

Abstract
Background
Aggregation of misfolded amyloid β (Aβ) in senile plaques causes oxidative stress and neuronal death in Alzheimer's disease (AD). Compounds possessing antiaggregation and antioxidant properties are promising candidate compounds for AD treatment.
Methods
We examined the potential of synthetic derivatives of licochalcone A and coumarin for inhibiting Aβ aggregation, scavenging reactive oxygen species (ROS), and providing neuroprotection by using biochemical assays and Tet‐On Aβ‐GFP 293/SH‐SY5Y cell models for AD.
Results
Among test compounds, LM‐031, a novel chalcone‐coumarin hybrid, inhibited Aβ aggregation and scavenged free oxygen radicals. LM‐031 markedly reduced Aβ misfolding and ROS as well as promoted neurite outgrowth and inhibited acetylcholinesterase in Tet‐On Aβ‐GFP 293/SH‐SY5Y cells. Mechanistic studies showed upregulation of the HSPB1 chaperone, NRF2/NQO1/GCLC pathway, and CREB/BDNF/BCL2 pathway. Decreased neurite outgrowth upon the induction of Aβ‐GFP was rescued by LM‐031, which was counteracted by knockdown of HSPB1, NRF2, or CREB.
Conclusion
Taken together, these findings demonstrate that LM‐031 exhibited antiaggregation, antioxidant, and neuroprotective effects against Aβ toxicity by enhancing HSPB1 and the NRF2‐related antioxidant pathway as well as by activating the CREB‐dependent survival and antiapoptosis pathway. These results imply that LM‐031 may be a new therapeutic compound for AD.
Keywords: Alzheimer’s disease, antioxidation, Aβ aggregation reduction, chalcone‐coumarin hybrid, neuroprotection, therapeutics
1. INTRODUCTION
Alzheimer's disease (AD), the most common type of dementia among the elderly, is characterized by the accumulation of amyloid β (Aβ) plaques and neurofibrillary tangles in the brain.1 Mutations in the Aβ precursor protein (APP) gene are a crucial genetic determinant of familial AD.2 APP is a cell surface receptor and transmembrane precursor protein that is proteolytically processed to generate distinct Aβ peptides containing 39‐43 amino acids.3 Among these peptides, the 42‐residue form of Aβ, Aβ42, is the predominant peptide deposited in amyloid plaques characteristic of AD.4 Aβ42 formation is favored by several APP mutations.5 Toxicity associated with aggregated Aβ42 is partly attributable to the overproduction of reactive oxygen species (ROS) and the disruption of oxidative stress pathways.6, 7, 8 In response to oxidative stress, cytosolic NRF2 (nuclear factor, erythroid 2 like 2) translocates to the nucleus and binds to the antioxidant response element (ARE) in the promoter region of antioxidant genes to initiate their transcription.9 NRF2‐mediated transcription is not induced in AD neurons despite the presence of oxidative stress.10 Both Aβ aggregation and NRF2 downregulation impair neuronal differentiation.11 Compounds possessing antiaggregation and antioxidant properties may modify the disease course in various AD models.12, 13, 14, 15
Licochalcone A (Lico A) is a major chalcone constituent obtained from the root of Glycyrrhiza inflata.16 Lico A activates the NRF2–ARE pathway, thereby reducing oxidative stress and polyglutamine aggregate formation in spinocerebellar ataxia type 3 cell models.17 In mouse RAW 264.7 macrophages, Lico A enhanced the NRF2‐mediated defense mechanism against oxidative stress and cell death.18 In addition, Lico A activated NRF2 in primary human fibroblasts and reduced oxidative stress in human skin.19 Coumarin, a natural product found in many green plants, also exhibits antioxidant activity.20, 21 In the APP/presenilin 1 (PS1) transgenic AD cell line, coumarin derivatives functioned as free radical scavengers to protect against H2O2‐induced oxidative stress.22 In addition, coumarin derivatives could prevent misfolded Aβ aggregation.23 These studies have suggested that Lico A and coumarin are promising lead compounds for the synthesis of novel AD therapeutics.
In the present study, we synthesized new derivatives of Lico A and coumarin and investigated their ability to prevent Aβ aggregation and oxidation as well as promote neuroprotection by using biochemical assays and Tet‐On Aβ‐GFP 293/SH‐SY5Y AD cells.24 Our findings indicate that the novel chalcone‐coumarin hybrid LM‐031 is a potential lead candidate for the development of AD therapeutics, owing to its ability to prevent Aβ aggregation and oxidation in neuronal cells.
2. MATERIALS AND METHODS
2.1. Test compounds
Lico A, coumarin, and curcumin were purchased from Sigma‐Aldrich, St. Louis, MO, USA. The Lico A derivatives LM‐004 and LM‐006 and the coumarin derivative LM‐016 were synthesized according to previous reports.25, 26 LM‐026 was synthesized using the procedure reported by Mazimba et al27 with some modifications, and LM‐031 was synthesized using the procedures reported by Dube et al28 and Tietze et al29 with some modifications. In a cell culture medium, LM‐004 and LM‐031 were soluble up to 1 mmol/L, whereas LM‐006, LM‐016, and LM‐026 were soluble up to 100 μmol/L.
2.2. Thioflavin T binding assay
To examine the inhibition of Aβ aggregation, the thioflavin T assay was performed. The Aβ42 (5 μmol/L; AnaSpec, Fremont, CA, USA) peptide was incubated with test compounds (5‐20 μmol/L) at 37°C for 48 hours to form aggregates. Subsequently, thioflavin T (10 μmol/L; Sigma‐Aldrich) was added and incubated for 5 minutes, and the fluorescence intensity of samples was recorded as described.24 The antiaggregation activity expressed as the half‐maximal effective concentration (EC50) was defined as the concentration of the extract or compound required for inhibiting Aβ aggregation by 50%.
2.3. 1,1‐Diphenyl‐2‐picrylhydrazyl assay
The free radical scavenging activity of the test compounds (10‐160 µmol/L) was determined using the stable 1,1‐diphenyl‐2‐picrylhydrazyl (DPPH, Sigma‐Aldrich) free radical assay as described previously.17 The radical scavenging activity was calculated using the following formula: [1 − (absorbance of the sample/absorbance of the control) × 100]. The antioxidant activity was expressed as EC50, which was defined as the concentration required for inhibiting DPPH radicals by 50%.
2.4. Cell culture and IC50 assay
Tet‐On Aβ‐GFP 293/SH‐SY5Y cells24 were maintained in Dulbecco's modified Eagle's medium (DMEM) (293) or DMEM‐F12 (SH‐SY5Y) containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 5 μg/mL of blasticidin, and 100 μg/mL of hygromycin (InvivoGen, San Diego, CA, USA). Cell viability was measured on the basis of the reduction of 3,(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT, Sigma‐Aldrich). Briefly, 5 × 104 cells were plated into 48‐well plates, grown for 20 hours, and treated with the test compounds. After a day, 20 µL of MTT (5 mg/mL in PBS, Sigma‐Aldrich) was added to the cells and incubated for 2 hours. The absorbance of the insoluble product was measured at 570 nm by using a spectrophotometer (µQuant, Bio‐Tek, Winooski, VT, USA). The half‐maximal inhibitory concentration (IC50) was defined as the concentration of a compound required for the reduction of 570‐nm signals by 50%.
2.5. Aβ‐GFP fluorescence assay
Tet‐On Aβ‐GFP 293 cells were seeded in 96‐well plates at a density of 8 × 103 cells/well. The test compounds were added the following day. After 8 hours in culture, doxycycline (2 µg/mL, Sigma‐Aldrich) was added to induce Aβ‐GFP expression. On day 5, the cells were stained with Hoechst 33342 (0.1 µg/mL, Sigma‐Aldrich) for 30 minutes, and images of the cells were automatically recorded at the wavelengths of 482 (excitation)/536 (emission) nm (ImageXpress Micro Confocal High‐Content Imaging System) and analyzed (MetaXpress Image Acquisition and Analysis Software) (Molecular Devices, Sunnyvale, CA, USA).
2.6. Aβ‐GFP RNA analysis
Tet‐On Aβ‐GFP 293 cells were plated at a density of 3 × 105 cells/well in 6‐well plates. Test compound treatment (1‐10 µmol/L) and Aβ‐GFP induction were performed as described. To measure the content of Aβ‐GFP RNA on day 5, total RNA was extracted and reverse‐transcribed to cDNA (SuperScript III Reverse Transcriptase; Invitrogen). Real‐time quantitative PCR experiments were performed using 100 ng of cDNA and the gene‐specific TaqMan fluorogenic probes PN4331348 (EGFP) and 4326321E (HPRT1) (StepOnePlus Real‐time PCR system, Applied Biosystems, Foster City, CA, USA). Fold change was calculated using the formula 2ΔCt, ΔCT = CT (control) − CT (target), in which CT indicates the cycle threshold.
2.7. ROS analysis
Tet‐On Aβ‐GFP 293 cells were plated at a density of 6 × 104 cells/well in a 12‐well plate. Test compound treatment (1‐10 µmol/L) and Aβ‐GFP induction were performed as described. To measure oxidative stress in the cells on day 5, a fluorogenic reagent (CellROX Deep Red, Molecular Probes, Waltham, MA, USA) was added at a final concentration of 5 μmol/L and incubated at 37°C for 30 minutes. After washing, the cells (104) were analyzed for red (ROS) fluorescence on a flow cytometer (Becton‐Dickinson, Franklin Lakes, NJ, USA) at excitation/emission wavelengths of 635/661 ± 8 nm.
2.8. Neurite outgrowth analysis
Tet‐On Aβ‐GFP SH‐SY5Y cells were seeded at a density of 3 × 104 cells/well in a 24‐well plate, and retinoic acid (10 µmol/L, Sigma‐Aldrich) was added at the seeding time. On day 2, the cells were treated with test compounds (1‐10 µmol/L) for 8 hours before doxycycline (2 µg/mL) was added to induce Aβ‐GFP expression. On day 8, the cells were fixed in 4% paraformaldehyde for 15 minutes, permeabilized in 0.1% Triton X‐100 for 10 minutes, and blocked in 1% bovine serum albumin (BSA) for 20 minutes. The primary TUBB3 antibody (1:1000, Covance, Princeton, NJ, USA) was used to stain cells at 4°C overnight, followed by a secondary goat antirabbit Alexa Fluor 555 antibody (1:1000, Molecular Probes) at room temperature for 3 hours. After staining nuclei with 4′,6‐diamidino‐2‐phenylindole, images of the cells were captured, and total neurite outgrowth was analyzed (neurite outgrowth application module, Molecular Devices).
2.9. Acetylcholinesterase activity assay
Tet‐On Aβ‐GFP SH‐SY5Y cells were seeded at a density of 4 × 105 cells/well in a 6‐well plate, and retinoic acid (10 µmol/L) was added at the seeding time. On the following day, the cells were treated with the test compounds (1 µmol/L), and Aβ‐GFP expression was induced as described. On day 8, the cells were collected and resuspended in PBS, followed by sonication and centrifugation at 14 000 g for 5 minutes at 4°C to collect the supernatant. Acetylcholinesterase (AChE) activity was determined by using 20 µg of cell extracts (AChE activity assay kit, Sigma‐Aldrich). The mixture was incubated for 2‐10 minutes at room temperature, and absorbance was read at 412 nm in a spectrophotometer (Multiskan GO, Thermo Scientific, Waltham, MA, USA).
2.10. Western blot analysis
Differentiated Tet‐On Aβ‐GFP SH‐SY5Y cells were treated with the test compounds (1 µmol/L). On day 8, the cells were collected, washed twice with ice‐cold PBS, and lysed in RIPA buffer containing a protease inhibitor cocktail (Sigma‐Aldrich) at 4°C for 30 minutes, followed by sonication. The cell lysate was centrifuged at 14 000 g for 20 minutes at 4°C, and the supernatant was stored at −70°C until further analysis. Protein samples (30 µg) were subjected to 10% SDS‐PAGE and subsequently transferred onto polyvinylidene difluoride membranes through reverse electrophoresis. After blocking, the membranes were incubated with primary antibodies, namely anti‐NRF2, anti‐CREB, anti‐BDNF, anti‐BCL2, anti‐HSPB1, anti‐GFP (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐NQO1 (1:500), anti‐β‐tubulin (1:5000) (Sigma‐Aldrich), anti‐GCLC (1:1000, Abcam, Cambridge, MA, USA), anti‐pCREB (Ser133; 1:500), anti‐β‐actin (1:5000) (Millipore, Billerica, MA, USA), anti‐BAX (1:500, Biovision, Milpitas, CA, USA), anti‐CASP3 (1:500, Cell Signaling, Danvers, MA, USA), and anti‐GAPDH (1:1000, MDBio, Taipei, Taiwan), at 4°C overnight. The resultant membranes were washed and incubated with the corresponding secondary antibodies coupled with a horseradish peroxidase‐conjugated goat antimouse, goat antirabbit, or donkey antigoat IgG antibody (1:5000, GeneTex, Irvive, CA, USA) at room temperature for 1 hour. Immunoreactive bands were detected using a chemiluminescent substrate (Millipore).
2.11. RNA interference
Lentiviruses containing short hairpin RNA (shRNA) targeting HSPB1 (TRCN0000008753, target sequence: CCGATGAGACTGCCGCCAAGT), NRF2 (TRCN0000007558, target sequence: CCGGCATTTCACTAAACACAA), and CREB (TRCN0000226466, target sequence: ACATTAGCCCAGGTATCTATG) and a negative control scrambled shRNA (TRC2.Void) were obtained from the National RNAi Core Facility, IMB/GRC, Academia Sinica. SH‐SY5Y Aβ‐GFP cells were plated at a density of 2 × 104/well (24‐well plates for neurite outgrowth analysis) or 6 × 105/well (6‐well plates for protein analysis) with retinoic acid (10 µmol/L) added on day 1 and transduced with lentivirus (multiplicity of infection: 3) in a medium containing 8 µg/mL of polybrene (Sigma‐Aldrich) on day 2. At 24 hours postinfection, the culture medium was changed and cells were pretreated with LM‐031 (1 µmol/L) for 8 hours, followed by the induction of Aβ‐GFP expression for 1 week. Subsequently, the cells were analyzed for neurite outgrowth as described or were collected for HSPB1, NRF2, and CREB protein analysis.
2.12. Statistical analysis
For each set of values, data were represented as the mean of three independent experiments. Differences between groups were evaluated using two‐tailed Student's t test or analysis of variance (ANOVA; one‐way, and two‐way) with post hoc LSD adjustment when appropriate. P values <0.05 were considered statistically significant.
3. RESULTS
3.1. Test compounds and IC50 cytotoxicity
The test compounds were classified into two groups: one is defined in terms of the structure of Lico A (LM‐004, LM‐006, and LM‐026) and the other is a coumarin‐chalcone hybrid (LM‐016 and LM‐031) generated through a ring closure reaction of chalcone (Figure 1A). Naturally occurring coumarin and Lico A possessing the antioxidant property17, 20, 21 were also included for comparison. The MTT assay was performed using uninduced Aβ‐GFP 293 and SH‐SY5Y cells following treatment with the test compounds (0.1‐100 μmol/L) for 24 hours (Figure 1B). The potent Aβ aggregate inhibitor curcumin30 reduced cell viability by 50% at a concentration of up to 10 μmol/L in uninduced Aβ‐GFP 293 and SH‐SY5Y cells. The IC50 values of Lico A, LM‐004, LM‐006, LM‐026, coumarin, LM‐016, and LM‐031 in uninduced 293/SH‐SY5Y cells were 51/47, 51/8, 51/10, 71/45, >100/>100, >100/55, and >100/>100 μmol/L, respectively. These results demonstrated the low cytotoxicity of the test compounds. In uninduced Aβ‐GFP 293/SH‐SY5Y cells, the IC50 value of LM‐031 (>100 μmol/L) was similar to that of coumarin (>100/>100 μmol/L), whereas the IC50 value of Lico A was lower (51/47 μmol/L).
Figure 1.

Cytotoxicity of test compounds. A, Structure, formula, and molecular weight of Lico A, coumarin, synthetic derivative compounds LM‐004, LM‐006, LM‐026, LM‐016, and LM‐031, and positive control curcumin. Below is the ring closure reaction of chalcone to generate the coumarin‐chalcone hybrid. B, Cytotoxicity of test compounds against Aβ‐GFP 293 and SH‐SY5Y cells using the MTT assay. Uninduced cells were treated with 0.1‐100 μmol/L Lico A, coumarin, synthetic derivative compounds, or 1‐10 μmol/L curcumin, and cell viability was measured the following day (n = 3). To normalize, the relative viability in untreated cells was set at 100%. IC50 values are presented
3.2. Inhibition of Aβ aggregation and radical scavenging activity of test compounds
The inhibition of Aβ aggregation and that of oxidative stress are considered important treatment approaches for AD. The inhibition of Aβ aggregation by the test compounds was measured on the basis of fluorescence generated by thioflavin T binding. As shown in Figure 2A, Lico A, LM‐004, LM‐006, LM‐026, coumarin, LM‐016, LM‐031, and curcumin had EC50 values of 18, 21, 12, 30, 27, 15, 11, and 27 μmol/L, respectively. LM‐031 showed the best activity in inhibiting Aβ aggregation among the test compounds.
Figure 2.
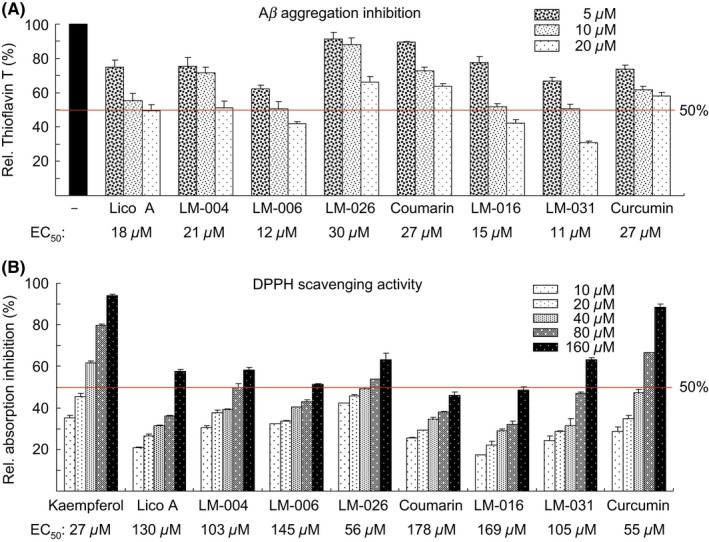
Prevention of aggregation and oxidation by test compounds in biochemical assays. A, Aβ aggregation inhibition of Lico A, coumarin, LM‐004, LM‐006, LM‐026, LM‐016, LM‐031, and curcumin (5‐20 μmol/L) (n = 3). To normalize, the relative thioflavin T fluorescence of Aβ42 without compound addition was set at 100%. Shown below are the EC50 values. B, DPPH radical scavenging activity of kaempferol, Lico A, coumarin, LM‐004, LM‐006, LM‐026, LM‐016, LM‐031, and curcumin (10‐160 μmol/L) (n = 3). Shown below are the EC50 values
The ROS scavenging activity of the test compounds was examined using the DPPH assay. Kaempferol, a natural flavonol with a strong antioxidant property,31 was also included as a positive control. As shown in Figure 2B, kaempferol, Lico A, LM‐004, LM‐006, LM‐026, coumarin, LM‐016, LM‐031, and curcumin had EC50 values of 27, 130, 103, 145, 56, 178, 169, 105, and 55 μmol/L, respectively. The number and position of the active OH group for trapping a free radical32 may account for the radical scavenging efficacy of test compounds.
3.3. Effects of test compounds on Tet‐On Aβ‐GFP 293 cells
We examined the potential of test compounds to prevent cellular Aβ aggregation and the associated oxidation. In Aβ‐GFP fusion protein, Aβ aggregates rapidly, resulting in misfolding of the fused GFP, thereby reducing the fluorescence intensity. The inhibition of Aβ aggregation may improve GFP folding, thus increasing the fluorescent signal in Aβ‐GFP‐expressing cells.33 Tet‐On Aβ‐GFP 293 cells were used to assess the effect of the test compounds on Aβ aggregation (Figure 3A). Figure 3B presents the representative fluorescent images of Aβ‐GFP cells with or without curcumin treatment. Relative GFP fluorescence (Figure 3C) was measured in wells showing >80% viability. As a positive control, curcumin at a concentration of 2.5‐5 μmol/L (effective dose) significantly increased Aβ‐GFP fluorescence (107%‐120%). Treatment with Lico A (0.001‐1 μmol/L, 105%‐115%), LM‐004 (0.001‐0.1 μmol/L, 103%‐111%), LM‐006 (0.001‐1 μmol/L, 105%‐112%), LM‐026 (0.01‐1 μmol/L, 102%‐105%), coumarin (0.01‐1 μmol/L, 103%‐109%), LM‐016 (0.001‐10 μmol/L, 103%‐117%), and LM‐031 (0.001‐10 μmol/L, 107%‐125%) also significantly increased Aβ‐GFP fluorescence. By contrast, treatment with Lico A (1 μmol/L), coumarin (1 μmol/L), LM‐031 (1‐10 μmol/L), or curcumin (5 μmol/L) did not significantly alter the Aβ‐GFP RNA level (24.2‐25.2 vs 25.0 times; Figure 3D). Among all the test compounds, the effective concentration of LM‐031 had the widest range.
Figure 3.
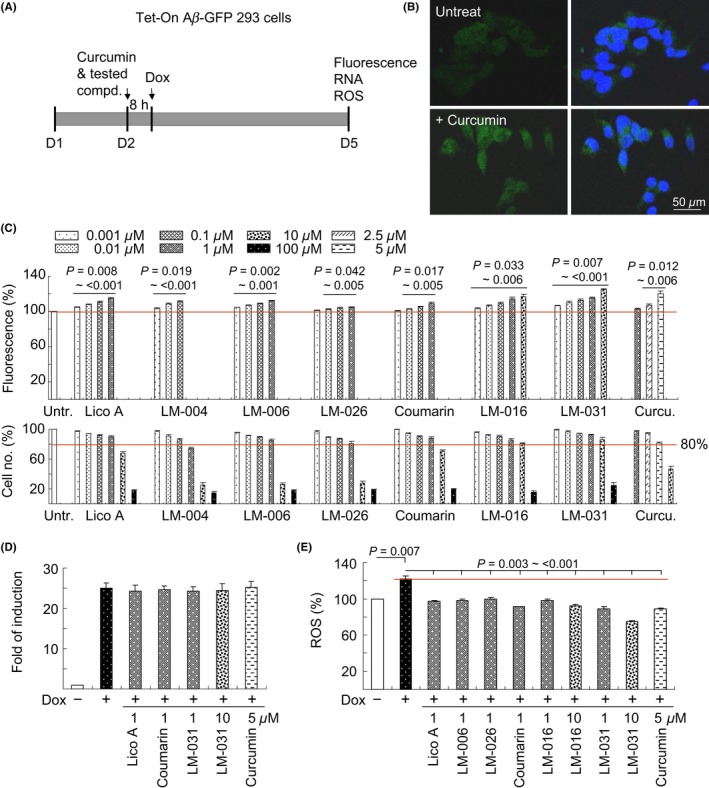
Prevention of aggregation and oxidation by test compounds in Tet‐On Aβ‐GFP 293 cells. A, Experiment flowchart. Cells were plated at day 1 and treated with curcumin or test compounds next day for 8 hours. Then, doxycycline (2 µg/mL) was added to the medium to induce Aβ‐GFP expression. GFP fluorescence, caspase 3 activity, and ROS were assessed on day 5. B, Fluorescent images of Aβ‐GFP cells with or without curcumin treatment. C, GFP fluorescence assay with curcumin (1‐10 µmol/L) or test compound (0.001‐100 µmol/L) treatment. To normalize, the relative GFP fluorescence of untreated cells was set at 100%, and P values were compared. Shown below are the relative cell numbers analyzed. GFP fluorescence was measured in wells containing at least 80% viable cells. D, Real‐time PCR quantification of Aβ‐GFP mRNA levels relative to HPRT1 mRNA in untreated and Lico A (1 µmol/L)‐, coumarin (1 µmol/L)‐, LM‐031 (1‐10 µmol/L)‐, or curcumin (5 µmol/L)‐treated cells (n = 3). E, ROS assay with 1 µmol/L Lico A, coumarin, LM‐006, LM‐026, 1‐10 µmol/L LM‐016, LM‐031, or 5 µmol/L curcumin treatment (n = 3). The relative ROS of uninduced cells was normalized to 100%. P values between induced and uninduced cells as well as between treated and untreated cells were compared
In addition, we examined the ROS level in Tet‐On Aβ‐GFP 293 cells following treatment with the test compounds. The ROS level (122%) significantly increased in cells with induced Aβ‐GFP expression compared with uninduced control cells (100%; Figure 3E). Treatment with the test compounds significantly reduced the ROS level (75%‐98% of the control) compared with no treatment (122%). LM‐031 at a concentration of 1‐10 μmol/L showed the best performance in reducing the ROS level (89%‐75% of the control) among the test compounds.
Aβ promotes oxidative stress, resulting in ROS formation, lipid peroxidation, protein oxidation, Ca2+ dysregulation, mitochondrial impairment, and other cellular responses that contribute to neuronal death.34, 35, 36 Anthocyanins protected PC‐12 cells from Aβ‐induced injury through the inhibition of oxidative damage, intracellular calcium influx, mitochondrial dysfunction, and apoptosis.37 The antioxidant activity represented by the scavenging of DPPH free radicals and the decrease in the ROS level in Aβ‐GFP 293 cells indicated that LM‐031 is a promising compound for the treatment of AD and other oxidative stress‐related neurodegenerative diseases.
3.4. Effects of test compounds on Tet‐On Aβ‐GFP SH‐SY5Y cells
To further evaluate the neuroprotective effect of test compounds, we applied these compounds to the Tet‐On Aβ‐GFP SH‐SY5Y cells (Figure 4A).38 The induction of Aβ‐GFP significantly upregulated the CASP3 expression level (168% of the control). Treatment of cells with Lico A, coumarin, LM‐006, LM‐026, LM‐016, LM‐031 (1 µmol/L), and curcumin (5 µmol/L) significantly attenuated the CASP3 expression level (92%‐104% vs 168%; Figure 4B). Aβ‐GFP induction significantly reduced the neurite length (87% of the control). Pretreatment with Lico A, coumarin, LM‐006, LM‐026 (1 µmol/L), LM‐016, LM‐031 (1‐10 µmol/L), and curcumin (5 µmol/L) reversed this negative effect (98%‐110% vs 87%; Figure 4C‐D). Among the test compounds, LM‐031 showed the best activity in promoting neurite outgrowth (104%‐110% in 1‐10 μmol/L).
Figure 4.
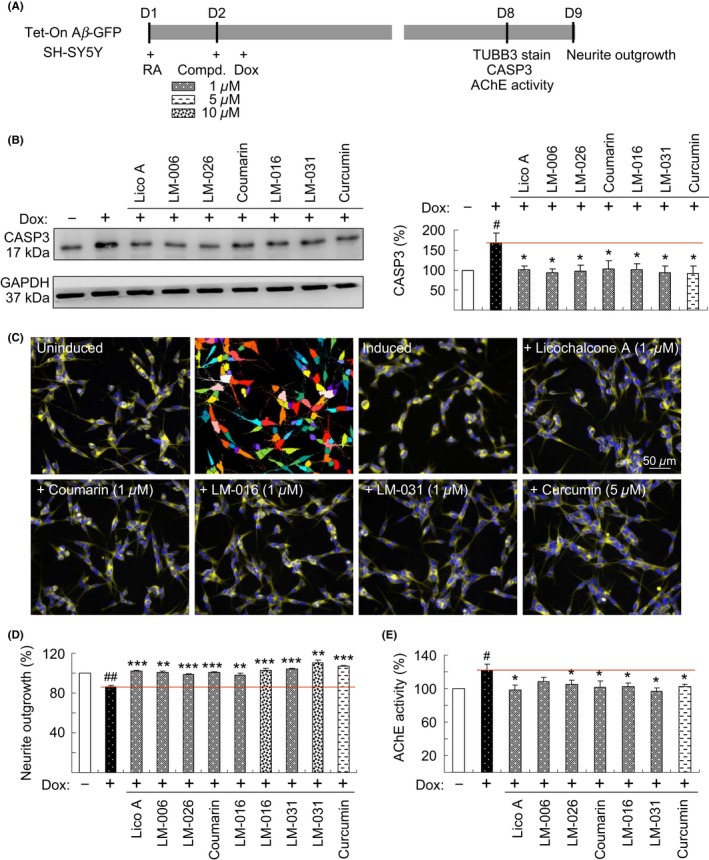
Neuroprotective effects of test compounds on Tet‐On Aβ‐GFP SH‐SY5Y cells. A, Experimental outline. Cells were plated with retinoic acid (RA, 10 µmol/L). On the following day, 1 µmol/L Lico A, coumarin, LM‐006, LM‐026, 1‐10 µmol/L LM‐016, LM‐031, or 5 µmol/L curcumin were added to cells for 8 hours, followed by the induction of Aβ‐GFP expression (+ Dox, 2 µg/mL) for 6 days. Neurite outgrowth was assessed after TUBB3 staining. In addition, the CASP3 level and AChE activity were assessed. B, CASP3 protein level was analyzed through immunoblotting using CASP3 and GAPDH (internal control) antibodies (n = 3). To normalize, the expression level in uninduced (−Dox) cells was set at 100%. P values between induced and uninduced cells as well as between treated and untreated cells were compared. C, Fluorescence microscopy images of uninduced or induced Aβ‐GFP cells treated with or without 1 µmol/L Lico A, coumarin, LM‐016, LM‐031, or 5 µmol/L curcumin. Shown next to the uninduced image is the image segmentation of uninduced cells with a multicolored mask to assign each outgrowth to a cell body for quantification. D, Quantification of neurite outgrowth with 1 µmol/L Lico A, coumarin, LM‐006, LM‐026, 1‐10 µmol/L LM‐016, LM‐031, or 5 µmol/L curcumin treatment (n = 3). To normalize, P values between induced and uninduced cells as well as between treated and untreated cells were compared, with the relative neurite outgrowth of uninduced cells as 100%. E, AChE activity assay with 1 µmol/L Lico A, coumarin, LM‐006, LM‐026, LM‐016, LM‐031, or 5 µmol/L curcumin treatment (n = 3). The relative AChE activity of uninduced cells was normalized to 100%. P values between induced and uninduced cells (# P < 0.05 and ## P < 0.01) as well as between treated and untreated cells (*P < 0.05, **P < 0.01, and ***P < 0.001) were compared
In the brains of patients with AD, Aβ aggregation colocalized with AChE, which accelerated Aβ misfolding.39 A monoclonal antibody against AChE reduced the formation of Aβ aggregation.40 Specific inhibitors of AChE provide an attractive possibility for treating AD.41 Thus, we assessed the potential of the test compounds to inhibit AChE in Aβ‐GFP SH‐SY5Y cells. After the induction of Aβ‐GFP for 6 days, AChE activity significantly increased (122% of the control). Treatment of cells with Lico A, LM‐006, LM‐026, coumarin, LM‐016, LM‐031 (1 μmol/L), and curcumin (5 μmol/L) attenuated the AChE activity induced by Aβ overexpression (97%‐109% vs 122%; Figure 4E). LM‐031 showed the best activity in reducing AChE activity (97% in 1 μmol/L) and thus was selected for further investigation.
3.5. Molecular targets of LM‐031, Lico A, and coumarin in Tet‐On Aβ‐GFP SH‐SY5Y cells
Small heat shock proteins may regulate Aβ aggregation and serve as antagonists of the biological action of misfolded Aβ.42 Among them, HSPB1 demonstrated the potential to protect cortical neurons against Aβ toxicity,43 and overexpression of HSPB1 reduced the amount of amyloid plaques in APP/PS1 mice.44 Thus, we examined the expression levels of HSPB1 and soluble Aβ‐GFP following treatment with LM‐031, Lico A, and coumarin in Tet‐on Aβ‐GFP SH‐SY5Y cells. As shown in Figure 5A, the induction of Aβ‐GFP in differentiated SH‐SY5Y cells significantly attenuated the expression of HSPB1 (77% of the control). This reduction could be rescued through treatment with LM‐031, Lico A, or coumarin (90%‐93% vs 77%), accompanied by an increased soluble Aβ‐GFP protein level (162%‐171% of the untreated cells). Previously, we reported that the synthetic indole/indolylquinoline compounds NC009‐1/NC009‐7 reduced Aβ and tau misfolding and aggregation by facilitating appropriate folding and enhancing HSPB1 expression.24, 37 Similar to the reported NC009 compounds, LM‐031 facilitated appropriate folding and enhanced HSPB1 expression to reduce Aβ misfolding and aggregation.
Figure 5.
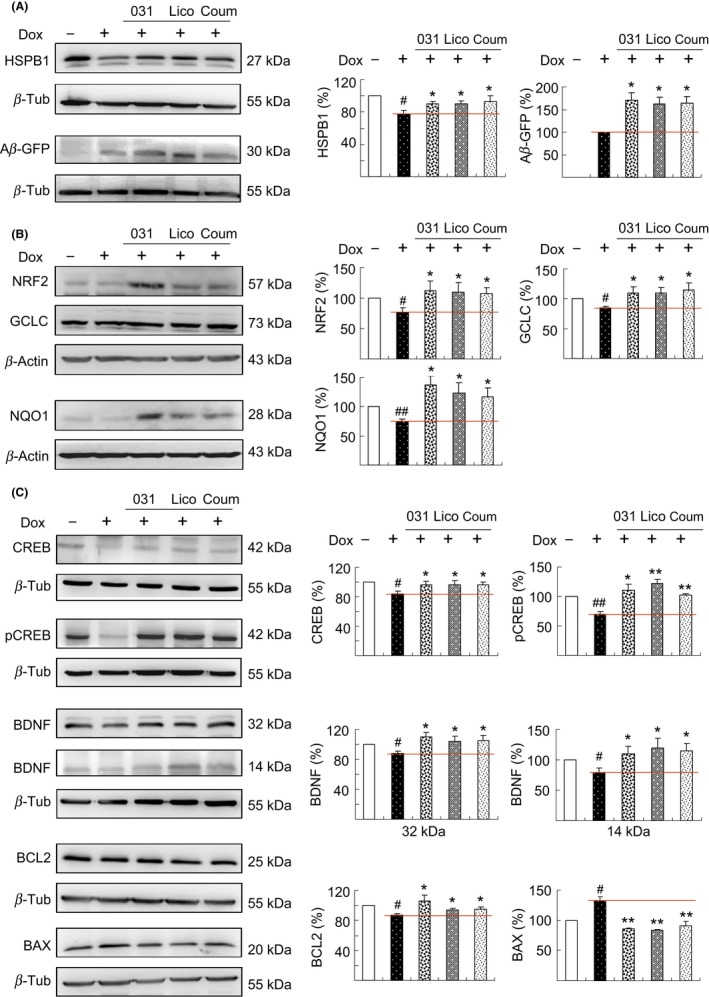
Enhanced expression of HSPB1, NRF2, and CREB pathways following test compound administration in Aβ‐GFP SH‐SY5Y cells. On day 2, differentiated SH‐SY5Y cells were pretreated with 1 μmol/L LM‐031, Lico A, or coumarin for 8 hours, and Aβ‐GFP expression was induced for 6 days. Relative A, HSPB1 and soluble Aβ‐GFP, B, NRF2, GCLC, and NQO1, and C, CREB, pCREB, BDNF, BCL2, and BAX protein levels were analyzed through immunoblotting using specific antibodies. Protein levels were normalized to β‐actin or β‐tubulin internal control. Relative protein levels are shown on the right side of the representative Western blot images. To normalize, the expression level in uninduced (−Dox) cells was set at 100%. For Aβ‐GFP, the soluble level in induced (+Dox) cells was set at 100%. P values between induced and uninduced cells (# P < 0.05 and ## P < 0.01) as well as between treated and untreated cells (*P < 0.05 and **P < 0.01) were compared
To determine molecular targets involved in preventing oxidation, we examined whether LM‐031, Lico A, and coumarin upregulated NRF2 and its downstream gene expression in Tet‐On Aβ‐GFP SH‐SY5Y cells, because NRF2 provides protection against Aβ‐induced neural stem/progenitor cell toxicity.11 As shown in Figure 5B, the induction of Aβ‐GFP for 6 days significantly attenuated the expression of NRF2 (76%), GCLC (84%), and NQO1 (75%) compared with that in uninduced control (100%). This reduction can be rescued by the addition of LM‐031, Lico A, or coumarin, which significantly increased the expression of NRF2 (108%‐112%), GCLC (110%‐115%), and NQO1 (117%‐137%) compared with that in untreated cells (76% for NRF2, 84% for GCLC, 75% for NQO1).
Accumulation of Aβ reduces CREB activation/phosphorylation,45 which plays a central role in synaptic dysfunction and memory impairment in AD.46 Following phosphorylation at Ser133, CREB upregulated BCL247 and BDNF48 gene expression through the recruitment of the coactivator CBP to promote cell survival and modulate synaptic activity. CREB‐dependent BDNF and BCL2 pathways are impaired in the hippocampus of APP transgenic mice, and the overexpression of CREB could protect against Aβ‐induced neuronal apoptosis in rat primary hippocampal neurons.49 Thus, we examined the protein levels of CREB/pCREB, BDNF, and BCL2 following treatment with LM‐031, Lico A, or coumarin in Tet‐On Aβ‐GFP SH‐SY5Y cells. The protein expression of CREB (84%), pCREB (70%), pro‐BDNF (88%), mature BDNF (93%), and BCL2 (87%) was attenuated after the induction of Aβ‐GFP for 6 days relative to the control (100%, Figure 5C). The addition of LM‐031, Lico A, or coumarin rescued the reduction of CREB (94% vs 84%), pCREB (103%‐122% vs 70%), pro‐BDNF (104%‐110% vs 88%), mature BDNF (99%‐106% vs 93%), and BCL2 (94%‐107% vs 87%). In response to the antiapoptotic BCL2 change, the expression of proapoptotic BAX increased after the induction of Aβ‐GFP (133% of the control), whereas addition of LM‐031, Lico A, or coumarin attenuated the BAX expression level (83%‐91% vs 133%).
Aβ deposition causes neuronal death through many possible mechanisms including oxidative stress, excitotoxicity, energy depletion, inflammation, and apoptosis.50 Catalpol protected primary cultured cortical neurons induced by Aβ42 through a mitochondrial‐dependent caspase pathway.51 Hyperbaric oxygen and a Ginkgo biloba extract (EGB761) improved escape latency in rats administered with Aβ25‐35 by increasing SOD and GSH expression and reducing BAX and caspase 3 expression.52 In our study, treatment with LM‐031 upregulated antioxidant NRF2 pathways and reduced BAX and CASP3 expression in Aβ‐GFP SH‐SY5Y cells, supporting the role of LM‐031 in treating AD by preventing oxidative stress and blocking mitochondria‐mediated apoptosis pathways. In addition, LM‐031 demonstrated its potential as an enhancer of CREB‐mediated pathways, further highlighting its possible role as a novel compound with multiple targets in AD treatment.
3.6. HSPB1, NRF2, and CREB as therapeutic targets in LM‐031‐treated Aβ‐GFP SH‐SY5Y cells
We explored the effect of silencing HSPB1, NRF2, and CREB genes on neurite outgrowth by using lentivirus‐mediated RNA interference. Aβ‐GFP SH‐SY5Y cells were infected with HSPB1‐specific, NRF2‐specific, CREB‐specific, or scramble (negative control) shRNA. On the following day, cells were pretreated with LM‐031 for 8 hours, followed by the induction of Aβ‐GFP expression by using doxycycline for 6 days (Figure 6A). In scramble shRNA‐infected cells, the induction of Aβ‐GFP reduced the expression of HSPB1 (79%), NRF2 (56%), and CREB (77%) relative to the control (100%). Treatment with LM‐031 significantly increased the expression of HSPB1 (125% vs 79%), NRF2 (136% vs 56%), and CREB (101% vs 77%), the effect of which was attenuated by HSPB1‐specific (75% vs 125%), NRF2‐specific (27% vs 136%), and CREB‐specific (68% vs 101%) shRNA (Figure 6B). In line with the expression of HSPB1, NRF2, and CREB, we observed an improvement in neurite outgrowth, including in its length (109% vs 86%), process (107% vs 87%), and branch (106% vs 73%; Figure 6C‐D), in scramble shRNA‐infected cells treated with LM‐031 compared with untreated cells. In addition, decreased neurite outgrowth (85%‐67% vs 109%), process (96%‐82% vs 107%), and branch (74%‐63% vs 106%) were observed in LM‐031‐treated cells infected with HSPB1, NRF2, or CREB shRNA compared with the scramble control.
Figure 6.
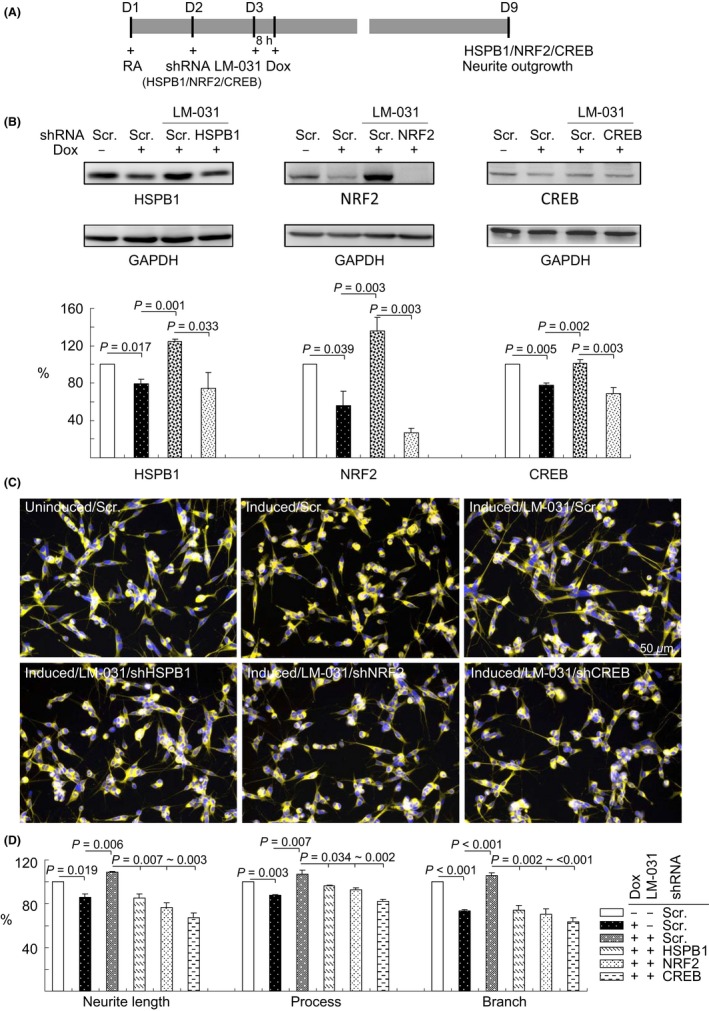
HSPB1, NRF2, and CREB as therapeutic targets in LM‐031‐treated Aβ‐GFP SH‐SY5Y cells. A, Experimental outline. Aβ‐GFP SH‐SY5Y cells were plated into 6‐well (for protein analysis) or 24‐well (for outgrowth analysis) plates with retinoic acid (RA, 10 µmol/L) added on day 1. On the following day, cells were infected with lentiviruses expressing shRNA (HSPB1, NRF2, CREB‐specific, or scrambled). At 24 hours postinfection, LM‐031 (1 µmol/L) was added to the cells for 8 hours, followed by the induction of Aβ‐GFP expression (+ Dox, 2 µg/mL) for 6 days. Then, the cells were collected for HSPB1, NRF2, or CREB protein analysis through immunoblotting (GAPDH as a loading control) and neurite outgrowth analysis through high‐content analysis. B, Western blot analysis of HSPB1, NRF2, and CREB protein levels in LM‐031‐treated cells infected with HSPB1, NRF2, CREB‐specific, or a negative control scramble shRNA. To normalize, the relative HSPB1/NRF2/CREB level of uninduced cells was set at 100%. P values: comparisons between induced vs uninduced cells, LM‐031‐treated vs untreated cells, or scramble vs HSPB1/NRF2/CREB shRNA‐infected cells (n = 3). C, Microscopic images of uninduced or induced Aβ‐GFP SH‐SY5Y cells infected with scramble shRNA or LM‐031 (1 μmol/L) treated cells infected with scramble or HSPB1/NRF2/CREB‐specific shRNA. Nuclei were counterstained with Hoechst 33342 (blue). D, Neurite outgrowth assay, including length, process, and branch, of LM‐031‐treated Aβ‐GFP SH‐SY5Y cells infected with HSPB1/NRF2/CREB‐specific or a scramble shRNA. To normalize, the relative neurite length/process/branch of scramble shRNA‐infected, uninduced cells without LM‐031 treatment was set at 100%. P values: comparisons between induced vs uninduced cells, LM‐031‐treated vs untreated cells, or scramble shRNA vs HSPB1/NRF2/CREB‐specific shRNA‐infected cells (n = 3)
4. CONCLUSION
Currently, no effective treatment exists for modifying or preventing AD progression. Aβ misfolding and aggregation cause oxidative stress, neuronal cell injury, and death. Novel synthetic LM‐031 exerted neuroprotective effects in Aβ‐GFP SH‐SY5Y cells by upregulating molecular chaperone HSPB1 and NRF2/NQO1/GCLC to reduce oxidative stress and by activating CREB‐dependent BDNF/AKT/ERK for cell survival and CREB‐dependent BCL2 for antiapoptosis. Because LM‐031 is soluble in the cell culture medium at a concentration up to 1 mmol/L, we are optimistic regarding the bioavailability of LM‐031. With low cytotoxicity in human cells and a wide range of the effective concentration for inhibiting Aβ aggregation and neuroprotection, novel LM‐031 has potential for being developed as an AD therapeutic. Additional in vivo and clinical studies are required to confirm the application of this multitargeted compound in modifying the progression of AD.
CONFLICT OF INTERESTS
The authors declare that there is no conflict of interest regarding the publication of this article.
ACKNOWLEDGMENTS
The authors thank the Molecular Imaging Core Facility of National Taiwan Normal University for the technical assistance. This work was supported by the grants 102‐2321‐B‐182‐013, 103‐2321‐B‐182‐008, and 105‐2325‐B‐003‐001 from the Ministry of Science and Technology, and the grant CMRPG3F1611‐1613 from Chang Gung Medical Foundation.
Lee S‐Y, Chiu Y‐J, Yang S‐M, et al. Novel synthetic chalcone‐coumarin hybrid for Aβ aggregation reduction, antioxidation, and neuroprotection. CNS Neurosci Ther. 2018;24:1286–1298. 10.1111/cns.13058
Contributor Information
Guey‐Jen Lee‐Chen, Email: t43019@ntnu.edu.tw.
Wenwei Lin, Email: wenweilin@ntnu.edu.tw.
Kuo‐Hsuan Chang, Email: gophy5128@cgmh.org.tw.
REFERENCES
- 1. Goedert M, Sisodia SS, Price DL. Neurofibrillary tangles and beta‐amyloid deposits in Alzheimer’s disease. Curr Opin Neurobiol. 1991;1:441‐447. [DOI] [PubMed] [Google Scholar]
- 2. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245‐4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roher AE, Lowenson JD, Clarke S, et al. β‐Amyloid‐(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:10836‐10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Storey E, Cappai R. The amyloid precursor protein of Alzheimer’s disease and the Aβ peptide. Neuropathol Appl Neurobiol. 1999;25:81‐97. [DOI] [PubMed] [Google Scholar]
- 6. Behl C. Amyloid beta‐protein toxicity and oxidative stress in Alzheimer’s disease. Cell Tissue Res. 1997;290:471‐480. [DOI] [PubMed] [Google Scholar]
- 7. Hensley K, Carney JM, Mattson MP, et al. A model for β‐amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270‐3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schubert D, Behl C, Lesley R, et al. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA. 1995;92:1989‐1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313‐322. [DOI] [PubMed] [Google Scholar]
- 10. Ramsey CP, Glass CA, Montgomery MB, et al. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kärkkäinen V, Pomeshchik Y, Savchenko E, et al. Nrf2 Regulates neurogenesis and protects neural progenitor cells against Aβ toxicity. Stem Cells. 2014;32:1904‐1916. [DOI] [PubMed] [Google Scholar]
- 12. Liu J, Qiu J, Wang M, et al. Synthesis and characterization of 1H‐phenanthro[9,10‐d]imidazole derivatives as multifunctional agents for treatment of Alzheimer’s disease. Biochim Biophys Acta. 2014;1840:2886‐2903. [DOI] [PubMed] [Google Scholar]
- 13. Pan Y, Chen Y, Li Q, Yu X, Wang J, Zheng J. The synthesis and evaluation of novel hydroxyl substituted chalcone analogs with in vitro anti‐free radicals pharmacological activity and in vivo anti‐oxidation activity in a free radical‐injury Alzheimer’s model. Molecules. 2013;18:1693‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao HF, Li N, Wang Q, Cheng XJ, Li XM, Liu TT. Resveratrol decreases the insoluble Aβ1–42 level in hippocampus and protects the integrity of the blood–brain barrier in AD rats. Neuroscience. 2015;310:641‐649. [DOI] [PubMed] [Google Scholar]
- 15. Zhong SZ, Ma SP, Hong ZY. Peoniflorin activates Nrf2/ARE pathway to alleviate the Abeta(1–42)‐induced hippocampal neuron injury in rats. Yao Xue Xue Bao. 2013;48:1353‐1357. [PubMed] [Google Scholar]
- 16. Wang QE, Lee FS, Wang X. Isolation and purification of inflacoumarin A and licochalcone A from licorice by high‐speed counter‐current chromatography. J Chromatogr A. 2004;1048:51‐57. [DOI] [PubMed] [Google Scholar]
- 17. Chen CM, Weng YT, Chen WL, et al. Aqueous extract of Glycyrrhiza inflata inhibits aggregation by upregulating PPARGC1A and NFE2L2–ARE pathways in cell models of spinocerebellar ataxia 3. Free Radic Biol Med. 2014;71:339‐350. [DOI] [PubMed] [Google Scholar]
- 18. Lv H, Ren H, Wang L, Chen W, Ci X. Lico A enhances Nrf2‐mediated defense mechanisms against t‐BHP‐induced oxidative stress and cell death via Akt and ERK activation in RAW 264.7 cells. Oxid Med Cell Longev. 2015;2015:709845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kühnl J, Roggenkamp D, Gehrke SA, et al. Licochalcone A activates Nrf2 in vitro and contributes to licorice extract‐induced lowered cutaneous oxidative stress in vivo. Exp Dermatol. 2015;24:42‐47. [DOI] [PubMed] [Google Scholar]
- 20. Kostova I, Bhatia S, Grigorov P, et al. Coumarins as antioxidants. Curr Med Chem. 2011;18:3929‐3951. [DOI] [PubMed] [Google Scholar]
- 21. Matos MJ, Vazquez‐Rodriguez S, Fonseca A, Uriarte E, Santana L, Borges F. Heterocyclic antioxidants in nature: coumarins. Curr Org Chem. 2017;21:311‐324. [Google Scholar]
- 22. Kontogiorgis CA, Xu Y, Hadjipavlou‐Litina D, Luo Y. Coumarin derivatives protection against ROS production in cellular models of Aβ toxicities. Free Radical Res. 2007;41:1168‐1180. [DOI] [PubMed] [Google Scholar]
- 23. Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg Med Chem. 2012;20:1175‐1180. [DOI] [PubMed] [Google Scholar]
- 24. Chang KH, Chiu YJ, Chen SL, et al. The potential of synthetic indolylquinoline derivatives for Aβ aggregation reduction by chemical chaperone activity. Neuropharmacology. 2016;101:309‐319. [DOI] [PubMed] [Google Scholar]
- 25. Lee CJ, Tsai CC, Hong SH, et al. Preparation of furo[3,2‐c]coumarins from 3‐cinnamoyl‐4‐hydroxy‐2H‐chromen‐2‐ones and acyl chlorides: A Bu3P‐mediated C‐acylation/cyclization sequence. Angew Chem. 2015;54:8502‐8505. [DOI] [PubMed] [Google Scholar]
- 26. Lee YT, Jang YJ, Syu SE, Chou SC, Lee CJ, Lin W. Preparation of functional benzofurans and indoles via chemoselective intramolecular Wittig reactions. Chem Commun. 2012;48:8135‐8137. [DOI] [PubMed] [Google Scholar]
- 27. Mazimba O, Masesane IB, Majinda RR. An efficient synthesis of flavans from salicylaldehyde and acetophenone derivatives. Tetrahedron Lett. 2011;52:6716‐6718. [Google Scholar]
- 28. Dube H, Kasumaj B, Calle C, et al. Probing hydrogen bonding to bound dioxygen in synthetic models for heme proteins: the importance of precise geometry. Chemistry. 2009;15:125‐135. [DOI] [PubMed] [Google Scholar]
- 29. Tietze LF, Ma L, Reiner JR, Jackenkroll S, Heidemann S. Enantioselective total synthesis of (‐)‐blennolide A. Chemistry. 2013;19:8610‐8614. [DOI] [PubMed] [Google Scholar]
- 30. Zhao LN, Long H, Mu Y, Chew LY. The toxicity of amyloid β oligomers. Int J Mol Sci. 2012;13:7303‐7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Żuk M, Kulma A, Dymińska L, et al. Flavonoid engineering of flax potentiate its biotechnological application. BMC Biotechnol. 2011;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bendary E, Francis RR, Ali H, Sarwat MI, El Hady S. Antioxidant and structure–activity relationships (SARs) of some phenolic and anilines compounds. Ann Agric Sci. 2013;58:173‐181. [Google Scholar]
- 33. Zhao T, Zeng Y, Kermode AR. A plant cell‐based system that predicts aβ42 misfolding: potential as a drug discovery tool for Alzheimer’s disease. Mol Genet Metab. 2012;107:571‐579. [DOI] [PubMed] [Google Scholar]
- 34. Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta‐peptide‐induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655‐664. [DOI] [PubMed] [Google Scholar]
- 36. Mancuso M, Orsucci D, Siciliano G, Murri L. Mitochondria, mitochondrial DNA and Alzheimer’s disease. What comes first? Curr Alzheimer Res. 2008;5:457‐468. [DOI] [PubMed] [Google Scholar]
- 37. Ye J, Meng X, Yan C, Wang C. Effect of purple sweet potato anthocyanins on β‐amyloid‐mediated PC‐12 cells death by inhibition of oxidative stress. Neurochem Res. 2010;35:357‐365. [DOI] [PubMed] [Google Scholar]
- 38. Chang KH, Lin CH, Chen HC, et al. The potential of indole/indolylquinoline compounds in tau misfolding reduction by enhancement of HSPB1. CNS Neurosci Ther. 2017;23:45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inestrosa NC, Alvarez A, Pérez CA, et al. Acetylcholinesterase accelerates assembly of amyloid‐β‐peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881‐891. [DOI] [PubMed] [Google Scholar]
- 40. Reyes AE, Perez DR, Alvarez A, et al. A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem Biophys Res Commun. 1997;232:652‐655. [DOI] [PubMed] [Google Scholar]
- 41. Inestrosa NC, Dinamarca MC, Alvarez A. Amyloid–cholinesterase interactions. FEBS J. 2008;275:625‐632. [DOI] [PubMed] [Google Scholar]
- 42. Wilhelmus MM, Boelens WC, Otte‐Höller I, Kamps B, de Waal RM, Verbeek MM. Small heat shock proteins inhibit amyloid‐β protein aggregation and cerebrovascular amyloid‐β protein toxicity. Brain Res. 2006;1089:67‐78. [DOI] [PubMed] [Google Scholar]
- 43. King M, Nafar F, Clarke J, Mearow K. The small heat shock protein Hsp27 protects cortical neurons against the toxic effects of β‐amyloid peptide. J Neurosci Res. 2009;87:3161‐3175. [DOI] [PubMed] [Google Scholar]
- 44. Tóth ME, Szegedi V, Varga E, et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones. 2013;18:759‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid β‐peptide inhibition of the PKA/CREB pathway and long‐term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217‐13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teich AF, Nicholls RE, Puzzo D, et al. Synaptic therapy in Alzheimer’s disease: a CREB‐centric approach. Neurotherapeutics. 2015;12:29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF‐dependent survival of sympathetic neurons. Science. 1999;286:2358‐2361. [DOI] [PubMed] [Google Scholar]
- 48. Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor‐dependent mechanism. Neuron. 1998;20:709‐726. [DOI] [PubMed] [Google Scholar]
- 49. Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB. Downregulation of CREB expression in Alzheimer’s brain and in Aβ‐treated rat hippocampal neurons. Mol Neurodegener. 2011;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parihar MS, Hemnani T. Alzheimer’s disease pathogenesis and therapeutic interventions. J Clin Neurosci. 2004;11:456‐467. [DOI] [PubMed] [Google Scholar]
- 51. Liang JH, Du J, Xu LD, et al. Catalpol protects primary cultured cortical neurons induced by Aβ1–42 through a mitochondrial‐dependent caspase pathway. Neurochem Int. 2009;55:741‐746. [DOI] [PubMed] [Google Scholar]
- 52. Tian X, Zhang L, Wang J, et al. The protective effect of hyperbaric oxygen and Ginkgo biloba extract on Aβ25–35‐induced oxidative stress and neuronal apoptosis in rats. Behav Brain Res. 2013;242:1‐8. [DOI] [PubMed] [Google Scholar]