

Abstract
Pregnancy‐related hypertension (PHTN) syndromes are a frequent and potentially deadly complication of pregnancy, while also negatively impacting the lifelong health of the mother and child. PHTN appears in women likely to develop hypertension later in life, with the stress of pregnancy unmasking a subclinical hypertensive phenotype. However, distinguishing between PHTN and chronic hypertension is essential for optimal management. Preeclampsia (PE) is linked to potentially severe outcomes and lacks effective treatments due to poorly understood mechanisms. Inadequate remodeling of spiral uterine arteries (SUAs), the cornerstone of PE pathophysiology, leads to hypoperfusion of the developing placenta. In normal pregnancies, extravillous trophoblast (EVT) cells assume an invasive phenotype and invade SUAs, transforming them into large conduits. Decidual natural killer cells play an essential role, mediating materno‐fetal immune tolerance, inducing early SUA remodeling and regulating EVT invasiveness. Notch signaling is important in EVT phenotypic switch and is dysregulated in PE. The hypoxic placenta releases antiangiogenic and proinflammatory factors that converge upon maternal endothelium, inducing endothelial dysfunction, hypertension, and organ damage. Hypoxia‐inducible factor 1‐α is upstream of such molecules, whereas endothelin‐1 is a major effector. We also describe important genetic links and evidence of incomplete materno‐fetal immune tolerance, with PE patients presenting with autoantibodies, lower Treg, and higher Th17 cells. Thus, PE manifestations arise as a consequence of mal‐placentation or/and because of a predisposition of the maternal vascular bed to excessively react to pathogenic molecules. From this pathophysiological basis, we provide current and propose future therapeutic directions for PE.
Keywords: Decidual Natural Killer Cells, Extravillous Trophoblast Cells, Hypoxia, Incomplete Materno‐Fetal Immune Tolerance, Preeclampsia
1. INTRODUCTION
Worldwide, 5% to 10% of women have elevated blood pressure (BP) during pregnancy, and hypertensive disorders are the second most frequent cause of pregnancy‐associated maternal mortality.1 The 2013 guidelines on hypertension in pregnancy2 distinguish between pregnancy‐related hypertension (PHTN) and chronic hypertension (HTN) (Table 1). This distinction is essential because of both mechanistic and prognostic considerations that heavily impact patient management; however, it can be challenging during clinical evaluation. Importantly, PHTN can have negative long‐term impacts on mother and child.
Table 1.
Classification of hypertensive disorders in pregnant women2
| Preeclampsia–eclampsia | New‐onset hypertension after 20 weeks' gestation in a previously normotensive woman + evidence of end‐organ damage (proteinuria, thrombocytopenia, hepatic or renal dysfunction, pulmonary edema or central nervous or visual disturbances) |
| Chronic hypertension | Hypertension diagnosed before pregnancy |
| Preeclampsia superimposed on chronic hypertension | New‐onset end‐organ dysfunction in a woman with chronic hypertension |
| Gestational hypertension | New‐onset hypertension after 20 weeks' gestation without signs of end‐organ dysfunction |
PHTN includes a spectrum of disorders ranging from gestational hypertension (GH) to preeclampsia (PE); the hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome; and eclampsia. PE occurs in 4% to 10% of pregnancies, and is a main cause of maternal and fetal morbidity and mortality worldwide. It is more frequent in women of African American or Hispanic heritage, and in those with traditional cardiovascular (CV) disease risk factors,3 with thrombophilia or family history of PE.4 Occurring in 6% to 7% of pregnancies, GH is generally less severe.1 However, it can lead to very high BP, evolve into PE, and/or be complicated by chronic hypertension after delivery. Thus, these women require intense monitoring during and after childbirth.2
2. DIAGNOSIS
The diagnosis of PHTN is made by extrapolating thresholds for normal BP in the general population (Table 2). However, adequacy of such criteria is debatable. Although BP physiologically declines 10 to 15 mmHg by the end of the first trimester and recovers to near‐normal levels in the third trimester, no large‐scale clinical trial has evaluated what the optimal BP should be in pregnancy. “Tight” (DBP target ≤85 mmHg) versus “less tight” (DBP target ≤100 mmHg) BP control in CHIPS (Control of Hypertension in Pregnancy Study) found no significant fetal adverse effects. However, women in the less tight BP control more frequently presented with severe HTN (≥160/110 mmHg). Prior meta‐analyses suggested lower (with antihypertensive therapy) versus higher BP may result in lower birth weight with increased risk of small‐for‐gestational‐age newborns, but CHIPS results do not support these concerns. The US Preventive Services Task Force recommends screening of all pregnant women by measuring BP at every prenatal visit. PE diagnosis requires 2 measurements at least 4 hours apart. However, the diagnosis must be swift when SBP ≥160 mmHg/DBP ≥110 mmHg to ensure prompt treatment. Proteinuria has been eliminated as a compulsory criterion for PE, which can be diagnosed when new‐onset HTN is accompanied by signs of end‐organ damage not explained by other pathologies (Table 2).2 Characteristics of severe PE are highlighted in Table 2.
Table 2.
Diagnosis of preeclampsia requires hypertension and a sign of end‐organ damage2
| Hypertension | SBP ≥140 mmHg or DBP ≥90 mmHg; Severe: SBP ≥160 mmHg or DBP ≥110 mmHg |
| Signs of end‐organ dysfunction | |
| Proteinuria | ≥300 mg proteins/24‐hour urinary volume or a ratio of protein to creatinine in a single voided urine ≥3.0 |
| Thrombocytopeniaa | Platelet counts <100,000/μL |
| Hepatic dysfunctiona | Liver transaminases 2× greater than normal levels or severe upper quadrant or epigastric pain |
| Renal insufficiencya | Serum creatinine >1.1 mg/dL or a 2‐fold increase above previous values, in the absence of other causes of renal impairment |
| Pulmonary edemaa | |
| Acute neurological dysfunction (including vision impairment)a | |
| The HELLP syndromea | Stands for: hemolysis, elevated liver enzymes and low platelets |
| Eclampsiaa | Grand‐mal seizures; premonitory signs: severe headaches, blurred vision, hyperreflexia, or altered mental status. |
Abbreviations: DBP, diastolic blood pressure; SBP, systolic blood pressure; PE, preeclampsia.
PE severity features.
Finally, PE is a disease of the placenta and does not require the presence of a fetus; thus, it can complicate pregnancies resulting in a hydatidiform mole. Also, PE can develop exclusively in the postpartum period, and women should be cautioned to contact their physician in case of severe headaches or epigastric pain.2
3. MATERNAL RISK FACTORS FOR PREECLAMPSIA AND EARLY DIAGNOSIS
A multisystemic disorder, PE stems from mal‐implantation of the developing placenta. The severity of PE for mother and fetus depends on symptom onset; 34 weeks' gestation defines early versus late PE. Early PE exposes the fetus to high risk of mortality and early delivery. However, prompt aspirin therapy can improve placentation and decrease PE risk. Although concerns regarding possible detrimental effects of aspirin on fetal development have been raised, benefits outweigh risks in women at risk of PE.2 Thus, there is an urgent need to accurately diagnose pregnancies at high risk of early PE.
According to American College of Obstetricians and Gynecologists (ACOG) guidelines, PE risk should be evaluated at first obstetrical visit by a history screening of risk factors (Table 3).2 Of note, a recent systematic review and meta‐analysis (>25 million cases) found women with antiphospholipid syndrome had the highest rate of PE (17.3%); prior PE had the greatest relative risk (8.4%), followed by chronic hypertension (5.1%).5 Women at high risk should be prescribed low‐dose aspirin.2 To date, no single test accurately predicts PE. Multivariate analysis of clinical (maternal characteristics), laboratory (pregnancy‐associated plasma protein A), soluble receptor for vascular endothelial growth factor (VEGF) (sFlt‐1) and placental growth factor (PIGF), and paraclinical investigations (uterine artery pulsatility index) have been proposed for PE prediction; however, none are currently endorsed by the ACOG.2
Table 3.
Risk factors for preeclampsia2
| Increased risk of placental mal‐implantation | Primiparity |
| Previous PE pregnancy | |
| Family history | |
| Multifetal pregnancy | |
| In vitro fertilization | |
| Advanced maternal age (>40 years) | |
| Maternal comorbidities associated with endothelial dysfunction | Chronic hypertension/renal disease |
| History of thrombophilia | |
| Type I/II diabetes mellitus | |
| Obesity | |
| Systemic lupus erythematosus |
Abbreviations: PE, preeclampsia.
Echocardiography of women at high and low risk of PE at midgestation indicates concentric hypertrophic remodeling in a third of the women who subsequently develop PE. This response compensates for the increased afterload, allowing normal wall stress in women with PE. However, hypertrophy was absent in both low‐ and high‐risk subgroups that did not develop PE.6
4. MECHANISMS OF PREECLAMPSIA
Normal placental development requires the spiral uterine arteries (SUAs) to be enlarged and transformed into capacitance vessels. Remodeling of the SUAs takes place in an early, trophoblast‐independent phase, followed by a later trophoblast‐dependent phase.7 In normal pregnancies, remodeling is complete by the beginning of the second trimester. In contrast, in PE the insufficient remodeling of SUAs generates a sustained pathologic ischemic milieu,8 with the dysfunctional placenta releasing pathogenic mediators into the maternal blood that induce generalized endothelial dysfunction (ED), perturbed coagulation, HTN, and organ dysfunction.
4.1. Early phase of SUA remodeling: trophoblast‐independent mechanisms
Early vascular changes associated with decidualization (such as endothelial vacuolation and disorganization and thinning of the media) occur prior to the presence of trophoblastic cells in the vicinity of SUAs. Leukocytes play a major role in cyclic remodeling of nonpregnant endometrium, by secreting matrix metalloproteinases (MMPs) and proinflammatory cytokines.9 They are abundant in human decidual stroma, with macrophages and decidual natural killer (dNK) cells amounting to 40%. dNKs differ in phenotype and surface marker expression from peripheral blood NKs (pbNK); they are regulatory NK cell (RNKs). Most dNKs stain positive for CD56brightCD16– and have marked secretory activity, with reduced cytotoxic activity (although they retain the ability to express perforin and granzyme). 10 Conversely, pbNKs are cytolytic, CD56dimCD16+ NK cells.11 RNKs play an important role in maintaining pregnancy, as they collaborate with CD4+ T cells to mediate tolerance toward fetal antigens. However, PE features decreased levels of Treg and increased levels of Th17 cells, which supports the long‐held belief that PE is a disease of partial fetal intolerance (Figure 1).12 DNKs are also involved in trophoblast‐independent SUA remodeling, which is severely impaired in the absence of dNKs (pregnant knockout mice model NK–T–B–). Reconstitution of dNK cells by bone marrow transplantation from severe combined immunodeficiency mice improves SUA remodeling. In samples of human placenta from pregnancies electively terminated between 8 and 12 weeks, involvement of leukocytes (CD56+ NKs and CD68+ macrophages) is described in the trophoblast‐independent phase of SUA remodeling. Leukocyte infiltration correlates with the stage of vessel wall remodeling (most leukocytes aggregating around actively remodeling vessels) and was associated with discrete vascular smooth muscle cell (VSMC) and endothelial cell apoptosis9 by activation of the Fas‐ligand pathway.13 Moreover, leucocytes produced extracellular matrix degrading enzymes MMP‐7 and MMP‐9 (Figure 1).9
Figure 1.
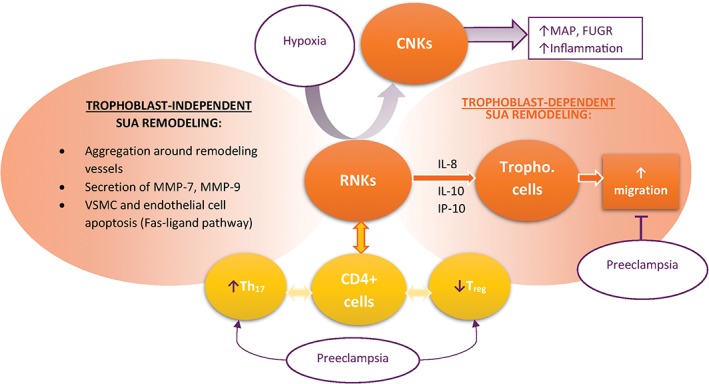
Role of decidual natural killer cells (DNKs) in placentation and preeclampsia pathology. In normal pregnancy, DNKs adopt a regulatory natural killer cell (RNK) phenotype to mediate spiral uterine artery (SUA) remodeling both directly and indirectly. In pathologic hypoxia, DNKs adopt a cytolytic phenotype. DNKs also mediate materno‐fetal immune tolerance by interacting with CD4+ cells. Preeclampsia is associated with increased Th17 cells and decreased T regulatory cells, compared with normal pregnancies. Abbreviations: CNK, cytolytic natural killer cell; FUGR, fetal intrauterine growth restriction; MAP, mean arterial pressure; MMP, matrix metalloproteinase; VSMC, vascular smooth muscle cell
DNKs also appear to set the stage for trophoblast‐dependent remodeling (Figure 1). Human NKs enhance migration of human trophoblast cells in vitro and in vivo by secreting interleukin (IL)‐8 and interferon gamma‐induced protein 10 (IP‐10), which bind receptors CXCR1 and CXCR3, respectively, on trophoblast cells.14 Wallace et al11 studied human dNKs in placentas from pregnancies terminated between 8 and 14 weeks. The extent of SUA remodeling and the risk of PE was evaluated indirectly, by measuring uterine artery Doppler resistance index (RI). DNK cells from all pregnancies secreted similar amounts of IL‐8, IL‐6, and IP‐10. However, dNK from pregnancies with high RI were unable to induce trophoblast chemotaxis and extravillous trophoblast outgrowth, which was associated with decreased signaling through the ERK1/2 and PI3K pathways11 or with decreased secretion of hepatocyte growth factor by dNK cells.13
Dysfunctional dNK cells may also become disease mediators under the stress of pathological placental hypoxia (Figure 1). A reduced uterine perfusion pressure rat model led to the conversion of tolerogenic dNKs to a cytolytic phenotype. This shift was associated with higher mean arterial pressure, fetal intrauterine growth restriction (FUGR), and increased inflammation, changes that were reversed by the depletion of NK cells.15
4.2. Inadequate remodeling of spiral uterine arteries by cytotrophoblast cells
The first observations of SUA remodeling in pregnancy came from placental biopsy specimens acquired during cesarean sections. The arteries were completely different from the SUAs described in nonpregnant endometrial samples, with large diameters and a media with scarce VSMCs and fibrinoid deposition. Further studies showed these changes took place in the presence of trophoblast cells in the vicinity.7 Presently, insufficient extravillous trophoblast (EVT) invasion of SUAs is considered to be at the cornerstone of PE pathophysiology.2
Throughout early pregnancy, EVTs transition from a proliferative to invasive phenotype is dependent on oxygen pressure. In the first trimester, the placenta is physiologically low in oxygen with PO2 averaging 20 mm HG, whereas the decidua is normally vascularized with a PO2 nearing 70 mm HG; these conditions upregulate factors such as hypoxia‐inducible factor‐1 α (HIF‐1α). This oxygen‐deprived environment induces trophoblast proliferation, protects trophoblast against DNA damage by lowering oxidative stress, and represses trophoblast invasion. Moreover, low oxygen is a prerequisite for normal pregnancy at this stage, with increased blood flow during the first trimester hampering placental development or even leading to spontaneous abortion.8
EVT proliferation pushes cells away from the chorionic villi's basal membrane toward the placenta and toward higher oxygen content, gradually increasing their invasive potential. Invasive trophoblasts will eventually invade the myometrium around week 5 postmenstruation.16 Between weeks 11 and 14 of gestation, EVTs remodel the SUA by inducing the loss of endothelial cells and the smooth muscle cells and elastic fibers of the media. This is a complex process, involving apoptosis, VSMC dedifferentiation, and degradation of extracellular matrix. Although the endothelium is completely regenerated by EVTs, the thick, elastic media is replaced by fibrinoid secreted by EVTs. These structural changes of SUA lead to an increased vessel caliber that allow an adequate blood supply to the developing fetus.7
In PE, EVTs fail to transition to a fully invasive phenotype, with decreased penetration into the decidua and the SUAs, and insufficient remodeling of these vessels. Trophoblastic invasion depends at least partially on the complex regulation of vascular adhesion molecules. Zhou et al. found that in PE, invading trophoblast cells have perturbed expression of αV‐family integrins. They retain expression of αVβ6, which in healthy controls is only expressed in remodeling epithelium and do not upregulate αVβ3, an integrin necessary for angiogenesis. Moreover, the expression of cadherins, a class of Ca2 + ‐dependent intercellular adhesion molecules, is markedly different in trophoblasts from PE patients by comparison to trophoblasts from controls. E‐cadherin mediates strong intercellular adhesion. Although it is poorly expressed on EVTs from normal pregnancies, EVTs from PE pregnancies richly express E‐cadherin and had a tendency to clump together. Conversely, both vascular endothelial‐cadherin (an endothelial junction molecule) and vascular cell adhesion molecule 1 are expressed by trophoblasts form normal pregnancies but are notably absent in pregnancies afflicted by PE.
The etiology of EVT malfunction in PE is unclear; however, incomplete immune tolerance for semiallogenic fetal antigens has been proposed as a major cause. Decreased exposure to paternal seminal fluid prior to conception is a clinical risk factor for PE.17 Moreover, immunocompromised women, such as those with human immunodeficiency virus have a decreased risk of PE, which normalizes upon restitution of immune competency.18 As previously mentioned, dNKs mediate early SUA remodeling and also cooperate with trophoblast cells in early placentation. This cooperation may become hindered when genotype AA killer immunoglobulin receptors (KIRs) on the surface of NKs encounter human leukocyte antigen‐C (HLA‐C) on EVTs, leading to mainly inhibitory signals toward dNKs.19 As these dNKs mediate tolerance to fetal antigens, their inhibition may partially explain incomplete materno‐fetal tolerance. Finally, KIR AA association with HLA‐C is also thought to mediate FUGR and recurrent miscariage.20
Notch signaling has been implicated as a key regulator of EVT invasiveness. Data in mice showed that inhibiting Notch signaling by deletion of Notch2 (only Notch receptor upregulated in mice EVTs) led to reduced SUA invasion and remodeling by EVTs, together with an absence of JAG1 expression in PE.21 These findings were confirmed on human trophoblast cells where expression of Notch2 and Notch3 increased EVT invasiveness and proliferation and decreased EVT proliferation, whereas downregulation of these receptors led to the opposite effects.22 In normal pregnancies, invading EVTs produce large amounts of proteolytic enzymes MMP‐2 and MMP‐9,23 a phenotype that can be recapitulated experimentally by Notch2 overexpression.22
5. PATHOGENESIS OF HYPERTENSION: LINKING PLACENTAL HYPOXIA TO GENERALIZED ENDOTHELIAL DYSFUNCTION
Regardless of underlying etiology, insufficient SUA remodeling by EVTs leads to a persistently hypoxic, dysfunctional placenta that releases pathogenic molecules into the maternal circulation (Figure 2). Exposing endothelial cells to serum from PE women leads to ED.24 Subsequently, many placental factors capable of inducing ED and the PE syndrome, such as antiangiogenic factors, microparticles (containing proinflammatory mediators), cell‐free nucleic acids, oxidized lipids, and free radicals, have been described.25 In the maternal circulation, they can induce second‐order mediators such as autoantibodies against angiotensin receptor 1 (AA‐AT1) or effectors such as endothelin‐1 (ET‐1) and superoxide.
Figure 2.
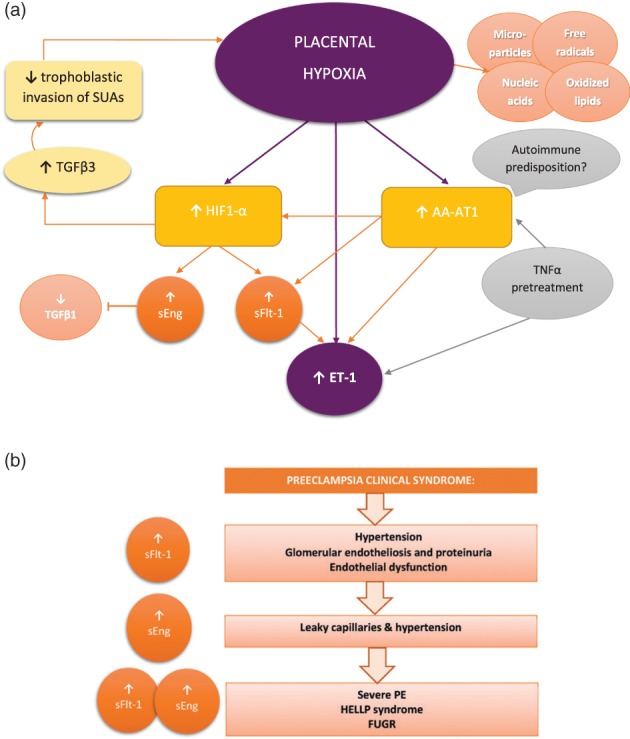
(a) Molecular pathways linking placental hypoxia to generalized endothelial dysfunction. (b) Infusing sFlt‐1, sEng, and the combination of sFlt‐1 and sEng recapitulates the clinical preeclampsia (PE) syndrome. Abbreviations: AA‐AT1, activating auto‐antibody against angiotensin 1 receptor; ET‐1, endothelin 1; FUGR, fetal intrauterine growth restriction; HELLP, hemolysis, elevated liver enzymes, and low platelets; HIF1‐ α, hypoxia‐inducible factor 1‐α; PE, preeclampsia; sEng, soluble endoglin; sFlt‐1, soluble receptor for VEGF and PIGF; SUA, spiral uterine artery; TGFβ, transforming growth factor β; TNFα, tumor necrosis factor α
The hypoxic placenta releases antiangiogenic molecules such as sFlt‐1 and soluble endoglin (sEng), the soluble receptor for transforming growth factor β (TGFβ), and endothelium‐derived vasoconstrictor (ET‐1). SFlt‐1 is elevated toward delivery in normal pregnancy but is constantly elevated in PE. It inhibits both VEGF and PIGF by sequestering circulating molecules and by blocking their common cellular receptor. In addition to their roles in angiogenesis, VEGF and PIGF participate in maintaining normal endothelial function. Increasing sFlt‐1 in pregnant rats leads to glomerular endotheliosis and proteinuria, whereas in pregnant mice it replicated the PE phenotype with hypertension, low platelets, and FUGR. Conversely, placental hypoperfusion in rats or baboons leads to hypertension, proteinuria, and increased sFlt‐1 levels. Moreover, sFlt‐1 levels are predictive of PE severity. sEng blocks TGFβ1 signaling, which inhibits vasodilatation in rats (resulting in HTN) and leads to leaky capillaries. Administering both sFlt‐1 and sEng to rats produces overt, severe PE, with HELLP syndrome and FUGR.18 ET‐1 is produced locally within tissues, and its level in different organs varies and is not correlated with the plasmatic concentration. It is increased in many experimental models of PE. More importantly, blocking endothelin‐A (ETA) receptors in these models alleviates HTN, suggesting that ET‐1 is the common pathway upon which pathogenic factors converge to induce HTN in PE.12
The renin‐angiotensin (AT)‐aldosterone (RAA) axis has also been involved in PE pathogenesis. Normal pregnancies are characterized by low vascular tone despite high levels of AT and aldosterone due to decreased sensitivity of vasculature to these molecules. However, PE patients have circulating activating AA‐AT1, which leads to excessive activation of the RAA axis. Exposure of pregnant mice to AA‐AT1 reproduces PE symptoms, including increased sFlt‐1 and sEng. Although AA‐AT1 are autoantibodies, they can also be induced by placental hypoxia and by tumor necrosis factor α treatment.12 This dualism suggests that, regardless of placentation efficiency, some women are more predisposed than others to develop PE. Moreover, the relationship between elevated AA‐AT1 and HIF‐1α is unclear; infusions of AA‐AT1 lead to increased HIF‐1α in the absence of placental hypoxia.26 Finally, AA‐AT1 levels can remain elevated after delivery, thus predisposing women to developing HTN later in life.18
Mounting evidence suggests that HIF‐1α is the main regulator of sFlt‐1, sEng, and of trophoblast invasiveness. Physiologic placental hypoxia early in gestation (before 9–11 weeks) is associated with increased expression of HIF‐1α and of its target, TGFβ3, which inhibits trophoblast differentiation and invasion. In PE, the sustained hypoxic environment creates a vicious circle, with high levels of HIF‐1α and TGFβ3 constantly repressing trophoblast invasiveness and leading to poor placentation and SUA remodeling.27
6. ZOOMING IN: MOLECULAR MECHANISMS OF ENDOTHELIAL DYSFUNCTION IN PE
The wide array of pathogenic molecules released by the insufficient placenta converge their effects on the maternal endothelium, disturbing the balance between endothelium‐derived relaxing and constricting factors (EDRF and EDCF, respectively). The main EDRFs are prostacyclin (PGI2) and nitric oxide (NO), whereas the main EDCFs are TXA2, ET‐1, and angiotensin.
The balance between TXA2 and PGI2 is a main local regulator of microvascular tone. Both TXA2 and PGI2 are synthetized from arachidonic acid by COX. Although PGI2 is synthetized in large quantities in endothelial cells, TXA2 is predominantly produced in platelets. Each of these cell types produces a low level of the corresponding, opposite‐action compound. In normal pregnancies, the balance is tipped in favor of the vasodilatatory PGI2. In pregnancies complicated by PE, lipid peroxidation activates COX but inhibits prostacyclin synthase, which tips the TXA2/PGI2 balance in favor of TXA2, with a rapid decrease of PGI2. These changes occur starting at 13 weeks of gestation. Epigenetic studies have shown that women with PE exhibit decreased methylation of the TBXAS1 gene promoter, which leads to increased TXA2 synthesis. Aspirin treatment inactivates COX by acetylation of a serine residue. Salicylic acid has a short half‐life, and nucleated cells are able to resynthesize COX. However, anuclear thrombocytes are unable to rebuild COX stores; thus, aspirin‐mediated inhibition of COX lasts for the entire life of platelets (7–10 days). Aspirin treatment normalizes the TXA2/PGI2 balance in 2 weeks. Furthermore, aspirin also helps to counteract the prothrombotic state induced by activation of platelet aggregation and adhesion, which is sustained by TXA2.28
7. GENETIC LINKS TO PREGNANCY‐RELATED HTN
Two genetic polymorphisms associated with familial PE are ACVR2A and STOX1.29 Women with pregnancies complicated by PE of different heritage (Iceland/Norway, Australia/New Zealand) shared susceptibility of the activin receptor type‐2 gene (ACVR2A) on chromosome 2q22, versus controls. The STOX1 gene was implicated in Dutch women and preclinical models have shown that STOX1 upregulation interferes with the epithelial‐to‐mesenchymal transition that underlies the trophoblast phenotypic switch from proliferative to invasive.30 A recent study found that TGFβR2 polymorphisms were significantly associated with both PE and GH.31
8. PREVENTION OF PREGNANCY‐RELATED HTN
8.1. Prevention of PE with aspirin prophylaxis—the ASPRE trial
Clinical trials and meta‐analyses evaluating low‐dose aspirin after 16 weeks' gestation in women at high‐risk of PE reduced the incidence of disease by 10%.32 However, a recent meta‐analysis suggested the incidence of PE could be halved by aspirin before 16 weeks' gestation.33 The ASPRE trial (Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia) showed that 150 mg of aspirin a day between the 11th/14th and 36th weeks of gestation was a more effective than placebo in preventing preterm PE in women at high‐risk of PE, without negative fetal outcomes. In contrast, 2013 ACOG guidelines recommend daily doses of 60 to 80 mg of aspirin in women with a history of prior early‐onset PE and preterm delivery before 34 weeks of gestation.2
8.2. Bed‐rest or physical exercise?
A recent meta‐analysis found regular aerobic physical exercise (30–60 minutes, 2–7 times a week) from early pregnancy leads to a lower incidence of GH but not of PE. Moreover, 16% less women needed urgent cesarean delivery in the exercise group versus controls.34 Although exercise as prevention is not currently a guideline recommendation, the ACOG states that bed rest or physical activity restriction is not recommended for primary prevention of PE and its complications.2
9. MANAGEMENT OF PREGNANCY‐RELATED HTN—ACOG RECOMMENDATIONS
In women with known, chronic hypertension, guidelines recommend antihypertensive treatment if SBP is >160 mmHg or DBP is >105 mmHg. In the absence of organ dysfunction, women with BP lower than these targets should not be treated.2 Moreover, the 2017 American College of Cardiology/American Heart Association hypertension lower threshold recommendations do not extend to the management of pregnancy‐related HTN.35 In women with nonsevere PE or with GH, ACOG recommends measuring BP at least 2 times/week. Administering antihypertensive medication to women with mild to moderate pregnancy‐related HTN is controversial, due to concerns of both efficacy and fetal safety. Specifically, antihypertensive therapy reduced the risk of developing severe HTN but did not alter progression to PE, eclampsia, pulmonary edema, or fetal death. However, β‐blockers were associated with an increased frequency of small‐for‐gestational‐age infants.36 Thus, the ACOG guidelines currently suggest that women with SBP <160 mmHg or DBP <110 mmHg should not be treated. Treatment is recommended in cases of severe HTN (systolic BP ≥160 mmHg or diastolic BP ≥110 mmHg).2 However, the CHIPS, in which labetolol was the recommended antihypertensive agent, may prompt changing target BP values in guidelines, as “tight control” was associated with reduced maternal risk of developing severe hypertension and no increase in fetal adverse effects.37 A recent retrospective cohort study found that in pregnancies complicated by PE, increased rates of antihypertensive drug administration were accompanied by decreased stroke rates.38
Safe, first‐line antihypertensive treatment in pregnancy includes labetalol, nifedipine, and methyldopa. Diuretics can be useful in women with known salt‐sensitive HTN; careful dose adjustments are needed to minimize hypokalemia. Blocking the RAA axis is discouraged, due to heavily documented adverse fetal effects. As 50% of pregnancies are unplanned, guidelines recommend such medication be avoided in women of reproductive age.2
Eclampsia or premonitory signs (Table 2) should prompt urgent treatment with magnesium sulfate for at least 24 hours after the last seizure. Magnesium sulfate is also recommended for severe PE (before, during, and after delivery) and for PE undergoing cesarean delivery (intraoperatory) to minimize the risk of progression to eclampsia. Delivery is the only curative treatment for severe PE.2
10. FUTURE THERAPEUTIC PERSPECTIVES IN PE
The antiangiogenic and pathogenic sFlt‐1 molecule may be an adequate target for future PE preventive strategies. A pilot study of plasmapheresis in pregnant women effectively reduced BP and proteinuria.39 In a rat preeclampsia model, nano‐scale artificial oxygen carriers (hemoglobin vesicles) reduced levels of antiangiogenic proteins and improved fetal intrauterine growth.40 Blocking ETA receptors could also prove effective in PE. However, ETA is important in fetal development during the first trimester; thus, such therapy needs to be administered later in pregnancy or in a compound that does not cross the placental barrier.12 Finally, cellular therapies with proven immune‐modulatory properties in various models of autoimmune diseases, such as mesenchymal stem cells, adipose‐derived stromal/stem cells, or stromal vascular fraction,41, 42, 43 could prove useful in restoring the balance between Treg and Th17 cells.
11. LONG‐TERM RISK FOR MOTHER AND CHILD
Pregnancy is a physiological stress that uncovers a woman's predisposition to disease; similarly, the importance of intrauterine development for the life‐long health of the fetus has been clearly established. Thus, pregnancy appears to be an ideal moment for intervention to reduce long‐term CV risk in both mother and child.
A history of PE increases the mother's CV risk to a magnitude similar to that of diabetes. A recent prospective cohort study found increased frequency of persistent hypertension after PE.44 Bearing in mind that PE has been included as a risk factor for CV disease by the AHA, women who have had PE should be followed by a cardiologist. Moreover, delivering a FUGR child, regardless of the underlying pathology, is also associated with increased maternal risk of developing ischemic heart disease, cerebrovascular disease, or CV insufficiency in later life.
In PE, the main determinant of fetal risk is FUGR. However, data regarding CV life‐long risk of children born from PE pregnancies have been extrapolated from studies evaluating adults that suffered FUGR due to maternal malnutrition. One study showed that maternal hunger during pregnancy correlates with high BP, high levels of low‐density lipoprotein, cholesterol, and fibrinogen, especially if nutrient deprivation occurred during the third trimester. Another study evaluating adults from a poor background, that subsequently led prosperous lives, found that these individuals had increased myocardial infarction risk. FUGR may also induce in utero cardiac remodeling that was persistent at 6 months after birth. Furthermore, adult women, rather than men, with low birth weights (under 2.5 kg) have a higher incidence of glucose intolerance, metabolic syndrome, and diabetes. Finally, PE can also lead to prematurity, which is associated with higher risk for HTN and insulin resistance in infancy.45
Around 20% of women with the HELLP syndrome during pregnancy exhibit concentric left ventricular hypertrophy, which is not present in PE or control patients. However, left ventricular concentric remodeling, diastolic dysfunction, and reduced left ventricular ejection fraction was noted at similar rates in patients with previous HELLP or PE.46 GH is believed to manifest in pregnancies of women who are predisposed to ultimately develop hypertension. Chronic hypertension is diagnosed when BP levels are persistently elevated beyond 12 weeks postpartum.47 GH can also progress to preeclampsia, especially if coupled with excessive weight gain (≥10 kg at 28 weeks' gestation vs 8 weeks' gestation), and grade III preterm placental calcification (at 28 weeks of pregnancy).48 Moreover, even if GH does not progress to PE, it can have more severe fetal consequences than mild PE by inducing premature delivery and small‐for‐gestational‐age infants more frequently.49
Finally, a recent study on the Women's Ischemia Syndrome Evaluation—Coronary Vascular Dysfunction (WISE‐CVD) cohort found PHTN correlated with lower coronary flow reserves, which is an indirect measurement of coronary microvascular dysfunction.50
Conflicts of interest
The authors declare no potential conflicts of interest.
Sava RI, March KL, Pepine CJ. Hypertension in pregnancy: Taking cues from pathophysiology for clinical practice. Clin Cardiol. 2018;41:221–228. 10.1002/clc.22892
Funding information Dr. Sava receives a Fulbright Student Scholarship from the Romanian‐American Fulbright Commission. Drs. Pepine and March receive support from the National Heart, Lung, and Blood Institute (NHLBI): Cardiovascular Cell Therapy Network (CCTRN) under cooperative agreement UM1 HL087318, and the University of Florida Regional Clinical Center CCTRN Training Core Supplement under UM1 HL087366; and from the US Department of Defense: Women's Ischemia Treatment Reduces Events in Non‐Obstructive CAD. Dr. Pepine receives support from the NHLBI: Brain‐Gut Microbiome‐Immune Axis in Hypertension, HL132448; the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine; National Institutes of Health, National Center for Advancing Translational Sciences‐University of Florida Clinical and Translational Science UL1TR001427; and PCORnet‐OneFlorida Clinical Research Consortium, Clinical Data Research Network‐1501‐26692
REFERENCES
- 1. Vest AR, Cho LS. Hypertension in pregnancy. Curr Atheroscler Rep. 2014;16:395. [DOI] [PubMed] [Google Scholar]
- 2. American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy . Report of the American College of Obstetricians and Gynecologists' Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122:1122–1131. [DOI] [PubMed] [Google Scholar]
- 3. Breathett K, Muhlestein D, Foraker R, et al. Differences in preeclampsia rates between African American and Caucasian women: trends from the National Hospital Discharge Survey. J Womens Health (Larchmt). 2014;23:886–893. [DOI] [PubMed] [Google Scholar]
- 4. Sibai B, Dekker G, Kupferminc M. Pre‐eclampsia. Lancet. 2005;365:785–799. [DOI] [PubMed] [Google Scholar]
- 5. Bartsch E, Medcalf KE, Park AL, et al. Clinical risk factors for pre‐eclampsia determined in early pregnancy: systematic review and meta‐analysis of large cohort studies. BMJ. 2016;353:i1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melchiorre K, Sutherland G, Sharma R, et al. Mid‐gestational maternal cardiovascular profile in preterm and term pre‐eclampsia: a prospective study. BJOG. 2013;120:496–504. [DOI] [PubMed] [Google Scholar]
- 7. Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. [DOI] [PubMed] [Google Scholar]
- 8. Huppertz B, Gauster M, Orendi K, et al. Oxygen as modulator of trophoblast invasion. J Anat. 2009;215:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith SD, Dunk CE, Aplin JD, et al. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am J Pathol. 2009;174:1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Inngjerdingen M, Kveberg L, Naper C, et al. Natural killer cell subsets in man and rodents. Tissue Antigens. 2011;78:81–88. [DOI] [PubMed] [Google Scholar]
- 11. Wallace AE, Host AJ, Whitley GS, et al. Decidual natural killer cell interactions with trophoblasts are impaired in pregnancies at increased risk of preeclampsia. Am J Pathol. 2013;183:1853–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palei AC, Spradley FT, Warrington JP, et al. Pathophysiology of hypertension in pre‐eclampsia: a lesson in integrative physiology. Acta Physiol (Oxf). 2013;208:224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fraser R, Whitley GS, Johnstone AP, et al. Impaired decidual natural killer cell regulation of vascular remodelling in early human pregnancies with high uterine artery resistance. J Pathol. 2012;228:322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanna J, Goldman‐Wohl D, Hamani Y, et al. Decidual NK cells regulate key developmental processes at the human fetal‐maternal interface. Nat Med. 2006;12:1065–1074. [DOI] [PubMed] [Google Scholar]
- 15. Elfarra J, Amaral LM, McCalmon M, et al. Natural killer cells mediate pathophysiology in response to reduced uterine perfusion pressure. Clin Sci (Lond). 2017;131:2753–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod. 2003;69:1–7. [DOI] [PubMed] [Google Scholar]
- 17. Saftlas AF, Rubenstein L, Prater K, et al. Cumulative exposure to paternal seminal fluid prior to conception and subsequent risk of preeclampsia. J Reprod Immunol. 2014;101‐102:104–110. [DOI] [PubMed] [Google Scholar]
- 18. Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation. 2011;123:2856–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiby SE, Walker JJ, O'Shaughnessy K M, et al. Combinations of maternal KIR and fetal HLA‐C genes influence the risk of preeclampsia and reproductive success. J Exp Med. 2004;200:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moffett A, Chazara O, Colucci F, et al. Variation of maternal KIR and fetal HLA‐C genes in reproductive failure: too early for clinical intervention. Reprod Biomed Online. 2016;33:763–769. [DOI] [PubMed] [Google Scholar]
- 21. Hunkapiller NM, Gasperowicz M, Kapidzic M, et al. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre‐eclampsia. Development. 2011;138:2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao WX, Wu ZM, Liu W, et al. Notch2 and Notch3 suppress the proliferation and mediate invasion of trophoblast cell lines. Biol Open. 2017;6:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halasz M, Szekeres‐Bartho J. The role of progesterone in implantation and trophoblast invasion. J Reprod Immunol. 2013;97:43–50. [DOI] [PubMed] [Google Scholar]
- 24. Roberts JM, Taylor RN, Musci TJ, et al. Preeclampsia: an endothelial cell disorder. Am J Obstet Gynecol. 1989;161:1200–1204. [DOI] [PubMed] [Google Scholar]
- 25. Goulopoulou S, Davidge ST. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol Med. 2015;21:88–97. [DOI] [PubMed] [Google Scholar]
- 26. Iriyama T, Wang W, Parchim NF, et al. Hypoxia‐independent upregulation of placental hypoxia inducible factor‐1alpha gene expression contributes to the pathogenesis of preeclampsia. Hypertension. 2015;65:1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tal R. The role of hypoxia and hypoxia‐inducible factor‐1alpha in preeclampsia pathogenesis. Biol Reprod. 2012;87:134. [DOI] [PubMed] [Google Scholar]
- 28. Atallah A, Lecarpentier E, Goffinet F, et al. Aspirin for prevention of preeclampsia. Drugs. 2017;77:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haram K, Mortensen JH, Nagy B. Genetic aspects of preeclampsia and the HELLP syndrome. J Pregnancy. 2014;2014:910751. 10.1155/2014/910751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Dijk M, van Bezu J, van Abel D, et al. The STOX1 genotype associated with pre‐eclampsia leads to a reduction of trophoblast invasion by alpha‐T‐catenin upregulation. Hum Mol Genet. 2010;19:2658–2667. [DOI] [PubMed] [Google Scholar]
- 31. Li X, Tan H, Chen M, et al. Transforming growth factor beta 1 related gene polymorphisms in gestational hypertension and preeclampsia: a case‐control candidate gene association study [published online November 23, 2017]. Pregnancy Hypertens. 10.1016/j.preghy.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 32. Poon LC, Nicolaides KH. Early prediction of preeclampsia. Obstet Gynecol Int. 2014;2014:297397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roberge S, Giguere Y, Villa P, et al. Early administration of low‐dose aspirin for the prevention of severe and mild preeclampsia: a systematic review and meta‐analysis. Am J Perinatol. 2012;29:551–556. [DOI] [PubMed] [Google Scholar]
- 34. Magro‐Malosso ER, Saccone G, Di Tommaso M, et al. Exercise during pregnancy and risk of gestational hypertensive disorders: a systematic review and meta‐analysis. Acta Obstet Gynecol Scand. 2017;96:921–931. [DOI] [PubMed] [Google Scholar]
- 35. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines [published online November 7, 2017]. J Am Coll Cardiol 10.1016/j.jacc.2017.11.006. [DOI] [PubMed]
- 36. Magee LA, Duley L. Oral beta‐blockers for mild to moderate hypertension during pregnancy. Cochrane Database Syst Rev. 2003;(3):CD002863. [DOI] [PubMed] [Google Scholar]
- 37. Magee LA, von Dadelszen P, Rey E, et al. Less‐tight versus tight control of hypertension in pregnancy. N Engl J Med. 2015;372:407–417. [DOI] [PubMed] [Google Scholar]
- 38. Cleary KL, Siddiq Z, Ananth CV, Wright JD, Too G, D'Alton ME, Friedman AM. Use of antihypertensive medications during delivery hospitalizations complicated by preeclampsia. Obstet Gynecol. 2018. Feb 5. 10.1097/AOG.0000000000002479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thadhani R, Kisner T, Hagmann H, et al. Pilot study of extracorporeal removal of soluble fms‐like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124:940–950. [DOI] [PubMed] [Google Scholar]
- 40. Li H, Ohta H, Tahara Y, et al. Artificial oxygen carriers rescue placental hypoxia and improve fetal development in the rat pre‐eclampsia model. Sci Rep. 2015;5:15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bowles AC, Wise RM, Gerstein BY, et al. Immunomodulatory effects of adipose stromal vascular fraction cells promote alternative activation macrophages to repair tissue damage. Stem Cells. 2017;35:2198–2207. [DOI] [PubMed] [Google Scholar]
- 42. Ghannam S, Pene J, Moquet‐Torcy G, et al. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol. 2010;185:302–312. [DOI] [PubMed] [Google Scholar]
- 43. Maccario R, Podesta M, Moretta A, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen‐specific immune response favors the differentiation of CD4+ T‐cell subsets expressing a regulatory/suppressive phenotype. Haematologica. 2005;90:516–525. [PubMed] [Google Scholar]
- 44. Ditisheim A, Wuerzner G, Ponte B, et al. Prevalence of hypertensive phenotypes after preeclampsia: a prospective cohort study. Hypertension. 2018;71:103–109. [DOI] [PubMed] [Google Scholar]
- 45. Bonamy AK, Parikh NI, Cnattingius S, et al. Birth characteristics and subsequent risks of maternal cardiovascular disease: effects of gestational age and fetal growth. Circulation. 2011;124:2839–2846. [DOI] [PubMed] [Google Scholar]
- 46. Orabona R, Vizzardi E, Sciatti E, et al. Maternal cardiac function after HELLP syndrome: an echocardiography study. Ultrasound Obstet Gynecol. 2017;50:507–513. [DOI] [PubMed] [Google Scholar]
- 47. Seely EW, Ecker J. Chronic hypertension in pregnancy. Circulation. 2014;129:1254–1261. [DOI] [PubMed] [Google Scholar]
- 48. Chen KH, Seow KM, Chen LR. Progression of gestational hypertension to pre‐eclampsia: a cohort study of 20,103 pregnancies. Pregnancy Hypertens. 2017;10:230–237. [DOI] [PubMed] [Google Scholar]
- 49. Buchbinder A, Sibai BM, Caritis S, et al. Adverse perinatal outcomes are significantly higher in severe gestational hypertension than in mild preeclampsia. Am J Obstet Gynecol. 2002;186:66–71. [DOI] [PubMed] [Google Scholar]
- 50. Park K, Quesada O, Cook‐Wiens G, et al. Adverse pregnancy outcomes are associated with reduced coronary flow reserve in women with signs and symptoms of ischemia without obstructive coronary disease. Circulation. 2017;136:A14676. [DOI] [PMC free article] [PubMed] [Google Scholar]