

Summary
Introduction
The neuroprotective effects of hypothermia in acute ischemic stroke are well documented. However, the mechanisms involved in the effects remain to be clearly elucidated and the role of hypothermia on long‐term white matter integrity after acute ischemic stroke has yet to be investigated.
Aims
To investigate the role of mild focal hypothermia on long‐term white matter (WM) integrity after transient cerebral ischemia.
Results
Mild focal hypothermia treatment immediately after ischemic stroke significantly promotes WM integrity 28 days after the occlusion of the middle cerebral artery (MCAO) in mice. Higher integrity of white matter, lower activation of total microglia, less infarct volume, and better neurobehavioral function were detected in hypothermia‐treated mice compared to normothermia‐treated mice. Furthermore, we found that hypothermia could decrease detrimental M1 phenotype microglia and promote healthy M2 phenotype microglia. In vitro, results also indicated that hypothermia promoted oligodendrocytes differentiation and maturation after oxygen glucose deprivation.
Conclusion
Hypothermia promotes long‐term WM integrity and inhibits neuroinflammation in a mouse model of ischemic brain injury.
Keywords: hypothermia, microglia, neuroprotection, stroke, white matter
1. INTRODUCTION
White matter (WM) is an essential component of the human brain accounting for nearly 40% of total volume.1 Functionally, WM plays an important role in neural signal transmission and communication within the brain. WM integrity is important in long‐term recovery after stroke and small‐vessel disease.2, 3 Unfortunately, WM injury is prevalent in nearly half of all cases of stroke and TBI.4, 5 Moreover, WM is extremely susceptible to ischemia as there is little collateral circulation and limited blood supply in this region. WM injury impairs sensorimotor function and induces severe neurobehavioral and cognitive impairments.6 Previous neuroprotective studies of brain injury have emphasized gray matter injury over WM injury, perhaps in part, due to the failures of neuroprotectants designed to target the pathological alterations in neurons.4, 7 Thus, we believe a more comprehensive study regarding WM injury in ischemic stroke is needed.
Although neuroprotectiveagents have shown robust efficacy in the laboratory, there have been, to date, no successful outcomes in clinical trials.8 Thus far, hypothermia has been shown to be the most promising neuroprotectant due to its multifaceted neuroprotective effects,9 including reduction in infarct volume10; attenuation of intracellular calcium influx; lowering of intracellular acidosis11; suppression of oxygen free radicals formation12; prevention of blood brain barrier breakdown13; depression of inflammatory cells infiltration14; delays proliferation of microglia15; and attenuation of production of microglial IFN‐β and nitric oxide.16 A recent study also showed that hypothermia partially protected against WM injury after global cerebral ischemia in neonatal sheep and enhanced axonal myelination after hypoxia‐ischemia in the immature rat brain.17, 18 However, the effects of hypothermia on WM injury after focal ischemic stroke in the adult brain have yet to be elucidated.
In this study, we use a mouse model of transient middle cerebral artery occlusion to investigate the effects of focal hypothermia on long‐time WM injury after ischemic stroke. We revealed for the first time that hypothermia improves long‐time WM integrity after focal ischemic stroke. In addition, results show that hypothermia promotes microglial differentiation into the beneficial M2 phenotype at the expense of the detrimental M1 phenotype.
2. METHODS
2.1. Ischemia model and hypothermia administration
Male 10‐ to 12‐week‐old C57BL/6 mice (Vital River Laboratory Animal Technology Co. Ltd, Beijing, China) were used in this study. Focal cerebral ischemia was induced byreversible intraluminal occlusion of the right middle cerebral artery (MCAO) for 60 minutes as previously described.19 In brief, mice were anesthetized with 1.5% enflurane in a 30% O2/70% N2O mixture after which a midline ventral neck incision was made, and right MCAO was induced by inserting a silicone‐coated nylon monofilament into the internal carotid artery via the right external carotid artery stump. This monofilament tip was positioned at a distance of 6 mm beyond the internal carotid/pterygopalatine artery bifurcation. The rectal temperature was maintained at 37.0 ± 0.5°C with a heating lamp and heating pad during surgery, ischemia and focal hypothermia, respectively. Using laser Doppler flowmetry, we measured regional cerebral blood flow and confirmed the success of ischemic occlusion. Mice that did not have a local cerebral blood flow reduction of <30% of baseline were excluded from further experimentation. Sham mice underwent the anesthesia and surgery but not MCAO.
Mice were randomly assigned to normothermia +sham (37°C + sham), focal hypothermia +sham (33°C + sham), normothermia +MCAO (37°C + MCAO), and focal hypothermia +MCAO (33°C + MCAO) groups, respectively. Focal hypothermia was initiated immediately after the end of MCAO by placing an ice pack under the skull of mouse, and the temperature of the temporalis muscle was monitored and maintained at 32 ‐34°C for 2 hours for each hypothermic group. Mice were euthanized at 5, 14 and 28 days after MCAO, and brains were removed for biochemical and histological assessments.
All experimental procedures were approved by the Animal Care and Use Committee at Capital Medical University.
2.2. Oligodendrocytes culture and hypothermia administration
Oligodendrocytes were cultured as previously described.20 Briefly, tissue was isolated from the cerebrums of 24‐hours‐old Sprague‐Dawley rat pups, triturated and filtrated before centrifugation. The supernatant was carefully separated, and the pelleted cells were resuspended in DMEM/F12 medium (containing 1% MEM Non‐Essential Amino Acids Solution, 1% Sodium Pyruvate, 1% GlutaMAXTM Supplement, 1% Penicillin‐Streptomycin, 10% Fatal Bovine Serum, all from Gibco, Carlsbad, CA, USA). The cellular suspension was seeded in T175 flasks (BD, Franklin Lakes, NJ, USA) with DMEM/F12 medium and cultured in a humidified 37°C incubator with 5% CO2. Cultures were ready for microglial removal (shaking) at 10 d after initial seeding. After the removal of microglia, T175 flasks were added in fresh medium and shaken continually overnight in order to obtain a pure population of oligodendrocytes. Oligodendrocytes were randomly divided into six groups: normothermic group (37°C), hypothermic group (33°C), normothermic‐oxygen glucose deprivation (OGD) group, hypothermic‐OGD group, normothermic‐OGD‐ triiodothyronine (T3) group, and hypothermic‐OGD‐T3 group. Each OGD group underwent 2 hours of OGD on day 1 after initial normothermic seeding. Concurrently, each hypothermic group underwent 48 hours of hypothermic treatment and was then returned to 37°C. Oligodendrocytes differentiation was induced by incubation with T3 (Sigma, St Louis, MO, USA; 20 ng/mL) for 8 days.21
2.3. Neurological function evaluation
2.3.1. Rotarod test
The rotarod test was performed to assess motor cooperative ability of experimental mice as previously described with small modifications.22 Mice were placed on a revolving rod with a rotating speed that was accelerated from 0 to 30 rounds/min (rpm) within 2 minutes and then maintained at this speed for the following 8 minutes. Mice were trained before MCAO until they could remain on the revolving rod for 10 minutes. Postsurgery tests were done on days 3, 5, 7, 9, 11, and 13, respectively, after MCAO and latency times of mice falling from the rotating rod were recorded. Three trials were performed per day, and the average latency times were used for analyses.
2.3.2. Morris water maze test
Spatial learning and memory ability of mice were assessed using the Morris water maze previously described.23, 24 Briefly, a black circular pool surrounded by visual spatial cues was located in a room and was filled with water (20 ± 1°C). An escape platform was submerged 2 cm below the water surface, and the location of this platform remained constant throughout all subsequent experiments. For the spatial learning phase, mice were placed into one of four locations (north, south, east, and west) and were given 60 seconds to find the escape platform. Each mouse was given four trials per day, and the average escape latency was used for statistical analysis. One day after the last spatial learning test, the escape platform was removed. Each mouse was released from the most distal location from the target quadrant and was given a single 60 seconds probe test. The time spent in the target quadrant and the swimming speed was recorded.
2.3.3. Infarct volume
Infarct volume was assessed by hematoxylin and eosin (H&E) staining. Mice were deeply anesthetized on day 5 after MCAO and perfused with saline and 4% paraformaldehyde (Sigma‐Aldrich, St Louis, MO, USA) in phosphate‐buffered saline (PBS). Brains were removed and fixed in 4% paraformaldehyde for 72 hours followed by 30% sucrose in 4% paraformaldehyde until brains submerged completely. Using a freezing microtome, brains were cut into 20‐μm‐thick sections. A series of six slices were chosen for subsequent H&E staining. Infarct volume was calculated using Image J software, a Java based public‐domain image processing program developed at the National Institutes of Health. The actual infarct volume was expressed by the following equation: (volume of contralateral hemisphere minus noninfarcted volume of ipsilateral hemisphere)/volume of contralateral hemisphere.25
2.4. Immunofluorescence staining
Brains were sectioned as above and blocked in 5% donkey serum albumin in PBS for 1 hours at room temperature after which sections were incubated overnight at 4°C using the following primary antibodies: rabbit anti‐Iba‐1 (Wako, Tokyo, Japan), rat anti‐ CD16 (BD biosciences, Franklin Lakes, NJ, USA), goat anti‐CD206 (R&D Systems, Minneapolis, MN, USA), mouse anti–nonphosphorylated neurofilament H (SMI32; Milipore, Billerica, MA, USA), and rabbit anti–myelin basic protein (MBP; Abcam, Cambridge, MA, USA). Sections were then incubated with fluorescent conjugated secondary antibodies. Images were captured using a fluorescence microscope (Olympus, Tokyo, Japan). The fluorescent intensity of MBP, SMI32, and the numbers of stained target cells was quantified using Image J software. Three random microscopic fields on the periphery of the infarct core of each section and three successive sections of each brain were analyzed. Immunostaining target cell counts were expressed as the average numbers of cells per square millimeter.
2.5. Western blot analysis
Brains isolated from mice were homogenized in RIPA lysis buffer containing protease inhibitors. The supernatants were collected after centrifugation, and the protein concentration of each sample was determined using the Pierce BCA protein assay kit (Bio‐Rad, Berkeley, CA, USA). Equivalent amounts of protein were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to cellulose acetate membranes. The membranes were blocked for 1 hours in 5% dried skimmed milk in Tris‐buffered saline solution, and proteins were probed overnight at 4°C with primary antibodies recognizing rabbit anti‐MBP (Abcam, Cambridge, MA, USA) or rabbit anti‐β‐actin (Santa Cruz, CA, USA). After three washes with Tris‐buffered saline, the membranes were incubated with horseradish peroxidase‐conjugated donkey anti‐rabbit secondary antibody (HRP, Abgent, San Diego, USA) for 1 hours at room temperature. Protein bands were detected using an enhanced chemiluminescence reagent (Millipore, Billerica, MA, USA). Membranes were scanned, and image capture was performed using computerized imaging software, the FluorChem HD2 AlphaView (ProteinSimple, San Jose, CA, USA). Protein gray values were calculated using AlphaEaseFC image analysis software and corrected with values of β‐actin in the same lane. The grays of proteins were showed as relative values.
2.6. Real‐time PCR (RT‐PCR)
RT‐PCR was carried out as previously described.26 Briefly, and according to the manufacturer's instructions, total RNA was extracted from brain tissue and purified using the RNeasy Mini Kit (Qiagen, Duesseldorf, Germany). The first strands of complementary DNA were synthesized using 2 μg of total RNA with the SuperScript First‐Strand Synthesis System for RT‐PCR (Invitrogen, Carlsbad, CA, USA). Quantitative RT‐PCR (qRT‐PCR) was carried out on the Rotor‐Gene Q (Qiagen) using SYBR gene PCR Master Mix (Qiagen) and primers previously described.26 GAPDH was used as the internal reference gene. The cycle time values of the genes of interest first normalized to GAPDH, and then the expression levels of the genes were calculated and expressed as fold changes versus sham control.
2.7. Immunofluorescence staining and RT‐PCR analysis of cultured oligodendrocytes
For immunofluorescence staining, cultured oligodendrocytes were seeded on coverslips in 24‐well plates. The coverslip cultures were fixed with 4% paraformaldehyde in PBS for 30 minutes at room temperature. The cell membranes were pierced with 1.0% Triton X‐100 for 20 minutes, and cells were blocked with 5% donkey serum for 60 minutes at room temperature. The coverslips were incubated overnight at 4°C with primary antibodies including mouse anti‐NG2 (Chemicon, Temecula, CA, USA) and rabbit anti‐MBP (Abcam, Cambridge, MA, USA). Subsequently, the coverslips were incubated with donkey anti‐mouse and donkey anti‐rabbit antibodies for 1 hours at room temperature the following day. Finally, the coverslips were transferred onto glass slides which were previously filled with DAPI (a nuclear marker; Thermo fisher, MA, USA). The glass slides were kept in the dark at 4°C for further confocal microscopy (Olympus, Tokyo, Japan) observation after natural drying.
For RT‐PCR analysis,oligodendrocytes were seeded in six‐well plates and then collected in 1.5‐mL centrifuge tubes. RNA was extracted, complementary DNA was synthesized, and RT‐PCR was performed using primers previously described.27, 28 GAPDH was used as the internal reference gene. The cycle time values of the interest genes were first normalized to GAPDH, and then the expression levels of the genes were calculated and expressed as fold changes versus normothermic group.
2.8. Statistical analysis
All assessments were carried out by investigators blinded to the experimental group conditions. All values were expressed as mean ± standard error of the mean. A two way analysis of variance followed by Bonferroni post hoc test was used for the analysis of neurological function evaluation. A one way analysis of variance followed by Tukey's post hoc test was used for the analysis of western blot, immunofluorescence, and qRT‐PCR data. Student's t test was used for two group comparisons. The analysis was performed using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). Differences were deemed statistically significant at P < 0.05.
3. RESULTS
3.1. Hypothermia reduces infarct volume and attenuates behavioral deficits after MCAO
Infarct volume was significantly reduced in mice maintained at 33°C compared to those kept at 37°C at day 5 after MCAO, as shown by H&E staining (P < 0.001; Figure 1A,B). Next, we used two neurobehavioral tests to evaluate whether hypothermia treatment could increase neurological functional recovery. The rotarod test was performed on days 3, 5, 7, 9, 11, and 13 after MCAO to assess sensorimotor functions. Hypothermia treatment significantly increased the latency time to fall off the rotating rod compared with normothermia on days 5, 7, 9, 11, and 13 after MCAO (P < 0.05 at day 11; P < 0.01 at day 5 and 9; P < 0.001 at day 7 and 13; Figure 1C). The Morris water maze test was carried out to evaluate cognitive performance on days 21‐25 after MCAO. Hypothermia‐treated mice used less time to find the hidden platform than normothermia‐treated mice in the spatial learning test (P < 0.01 at day 23 and 25; P < 0.001 at day 24; Figure 1E). Results indicated that hypothermia facilitated spatial learning function recovery after MCAO. Furthermore, hypothermia‐treated mice also spent more time in the target quadrant than normothermia‐treated mice when the escape platform was removed (P < 0.01; Figure 1G), suggesting that hypothermia can alleviate memory deficits after MCAO. By comparison, the swimming speed of the both groups was not statistically different (Figure 1F), indicating that these differences in spatial learning and memory could not be due to variations in motor skills.
Figure 1.
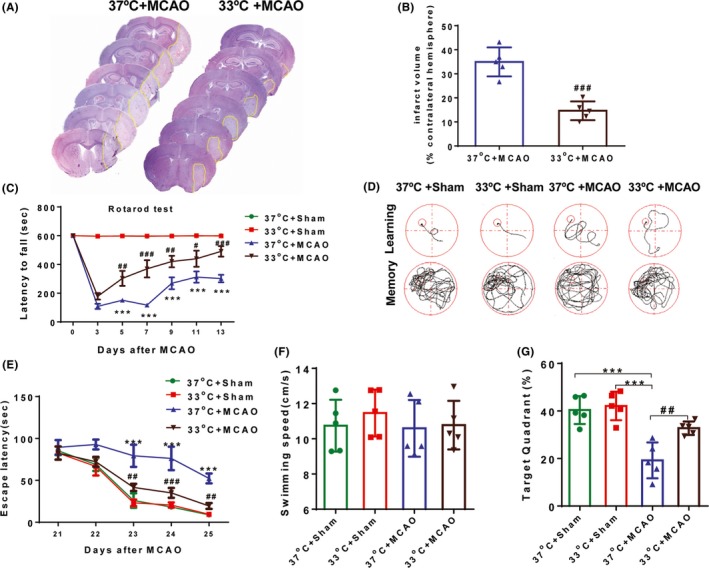
Hypothermia reduces infarct volume and improves behavioral functions after ischemic stroke. A, Representative H&E‐stained coronal sections at day 5 after MCAO in 37 and 33°C mice, respectively. Yellow lines indicate infarct areas. B, Infarct volumes were significantly reduced in 33°C mice compared with 37°C mice. C, The rotarod test showed 33°C + MCAO mice exhibited improved performance compared to 37°C + MCAO mice at 5, 7, 9, 11, and 13 days after MCAO. D, Representative images of the swimming paths in the Morris water maze test when the submerged platform was present and after it was removed. E, Time to find the submerged platform was measured at days 21‐25 after MCAO or sham surgery. F, Swimming speed was comparable among the four groups. G, The percentage of time spent in the goal quadrant after the submerged platform was removed was measured in both the MCAO and sham groups at day 25. All data are expressed as mean ± SEM, n = 5‐6 mice/group. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham groups; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus 37°C + MCAO
3.2. Hypothermia promotes WM integrity in ischemic mice
WM integrity is essential for normal neurological function, and disruption to this integrity is partially responsible for behavioral deficits after stroke.29, 30 Thus, we examined the effects of hypothermia on WM injury after MCAO. To accomplish this goal, we assessed two WM markers using double‐labeling immunofluorescence for SMI32, a marker of axonal damage, and MBP, a marker of myelin integrity.31 A marked reduction in MBP intensity and increased SMI‐32 intensity were detected in the WM‐enriched striatum (Figure 2A) and corpus callosum (Figure 2B) in the normothermia‐treated brains at day 28 after MCAO. Treatment with hypothermia significantly protected WM integrity after MCAO, as quantified by decreased SMI32 intensity (P < 0.001; Figure 2A,B) and increased MBP intensity (P < 0.05; Figure 2A,B) relative to normothermia‐treated controls. To confirm the protective effects of hypothermia in WM, we further assessed changes in MBP using western blotting at 28 day after MCAO (Figure 2C). Quantitative data showed that MBP was significantly higher in hypothermia‐treated mice than in normothermia mice (P < 0.05; Figure 2D).
Figure 2.
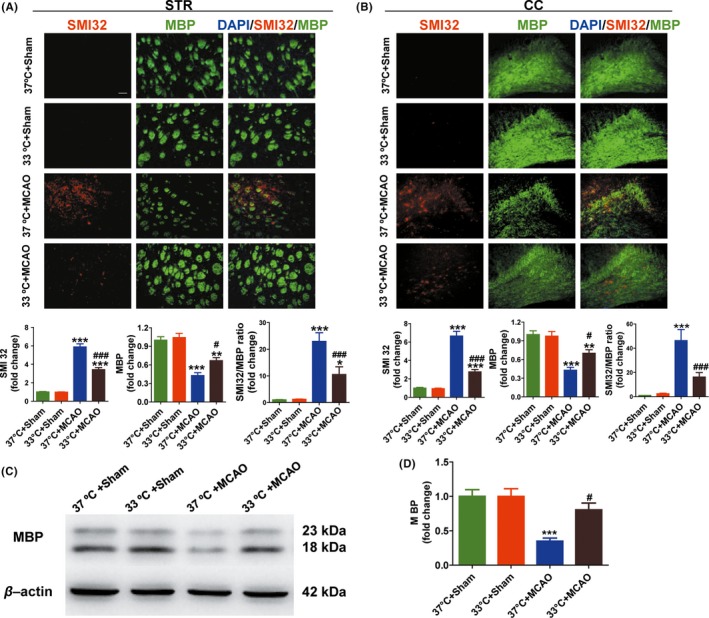
Hypothermia promotes WM integrity and increases MBP protein levels at day 28 after MCAO. A and B, Representative images and quantitative data of MBP (green) and SMI32 (red) immunostaining in the striatum (STR, A) and corpus callosum (CC, B) in the ipsilateral hemisphere. Scale bar: 50 μm. Degree of WM injury was expressed as the fold increase in SMI32 (A & B, lower, left), decrease in MBP (A & B, lower, middle), and the relative ratio of SMI‐32 to MBP (A & B, lower, right) immunostaining intensity. C, Representative images of western blot performed using the ipsilateral brain tissues at day 28 in MCAO and sham groups. β‐actin was used as an internal control. D, Quantitative data showed that the MBP was significantly higher in hypothermia‐treated mice (33°C) than in normothermia mice (37°C); n = 5 per group. Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham groups, #P < 0.05, ###P < 0.001 versus 37°C + MCAO group
3.3. Hypothermia attenuates M1 and promotes M2 polarization of microglia after MCAO
The differentiation of oligodendrocyte lineage cells is partially regulated by microglia. For example, the beneficial M2 phenotype microglia drive oligodendrocytes differentiation and enhance remyelination.32 Here, we tested whether hypothermia could regulate microglial phenotype after MCAO. Consistent with previous studies, Iba‐1 (a marker of microglia/macrophage) immunostaining demonstrated that hypothermia attenuated the proliferation of microglia in the peri‐infarct area of ischemic brains at 14 days after MCAO (P < 0.05; Figure 3C, right). Additionally, we found that hypothermia treatment significantly decreased M1 microglia (P < 0.001; Figure 3C, left), while at the same time significantly increased M2 microglia (P < 0.01; Figure 3C, middle), as found by decreased CD16+ (a marker of M1 microglia) and increased CD206+ (a marker of M2 microglia), respectively. Consistent with immunofluorescence results, mRNA expression of M1 microglia markers in hypothermia mice, including CD11b, iNOS, CD32, CD16, and CD86, was lower compared with normothernia mice (Figure 4A). By comparison, the levels of M2‐type genes (CD206, Arg1, IL‐10, and TGF‐β) in hypothermia mice were higher than those in normothermia mice (Figure 4B). In summary, these findings indicate that hypothermia can attenuate the harmful M1 phenotype and at the same time promote beneficial M2 phenotype after ischemia.
Figure 3.
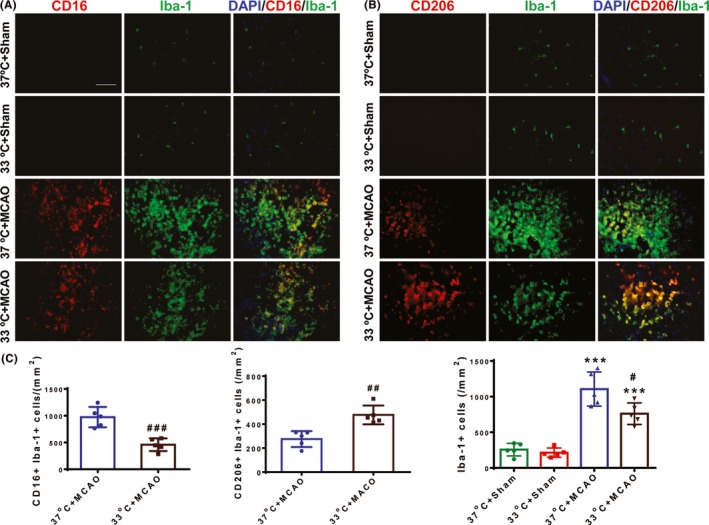
Hypothermia decreases the M1 microglial phenotype, increases the M2 phenotype, and decreases total microglia at 14 d after ischemic stroke. A, Representative double‐staining immunofluorescence of CD16 (red) and Iba‐1 (green) in the peri‐infarct areas of the ischemic and sham brain sections. B, Representative double‐staining immunofluorescence of CD206 (red) and Iba‐1 (green) in the peri‐infarct areas of the ischemic and sham brain sections. Scale bar: 50 μm. C, Quantification of CD16+/Iba‐1+ (left), CD206+/Iba‐1+ (middle), and Iba‐1+ (right) cells in the peri‐infarct areas of the ischemic brains; n = 5 per group. ***P < 0.001 versus sham groups; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus 37°C + MCAO group
Figure 4.
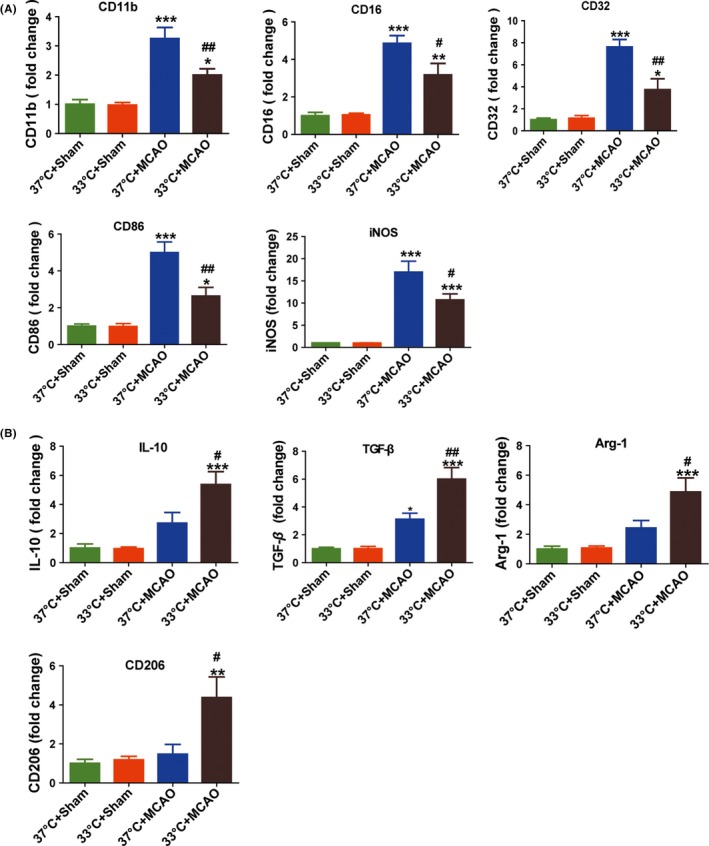
Hypothermia induces changes in mRNA expression of M1 and M2 phenotypic markers after MCAO. Total RNA was extracted from ischemic ipsilateral and sham brains at day 14 after MCAO and processed for RT‐PCR. A, Expression levels of mRNA for M1 markers. B, Expression levels of mRNA for M2 markers. Data are expressed as fold change versus 37°C + sham group; n = 5 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham groups; #P < 0.05 and ##P < 0.01 versus 37°C + MCAO
3.4. Hypothermia promotes oligodendrocytes differentiation after OGD
Hypothermia is known to promoteoligodendrocytes maturation in the hypoxic‐ischemic neonatal rat brain.21 In this study, we evaluated the effects of hypothermia on oligodendrocytes differentiation and maturation in vitro. The protocol for oligodendrocytes culture and hypothermia administration was shown in Figure 5B. Immunocytochemical characterization of mature oligodendrocytes and oligodendrocyte progenitor cells (OPCs) was analyzed using MBP and NG2 (a specific marker of OPC), respectively. The representative images of oligodendrocytes differentiation and maturation in every group at 8 days of T3 treatment are shown in Figure 5D. Changes in levels of NG2 and MBP mRNA in each group were found using qRT‐PCR. Hypothermia promoted oligodendrocytes differentiation after OGD, as found by decreased NG2 mRNA and increased MBP mRNA at day 8 of T3 treatment compared to controls (P < 0.05; Figure 5C).
Figure 5.
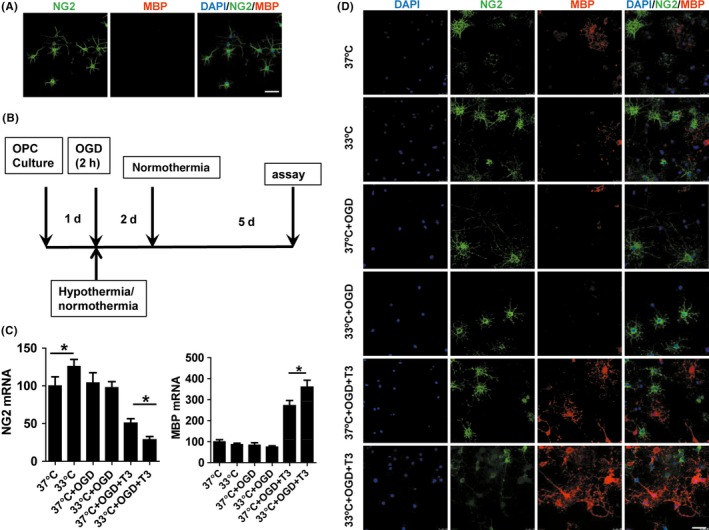
Cultured oligodendrocytes and the effects of hypothermia on oligodendrocytes differentiation. A, Almost all of the cultured oligodendrocytes were stained by NG2 but not MBP at 0 d. B, Protocol for oligodendrocytes culture and hypothermia administration. C, Quantitative RT‐PCR was performed to detect NG2 and MBP mRNA changes in the different groups. Hypothermia promotes oligodendrocytes differentiation, as manifested by decreased NG2 mRNA and increased MBP mRNA after oligodendrocytes differentiation was induced by T3 for 8 d. Data are expressed as fold change versus 37°C group. *P < 0.05. D, Representative immunofluorescence images of stage‐specific oligodendrocytes markers by NG2 and MBP at 8 d after culture in each group. Scale bar: 50 μm
4. DISCUSSION
Hypothermia is defined as an intentionally controlled reduction from a normal body temperature to temperatures in the range of 32‐35°C (mild), 28‐32°C (moderate), and 20‐28°C (deep), and mild hypothermia (32‐35°C) is the most promising neuroprotectant due to its multifaceted neuroprotective effects and is similarly protective as moderate to deep hypothermia.9 WM injury is an inevitable complication of ischemic stroke and an important factor of sensorimotor and cognitive impairment. However, the long‐term effects of mild hypothermia on WM injury after acute ischemic stroke have been, to date, rarely assessed. In the current study, we found that the mild hypothermia reduced WM injury and improved neurobehavioral functions 28 days after MCAO. WM damage, including loss of myelin and axonal degeneration, is known to induce signal transmission disturbances between different brain areas and results in severe neurobehavioral function deficits.33 In accordance with a previous study,34 we found that focal cerebral ischemia resulted in serious myelin damage in the WM‐enriched striatum and corpus callosum, which were significantly reduced by hypothermia. Thus, we believe that the preservation or restoration of WM integrity may enhance sensorimotor and cognitive functions after ischemic stroke.
Oligodendrocytes are essential for remyleniation of damaged WM and play a critical role in the restoration of neurobehavioral functions after ischemic stroke.35 OPCs are immature oligodendrocytes which have been shown to differentiate into mature oligodendrocytes and decrease the rates of demyelination after brain injury.36 Indeed, hypothermia has been shown to promote oligodendrocytes maturation in hypoxic‐ischemic neonatal rat brains.21 In this study, results demonstrated that hypothermia promoted the differentiation of OGD‐induced OPCs into mature oligodendrocytes. These mature oligodendrocytes produced MBP, which may have promoted WM recovery. Therefore, the enhanced differentiation of oligodendrocytes by hypothermia may be an important mechanism for the restoration of damaged WM.
Recent studies have emphasized the critical role of microglia in WM injury.37, 38 Neuroinflammation in the central nervous system involves the activation of resident microglia has been shown to induce secondary damage after initial traumatic brain injury.39, 40 Thus, current research has focused on finding novel agents to prevent, or at least, ameliorate this secondary damage after neural injury.41, 42 However, to date, findings have indicated that many of these antiinflammatory drugs have no evident role in the prevention or amelioration of neural inflammatory conditions.42 One of these failed factors in combating neuroinflammation has been the blanketed suppression of microglia, which are most likely deprived the brain of normal physiological functions associated with microglia. Although activated microglia have been shown to produce proinflammatory cytokines that result in neural damage,43, 44 other studies have shown that activated microglia are beneficial to the injured brain via the suppression of inflammation and removal of cellular debris.45, 46 It is now widely accepted in the scientific field that microglia have two distinct phenotypes, a beneficial M2 type and a destructive M1 type.26, 47, 48, 49 The M2 phenotype has been shown to drive oligodendrocyte differentiation and enhance remyelination,32 while the M1 phenotype was shown to exacerbate OGD‐induced oligodendrocytes cell death and WM damage after brain injury.38 In this study, we demonstrated that hypothermia attenuated the activation of microglia around the ischemic zone which was consistent with previous research.50 Intriguingly, we also found that hypothermia attenuated the polarization of M1 microglia and at the same time increased the polarization of M2 microglia, which likely promoted oligodendrocytes differentiation and increased mature oligodendrocytes after ischemic stroke. Collectively, these findings suggest that hypothermia can suppress inflammation mediated by activated microglia and promote microglial polarization to the beneficial M2 phenotype. Therefore, hypothermia most likely modulates the differentiation of oligodendrocytes indirectly through the regulation of microglial polarization after ischemic stroke.
In summary, our study concludes that mild focal hypothermia treatment immediately after ischemic stroke significantly promotes WM integrity 28 days after MCAO and improves neurobehavioral function. Hypothermia can also suppress inflammation mediated by activated microglia and promotes microglia polarization to the beneficial M2 phenotype. These findings likely contribute to the partial mechanisms involved in neuroprotection against acute cerebral ischemic stroke. Further studies are warranted to confirm the underlying mechanisms of hypothermia on microglia polarization and the differentiation of oligodendrocytes after ischemic stroke.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation for Distinguished Young Scholars (81325007), the National Natural Science Foundation of China (81641055 and 81471209), the Distinguished Professor of Cheung Kong Scholars program (T2014251), and the Beijing Municipal Administration of Hospitals' Mission Plan (SML20150802). We thank the CNS Neuroscience and Therapeutics editors for constructive advices to improve our manuscript.
Liu L‐Q, Liu X‐R, Zhao J‐Y, et al. Brain‐selective mild hypothermia promotes long‐term white matter integrity after ischemic stroke in mice. CNS Neurosci Ther. 2018;24:1275–1285. 10.1111/cns.13061
Liu and Liu equally contributed to this work.
REFERENCES
- 1. Pausova Z, Paus T, Abrahamowicz M, et al. Genes, maternal smoking, and the offspring brain and body during adolescence: design of the Saguenay Youth Study. Hum Brain Mapp. 2007;28:502‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ding G, Chen J, Chopp M, et al. White matter changes after stroke in type 2 diabetic rats measured by diffusion magnetic resonance imaging. J Cereb Blood Flow Metab. 2017;37:241‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muñoz Maniega S, Chappell FM, Valdés Hernández MC, et al. Integrity of normal‐appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small‐vessel disease. J Cereb Blood Flow Metab. 2017;37:644‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ho PW, Reutens DC, Phan TG, et al. Is white matter involved in patients entered into typical trials of neuroprotection? Stroke. 2005;36:2742‐2744. [DOI] [PubMed] [Google Scholar]
- 5. Hulkower MB, Poliak DB, Rosenbaum SB, Zimmerman ME, Lipton ML. A decade of DTI in traumatic brain injury: 10 years and 100 articles later. Am J Neuroradiol. 2013;34:2064‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Desmond DW. Cognition and white matter lesions. Cerebrovasc Dis. 2002;13(Suppl 2):53‐57. [DOI] [PubMed] [Google Scholar]
- 7. Wang CX, Shuaib A. Neuroprotective effects of free radical scavengers in stroke. Drugs Aging. 2007;24:537‐546. [DOI] [PubMed] [Google Scholar]
- 8. Moretti A, Ferrari F, Villa RF. Neuroprotection for ischaemic stroke: Current status and challenges. Pharmacol Therapeut. 2015;146:23‐34. [DOI] [PubMed] [Google Scholar]
- 9. Kim JY, Yenari MA. Hypothermia for treatment of stroke. Brain Circ. 2015;1:14‐25. [Google Scholar]
- 10. Rewell SS, Jeffreys AL, Sastra SA, et al. Hypothermia revisited: Impact of ischaemic duration and between experiment variability. J Cereb Blood Flow Metab. 2017;37:3380‐3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia‐induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904‐910. [DOI] [PubMed] [Google Scholar]
- 12. Globus MY, Alonso O, Dietrich WD, Busto R, Ginsberg MD. Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem. 1995;65:1704‐1711. [DOI] [PubMed] [Google Scholar]
- 13. Smith SL, Hall ED. Mild pre‐ and posttraumatic hypothermia attenuates blood‐brain barrier damage following controlled cortical impact injury in the rat. J Neurotrauma. 1996;13:1‐9. [DOI] [PubMed] [Google Scholar]
- 14. Deng H, Han HS, Cheng D, Sun GH, Yenari MA. Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke. 2003;34:2495‐2501. [DOI] [PubMed] [Google Scholar]
- 15. Silasi G, Colbourne F. Therapeutic hypothermia influences cell genesis and survival in the rat hippocampus following global ischemia. J Cereb Blood Flow Metab. 2011;31:1725‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsui T, Motoki Y, Yoshida Y. Hypothermia reduces toll‐like receptor 3‐activated microglial interferon‐β and nitric oxide production. Mediat Inflamm. 2013;2013:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davidson JO, Yuill CA, Zhang FG, Wassink G, Bennet L, Gunn AJ. Extending the duration of hypothermia does not further improve white matter protection after ischemia in term‐equivalent fetal sheep. Sci Rep. 2016;6:25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiong M, Chen L, Ma S, Yang Y, Zhou W. Short‐term effects of hypothermia on axonal injury, preoligodendrocyte accumulation and oligodendrocyte myelination after hypoxia‐ischemia in the hippocampus of immature rat brain. Dev Neurosci. 2013;35:17‐27. [DOI] [PubMed] [Google Scholar]
- 19. Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, et al. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mouse. J Cereb Blood Flow Metab. 2000;20:112‐118. [DOI] [PubMed] [Google Scholar]
- 20. Zhang SC, Lundberg C, Lipsitz D, O'Connor LT, Duncan ID. Generation of oligodendroglial progenitors from neural stem cells. J Neurocytol. 1998;27:475‐489. [DOI] [PubMed] [Google Scholar]
- 21. Xiong M, Li J, Ma S, Yang Y, Zhou W. Effects of hypothermia on oligodendrocyte precursor cell proliferation, differentiation and maturation following hypoxia ischemia in vivo and in vitro. Exp Neurol. 2013;247:720‐729. [DOI] [PubMed] [Google Scholar]
- 22. Wang J, Xia J, Zhang F, et al. Galectin‐1‐secreting neural stem cells elicit long‐term neuroprotection against ischemic brain injury. Sci Rep. 2015;5:9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bouët V, Freret T, Toutain J, Divoux D, Boulouard M, Schumann‐Bard P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol. 2007;203:555‐567. [DOI] [PubMed] [Google Scholar]
- 24. Wang G, Jiang X, Pu H, et al. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway. Neurotherapeutics. 2013;10:124‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang M, Wang S, Mao L, et al. Omega‐3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J Neurosci. 2014;34:1903‐1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063‐3070. [DOI] [PubMed] [Google Scholar]
- 27. Dehn D, Burbach GJ, Schäfer R, Deller T. NG2 upregulation in the denervated rat fascia dentata following unilateral entorhinal cortex lesion. Glia. 2006;53:491‐500. [DOI] [PubMed] [Google Scholar]
- 28. Mozafari S, Sherafat MA, Javan M, Mirnajafi‐Zadeh J, Tiraihi T. Visual evoked potentials and MBP gene expression imply endogenous myelin repair in adult rat optic nerve and chiasm following local lysolecithin induced demyelination. Brain Res. 2010;1351:50‐56. [DOI] [PubMed] [Google Scholar]
- 29. Wang Y, Liu G, Hong D, Chen F, Ji X, Cao G. White matter injury in ischemic stroke. Prog Neurogibol. 2016;141:45‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suenaga J, Hu X, Pu H, et al. White matter injury and microglia/macrophage polarization are strongly linked with age‐related long‐term deficits in neurological function after stroke. Exp Neurol. 2015;272:109‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sozmen EG, Kolekar A, Havton LA, Carmichael ST. A white matter stroke model in the mouse: axonal damage, progenitor responses and MRI correlates. J Neurosci Methods. 2009;180:261‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miron VE, Boyd A, Zhao J, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mifsud G, Zammit C, Muscat R, Di Giovanni G, Valentino M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neurosci Ther. 2014;20:603‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McIver SR, Muccigrosso M, Gonzales ER, et al. Oligodendrocyte degeneration and recovery after focal cerebral ischemia. Neuroscience. 2010;169:1364‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang R, Chopp M, Zhang ZG. Oligodendrogenesis after cerebral ischemia. Front Cell Neurosci. 2013;7:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gregersen R, Christensen T, Lehrmann E, Diemer NH, Finsen B. Focal cerebral ischemia induces increased myelin basic protein and growth‐associated protein‐43 gene transcription in peri‐infarct areas in the rat brain. Exp Brain Res. 2001;138:384‐392. [DOI] [PubMed] [Google Scholar]
- 37. Wang G, Shi Y, Jiang X, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA. 2015;112:2853‐2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang G, Zhang J, Hu X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864‐1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosi S. A polarizing view on posttraumatic brain injury inflammatory response. Brain Circ. 2016;2:126‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Truettner JS, Bramlett HM, Dietrich WD. Posttraumatic therapeutic hypothermia alters microglial and macrophage polarization toward a beneficial phenotype. J Cereb Blood Flow Metab. 2017;37:2952‐2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang S, Wang H, Turlova E, et al. GSK‐3β inhibitor TDZD‐8 reduces neonatal hypoxic‐ischemic brain injury in mice. CNS Neurosci Ther. 2017;23:405‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nimmo AJ, Vink R. Recent patents in CNS drug discovery: the management of inflammation in the central nervous system. Recent Pat CNS Drug Discov. 2009;4:86‐95. [DOI] [PubMed] [Google Scholar]
- 43. Dang DD, Saiyin H, Yu Q, et al. Effects of sevoflurane preconditioning on microglia/macrophage dynamics and phagocytosis profile against cerebral ischemia in rats. CNS Neurosci Ther. 2018;24:564‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lalancette‐Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596‐2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanisch U, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387‐1394. [DOI] [PubMed] [Google Scholar]
- 47. Ghanekar S, Corey S, Stonesifer C, et al. Current challenges in regenerative medicine for central nervous system disorders. Brain Circ. 2016;2:105‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He Y, Ma X, Li D, et al. Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF‐κB p65 signaling. J Cereb Blood Flow Metab. 2017;37:2938‐2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang M, Wan Y, Mao L, et al. Inhibiting the migration of M1 microglia at hyperacute period could improve outcome of tMCAO rats. CNS Neurosci Ther. 2017;23:222‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Drabek T, Tisherman SA, Beuke L, et al. Deep hypothermia attenuates microglial proliferation independent of neuronal death after prolonged cardiac arrest in rats. Anesth Analg. 2009;109:914‐923. [DOI] [PubMed] [Google Scholar]