

Summary
Aim
Multiple sclerosis (MS) is a neurological autoimmune disorder characterized by mistaken attacks of inflammatory cells against the central nervous system (CNS), resulting in demyelination and axonal damage. Kv1.3 channel blockers can inhibit T‐cell activation and have been designed for MS therapy. However, little is known about the effects of Kv1.3 blockers on protecting myelin sheaths/axons in MS. This study aimed at investigating the neuroprotection efficacy of a selective Kv1.3 channel blocker ImKTx88 (ImK) in MS animal model.
Methods
Experimental autoimmune encephalomyelitis (EAE) rat model was established. The neuroprotective effect of ImK was assessed by immunohistochemistry and transmission electron microscopy (TEM). In addition, the antiinflammatory effect of ImK by suppressing T‐cell activation was assessed by flow cytometry and ELISA in vitro.
Results
Our results demonstrated that ImK administration ameliorated EAE clinical severity. Moreover, ImK increased oligodendrocytes survival, preserved axons, and myelin integrity and reduced the infiltration of activated T cells into the CNS. This protective effect of the peptide may be related to its suppression of autoantigen‐specific T‐cell activation via calcium influx inhibition.
Conclusion
ImK prevents neurological damage by suppressing T‐cell activation, suggesting the applicability of this peptide in MS therapy.
Keywords: EAE, immunomodulation, Kv1.3 channel, multiple sclerosis, neuroprotection
1. INTRODUCTION
MS is an autoimmune disorder of the CNS characterized by blood‐brain barrier (BBB) destruction, inflammatory cell infiltration, demyelination, and axonal damage.1 Approximately 2.5 million young people aged from 20 to 40 years suffer from MS worldwide.2 Currently, most immunotherapeutic drugs targeting the peripheral immune system can inhibit further development of autoinflammation, while the neurodegenerative process caused by neuroaxonal injury remains a problem.3, 4, 5 During the progression of the disease, the death of oligodendrocytes and the continued loss of myelin can expose neuron axons, leading to nerve conduction block.6 In addition, large numbers of tissue fragments cannot be promptly cleared, resulting in harmful substance deposition and neurological dysfunction.7 Therefore, the discovery of new potent drugs inhibiting neurological lesion accumulation is required for MS therapeutics.
Kv1.3 blockers have been used for the treatments in MS and related animal models through the suppression of T‐cell activation and proliferation.8, 9 The underlying mechanism targets the regulation of calcium signaling in effector memory T (TEM) cells, which plays an important role in the chronic progression of MS and related animal models.10, 11, 12 However, the alterations of demyelinated plaques in the white matter of the brain and spinal cord in the disease are not fully studied. Whether Kv1.3 blockers can alleviate diffuse myelin reduction and axonal injury should be further investigated.
Besides, the unsatisfactory selectivity of blockers on Kv1.3 over other potassium channels, such as Kv1.1 or Kv1.2, may result in neurological side effects, which limit their clinical applications.13 For example, 4‐AP, a chemical Kv1.3 channel blocker, has been used clinically to improve walking impairment in MS patients.14 Meanwhile, 4‐AP may impact neuronal excitability in MS, resulting in some side effects, such as headaches and seizures.15, 16 This indicates that Kv1.3 blockers may ameliorate impaired neurological function, but neurological dysfunction caused by poor selectivity should be improved.
In previous work, we discovered a novel Kv1.3 peptide blocker ImK, displayed high selectivity toward the Kv1.3 channel over Kv1.1 and Kv1.2,17 which effectively stabilized the BBB integrity.18 Compared with other identified Kv1.3 channel blockers, such as ADWX‐119 and ShK (L5),20 the ImK peptide has much higher Kv1.3 selectivity, which may provide applicable safety for future treatment. Here, we used ImK to investigate its neuroprotective roles in EAE rats, specifically whether it can ameliorate myelin loss and axonal damage through inhibiting T‐cell activation.
2. MATERIALS AND METHODS
2.1. Peptide
ImK peptide was expressed and purified as described previously.17 Escherchia coli Rosetta (DE3) cells containing plasmid pGEX‐6p‐1‐ImK were cultured at 37°C, and protein expression was induced by IPTG at 28°C for 4 hours. The fusion protein from the cell extract was purified through GSH affinity chromatography and was then cleaved by enterokinase. ImK peptide was separated from the mixture through RP‐HPLC and then lyophilized.
2.2. Animal
All animals were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Female Sprague‐Dawley (SD) rats weighing from 200 to 220 g were used to establish EAE model, and P0‐2 SD rat pups were used to obtain primary neural cells. All the experiments were performed in accordance with the National Institutes of Health guide for the care and use of laboratory animals, with approval from the Institutional Animal Care and Use Committee of Wuhan University.
2.3. EAE induction and ImK treatment
EAE was induced with an emulsion of rat spinal cord homogenate in phosphate‐buffered saline (PBS, 50% w/v) mixed with an equal volume of complete Freund's adjuvant (Sigma‐Aldrich, St. Louis, MO) supplemented with 5 mg/mL Mycobacterium tuberculosis. Each rat was immunized by subcutaneous injection of antigen emulsion at a dose of 0.75 mL/kg into its footpad, followed by an auxiliary injection of 10 μg/kg diluted in 0.5 mL pertussis toxin in the dorsum of the foot. The second immunization was administered 7 days after the first immunization. Clinical signs of EAE were monitored daily. Beginning on the day when first neurological signs appeared, animals were subcutaneously injected with ImK (100 μg/kg) or PBS once daily.
2.4. Clinical evaluation of EAE
Following the immunization, neurological signs were scored daily for each rat according to the following procedure, as previously described: 0, no clinical signs; 0.5, partial loss of tail tone; 1, affected tail tonus; 2, paresis of hind legs; 3, complete paralysis of the hind legs; 4, complete hind leg paralysis and foreleg paresis; and 5, death due to EAE. These criteria were established and modified according to previous clinical scale systems.19
2.5. Histological analysis
After ImK treatment, the rats from each group were killed at the 23rd day after the first immunization. After anesthetization with 10% choral hydrate (350 mg/kg, i.p.), the rats were intracardially perfused with PBS, followed by 4% paraformaldehyde in PBS. The rats were euthanized, and the lumbar spinal cords were removed and immersed for 48 hours in 4% paraformaldehyde for fixation. Four‐micrometer‐thick (4 μm) transverse sections taken from embedded blocks of spinal cord were deparaffinized and stained with Luxol fast blue (LFB) to assess demyelination. Demyelination score was evaluated using the following scale described by Wraith et al (0 = no demyelination; 1 = a few, scattered naked axons; 2 = small groups of naked axons; 3 = large groups of naked axons; and 4 = confluent foci of demyelination).21
2.6. Immunohistochemistry
For immunohistochemistry staining, tissue sections of the lumbar segment were incubated with primary antibodies overnight at 4°C. The primary antibodies used were as follows: CD3 (1:200, DAKO, Carpinteria, CA); NG2 (1:100, Millipore, Bedford, USA); CC‐1(1:250, Calbiochem, San Diego, CA, USA); MBP (1:1000, Millipore, Bedford, USA); NF200 (1:50, NeoMarkers, Fremont, CA); and rabbit anti‐β‐APP (1:200, Boster Bio‐engineering, Wuhan, China). For NG2 fluorescent staining, nuclei were stained with DAPI (ThermoScientific, Waltham, MA). PBS was used instead of the primary antibody as the negative control.
To identify the quantity of CD3+Kv1.3+ cells in the lesion of white matter, quantum dot (QD)‐based double immunofluorescent staining was performed on 3‐μm‐thick paraffin sections using a specific secondary antibody.22 The paraffin‐embedded sections were pretreated as described above and then incubated with a rat monoclonal anti‐CD3 antibody (1:200, DAKO, Carpinteria, CA) and a rabbit polyclonal Kv1.3 antibody (1:100, NeuroLab, St Louis, MO, USA) at 4°C overnight. After being washed with PBS three times, the sections were incubated with the biotinylated secondary antibodies to the QDs for 1 hour at 37°C and then with the corresponding antibodies of goat anti‐mouse QDs‐525 (1:100, Invitrogen, Eugene, USA) and goat anti‐rabbit QDs‐605 (1:300, Invitrogen, Eugene, USA). The sections were assessed using an Olympus BX51 microscope after washing. Images were collected and analyzed using a CRi Nuance multispectral imaging system (Cambridge Research and Instrumentation, Inc., USA).
2.7. Transmission electron microscopy
After anesthesia, the rats were killed by transcardial perfusion with physiological saline. The lumbar spinal cord was removed and immersed in 2.5% glutaraldehyde fixative for 2 hours. Tissue samples were rinsed 3 times in 0.1 M sodium cacodylate buffer, treated with 1% OsO4, dehydrated in a graded series of ethanol and propylene oxide, and embedded in Epon. Thin sections of the Epon‐embedded blocks were stained with a solution of uranyl acetate and lead citrate. The stained sections were observed by TEM (Hitachi, Tokyo, Japan) at 80 kV. For analysis of myelin pathology, the number of nerve fibers, myelin thickness, and nerve fiber diameter were measured using Image‐Pro Plus software. The g‐ratio is defined as the ratio of the diameter of a given axon to the diameter of the whole nerve fiber.23, 24, 25 Axons smaller than 0.3 μm in diameter were excluded from the quantitative analysis.26, 27 Quantification of the g‐ratio was performed in micrographs (n = 2 rats/group).
2.8. PBMC isolation and stimulation
Blood was collected from abdominal veins of anesthetized rats. Lymphocytes were diluted with PBS, followed by enrichment based on 70% Percoll gradients and centrifugation for 30 minutes at 800 g. For stimulation, lymphocytes from WT rats were treated with 100 μg/mL ConA (Sigma‐Aldrich, St. Louis, MO), and lymphocytes from EAE rats were treated with 100 μg/mL MOG35‐55 (Sigma‐Aldrich, St. Louis, MO). ImK was added 1 hour before stimulation.
2.9. Flow cytometry
Lymphocyte suspensions of rat blood were prepared by ACK lysis buffer or collected from 6‐well culture plates. The following Abs were used for cell surface staining and intracellular staining: PE‐CD3 (mouse anti‐rat, BD, Breda, The Netherlands), CCR7 (goat anti‐rat, Novus, Littleton, USA), APC anti‐goat (donkey, Novus, Littleton, USA), Fluo‐3/AM (Beyotime Biotechnology, Shanghai, China), and CFSE (Beyotime Biotechnology, Shanghai, China). The cells were analyzed on an FACS Aria III flow cytometer (BD Bioscience, Breda, The Netherlands).
2.10. Enzyme‐linked immunosorbent assay (ELISA)
Peripheral blood and cerebrospinal fluid (CSF) samples were collected at the peak of EAE. The levels of cytokines IL‐2 (DAKAWE, Beijing, China) and IFN‐γ (DAKAWE, Beijing, China) in each sample were measured using the Bradford method according to the manufacturer's instructions.
2.11. Primary neural cell cultures
Purified cultures of oligodendrocytes were prepared as previously described by Chen et al with minor modifications.28 To promote proliferation, isolated oligodendrocyte progenitor cells (OPCs) were seeded on 96‐well plates in Neurobasal media (Gibco, Grand Island, NY, USA) with PDGF‐AA (20 ng/mL, PeproTech, Rocky Hill, NJ, USA); bFGF (20 ng/mL, PeproTech, Rocky Hill, NJ, USA); 2% B27 (Gibco, Grand Island, NY, USA); and 1% penicillin/streptomycin at a density of 2 × 104/well. After 2 days, cells were switched to Neurobasal media with 40 ng/mL triiodothyronine (T3) (Sigma‐Aldrich, St. Louis, MO) and 2% B27 to promote differentiation for 7 days. Purified cultures of microglia were prepared as previously described.29 Isolated microglia were seeded on 96‐well plates in DMEM/F12 (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin at a density of 1 × 104/well. Purified cultures of astrocytes were prepared as previously described.30 Isolated astrocytes were seeded on 96‐well plates in DMEM (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum and 1% penicillin/streptomycin at a density of 1 × 104/well. Primary cultures of neurons were prepared as previously described.31 Cerebral cortical cells from a P0‐2 SD rat were seeded on 96‐well plates in Neurobasal media (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum, 1% L‐glutamine and 2% B27 and 1% penicillin/streptomycin at a density of 1 × 104/well. Neurons were cultured for 7‐9 days before being used.
2.12. Neural cytotoxicity assay
Neural cell viability was measured with a Cell Counting Kit‐8 (CCK‐8, Dojindo, Tokyo, Japan) according to the manufacturer's instructions. Ten microliters of the CCK‐8 reagent were added into each well and incubated for another 1‐4 hours. The absorbance at 450 nm was measured using a scanning microplate reader.
2.13. Statistical analysis
Statistical significance between groups was determined using one‐way analysis of variance (one‐way ANOVA), followed by Newman‐Keuls comparison. The EAE clinical scores were analyzed using generalized estimating equations. Incidence and lethality rate of EAE were analyzed by chi‐square test. Student's t‐tests were performed for parametric two‐group comparisons. Data are shown as the mean ± SEM or SD. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. ImK attenuates EAE clinical severity
To investigate the efficient treatment of ImK on EAE clinical severity, the rats were immunized with myelin antigens in CFA to induce EAE and randomly treated with ImK or vehicle alone at the onset of EAE. The clinical scores of each rat from different groups were observed longitudinally. As shown in Figure 1A, autoantigen‐induced rats began to develop clinical symptoms on day 12 after immunization. The maximal average clinical score in the EAE group was 2.8 and 1.3 in the ImK‐treated EAE group. In addition, ImK decreased the incidence rate (Figure 1B) from 85.7% to 37.5% and the lethality rate (Figure 1C) from 56.3% to 12.5%. These showed that ImK ameliorated the clinical signs of EAE rats.
Figure 1.
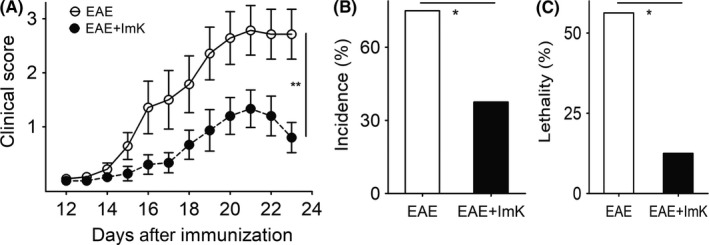
ImK significantly reduces clinical score, incidence, and lethality of Experimental autoimmune encephalomyelitis (EAE) rats. Clinical score (A), incidence (B), and lethality (C) of EAE rats were determined following EAE induction with or without ImK treatment (n = 16; *P < 0.05, **P < 0.01)
3.2. ImK diminishes demyelination and prevents oligodendrocyte loss in EAE rats
Luxol fast blue staining and immunohistochemistry for myelin basic protein (MBP) were employed to evaluate the severity of demyelination in the white matter of spinal cords. Control spinal cords showed uniform LFB staining of the white matter (Figure 2A). In contrast, focal areas surrounding the vessels, which were devoid of LFB staining, were more frequently observed in the EAE spinal cord white matter. The ImK‐treated group showed fewer LFB‐unstained areas than did the EAE group, suggesting a less severe demyelination. The mean demyelination score of EAE group was 2.50 ± 0.50, whereas the mean score of ImK‐treated group was 1.00 ± 0.26. MBP expression is also a pathological index used for demyelination assessment. We quantified the demyelination areas of MBP immunoreactivity in the EAE and ImK‐treated groups (Figure 2B). Similar to the LFB staining result, compared with the EAE group, the ImK‐treated EAE group exhibited remarkably reduced demyelination areas.
Figure 2.
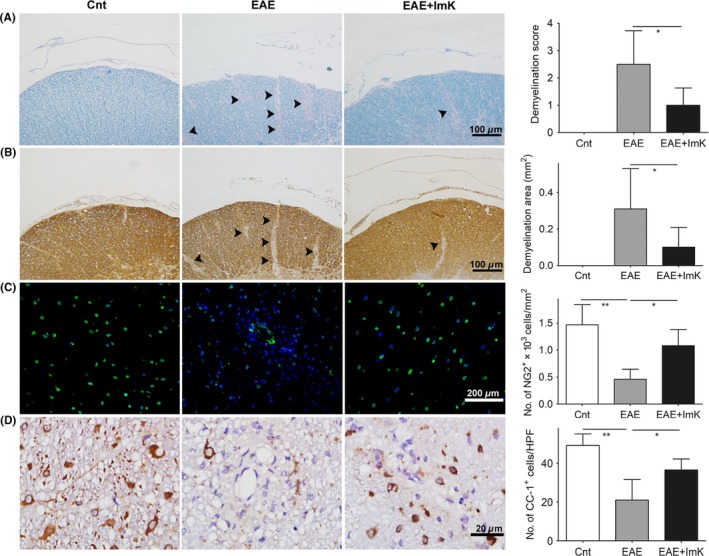
The prevention of ImK on demyelination and oligodendrocyte loss in the Experimental autoimmune encephalomyelitis group. Luxol fast blue (A), immature oligodendrocyte (NG2) (B), and mature oligodendrocyte marker (CC‐1, MBP) (C, D) were stained in each group. (A, B): Arrows point to the border of the demyelinating area in the white matter. Quantification of demyelination scores (A, n = 6, *P < 0.05), demyelination area (μm2) (B, n = 8, *P < 0.05), and density of NG2+ OPCs (C, n = 6, *P < 0.05, **P < 0.01) and CC‐1+ oligodendrocytes (D, n = 6, *P < 0.05) were analyzed in the lumbar spinal cord white matter. HPF: 1000× high‐power field
We next investigated the effects of ImK on OPCs and mature oligodendrocytes using immunostaining for NG2 and CC‐1 (Figure 2C, D), respectively. As expected, NG2+ cell density was reduced in the EAE group. Compared with the EAE spinal cords, the ImK‐treated spinal cords showed a 2.35‐fold higher density of NG2+ cells. Similarly, the number of mature oligodendrocytes was more significantly elevated in the ImK‐treated group than that in the EAE group, even though the CC‐1+ cell density in the ImK‐treated group remained below the control level.
3.3. ImK preserves axon and myelin integrity in the spinal cord of the EAE rats
For axon and myelin fine structure observation, spinal cord ultrathin sections from control, EAE, and ImK‐treated groups were analyzed by TEM. Figure 3A reveals a normal myelin structure in the control group, but the EAE group exhibited myelin disorganization and a significantly decreased number of myelin. Myelin organization in ImK‐treated group was more uniform than that in the EAE group. Statistical results (Figure 3B) showed that the mean g‐ratio of the EAE group (0.78 ± 0.06) was higher than that of the control group (0.64 ± 0.08), whereas g‐ratios of the ImK‐treated EAE group (0.68 ± 0.07) were lower than those of the EAE group. Additionally, axons of the EAE group had significantly thinner myelin. Further analysis was performed to examine the impact of ImK on the number of axons/mm2.The number of axons was significantly decreased in the EAE group while only slightly decreased in the ImK‐treated EAE group. To assess the integrity of axons, spinal cord sections were stained for NF200, an antibody that recognized phosphorylated neurofilament protein in healthy axon.32, 33 NF200 staining revealed that ImK treatment preserved axon structure in the white matter of lumber spinal cords (Figure 3C). The mean optical density analysis of NF200 in each group showed that there were significantly more NF200‐positive axons in the white matter of the ImK‐treated group than in that of the EAE group (Figure 3D). In addition, we stained the sections with β‐APP, a marker of acute axonal damage. As demonstrated in Figure 3C, the control group did not show any β‐APP expression, indicating no axonal injury. In the EAE group, many β‐APP+ spheroids within white matter were found in demyelinating lesions. However, compared with EAE, ImK treatment resulted in fewer β‐APP+ spheroids (Figure 3D). These findings pointed to a significant beneficial effect of therapeutic ImK in preventing CNS axon damage and myelin loss in the EAE group.
Figure 3.
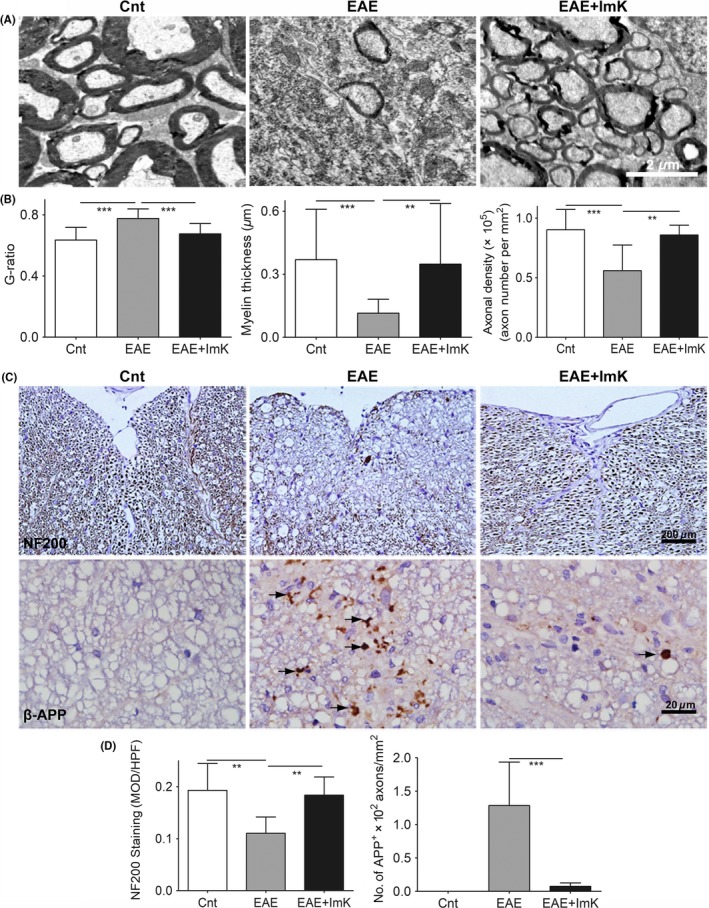
ImK preserves axon and myelin integrity. (A) Representative electron microscopy images illustrated axons and myelin in the lumbar spinal cord of each group. (B) The g‐ratio, axon diameter, and axonal density of each group were analyzed using Image‐Pro Plus software. (C) Immunostaining for NF200 and β‐APP in the white matter of lumbar spinal cord in Experimental autoimmune encephalomyelitis rats (arrows). (D) Quantification of the immunoreactivity in each group is shown (n = 6 for NF200, **P < 0.01; n = 8 for APP, ***P < 0.001). MOD: mean optical density
3.4. ImK reduces T‐cell infiltration into the CNS and inflammatory cytokine production in the CNS
CD3 immunostaining was totally negative in any area of the spinal cord in the control group. Although CD3+ T cells were detected in both gray and white matter areas of EAE rats, they were largely distributed to axonal tracts in the white matter (Figure 4A). However, compared with the EAE group, the ImK‐treated group displayed significantly reduced density of CD3+ T cells. We next confirmed colocalization of Kv1.3+ and CD3+ using double‐fluorescent immunostaining (Figure 4B). Quantification of CD3+Kv1.3+ cells demonstrated lesions with lesser pathogenic T‐cell infiltration after ImK treatment (Figure 4C). To confirm whether ImK modulated infiltrated T‐cell immune response, we further evaluated inflammatory cytokines in rats’ CSF. IL‐2 and IFN‐γ levels were increased in the EAE group compared with those in the control group. IL‐2 and IFN‐γ levels, by contrast, were significantly reduced in the ImK‐treated EAE group compared with those in the EAE group (Figure 4D, E).
Figure 4.
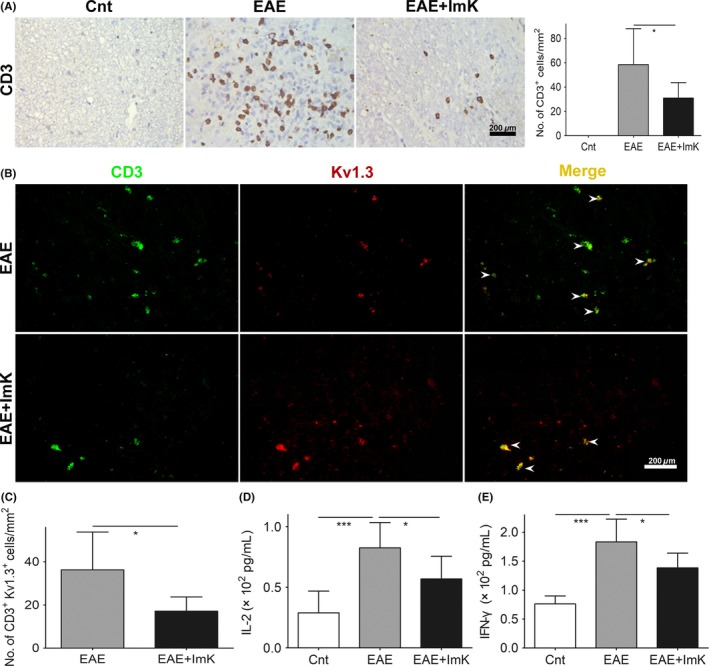
Suppressive effect of ImK on T‐cell infiltration and inflammatory cytokine production in the CNS of the Experimental autoimmune encephalomyelitis group. CD3+ cell infiltration (A) was displayed in the white matter of lumbar spinal cord (n = 8, *P < 0.05). Double‐fluorescent immunostaining demonstrated CD3+Kv1.3+ cells (arrowheads) (B,C) infiltration in lesions (n = 6, *P < 0.05). The concentrations of IL‐2 (D) and IFN‐γ (E) in CSF were measured using ELISA (n = 6, *P < 0.05, ***P < 0.001)
3.5. ImK suppresses peripheral T‐cell activation and cytokine release
It has been reported that Kv1.3 blocking represses calcium signaling and may affect immunological functions in TEM cells.34 ImK suppressed the activation of T cells by downregulating Ca2+ signals and reducing inflammatory cytokine release in vitro (Figure S1). Based on the results of inhibiting T‐cell activation by ImK, we then investigated the immunosuppression effects of ImK on the peripheral blood from EAE rats. At the peak of EAE, CD3+CCR7− cells increased in rat peripheral blood. Subcutaneous injection of ImK reduced CD3+CCR7−cells (Figure 5A), suggesting a selective inhibition of ImK for TEM cells. The concentrations of cytokines IL‐2 (Figure 5B) and IFN‐γ (Figure 5C) in the serum showed a significant drop treated with ImK.
Figure 5.
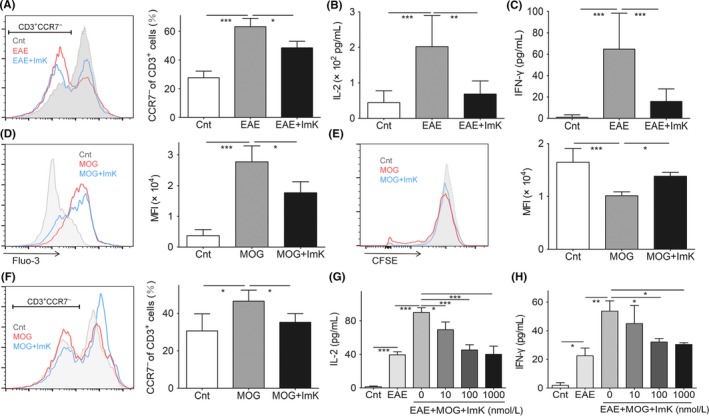
Effects of ImK on peripheral T‐cell activation and cytokine release. CD3+ CCR7− cells (A) and serum IL‐2 (B) or IFN‐γ (C) from peripheral blood of each group after ImK treatment. The intracellular calcium concentration (D), proliferation (E), and CCR7− proportion (F) of T cells were quantified from restimulated PBMCs of Experimental autoimmune encephalomyelitis (EAE) rats by with or without ImK pretreatment. IL‐2 (G) or IFN‐γ (H) in the supernatant of control, EAE, or EAE PBMC restimulated by MOG 35‐55 with different concentrations of ImK was detected using ELISA. (Each group n = 3, *P < 0.05,**P < 0.01, ***P < 0.001)
Furthermore, PBMCs were isolated from EAE rat blood to test whether ImK inhibited autoreactive T cells in vitro. During isolation, we observed a significant increase in leukocytes (data not shown). With autoantigen MOG35‐55 stimulation, T cells receive a Ca2+ influx to initiate downstream signals, while ImK suppressed reactivated T‐cell intracellular Ca2+ increase (Figure 5D). Autoreactive T cells proliferated for a potent immune response, which was suppressed by ImK (Figure 5E). After restimulation, more T cells differentiated to TEM cells. ImK reduced CCR7− cells to a low degree (Figure 5F). Finally, IL‐2 and IFN‐γ release were also controlled by ImK in a concentration‐dependent manner (Figure 5G, H). These data suggested that ImK selectively inhibited autoantigen‐reactive T cells in vivo and in vitro.
3.6. ImK shows no neurotoxicity and reduces inflammatory damages in neural cells
To explore whether the inhibition of Kv1.3 channels is associated with neurotoxicity, we evaluated the cytotoxic effects of ImK at various concentrations in a standardized cytotoxicity assay in the following primary cells: neurons, oligodendrocytes, astrocytes, and microglia (Figure 6A). Within 48 hours, compared with the control, ImK failed to induce any type of neural cell death even at concentrations of 1 μM. Based on the above results, we next investigated ImK‐regulated neuroprotection in vitro. Inflammatory supernatants were harvested from cultured rats’ PBMCs after stimulation for 24 hours with different concentrations of ImK. Then, neurons or oligodendrocytes were exposed to various inflammatory supernatants for 48 hours. Data showed that ImK above 100 nM significantly reduced neurological damage (Figure 6B), suggesting that ImK protected neuronal basal cells probably by inflammatory cytokine inhibition.
Figure 6.
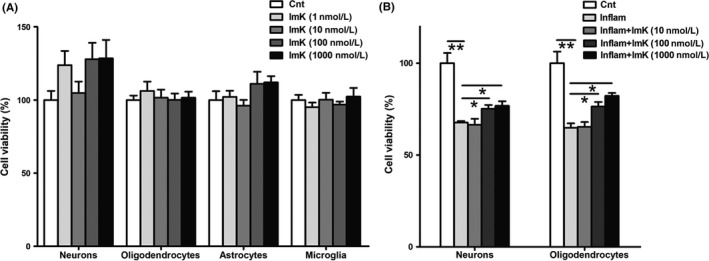
Neural cell viability after exposure to ImK and ImK‐treated inflammatory supernatant. (A) Primary neurons, oligodendrocytes, astrocytes, and microglia viabilities were determined following treatment with varying concentrations of ImK ranging from 1 nM to 1 μM for 48 h (n = 4 for neurons, n = 6 for oligodendrocytes, n = 3 for astrocytes, n = 4 for microglia, P > 0.05 compared with the control). Neuron and oligodendrocyte viability (B) were quantified with exposure to different concentrations of ImK‐treated inflammatory supernatant for 48 h (n = 3, *P < 0.05,**P < 0.01)
4. DISCUSSION
Although Kv1.3 channel blockers are considered potent for the therapy of autoimmune diseases, a poor selectivity may cause side effects toward CNS that limits their applications for neuroprotection in MS. Our previous work reported that ImK exhibited good selectivity and affinity toward the Kv1.3 channel, displaying 4200‐fold and 93 000‐fold selectivity for Kv1.3 over homologous potassium channels Kv1.1 and Kv1.2, respectively.17
According to the antiinflammatory effects of ImK in vitro (Figure S1) and behavioral improvement by ImK treatment in EAE (Figure 1), we investigated the antiinflammatory and neuroprotective effects of ImK on EAE. CNS suffers from autoreactive T‐cell infiltration in MS and EAE.35 ImK significantly reduced activated T cells in the CNS of EAE rats. In addition, the detection of cytokine levels in CSF revealed that ImK decreased the Th1 immune response. Several studies demonstrated that high IFN‐γ production by Th1 cells correlated with the seriousness of EAE and led to CNS demyelination.36, 37 The blockade of inflammatory responses in circulating CSF and CNS parenchyma indicated the indirect neuroprotective effect of ImK.
Neuroaxonal injuries may get alleviated due to the reduction in inflammatory infiltration. The depletion of oligodendrocytes is a recognized feature of EAE lesions, induced by inflammatory damage and becoming more apparent as the disease evolves. Extensive loss of myelin in EAE rats was observed by LFB and MBP staining. Furthermore, OPCs labeled with the early marker NG2 and mature oligodendrocytes expressing CC‐1 declined in the EAE group, demonstrating an impairment of the oligodendrocyte lineage. Meanwhile, significant decreases of demyelination areas and axonal damage were detected in the ImK‐treated EAE group. Besides, cumulative studies suggested that Kv1.3 channels took part in the regulation of oligodendrocyte differentiation and maturation.38, 39, 40 Some Kv1.3 blockers exhibited neuroprotective effects as MBP mRNA decay was completely blocked by the inhibition of Kv1.3 expression.40, 41, 42, 43 Together, these previous findings indicated that ImK affected the attenuation of oligodendrocyte damage and probably the enhancement of their survival and remyelination.
The transport of APP from neuron cell bodies to axon terminals is disturbed when axons enwrapped by myelin sheaths suffer pathological damage.44 Detection of APP in swollen axons is a sign of defects in axonal transport,45, 46 suggesting acute axonal damage in EAE models.47, 48 NF200 is the heavy molecular weight microfilament subunit, and it is a marker of axonal damage in MS or EAE due to the decrease in its immunoreactivity.49, 50 In our study, we performed immunohistochemistry for β‐APP supplemented with NF200 to assess the extent of neuronal damage. Axonal loss and β‐APP deposition in lesions were significantly decreased in the ImK‐treated group compared with those in the EAE group but were negative in control rats. Furthermore, a previous study demonstrated that when the g‐ratio was nearly 0.6,the spread of current from one node of Ranvier to the next was the best.51 Conversely, an elevated g‐ratio indicated myelin impairments. TEM analysis showed that rats that received ImK treatment presented a nonsignificant decrease in axonal density, myelin thickness, and mildly elevated g‐ratios relative to the control group, while rats in the EAE group exhibited significant damage in axons. The decline in the g‐ratio of the ImK‐treated group compared with that of the EAE group indicated myelinated axon enhancement, consistent with the improvement in rats’ behavioral performance in the ImK‐treated group. Taken together, ImK exhibited antiinflammatory properties, as well as neuroprotective effects, taking advantages over most immune‐modulating drugs,52 for example, IFN‐β,53 which do not halt progression of neurodegeneration.
In addition, we identified the cytotoxic effect of ImK on CNS cells in vitro. Although some studies reported that Kv1.3 blockers can prevent proliferation of oligodendrocytes and microglia,38, 54, 55 we did not observe any direct cytotoxicity of ImK on primary cultured CNS cells, even at a high concentration (1 μM). The high selectivity of ImK on Kv1.3 might guarantee the safety for nervous system applications. Besides, proinflammatory cytokines secreted by T cells, such as IFN‐γ and IL‐2, were recognized as major cytokines contributing to MS pathology.56 Our study showed that ImK preserved neurons and oligodendrocyte viabilities by inhibiting cytokine release in vitro, demonstrating the neuroprotective role of ImK without any neurotoxicity.
5. CONCLUSION
In summary, we show that ImK, a highly selective blocker for the Kv1.3 potassium channel, prevents oligodendrocyte loss and maintains the integrity of axons by suppressing autoreactive T‐cell response via calcium influx inhibition in EAE rats. Our work indicates that ImK has the therapeutic effects of neuroprotection through immunomodulation and is a promising drug candidate for MS therapy.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Natural Sciences Foundation of China (Nos. 81171127, 81371422, 81401230, and 81401241) and the Natural Science Foundation of Hubei Province of China (2017CFA017).
Yuan X‐L, Zhao Y‐P, Huang J, et al. A Kv1.3 channel‐specific blocker alleviates neurological impairment through inhibiting T‐cell activation in experimental autoimmune encephalomyelitis. CNS Neurosci Ther. 2018;24:967–977. 10.1111/cns.12848
The first two authors contributed equally to this work.
Contributor Information
Song Han, Email: hansong@whu.edu.cn.
Xiao‐Hua He, Email: hexiaohua@whu.edu.cn.
REFERENCES
- 1. Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med. 2006;354:942‐955. [DOI] [PubMed] [Google Scholar]
- 2. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14:406‐419. [DOI] [PubMed] [Google Scholar]
- 3. Haghikia A, Hohlfeld R, Gold R, Fugger L. Therapies for multiple sclerosis: translational achievements and outstanding needs. Trends Mol Med. 2013;19:309‐319. [DOI] [PubMed] [Google Scholar]
- 4. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545‐558. [DOI] [PubMed] [Google Scholar]
- 5. Feinstein A, Freeman J, Lo AC. Treatment of progressive multiple sclerosis: what works, what does not, and what is needed. Lancet Neurol. 2015;14:194‐207. [DOI] [PubMed] [Google Scholar]
- 6. Baker D, Gerritsen W, Rundle J, Amor S. Critical appraisal of animal models of multiple sclerosis. Mult Scler. 2011;17:647‐657. [DOI] [PubMed] [Google Scholar]
- 7. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125(Pt 10):2202‐2212. [DOI] [PubMed] [Google Scholar]
- 8. Bever CT Jr. The current status of studies of aminopyridines in patients with multiple sclerosis. Ann Neurol. 1994;36:S118‐S121. [DOI] [PubMed] [Google Scholar]
- 9. Beeton C, Barbaria J, Giraud P, et al. Selective blocking of voltage‐gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol. 2001;166:936‐944. [DOI] [PubMed] [Google Scholar]
- 10. Wulff H, Calabresi PA, Allie R, et al. The voltage‐gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rus H, Pardo CA, Hu L, et al. The voltage‐gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A. 2005;102:11094‐11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rangaraju S, Chi V, Pennington MW, Chandy KG. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin Ther Targets. 2009;13:909‐924. [DOI] [PubMed] [Google Scholar]
- 13. Ovsepian SV, LeBerre M, Steuber V, O'Leary VB, Leibold C, Oliver DJ. Distinctive role of KV1.1 subunit in the biology and functions of low threshold K+ channels with implications for neurological disease. Pharmacol Ther. 2016;159:93‐101. [DOI] [PubMed] [Google Scholar]
- 14. Blight AR, Henney HR 3rd, Cohen R. Development of dalfampridine, a novel pharmacologic approach for treating walking impairment in multiple sclerosis. Ann N Y Acad Sci. 2014;1329:33‐44. [DOI] [PubMed] [Google Scholar]
- 15. Davis FA, Stefoski D, Rush J. Orally administered 4‐aminopyridine improves clinical signs in multiple sclerosis. Ann Neurol. 1990;27:186‐192. [DOI] [PubMed] [Google Scholar]
- 16. Zhao N, Dong Q, Qian C, et al. Lovastatin blocks Kv1.3 channel in human T cells: a new mechanism to explain its immunomodulatory properties. Sci Rep. 2015;5:17381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han S, Hu Y, Zhang R, et al. ImKTx88, a novel selective Kv1.3 channel blocker derived from the scorpion Isometrus maculates. Toxicon. 2011;57:348‐355. [DOI] [PubMed] [Google Scholar]
- 18. Huang J, Han S, Sun Q, et al. Kv1.3 channel blocker (ImKTx88) maintains blood‐brain barrier in experimental autoimmune encephalomyelitis. Cell Biosci. 2017;7:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han S, Yi H, Yin SJ, et al. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J Biol Chem. 2008;283:19058‐19065. [DOI] [PubMed] [Google Scholar]
- 20. Chi V, Pennington MW, Norton RS, et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon. 2012;59:529‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wraith DC, Pope R, Butzkueven H, et al. A role for galanin in human and experimental inflammatory demyelination. Proc Natl Acad Sci U S A. 2009;106:15466‐15471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sweeney E, Ward TH, Gray N, et al. Quantitative multiplexed quantum dot immunohistochemistry. Biochem Biophys Res Commun. 2008;374:181‐186. [DOI] [PubMed] [Google Scholar]
- 23. Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372‐381. [DOI] [PubMed] [Google Scholar]
- 24. Forrest AD, Beggs HE, Reichardt LF, Dupree JL, Colello RJ, Fuss B. Focal adhesion kinase (FAK): a regulator of CNS myelination. J Neurosci Res. 2009;87:3456‐3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fyffe‐Maricich SL, Schott A, Karl M, Krasno J, Miller RH. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J Neurosci. 2013;33:18402‐18408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sullivan GM, Mierzwa AJ, Kijpaisalratana N, et al. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J Neuropathol Exp Neurol. 2013;72:1106‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ritter J, Schmitz T, Chew LJ, et al. Neonatal hyperoxia exposure disrupts axon‐oligodendrocyte integrity in the subcortical white matter. J Neurosci. 2013;33:8990‐9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y, Balasubramaniyan V, Peng J, et al. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc. 2007;2:1044‐1051. [DOI] [PubMed] [Google Scholar]
- 29. Vazquez‐Villoldo N, Domercq M, Martin A, Llop J, Gomez‐Vallejo V, Matute C. P2X4 receptors control the fate and survival of activated microglia. Glia. 2014;62:171‐184. [DOI] [PubMed] [Google Scholar]
- 30. Ponath G, Ramanan S, Mubarak M, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain. 2017;140(Pt 2):399‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Z, Zhang Y, Fang J, et al. Decreased methylation level of H3K27me3 increases seizure susceptibility. Mol Neurobiol. 2017;54:7343‐7352. [DOI] [PubMed] [Google Scholar]
- 32. Quintanar JL, Salinas E. Neurofilament expression in cultured rat adenohypophysial cells. Cell Physiol Biochem. 2001;11:27‐32. [DOI] [PubMed] [Google Scholar]
- 33. Haile Y, Carmine‐Simmen K, Olechowski C, Kerr B, Bleackley RC, Giuliani F. Granzyme B‐inhibitor serpina3n induces neuroprotection in vitro and in vivo. J Neuroinflammation. 2015;12:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690‐702. [DOI] [PubMed] [Google Scholar]
- 35. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chew LJ, King WC, Kennedy A, Gallo V. Interferon‐γ inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia. 2005;52:127‐143. [DOI] [PubMed] [Google Scholar]
- 37. Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chittajallu R, Chen Y, Wang H, et al. Regulation of Kv1 subunit expression in oligodendrocyte progenitor cells and their role in G1/S phase progression of the cell cycle. Proc Natl Acad Sci U S A. 2002;99:2350‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vautier F, Belachew S, Chittajallu R, Gallo V. Shaker‐type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. Glia. 2004;48:337‐345. [DOI] [PubMed] [Google Scholar]
- 40. Tegla CA, Cudrici C, Rozycka M, et al. C5b‐9‐activated, Kv1.3 channels mediate oligodendrocyte cell cycle activation and dedifferentiation. Exp Mol Pathol. 2011;91:335‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koeberle PD, Wang Y, Schlichter LC. Kv1.1 and Kv1.3 channels contribute to the degeneration of retinal ganglion cells after optic nerve transection in vivo. Cell Death Differ. 2010;17:134‐144. [DOI] [PubMed] [Google Scholar]
- 42. Peng Y, Lu K, Li Z, et al. Blockade of Kv1.3 channels ameliorates radiation‐induced brain injury. Neuro Oncol. 2014;16:528‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reeves TM, Trimmer PA, Colley BS, Phillips LL. Targeting Kv1.3 channels to reduce white matter pathology after traumatic brain injury. Exp Neurol. 2016;283(Pt A):188‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoflich KM, Beyer C, Clarner T, et al. Acute axonal damage in three different murine models of multiple sclerosis: a comparative approach. Brain Res. 2016;1650:125‐133. [DOI] [PubMed] [Google Scholar]
- 45. Pfeifenbring S, Bunyan RF, Metz I, et al. Extensive acute axonal damage in pediatric multiple sclerosis lesions. Ann Neurol. 2015;77:655‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schirmer L, Merkler D, Konig FB, Bruck W, Stadelmann C. Neuroaxonal regeneration is more pronounced in early multiple sclerosis than in traumatic brain injury lesions. Brain Pathol. 2013;23:2‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Herrero‐Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30:162‐173. [DOI] [PubMed] [Google Scholar]
- 48. Nikic I, Merkler D, Sorbara C, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495‐499. [DOI] [PubMed] [Google Scholar]
- 49. Gresle MM, Shaw G, Jarrott B, et al. Validation of a novel biomarker for acute axonal injury in experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86:3548‐3555. [DOI] [PubMed] [Google Scholar]
- 50. Kuhle J, Regeniter A, Leppert D, et al. A highly sensitive electrochemiluminescence immunoassay for the neurofilament heavy chain protein. J Neuroimmunol. 2010;220:114‐119. [DOI] [PubMed] [Google Scholar]
- 51. Rushton WA. A theory of the effects of fibre size in medullated nerve. J Physiol. 1951;115:101‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maghzi AH, Minagar A, Waubant E. Neuroprotection in multiple sclerosis: a therapeutic approach. CNS Drugs. 2013;27:799‐815. [DOI] [PubMed] [Google Scholar]
- 53. Shirani A, Zhao Y, Karim ME, et al. Association between use of interferon beta and progression of disability in patients with relapsing‐remitting multiple sclerosis. JAMA. 2012;308:247‐256. [DOI] [PubMed] [Google Scholar]
- 54. Beeton C, Chandy KG. Potassium channels, memory T cells, and multiple sclerosis. Neuroscientist. 2005;11:550‐562. [DOI] [PubMed] [Google Scholar]
- 55. Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci. 1999;19:10680‐10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallo P, Piccinno MG, Tavolato B, Siden A. A longitudinal study on IL‐2, sIL‐2R, IL‐4 and IFN‐γ in multiple sclerosis CSF and serum. J Neurol Sci. 1991;101:227‐232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials