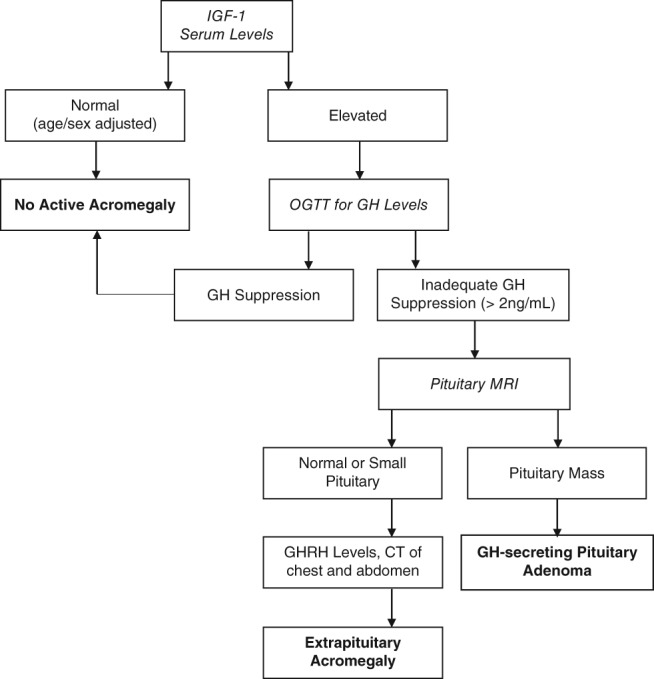
Diagnostic algorithm for acromegaly.40 Abbreviations: CT, computed tomography; GH, growth hormone; GHRH, growth hormone–releasing hormone; IGF‐1, insulin‐like growth factor 1; MRI, magnetic resonance imaging; OGTT, oral glucose tolerance test
6. MANAGEMENT
The goal of management in patients with acromegalic cardiomyopathy is to cure or minimize the parent condition—acromegaly. Different strategies for management can broadly be categorized as either endocrine or cardiac related (see Supporting Figure 1 in the online version of this article).
6.1. Endocrine
As the vast majority of acromegaly is related to a pituitary adenoma, transsphenoidal resection is the most cost‐effective and permanent treatment.25 Surgical cure rates are highly dependent on the tumor characteristics and the experience of the surgeon. More invasive macroadenomas may require a craniotomy. Patients with severe acromegalic cardiomyopathy can potentially benefit from preoperative somatostatin receptor ligands (SRLs) to reduce perioperative morbidity, but this approach has not been well studied. Postoperatively, medical and other adjunctive therapy (eg, stereotactic radiotherapy) may be needed for persistent disease.
Ultimately, the goal is to lower IGF‐1 concentrations to the normal gender‐ and age‐specific reference ranges and achieve a random GH concentration < 1 ng/mL. With a successful resection, GH levels can normalize within hours of surgery, but IGF‐1 levels require weeks to months to normalize. Fluid retention and hyperglycemia may resolve within days, whereas the cardiac complications require months of treatment before improvement. Overall, about 60% of surgically corrected patients can be considered cured.26
For those who are not surgical candidates, SRLs can inhibit GH secretion and may lead to moderate tumor shrinkage. It is important to note that SRLs are costly and can have side effects, including nausea and hyperglycemia. Medical therapy alone can relieve acromegalic symptoms and control tumor growth in around 50% to 60% of patients.27 A dopamine agonist (DA), such as cabergoline, may be added in conjunction with an SLR or used alone in milder cases. The newest medical therapy is pegvisomant, a GH analogue, which functions as a competitive GH receptor antagonist. Whereas SRLs and DAs inhibit GH secretion, pegvisomant prevents GH‐stimulated production of IGF‐1.26 Theoretically, the higher GH levels could lead to an increase in tumor size, but this is uncommon. However, tumor size should be monitored at regular intervals to ensure detection of any tumor growth.
Achieving control of GH levels has been successful in improving prognosis; reduction of serum GH to less than 1 μg/L projects a mortality rate almost identical to age‐matched controls.28 IGF‐1 is not an independent prognostic indicator of mortality in acromegaly, though normalization of serum IGF‐1 is weakly correlated with a reduction in mortality rate.28
6.2. Cardiac
The cardiovascular benefits of controlling and normalizing GH and IGF‐1 levels are notable. Successful surgery or pharmacological therapy have been shown to significantly decrease left ventricular hypertrophy and improve left ventricular filling.29 Despite mitigation of cardiac hypertrophy, cardiac systolic function is reported to be unaffected by GH/IGF‐1 suppression.30 Nevertheless, more recent studies have shown that approximately half of patients who achieve GH/IGF‐1 control will reobtain a normal left ventricular ejection fraction response at peak exercise.31
Successful case reports have supported the use of traditional heart failure therapy to further improve cardiovascular function, as GH/IGF‐1 control alone is insufficient. Treatment of CHF, present in around 3% of late‐stage acromegalic cardiomyopathy patients, and involves strict volume and pressure regulation.32 These regimens, consisting of angiotensin receptor blockers (ARBs), β‐adrenergic antagonists, diuretics, and calcium channel blockers, have normalized blood pressure and increased left ventricular ejection fraction in acromegalic patients.33 Angiotensin‐converting enzyme inhibitors (ACEi) and β‐adrenergic antagonists specifically limit fibrotic cardiac remodeling.33 Additionally, a recent preliminary study has reported that ACEi/ARBs improve end‐diastolic and end‐systolic volumes in acromegalic patients.34 More severe heart failure may require inotropic agents, parenteral diuretics, or cardiac resynchronization therapy, though the use of these interventions has not been adequately studied in acromegalic heart failure.35 Although these treatments have been effective in improving short‐term cardiovascular status in nonacromegalic decompensated cardiomyopathy, the long‐term prognosis of CHF in acromegalic patients remains poor; the mortality rate is 25% at 1 year and over 35% at 5 years.36
To combat the potentially life‐threatening arrhythmias present in acromegalic cardiomyopathy, implantable cardioverter‐defibrillators (ICDs) have been implemented for both primary and secondary prevention of sudden cardiac death.37 However, due to the relatively recent incorporation of ICDs as primary and secondary preventative measures in acromegalic cardiomyopathy, there are no guidelines regarding cardiac magnetic resonance imaging (CMR) in risk stratification for ICDs. Given the lack of specific findings on CMR that distinguishes acromegalic cardiomyopathy, its utility for risk stratification may be limited. Nevertheless, ICD implantation should occur for secondary prevention because the source of the ventricular tachycardia often seen in these patients is myocardial fibrosis and scar as a result of the underlying acromegalic pathology. Whether implementation of heart failure or endocrine therapy reduces the future risk in patients with aborted sudden cardiac arrest is unknown.
7. PROGNOSIS
Due to the vast array of comorbidities associated with acromegaly, the mortality rates of acromegalic patients are increased 2‐ to 3‐fold compared to healthy controls, and life expectancy is decreased an average of 10 years.38 Cardiovascular disease is the most significant contributor to increasing morbidity and mortality in this population (>1.7 increase in standard mortality rate).39 With acromegaly often diagnosed late in its progression, the related cardiac complications may contribute to the death of up to 60% of patients, and almost all patients presenting with a history of cardiovascular disease die within 15 years.38
8. CONCLUSION
Acromegalic cardiomyopathy, though rare, is the leading cause of morbidity and all‐cause mortality in patients with acromegaly. Although it affects the entire cardiovascular system, this condition is often a challenge for clinicians, as there are few specifically identifiable characteristics that allow discrimination of acromegalic heart failure from other etiologies of cardiac decompensation. Current recommendations for diagnosis and management include both endocrine and cardiac approaches. Re‐establishing age‐ and sex‐appropriate levels of GH and IGF‐1, in addition to addressing relevant cardiovascular risk factors, are required to improve outcomes of these patients.
Conflicts of interest
The authors declare no potential conflicts of interest.
Supporting information
SUPPORTING FIGURE 1 Treatment of acromegalic cardiomyopathy.
Sharma AN, Tan M, Amsterdam EA, Singh GD. Acromegalic cardiomyopathy: Epidemiology, diagnosis, and management. Clin Cardiol. 2018;41:419–425. 10.1002/clc.22867
References
- 1. Castellano G, Affuso F, Di Conza P, et al. The GH/IGF‐1 Axis and Heart Failure. Curr Cardiol Rev. 2009;5:203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lugo G, Pena L, Cordido F. Clinical manifestations and diagnosis of acromegaly. Int J Endocrinol. 2012;2012:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary 1999;2:29–41. [DOI] [PubMed] [Google Scholar]
- 4. Mantri NM, Amsterdam E, Tan M, et al. Power failure: acromegalic cardiomyopathy. Am J Med. 2016;129:674–7. [DOI] [PubMed] [Google Scholar]
- 5. Nixon J V. The AHA Clinical Cardiac Consult Lippincott Williams & Wilkins; 2010.
- 6. Nascimento GC, Oliveira D, Torres M, et al. Acromegalic cardiomyopathy in an extensively admixed population: is there a role for GH/IGF‐I axis? Clin Endocrinol (Oxf). 2013;78:94–101. [DOI] [PubMed] [Google Scholar]
- 7. Lopes MBS. Growth hormone‐secreting adenomas: pathology and cell biology. Neurosurg Focus. 2010;29:E2. [DOI] [PubMed] [Google Scholar]
- 8. Grey A, Chen Q, Xu X, et al. Parallel phosphatidylinositol‐3 kinase and p42/44 mitogen‐activated protein kinase signaling pathways subserve the mitogenic and antiapoptotic actions of insulin‐like growth factor I in osteoblastic cells. Endocrinology. 2003;144:4886–93. [DOI] [PubMed] [Google Scholar]
- 9. Timsit J, Riou B, Bertherat J, et al. Effects of chronic growth hormone hypersecretion on intrinsic contractility, energetics, isomyosin pattern, and myosin adenosine triphosphatase activity of rat left ventricle. J Clin Invest. 1990;86:507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saccà L, Cittadini A, Fazio S. Growth Hormone and the Heart. Endocr Rev. 1994;15:555–73. [DOI] [PubMed] [Google Scholar]
- 11. Frasca F, Pandini G, Sciacca L, et al. The role of insulin receptors and IGF‐I receptors in cancer and other diseases. Arch. Physiol Biochem. 2008;114:23–37. [DOI] [PubMed] [Google Scholar]
- 12. McMullen JR, Shioi T, Huang W‐Y, et al. The insulin‐like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3‐kinase(p110alpha) pathway. J Biol Chem. 2004;279:4782–93. [DOI] [PubMed] [Google Scholar]
- 13. Kamenicky P, Blanchard A, Frank M, et al. Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J Clin Endocrinol Metab. 2011;96:2127–35. [DOI] [PubMed] [Google Scholar]
- 14. Romero MJ, Lucas R, Dou H, et al. Role of growth hormone‐releasing hormone in dyslipidemia associated with experimental type 1 diabetes. Proc Natl Acad Sci U S A. 2016;113:1895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Epstein FH, Hunter JJ, Chien KR. Signaling Pathways for Cardiac Hypertrophy and Failure. N Engl J Med. 1999;341:1276–83. [DOI] [PubMed] [Google Scholar]
- 16. Hradec J, Marek J, Kral J, et al. Long‐term echocardiographic follow‐up of acromegalic heart disease. Am J Cardiol. 1993;72:205–10. [DOI] [PubMed] [Google Scholar]
- 17. Colao A, Marzullo P, Di Somma C, et al. Growth hormone and the heart. Clin Endocrinol (Oxf). 2001;54:137–54. [DOI] [PubMed] [Google Scholar]
- 18. López‐Velasco R, Escobar‐Morreale HF, Vega B, et al. Cardiac Involvement in Acromegaly: Specific Myocardiopathy or Consequence of Systemic Hypertension? J Clin Endocrinol Metab. 1997;82:1047–53. [DOI] [PubMed] [Google Scholar]
- 19. Sicolo N, Bui F, Sicolo M, et al. Acromegalic cardiopathy: A left ventricular scintigraphic study. J Endocrinol Invest. 1993;16:123–7. [DOI] [PubMed] [Google Scholar]
- 20. Fazio S, Cittadini A, Sabatini D, et al. Evidence for biventricular involvement in acromegaly: a Doppler echocardiographic study. Eur Heart J. 1993;14:26–33. [DOI] [PubMed] [Google Scholar]
- 21. Pereira AM, van Thiel SW, Lindner JR, et al. Increased prevalence of regurgitant valvular heart disease in acromegaly. J Clin Endocrinol Metab. 2004;89:71–5. [DOI] [PubMed] [Google Scholar]
- 22. Stoffel‐Wagner B, Springer W, Bidlingmaier F, et al. A comparison of different methods for diagnosing acromegaly. Clin Endocrinol (Oxf). 1997;46:531–7. [DOI] [PubMed] [Google Scholar]
- 23. Iranmanesh A, Grisso B, Veldhuis JD. Low basal and persistent pulsatile growth hormone secretion are revealed in normal and hyposomatotropic men studied with a new ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab. 1994;78:526–35. [DOI] [PubMed] [Google Scholar]
- 24. Carmichael JD, Bonert VS, Mirocha JM, et al. The utility of oral glucose tolerance testing for diagnosis and assessment of treatment outcomes in 166 patients with acromegaly. J Clin Endocrinol Metab. 2009;94:523–7. [DOI] [PubMed] [Google Scholar]
- 25. Zada G, Kelly DF, Cohan P, et al. Endonasal transsphenoidal approach to treat pituitary adenomas and other sellar lesions: an assessment of efficacy, safety, and patient impressions of the surgery. J Neurosurg. 2003;98:350–8. [DOI] [PubMed] [Google Scholar]
- 26. Trainer PJ, Drake WM, Katznelson L, et al. Treatment of Acromegaly with the Growth Hormone–Receptor Antagonist Pegvisomant. N Engl J Med. 2000;342:1171–7. [DOI] [PubMed] [Google Scholar]
- 27. Chanson P. Medical treatment of acromegaly with dopamine agonists or somatostatin analogs. Neuroendocrinology 2015;103:50–8. [DOI] [PubMed] [Google Scholar]
- 28. Holdaway IM, Rajasoorya RC, Gamble GD. Factors Influencing Mortality in Acromegaly. J Clin Endocrinol Metab. 2004;89:667–74. [DOI] [PubMed] [Google Scholar]
- 29. Lim MJ, Barkan AL, Buda AJ. Rapid reduction of left ventricular hypertrophy in acromegaly after suppression of growth hormone hypersecretion. Ann Intern Med. 1992;117:719–26. [DOI] [PubMed] [Google Scholar]
- 30. Thuesen L, Christensen SE, Weeke J, et al. The Cardiovascular Effects of Otreotide Treatment in Acromegaly: An Echocardiographic Study. Clin Endocrinol (Oxf). 1989;30:619–25. [DOI] [PubMed] [Google Scholar]
- 31. Colao A, Cuocolo A, Marzullo P, et al. Is the acromegalic cardiomyopathy reversible? Effect of 5‐year normalization of growth hormone and insulin‐like growth factor I levels on cardiac performance. J Clin Endocrinol Metab. 2001;86:1551–7. [DOI] [PubMed] [Google Scholar]
- 32. Dal J, Feldt‐Rasmussen U, Andersen M, et al. Acromegaly incidence, prevalence, complications and long‐term prognosis: a nationwide cohort study. Eur J Endocrinol. 2016;175:181–90. [DOI] [PubMed] [Google Scholar]
- 33. Anumah F, Danbauchi S, Garko S. Acromegaly presenting as cardiac failure. Ethn Dis. 2008;18:104–6. [PubMed] [Google Scholar]
- 34. Thomas J, Dattani A, Zemrak F, et al. Renin‐Angiotensin System Blockade Improves Cardiac Indices in Acromegaly Patients. Exp Clin Endocrinol Diabetes. 2017;125:365–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwarz ER, Jammula P, Gupta R, et al. A case and review of acromegaly‐induced cardiomyopathy and the relationship between growth hormone and heart failure: cause or cure or neither or both? J Cardiovasc Pharmacol Ther. 2006;11:232–44. [DOI] [PubMed] [Google Scholar]
- 36. Bihan H, Espinosa C, Valdes‐Socin H, et al. Long‐term outcome of patients with acromegaly and congestive heart failure. J Clin Endocrinol Metab. 2004;89:5308–13. [DOI] [PubMed] [Google Scholar]
- 37. Bardy GH, Smith WM, Hood MA, et al. An entirely subcutaneous implantable cardioverter–defibrillator. N Engl J Med. 2010;363:36–44. [DOI] [PubMed] [Google Scholar]
- 38. Colao A. 5 Long‐term acromegaly and associated cardiovascular complications: a case‐based review. Best Pract. Res Clin Endocrinol Metab. 2009;23:S31–8. [DOI] [PubMed] [Google Scholar]
- 39. Orme SM, McNally RJQ, Cartwright RA, et al. Mortality and Cancer Incidence in Acromegaly: A Retrospective Cohort Study. J Clin Endocrinol Metab. 1998;83:2730–4. [DOI] [PubMed] [Google Scholar]
- 40. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119:3189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iida K, Koide Y, Sugishita Y, et al. Follow‐up study of the heart in acromegaly: pre‐ and post‐operative evaluation. Jpn J Med. 1990;29:22–6. [DOI] [PubMed] [Google Scholar]
- 42. Mercuro G, Zoncu S, Colonna P, et al. Cardiac dysfunction in acromegaly: evidence by pulsed wave tissue Doppler imaging. Eur J Endocrinol. 2000;143:363–9. [DOI] [PubMed] [Google Scholar]
- 43. Twardowski R, Mandecki T, Mizia‐Stec K, et al. [Left and right ventricular function in acromegalic patients]. Pol Arch Med Wewn. 2001;106:669–74. [PubMed] [Google Scholar]
- 44. Damjanovic SS, Neskovic AN, Petakov MS, et al. High output heart failure in patients with newly diagnosed acromegaly. Am J Med. 2002;112:610–6. [DOI] [PubMed] [Google Scholar]
- 45. Herrmann BL, Bruch C, Saller B, et al. Acromegaly: evidence for a direct relation between disease activity and cardiac dysfunction in patients without ventricular hypertrophy. Clin Endocrinol (Oxf). 2002;56:595–602. [DOI] [PubMed] [Google Scholar]
- 46. Vianna CB, Vieira MLC, Mady C, et al. Treatment of acromegaly improves myocardial abnormalities. Am Heart J. 2002;143:873–6. [DOI] [PubMed] [Google Scholar]
- 47. Galderisi M, Vitale G, Bianco A, et al. Pulsed tissue Doppler identifies subclinical myocardial biventricular dysfunction in active acromegaly. Clin Endocrinol (Oxf). 2006;64:390–7. [DOI] [PubMed] [Google Scholar]
- 48. Colao A, Pivonello R, Spinelli L, et al. A retrospective analysis on biochemical parameters, cardiovascular risk and cardiomyopathy in elderly acromegalic patients. J Endocrinol Invest. 2007;30:497–506. [DOI] [PubMed] [Google Scholar]
- 49. van der Klaauw AA, Bax JJ, Smit JWA, et al. Increased aortic root diameters in patients with acromegaly. Eur J Endocrinol. 2008;159:97–103. [DOI] [PubMed] [Google Scholar]
- 50. Kırış A, Erem C, Turan OE, et al. Left ventricular synchronicity is impaired in patients with active acromegaly. Endocrine 2013;44:200–6. [DOI] [PubMed] [Google Scholar]
- 51. Unubol M, Guney E, Ture M, et al. Mean platelet volume and arterial stiffness in patients with acromegaly. Anadolu Kardiyol Dergisi/The Anatol J Cardiol. 2014;14:456–63. [DOI] [PubMed] [Google Scholar]
- 52. Guo X, Gao L, Zhang S, et al. Cardiovascular System Changes and Related Risk Factors in Acromegaly Patients: A Case‐Control Study. Int J Endocrinol. 2015;2015:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Orosz A, Csajbók É, Czékus C, et al. Increased Short‐Term Beat‐To‐Beat Variability of QT Interval in Patients with Acromegaly. PLoS One 2015;10:e0125639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanchez‐Ortiga R, Climent V, Sanchez‐Tejada L, et al. Severe sleep apnea–hypopnea syndrome is related to left ventricle dysfunction and hypertrophy in acromegalic patients. Endocrinol Y Nutr. 2015;62:366–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING FIGURE 1 Treatment of acromegalic cardiomyopathy.