

Summary
Aims
We aimed to identify a clinically useful biomarker using DNA methylation‐based information to optimize individual treatment of patients with glioblastoma (GBM).
Methods
A six‐CpG panel was identified by incorporating genome‐wide DNA methylation data and clinical information of three distinct discovery sets and was combined using a risk‐score model. Different validation sets of GBMs and lower‐grade gliomas and different statistical methods were implemented for prognostic evaluation. An integrative analysis of multidimensional TCGA data was performed to molecularly characterize different risk tumors.
Results
The six‐CpG risk‐score signature robustly predicted overall survival (OS) in all discovery and validation cohorts and in a treatment‐independent manner. It also predicted progression‐free survival (PFS) in available patients. The multimarker epigenetic signature was demonstrated as an independent prognosticator and had better performance than known molecular indicators such as glioma‐CpG island methylator phenotype (G‐CIMP) and proneural subtype. The defined risk subgroups were molecularly distinct; high‐risk tumors were biologically more aggressive with concordant activation of proangiogenic signaling at multimolecular levels. Accordingly, we observed better OS benefits of bevacizumab‐contained therapy to high‐risk patients in independent sets, supporting its implication in guiding usage of antiangiogenic therapy. Finally, the six‐CpG signature refined the risk classification based on G‐CIMP and MGMT methylation status.
Conclusions
The novel six‐CpG signature is a robust and independent prognostic indicator for GBMs and is of promising value to improve personalized management.
Keywords: bevacizumab, DNA methylation, glioblastomas, prognostication, risk‐score signature
1. INTRODUCTION
Glioblastomas (GBMs) are the most frequent and vicious subtype of all gliomas.1, 2 Molecular and clinical heterogeneity critically hindered better treatment outcomes for this deadly disease. The development of clinical informative biomarkers would be helpful for improving the current management of GBMs.
DNA methylation marks have long been the leading candidates for cancer biomarker discovery.3 Human cancers including GBMs were commonly developed with global hypomethylation of gene‐poor DNA repeats and large hypomethylated blocks of gene regions concurrent with relevant CpGs island (CGI) hypermethylation.4 Those epigenetic abnormalities played crucial roles in determining tumor phenotypic behaviors via regulating gene expression and chromatin organization.5 Early studies with candidate‐gene approaches have identified numerous DNA methylation alterations in key genes (eg, TIMP3, RASSF1A, and p16INK4a) with potential clinical values for GBMs.6 Promoter methylation status of the O‐6‐methylguanine‐DNA methyltransferase (MGMT), encoding a DNA repair enzyme that confers resistance to alkylating agents, represented the most promising one with robust predictive ability for temozolomide (TMZ) outcome.7 Unfortunately, the single‐gene‐based epigenetic biomarkers including MGMT had limited roles in guiding clinical decision and failed to warrant a change in routine testing.7 In recent years, there has been an increasing number of high‐throughput techniques devoted to accomplishing genome‐wide assessment of cancer epigenomes.4, 5, 8 The application of those latest approaches may be helpful for identifying more powerful biomarkers based on multimarker epigenetic signatures.
In this study, by integrating genome‐wide DNA methylation microarray data and clinical information, we reported a novel biologically relevant six‐CpG signature for GBMs. The signature robustly predicted survival of patients with GBM in a treatment‐independent manner and was of promising value to improve current patient management.
2. MATERIALS AND METHODS
2.1. Patient cohorts
Seventy‐nine adult patients (aged ≥18 years old) with newly diagnosed GBMs were collected between 2004 and 2013 from the Neurosurgery Departments of Rennes and Angers University Hospitals (RAUH_450k). Initial histological diagnoses were confirmed by a central review panel including at least two neuropathologists. All patients were homogenously treated with Stupp regimen.9 The median follow‐up period was 53 months, with a range of 8‐113 months.10 Snap‐frozen samples were collected at the time of surgery, following informed consent, in accordance with the French regulations and the Helsinki Declaration. DNA was extracted using the NucleoSpin TissueKit (Macherey Nagel). The quality of DNA samples was assessed by electrophoresis in a 1% agarose gel. DNA methylation profiling was performed by the Infinium HumanMethylation450k platform (Illumina Inc., San Diego, CA, USA) according to the manufacturer's instructions. Image processing and intensity data extraction were performed within Genome Studio (Illumina Inc.). The novel BMIQ (Beta MIxture Quantile dilation) algorithm was used for intra‐array normalization.11 Methylation level of each CpGs locus is summarized as β value, ranging from 0 (completely unmethylated) to 1 (completely methylated). Methylation data have been submitted in The ArrayExpress under accession number “E‐MTAB‐4969”.
A published cohort of fifty GBMs and three nontumor brains (NBs) from the Neurosurgery Departments of Rennes and Angers University Hospitals was also included (GSE22867; RAUH_27k),12 with microarray data by Infinium 27k platform (Illumina Inc.).
Public GBM datasets with DNA methylation data were obtained from The Cancer Genome Atlas (TCGA) including TCGA_27k (GBMs, n = 282; NBs, n = 4) and TCGA_450k (GBMs, n = 113)13; from Chinese Glioma Genome Atlas (CGGA) (CGGA_27k; GBMs, n = 30)14; and from the Gene Expression Omnibus (GEO) repository including GSE50923_27k (GBMs, n = 54; NBs, n = 24),15 GSE60274_450k (GBMs, n = 64; NBs, n = 5),16 and GSE36278_450k (GBMs, n = 57; tumors harboring mutations in H3F3A and those from TCGA were excluded).17 Patient characteristics of the included GBM datasets are summarized in Table 1. Molecular datasets of lower‐grade gliomas (LGG, grade II to III) were also used for additional validation, including TCGA‐LGG_450k (n = 482), CGGA‐LGG_27k (n = 109), and GSE48462_450k (n = 117). All NBs were obtained from apparently healthy individuals or patients with chronic epilepsy without pathological evidence of other neurological or psychiatric diseases in each dataset. Among the datasets with gliomas of all grades and ages, only those aged ≥18 years old, and with a histological diagnosis of GBMs, were included in this study, and patients with a follow‐up time ≥1 month were kept for survival analysis.
Table 1.
Patient characteristics of all discovery and validation sets of GBMs with DNA methylation data
| Variables | Discovery sets | Independent validation sets | ||||||
|---|---|---|---|---|---|---|---|---|
| RAUH_27k | TCGA_27ka | GSE50923_27k | RAUH_450k | TCGA_450ka | GSE60274_450k | GSE36278_450k | CGGA_27k | |
| Sample size | 50 | 282 | 54 | 79 | 113 | 64 | 57 | 30 |
| Age | ||||||||
| Mean | 56.9 | 58 | 57.6 | 58.9 | 60.6 | 50.8 | 42.2 | 45.7 |
| Range | 26‐80 | 18‐89 | 22‐82 | 36‐75 | 21‐85 | 26‐70 | 18‐57 | 18‐70 |
| Preoperative KPS | ||||||||
| Median | 80 | 80 | 80 | 90 | 80 | NA | NA | 80 |
| Range | 40‐100 | 40‐100b | 50‐90 | 50‐100 | 60‐100b | NA | NA | 50‐100 |
| Gender | ||||||||
| Male | 25 | 168 | 32 | 56 | 65 | 50 | 34 | 18 |
| Female | 24 | 114 | 22 | 23 | 48 | 14 | 23 | 12 |
| Unknown | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Extent of surgery | ||||||||
| Surgery (total/partial) | 45 (36/9) | 238 (NA/NA) | 52 (32/20) | 74 (57/17) | 112 (NA/NA) | 0 | 0 | 0 |
| Biopsy | 1 | 42 | 2 | 4 | 1 | 0 | 0 | 0 |
| Unknown | 4 | 2 | 0 | 1 | 0 | 64 | 57 | 30 |
| Adjuvant treatments | ||||||||
| RT and TMZ | 50 | 191 | 54 | 79 | 72 | 36 | 0 | 0 |
| RT | 0 | 53 | 0 | 0 | 17 | 28 | 0 | 0 |
| Others or unknown | 0 | 38 | 0 | 0 | 24 | 0 | 57 | 30 |
KPS, Karnofsky performance score; RT, radiotherapy; TMZ, temozolomide; NA, not available.
TCGA (27k and 450k collectively) included secondary, recurrent, or previously treated samples (n = 30).
KPS were available for only a small subset of patients from TCGA_27k (n = 55) and TCGA_450k (n = 25).
2.2. Probe selection and risk‐score model construction
Prior probe selection was performed by removing those not interrogated on both platforms, those targeting the sex chromosomes, those containing a single‐nucleotide polymorphism (SNP) within five base pairs of the probes, and those not annotated with any protein‐coding or non‐protein‐coding genes. Finally, 21248 CpGs were kept for analysis. Differentially methylated CpGs were computed by two‐sample Wilcoxon sum rank test (samr R package). GBM‐specific CpGs were defined as those having a median β difference ≥0.2 between tumors and controls and a false discovery rate (FDR) q‐value ≤0.05. Correlation of DNA methylation with OS was evaluated by univariate Cox regression analysis with permutation test by Biometric Research Branch (BRB)‐Array Tools. Prognostic CpGs were those with a permutation P‐value ≤0.05. Batch effects between each platform and dataset were adjusted by M‐value transformation and the empirical Bayes approach (ber R package).18, 19 Missing β values were imputed by impute R package. The discovery‐validation approach was employed to develop a risk‐score model which is the sum of the methylation levels of each CpGs weighted by their univariate Cox coefficients (Figure 1A). The discovery phase was performed in TCGA_27k, RAUH_27k, and GSE50923_27k. Cox coefficients were calculated from RAUH_27k; optimal cutoff for stratifying low‐risk and high‐risk tumors was determined by maxstat R package from all the discovery sets.20 The validation phase was performed in five GBM cohorts and two LGG cohorts. The risk‐score signature was also assessed with PFS outcome.
Figure 1.
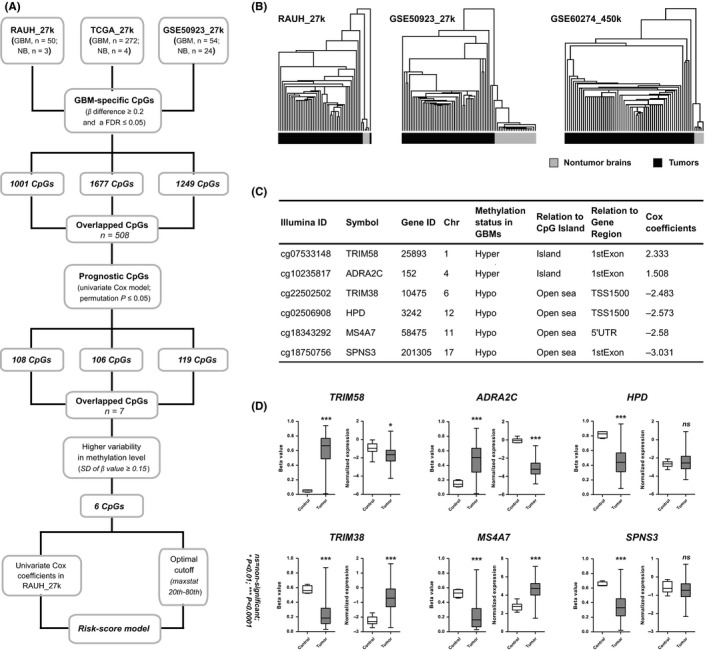
The development of the six‐CpG prognostic signature; (A) the study workflow for the risk‐score signature construction; (B) hierarchical clustering on the 508 CpGs that were commonly identified in all the three discovery sets accurately distinguished GBMs from nontumor brain tissues in two discovery sets (RAUH_27k and GSE50923_27k) and an independent validation set (GSE60274_450k), with the similarity metric “Euclidean distance” and the clustering method “Centroid linkage”; TCGA_27k was not tested due to too few nontumor controls (n = 4) relative to the large number of GBMs (n = 282); (C) the characteristics of the six‐CpG panel; open sea loci refer to CpGs that are more than 4000 bp away from CpGs island; (D) the DNA methylation of the six CpGs and the expression levels of the relevant genes between GBMs (n = 279) and nontumor brain tissues (n = 10) from TCGA_27k; P‐values for Wilcoxon sum rank test and standard t test were, respectively, indicated for DNA methylation and expression data. *P < 0.01, ***P < 0.001, ns, nonsignificant
2.3. Indirect validation based on differential gene expression prediction
To add another layer of prognostic validation, we used the Support Vector Machines (SVM) model based on the differential expressed genes (4201 genes) between each risk subgroups from TCGA_27k, to predict the risk classification of our 6‐CpG signature. The prediction accuracy rate of the SVM model was 87% in TCGA_27k. Public gene expression datasets of GBMs were downloaded for indirect validation, including REMBRANDT (the Repository of Molecular Brain Neoplasia Data, n = 181) 21 and GSE16011 (n = 147).22
2.4. Bioinformatic analysis
To gain biologically insightful view of the risk‐score signature, an integrative analysis of multidimensional molecular data was performed within TCGA samples; (i) level 3 gene expression data from the Agilent G4502A Microarray (n = 386) were analyzed by gene set enrichment analysis (GSEA) to evaluate the functional profiles between the risk subgroups on the gene sets of Gene Ontology Biological Processes and Kyoto Encyclopedia of Genes and Genomes (KEGG) from The Molecular Signatures Database (MSigDB)23; (ii) level 3 copy number data from Affymetrix Genome‐Wide Human SNP6.0 Array (n = 382) were analyzed by GISTIC2.0 with amplitude threshold being ±0.224; (iii) level 2 somatic mutation data from Whole Exome sequencing (n = 245) were analyzed by MutSigCV to identify significantly mutated genes, with a FDR q‐value ≤0.05 being significant; and (iv) level 3 microRNA data from Agilent 8 × 15K Human miRNA‐specific Microarray (n = 386) and level 3 protein data from Reverse Phase Protein Array (n = 171) were both computed by two‐sample t test to identify differentially expressed targets, with confidence level of FDR assessment = 80% and maximum allowed proportion of false‐positive genes = 0.1. The DNA methylation clusters were determined by k‐means (k = 3) clustering on the 1503 probes reported by Noushmehr et al.25 The gene expression subtypes were predicted using the binary tree classification on expression data of the 840 classifiers reported by Verhaak et al26 MGMT promoter methylation status was determined using a logistic regression model based on two probes, that is, cg12434587 and cg12981137.27
2.5. Statistical analysis
Hierarchical clustering analysis was performed within GenePattern. The distribution of molecular features with respect to each risk subgroup was tested by Fisher's exact test or chi‐square test. Overall survival (OS) was defined as the interval from the date of diagnosis to the date of death or last follow‐up; progression‐free survival (PFS) was the interval to the date of progression according to clinical and imaging criteria,28 or to the date of death or last follow‐up without progression. Survival data were estimated by the Kaplan‐Meier Method and compared by log‐rank test. Univariate and multivariate Cox regression models were used to evaluate the correlation and independence of potential prognosticators. Meta‐analysis was performed by the inverse‐variance method where application of either fixed‐ or random effect models were based on the statistical heterogeneity, with P‐value for chi‐square test ≤0.05 for significance. The prognostic performance was evaluated by time‐dependent receiver operating characteristic (ROC) curve (survcomp R package).29 Interaction analysis was conducted between the risk subgroups and paired treatments. All the calculations were carried out within SPSS Statistics and R software, and P‐values ≤0.05 for significance were used.
3. RESULTS
3.1. Identification of a novel GBM‐specific six‐CpG panel for risk‐score modeling
GBM‐specific CpGs were, respectively, calculated from RAUH_27k, TCGA_27k, and GSE50923_27k (Figure 1A and Table S1). Given the limitations in computing differential methylation for GBMs in each dataset (eg, a few number of NB samples, nonmatched controls, and inability for adjusting age and brain location), we used the overlap of 508 CpGs from all discovery sets to generate a representative list. Hierarchical clustering on the 508‐CpG signature accurately distinguished GBMs from NBs in two discovery sets and an independent validation cohort with only five tumors (1.8% of 168) being misclassified into the NB group, supporting the robustness of the list as differential methylation for GBMs (Figure 1B). Then by correlating methylation levels with OS, we identified an overlap of seven CpGs with high prognostic value (permutation P ≤ 0.05) from the discovery sets. A panel of six CpGs with highly variable methylation patterns across tumors (standard deviation of β value ≥0.15) was used for risk‐score modeling (Figure 1C). Among the panel, two CpGs were from CGIs and hypermethylated, while the others were outside CGI and hypomethylated in GBMs. The two CGI CpGs were regarded as risky for prognosis as their DNA methylation levels showed inverse correlation with OS; the four open sea CpGs were all protective with a positive correlation (Figure 1C). Moreover, four locus‐related genes were associated with epigenetic silencing (TRIM58 and ADRA2C) or re‐expression (TRIM38 and MS4A7) in GBMs (Figure 1D). In addition, despite not associating with differential expression status in GBMs (HPD and SPNS3), two open sea CpGs were essential for optimal prognostication as indicated by additional analyses (Figure S1 and Table S2). Collectively, the risk‐score formula was constructed as follows: risk score = (2.333 × β value of cg07533148) + (1.508 × β value of cg10235817) + (−2.483 × β value of cg22502502) + (−2.573 × β value of cg02506908) + (−2.580 × β value of cg18343292) + (−3.031 × β value of cg18750756), with the optimal cutoff of −2.485 (around the 20th percentile risk value from the discovery cohorts) for stratifying low‐risk and high‐risk patients (Figure 1A).
3.2. The prognostic value of the six‐CpG signature in the discovery and independent validation cohorts
Patients were divided to low‐risk groups (with lower risk scores) and high‐risk groups (with higher risk scores) in the discovery cohorts, where low‐risk patients were consistently associated with longer OS than high‐risk ones (Figure 2A). The epigenetic signature had been further validated in five independent validation cohorts of heterogeneous population; it accurately predicted OS not only for patients with combined radiation (RT) and TMZ but also for those with heterogeneous or unknown treatments (Figure 2B). Moreover, risk classification on differential gene expression profiles yielded significant OS difference between the predicted low‐risk and high‐risk subgroups in REMBRANDT and GSE16011 (Figure S2). Finally, the six‐CpG signature was successfully validated in different datasets of LGGs and in particular the subtype with wide‐type IDH and intact chromosome 1p/19q, which is reported to be molecularly resemble with GBMs (Figure S3).30 The six‐CpG signature also predicted PFS in available GBM cohorts (Figure S4).
Figure 2.
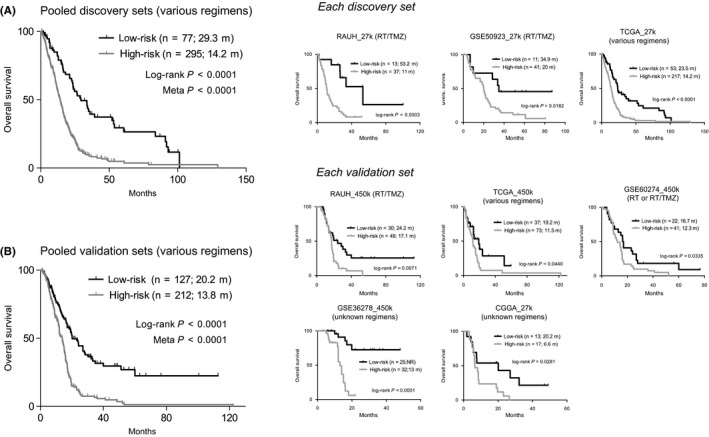
The prognostic performance of the six‐CpG signature on overall survival (A) in the pooled discovery cohorts (left) and each discovery set (right); and (B) in the pooled validation cohorts (left) and each validation set (right); P‐values from log‐rank test and meta‐analysis were indicated; RT, radiation; TMZ, temozolomide
3.3. The six‐CpG signature was an independent and superior prognostic factor for GBMs
Within all RAUH samples (27k and 450k collectively), univariate Cox regression model revealed that age, MGMT promoter methylation status, and the six‐CpG signature were significantly correlated with OS (Table 2). Multivariate Cox model further demonstrated that the six‐CpG signature was an independent prognostic indicator (Table 2). Cox regression analyses yielded similar results with all TCGA patients (Table 2). The consistent prognostic value in stratified cohorts by different treatments supported that the six‐CpG signature was not a predictive indicator for specific treatment, but a prognostic factor for GBMs, which provides information on the likely outcome of cancer diseases independent of treatment (Figure S5).
Table 2.
Results for Cox regression models of the six‐CpG signature
| Variables | Univariate Cox model | Multivariate Cox modela | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| RAUH (n = 129) | ||||||
| Patient age | 1.028 | 1.008‐1.049 | 0.005 | 1.028 | 1.007‐1.050 | 0.009 |
| KPS | 1.003 | 0.990‐1.016 | 0.676 | |||
| Six‐CpG signature | 2.772 | 1.743‐4.412 | <0.001 | 2.502 | 1.552‐4.032 | <0.001 |
| MGMT methylation status | 2.661 | 1.732‐4.087 | <0.001 | 2.669 | 1.711‐4.161 | <0.001 |
| G‐CIMP status | 6.009 | 0.837‐43.145 | 0.075 | |||
| Proneural subtype | 1.430 | 0.874‐2.338 | 0.154 | |||
| Gender | 0.882 | 0.586‐1.327 | 0.547 | |||
| Extent of surgery | 0.799 | 0.577‐1.108 | 0.179 | |||
| TCGA (n = 380)b | ||||||
| Patient age | 1.034 | 1.024‐1.044 | <0.001 | 1.019 | 1.006‐1.032 | 0.003 |
| Treatments (RT vs RT/TMZ) | 2.880 | 2.095‐3.958 | <0.001 | 2.803 | 1.997‐3.933 | <0.001 |
| Histological typesc | 0.465 | 0.304‐0.711 | <0.001 | 0.568 | 0.319‐1.013 | 0.055 |
| Six‐CpG signature | 0.480 | 0.353‐0.653 | <0.001 | 0.670 | 0.449‐1.000 | 0.050 |
| MGMT methylation status | 1.216 | 0.955‐1.547 | 0.112 | |||
| G‐CIMP status | 3.060 | 1.881‐4.979 | <0.001 | 1.581 | 0.769‐3.249 | 0.213 |
| Proneural subtype | 1.518 | 1.148‐2.009 | 0.003 | 1.165 | 0.831‐1.633 | 0.376 |
| Gender | 0.672 | 0.524‐0.861 | 0.002 | 0.613 | 0.461‐0.815 | 0.001 |
HR, hazard ratio; CI, confidence interval; KPS, Karnofsky performance score; RT, radiotherapy; TMZ, temozolomide; G‐CIMP, glioma‐CpG island methylator phenotype.
In bold are significant results for Cox regression models.
Multivariate Cox regression analysis was performed by incorporating significant factors in the univariate Cox model.
TCGA patients with a follow‐up time <1 mo (n = 15) were excluded for Cox regression analysis.
Histological types for primary de novo GBMs vs other types (eg, secondary, recurrent, or previously treated cases).
Time‐dependent ROC analysis reported that the six‐CpG signature was associated with larger area under the curve (AUC) values than G‐CIMP+ and proneural subtype at each time point, suggesting its superiority in survival prediction (Figure S6). In addition, among patients treated with RT/TMZ, the six‐CpG signature and MGMT methylation status showed similar integrated AUC values, but had distinct evolution with respect to time; MGMT status and the six‐CpG signature, respectively, had larger AUC values at earlier and later time points (Figure S6). This finding suggested a possibility of the combination of the two indicators for optimal prognostication.
3.4. Molecular characterization of the six‐CpG signature using TCGA data
Correlation with established molecular subgroups showed that the low‐risk group included all C‐GIMP+ tumors and was enriched with proneural subtypes, while the high‐risk group was enriched with DNA methylation cluster#2 tumors described by Noushmehr et al25 and classical and mesenchymal subtypes (Figure 3A). Secondary GBMs were enriched in the low‐risk group (Figure 3A). The risk subgroups were also associated with different ages even after excluding patients with C‐GIMP+ tumors, which were known for younger ages at diagnosis (mean ages for G‐CIMP‐ low‐risk vs high‐risk tumors: 54.0 vs 61.6 years old, P < 0.0001). Somatic copy number variation (SCNV) analysis showed that the risk subgroups were associated with distinct chromosomal alterations: Gain of Chr.7, Chr.19, and Chr.20 and loss of Chr.7 were more frequently seen in high‐risk tumors (Figures 3A and S7). We also found that high‐risk tumors harbored more significantly regional SCNVs (Figure S7). Accordingly, at gene level, high‐risk tumors were associated with more SCNVs in known cancer genes such as EGFR, PDGFA, PTEN, and CDNK2A/B (Figure 3A). Somatic mutation analysis showed that the significantly mutated genes were much more in high‐risk vs low‐risk tumors (227 vs 11; Table S3), among which mutations in EGFR, COL6A3, PFAS, and WDR92 were more frequently seen in high‐risk tumors while mutations in TP53, ATRX, and IDH1 were enriched in low‐risk tumors (Figure 3A). Of note, despite that some of the observed features (eg, secondary cases, SCNVs in chr20, and mutations in TP53, ATRX, and IDH1) were exclusively attributed to the enrichment of G‐CIMP+ tumors in the low‐risk group, the majority remained significant in the comparison of G‐CIMP‐ low‐risk and high‐risk cases (Figure 3A).
Figure 3.
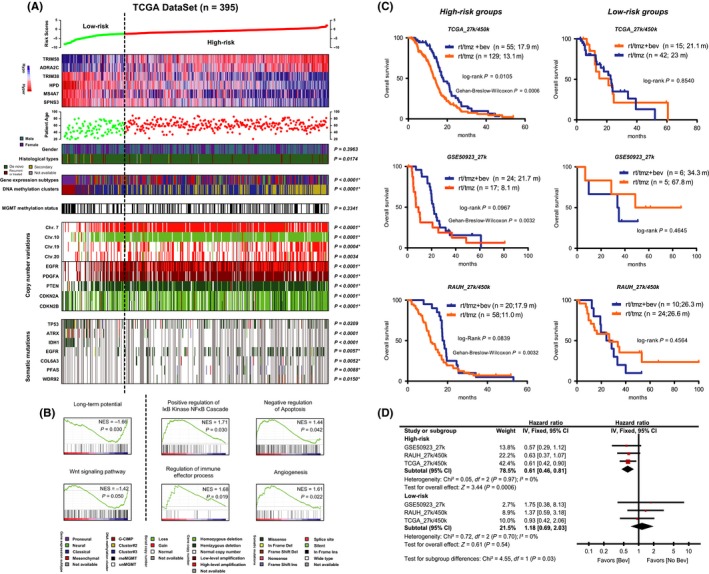
Molecular characterization of the six‐CpG signature using the multidimensional TCGA data; (A) heat maps of methylation levels of the six CpGs; each row represents a CpGs; each column represents a sample which is ordered by the assigned risk scores; patient age, clinical features, molecular subgroups, copy number variations, and mutational status are indicated for each sample (n = 395); regarding distribution, P‐values for chi‐square or fisher's exact tests were indicated; *indicated significant distribution features event after excluding G‐CIMP+ tumors; (B) GSEA enrichment plots of representative gene sets for low‐risk and high‐risk tumors; (C) the potential links of the defined risk subgroups to differential outcomes of bevacizumab in patients with combination of RT and TMZ; the usage of bevacizumab conferred a clear benefit in OS and especially short‐term survival to high‐risk patients, but appeared to be associated with similar OS in low‐risk ones in TCGA_27k/450k (either first‐line or at progression; upper panel), GSE50923_27k (at progression; middle panel), and RAUH_27k/450k (at progression; bottom panel); secondary, recurrent, and treated samples from TCGA were excluded for this analysis; (D) meta‐analysis of each cohort confirmed the differential bevacizumab outcomes with respect to each risk subgroup by yielding a significant result for subgroup difference test (P = 0.03); Bev, bevacizumab
As for functional profiles, GSEA on transcriptome data showed that low‐risk tumors were enriched in signatures relating to normal brain function and developmental process, while high‐risk tumors were enriched with cancer‐promoting signatures relating to immune response, NF‐κB activation, apoptosis, and angiogenesis (Figure 3B and Table S4). Consistent with transcriptome data, the risk subgroups were also associated with distinct functional profiles at microRNAs and protein levels, among which high‐risk tumors were mostly featured by elevation of proangiogenic signaling (Figure S8).
3.5. Potential links to differential outcomes of bevacizumab therapy
The multiplatform molecular profiling revealed concordant activation of proangiogenic signaling in high‐risk tumors and thus suggested possible better outcomes for antiangiogenic therapy in this subgroup. We observed that, among TCGA patients who were treated with combined RT/TMZ, the utility of bevacizumab (either first‐line or at progression; a humanized monoclonal antibody against VEGFA31, 32) did confer a clear OS benefit to high‐risk patients, but was associated with similar outcome in the low‐risk group (Figure 3B). Similar benefits were also observed in two independent sets on bevacizumab at progression (Figure 3B). Meta‐analysis confirmed the significant differential outcomes by bevacizumab‐contained therapy within each risk subgroup (test for subgroup differences, P = 0.03; Figure 3D). Gehan‐Breslow‐Wilcoxon test further indicated that bevacizumab‐contained therapy may be more useful for improving shorter‐term survival for high‐risk patients (Figure 3A).
3.6. The six‐CpG signature in stratified cohorts by known epigenetic markers
We also tested the prognostic interrelationship of the six‐CpG signature with known epigenetic indicators. Despite the enrichment of favorable G‐CIMP+ tumors in the low‐risk groups, the six‐CpG signature still showed great discriminating value for prognosis in the majority of GBMs without G‐CIMP (Figures 4A and S9A).
Figure 4.
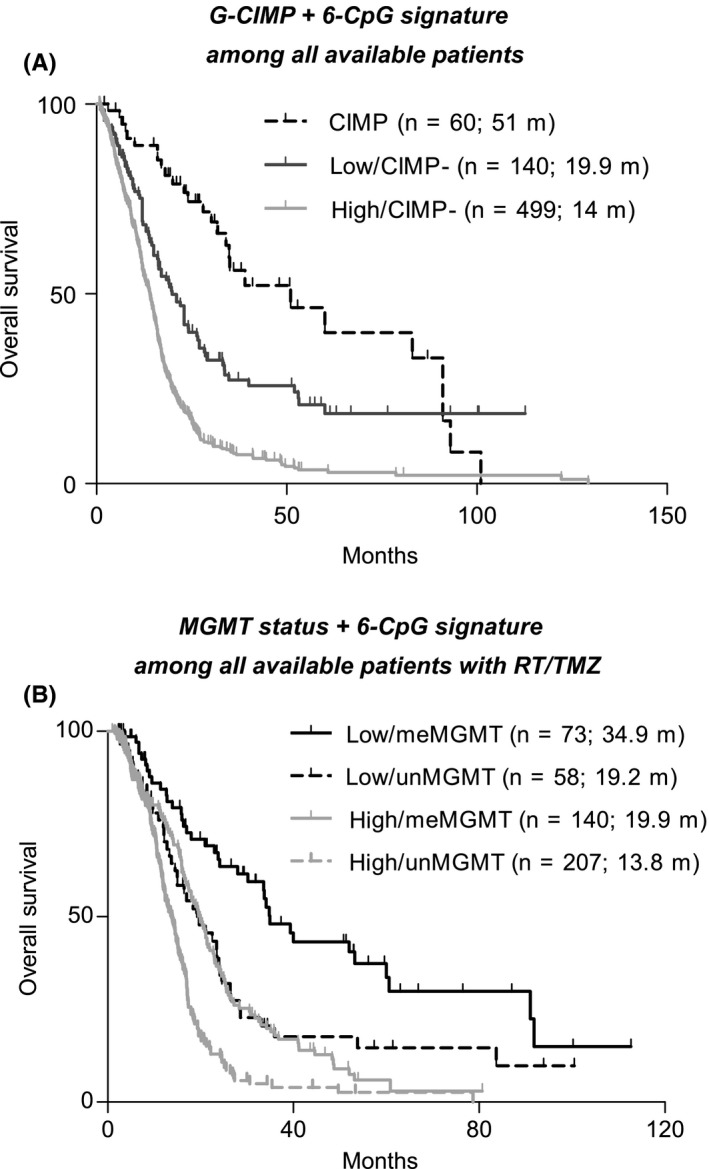
The prognostic performance of the six‐CpG signature in stratified cohorts by known epigenetic biomarkers; (A) risk classification of the six‐CpG signature and G‐CIMP status in a pooled survival analysis of all available patients; P‐values from meta‐analysis <0.01 for each pairwise comparison; (B) risk classification of the six‐CpG signature and G‐CIMP status in a pooled survival analysis of all available patients with combination of RT and TMZ; P‐values from meta‐analysis <0.01 for each pairwise comparison, except for the comparison of low‐risk and MGMT unmethylated tumors vs high‐risk and unmethylated ones (P = 0.540)
As encouraged by time‐dependent ROC analysis, we also employed the combination of the six‐CpG signature and MGMT methylation status to stratify patients who were treated with RT and TMZ, which yielded four distinct subgroups; patients with low‐risk and MGMT methylated tumors had the best OS, followed by two subgroups with only one favorable mark, while those with high‐risk and unmethylated tumors had the worst survival (Figures 4B and S9B).
Finally, time‐dependent ROC analysis confirmed the refined risk classification with the addition of our signature (Figure S6).
4. DISCUSSION
Clinically informative biomarkers played crucial roles in precision oncology.33 Historically, RNA‐ or protein‐based information had been the mainstream for biomarker discovery and, indeed, brought clinical benefits to patients with cancer.3 However, the expression‐based biomarkers had critical drawbacks for clinical utility—the information provided was unstable and sometimes misleading due to the highly dynamic nature of RNAs and proteins in cancer biology and the vulnerable physic‐chemical nature in biological specimens.3 In this respect, DNA methylation‐based information was much reliable because cancer‐linked DNA methylation patterns were relatively stable over time and DNA was considerably more stable than RNAs or proteins in archived materials.3 The epigenetic marks also have advantages over the stable genetic alterations (eg, somatic mutations, SCNVs, and SNPs) such as tolerance of nontumor cell contamination of samples, hints of tumor cells of origin, and allowance of a quantitative test.34 Moreover, aberrant DNA methylation changes usually preceded genetic defects and abnormal expression and represented very early events during carcinogenesis.35, 36 The assessment of DNA methylation could render a more timely and accurate molecular profiling of a given tumor. Finally, the availability of drugs that reverse epigenetic modifications (eg, DNA methyltransferase inhibitors and histone deacetylase inhibitors) makes DNA methylation analysis more therapeutically useful.3, 34 Collectively, all those advantages had made more appealing the development of a powerful DNA methylation signature for GBM prognostication.
In this study, by focusing on differential DNA methylation in GBMs, we developed a novel six‐CpG panel for prognostication. The six‐CpG signature had been demonstrated to be a robust and independent prognostic factor for GBMs and was better than other molecular indicators such as G‐CIMP status and proneural subtype. Another major advantage of the epigenetic signature is its biological implications. Among the locus‐specific genes, ADRA2C and TRIM58 were epigenetically silenced, and TRIM38 and MS4A7 were upregulated in GBMs. ADRA2C is a subtype of alpha‐2‐adrenergic receptors and has critical roles in normal brains function.37 The dramatic decrease in ADRA2C expression indicated the disruption of normal brain function in GBMs. Interestingly, the other three transcriptionally altered genes were all related to immune system. TRIM38 and TRIM58 belong to the E3 ubiquitin ligase superfamily.37 TRIM38 was reported to be a negative regulator of innate immunity and inflammatory response.38, 39, 40 TRIM58 was involved in the regulation of pathogen‐recognition and innate response.41 MS4A7 was associated with mature cellular function in the monocytic lineage.37 Previous studies showed that epigenetic modulation of immune‐related genes is often taken advantage by neoplastic cells to promote immune escape by impairing their immunogenicity and immune recognition and establishing immunosuppressive microenvironments.42 In this study, we found that the defined risk subgroups were highly associated with differential enrichments in immune‐relevant gene sets. Therefore, we proposed that the epigenetic panel may have implications in regulating GBM‐specific immune response. Of note, two hypomethylated CpGs were not associated with apparent expression alteration in GBMs, but were essential for optimal prognostication. Recent studies suggested that, instead of a direct linkage to altered expression, cancer‐specific DNA hypomethylation may also have functional impacts via contributing to disrupted heterochromatin, leading to loss of both epigenetic and transcriptional regulation, and resulting in hypervariability of expression, and even have interactions with important genetic domains in cancers.36
Bevacizumab has been the most promising antiangiogenic agents for treating GBMs and especially recurrent cases.2 Unfortunately, two recent Phase III trials failed to yield clear OS benefits with the addition of bevacizumab to Stupp regimen in newly diagnosed GBMs, making more necessary the search of powerful predictive indicator for bevacizumab outcomes.31, 32 Intriguingly, in this study, we observed the differential angiogenic profiles and survival outcomes of antiangiogenic therapy within each risk subgroup; high‐risk patients seemed to benefit more from bevacizumab‐contained therapy than low‐risk ones. Therefore, the six‐CpG signature may have potential value to guide the usage of bevacizumab especially for high‐risk patients, and thus be helpful for sparing low‐risk ones who are molecularly unlikely to benefit from the aggressive therapy of higher cost and potential toxicity. Of note, the finding was encouraging but should be conservatively interpreted due to study limitations (eg, incomplete drug data, second‐line bevacizumab in most cases, and retrospective design). Prospective validation in randomized trials of first‐line bevacizumab will be needed for definitive conclusion.
The epigenetic signature also had potential to improve the current risk classification. The six‐CpG signature showed no discriminating value in the small subgroup of GBMs with G‐CIMP (about 10%), characterized by mutations in IDH and favorable OS,25 as all G‐CIMP+ tumors were low‐risk. However, it was useful for identifying patients with different prognoses among the majority of GBMs without G‐CIMP. The six‐CpG signature also showed great discriminating value in stratified RT/TMZ cohorts with each MGMT methylation status. MGMT methylation status had been by far the most informative biomarker for GBMs. However, its clinical value was much compromised due to lack of a direct linkage between MGMT testing and TMZ usage especially in unmethylated tumors.7 Our six‐CpG signature could refine the MGMT‐based risk classification and be helpful for improving current clinical choice on TMZ for GBMs.
There have been fewer multimarker epigenetic biomarkers for GBM prognostication. Shukla et al reported a nine‐CpG prognostic signature with implications of abnormal activation of NF‐kB signaling.43 Lai et al15 reported a hypermethylated signature of human embryonic stem cell (hESC)‐associated genes. The signatures both had greatly expanded our knowledge of the epigenetic features for GBMs. Unfortunately, they had limitations as clinically useful biomarkers in some crucial respects such as the employment of single discovery set with small sample size, the inclusion of CpGs associating SNPs, and the insufficient external validation. Our six‐CpG signature had been carefully developed with a particular focus on those issues. However, limitations still existed in this study. First, the selection of prognostic CpGs was mainly focused on CpGs that were differentially methylated in GBMs, which may result in the exclusion of other probes with high informative value. Second, our six‐CpG signature was more useful to identify a relatively small subset of clinically favorable patients, but showed limited capacity to provide information on the rest block of cases that are in the middle of the clinical spectrum. Third, despite that the six‐CpG signature showed robust prognostic value in G‐CIMP‐ tumors, the model was not developed for that purpose. More powerful prognostic signatures for that tumor subset are still much needed and should be developed with exclusion of G‐CIMP+ tumors. Finally, lack of functional and mechanism studies hindered better application of the epigenetic biomarker.
Collectively, our six‐CpG signature represented a promising tool for prognostication and was of promising value for optimizing personalized management toward GBMs.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
All procedures performed in studies involving human were in accordance with the ethical standards of the institutional research committee of Rennes and Angers University Hospitals (France) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
INFORMED CONSENT
Informed consent was obtained from all participants from the Neurosurgery Departments of Rennes and Angers University Hospitals.
Supporting information
ACKNOWLEDGEMENTS
We gratefully acknowledge all the patients who agreed to participate in this study and to those who have provided their medical care. We gratefully acknowledge Pr Menei, Dr Le Reste, Dr. Vauleon, Dr. Quillien, and the Tumor Banks from Angers and Rennes for their constant support in the collection, processing, and histological analysis of tumor samples. We also gratefully thank those who are willing to share their valuable scientific data (Prof. Rose K Lai for clinical data). I (A.Y.) thank my fiancée (Dr. Yu Dong) for her great support and would you marry me? The results published here are in part based upon data generated by The Cancer Genome Atlas, German Cancer Research Center, and the research teams mentioned in our study. This work was supported by grants from National Natural Science Foundation of China (No. 81402049, No. 81471266, and No. 81372457) and by grants from the Brittany Region (France) et the FEDER (Europe).
Yin A‐A, Lu N, Etcheverry A, et al. A novel prognostic six‐CpG signature in glioblastomas. CNS Neurosci Ther. 2018;24:167–177. 10.1111/cns.12786
The first three authors contribute equally to this work.
Contributor Information
Yu‐He Liu, Email: lyhws@sina.com.
Ya‐Long He, Email: heyl.fmmu@hotmail.com.
REFERENCES
- 1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492‐507. [DOI] [PubMed] [Google Scholar]
- 2. Yin AA, Cheng JX, Zhang X, et al. The treatment of glioblastomas: A systematic update on clinical Phase III trials. Crit Rev Oncol Hematol. 2013;87:265‐282. [DOI] [PubMed] [Google Scholar]
- 3. Issa JP. DNA methylation as a clinical marker in oncology. J Clin Oncol. 2012;30:2566‐2568. [DOI] [PubMed] [Google Scholar]
- 4. Baylin SB, Jones PA. A decade of exploring the cancer epigenome ‐ biological and translational implications. Nat Rev Cancer. 2011;11:726‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriguez‐Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330‐339. [DOI] [PubMed] [Google Scholar]
- 6. Malzkorn B, Wolter M, Riemenschneider MJ, et al. Unraveling the glioma epigenome: from molecular mechanisms to novel biomarkers and therapeutic targets. Brain Pathol. 2011;21:619‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin AA, Zhang LH, Cheng JX, et al. The predictive but not prognostic value of MGMT promoter methylation status in elderly glioblastoma patients: a meta‐analysis. PLoS ONE. 2014;9:e85102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin AA, Etcheverry A, He YL, et al. Integrative analysis of novel hypomethylation and gene expression signatures in glioblastomas. Oncotarget. 2017;8:89607‐89619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987‐996. [DOI] [PubMed] [Google Scholar]
- 10. Altman DG, De Stavola BL, Love SB, et al. Review of survival analyses published in cancer journals. Br J Cancer. 1995;72:511‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teschendorff AE, Marabita F, Lechner M, et al. A beta‐mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Etcheverry A, Aubry M, de Tayrac M, et al. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genom. 2010;11:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang W, Yan W, You G, et al. Genome‐wide DNA methylation profiling identifies ALDH1A3 promoter methylation as a prognostic predictor in G‐CIMP‐ primary glioblastoma. Cancer Lett. 2013;328:120‐125. [DOI] [PubMed] [Google Scholar]
- 15. Lai RK, Chen Y, Guan X, et al. Genome‐wide methylation analyses in glioblastoma multiforme. PLoS ONE. 2014;9:e89376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurscheid S, Bady P, Sciuscio D, et al. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX‐signature in glioblastoma. Genome Biol. 2015;16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425‐437. [DOI] [PubMed] [Google Scholar]
- 18. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118‐127. [DOI] [PubMed] [Google Scholar]
- 19. Du P, Zhang X, Huang CC, et al. Comparison of Beta‐value and M‐value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hothorn T, Zeileis A. Generalized maximally selected statistics. Biometrics. 2008;64:1263‐1269. [DOI] [PubMed] [Google Scholar]
- 21. Madhavan S, Zenklusen JC, Kotliarov Y, et al. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7:157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gravendeel LA, Kouwenhoven MC, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065‐9072. [DOI] [PubMed] [Google Scholar]
- 23. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol. 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bady P, Sciuscio D, Diserens AC, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP‐status. Acta Neuropathol. 2012;124:547‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Macdonald DR, Cascino TL, Schold SJ, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277‐1280. [DOI] [PubMed] [Google Scholar]
- 29. Heagerty PJ, Lumley T, Pepe MS. Time‐dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337‐344. [DOI] [PubMed] [Google Scholar]
- 30. Ceccarelli M, Barthel FP, Malta TM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse Glioma. Cell. 2016;164:550‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy‐temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709‐722. [DOI] [PubMed] [Google Scholar]
- 32. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin. 2016;66:75‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu J, Yao X. Use of DNA methylation for cancer detection: promises and challenges. Int J Biochem Cell Biol. 2009;41:147‐154. [DOI] [PubMed] [Google Scholar]
- 35. Fleischer T, Frigessi A, Johnson KC, et al. Genome‐wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timp W, Bravo HC, McDonald OG, et al. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014;6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pruitt KD, Tatusova T, Brown GR, et al. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130‐D135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu MM, Xie XQ, Yang Q, et al. TRIM38 negatively regulates TLR3/4‐mediated innate immune and inflammatory responses by two sequential and distinct mechanisms. J Immunol. 2015;195:4415‐4425. [DOI] [PubMed] [Google Scholar]
- 39. Hu MM, Yang Q, Zhang J, et al. TRIM38 inhibits TNFalpha‐ and IL‐1beta‐triggered NF‐kappaB activation by mediating lysosome‐dependent degradation of TAB 2/3. Proc Natl Acad Sci USA. 2014;111:1509‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao W, Wang L, Zhang M, et al. Tripartite motif‐containing protein 38 negatively regulates TLR3/4‐ and RIG‐I‐mediated IFN‐beta production and antiviral response by targeting NAP1. J Immunol. 2012;188:5311‐5318. [DOI] [PubMed] [Google Scholar]
- 41. Thom CS, Traxler EA, Khandros E, et al. Trim58 degrades Dynein and regulates terminal erythropoiesis. Dev Cell. 2014;30:688‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maio M, Covre A, Fratta E, et al. Molecular pathways: at the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res. 2015;21:4040‐4047. [DOI] [PubMed] [Google Scholar]
- 43. Shukla S, Pia PI, Thinagararjan S, et al. A DNA methylation prognostic signature of glioblastoma: identification of NPTX2‐PTEN‐NF‐kappaB nexus. Cancer Res. 2013;73:6563‐6573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials