

Summary
Aim
Multifactors contribute to the development of postoperative cognitive dysfunction (POCD), of which the most important mechanism is neuroinflammation. Prostaglandin E2 (PGE2) is a key neuroinflammatory molecule and could modulate hippocampal synaptic transmission and plasticity. This study was designed to investigate whether PGE2 and its receptors signaling pathway were involved in the pathophysiology of POCD.
Methods
Sixteen‐month old male C57BL/6J mice were exposed to laparotomy. Cognitive function was evaluated by fear conditioning test. The levels of PGE2 and its 4 distinct receptors (EP1‐4) were assessed by biochemical analysis. Pharmacological or genetic methods were further applied to investigate the role of the specific PGE2 receptors.
Results
Here, we found that the transcription and translation level of the EP3 receptor in hippocampus increased remarkably, but not EP1, EP2, or EP4. Immunofluorescence results showed EP3 positive cells in the hippocampal CA1 region were mainly neurons. Furthermore, pharmacological blocking or genetic suppression of EP3 could alleviate surgery‐induced hippocampus‐dependent memory deficits and rescued the expression of plasticity‐related proteins, including cAMP response element‐binding protein (CREB), activity‐regulated cytoskeletal‐associated protein (Arc), and brain‐derived neurotrophic factor (BDNF) in hippocampus.
Conclusion
This study showed that PGE2‐EP3 signaling pathway was involved in the progression of POCD and identified EP3 receptor as a promising treatment target.
Keywords: hippocampus, neuroinflammation, postoperative cognitive dysfunction, Prostaglandin E2, synaptic plasticity‐related protein
1. INTRODUCTION
Postoperative cognitive dysfunction (POCD) was defined as impairments following exposure to surgery and anesthesia in a broad spectrum of abilities referred to as cognition, including learning and memory, attention, and language comprehension.1, 2 Adverse postoperative cognitive effects have seriously reduced the quality of life, resulting in long‐term morbidity and increased mortality.3, 4, 5 According to multiple clinical studies, the prevalence of POCD ranges from 8.9% to 46.1%.6 Patients aged over 65 years are more at risk and some of them even develop dementia 3‐5 years later.7 Emerging preclinical data suggest that hippocampal‐dependent cognition is especially vulnerable to surgery‐induced memory impairment in aged animals.8, 9 However, the precise molecular mechanisms of POCD remain largely unknown, and few therapies exist to improve memory performances in this situation.
Multifactors contribute to the development of POCD. One of the most important mechanisms is neuroinflammation.10, 11 Preclinical evidence showed that surgical trauma resulted in robust immune cell infiltration, inflammatory cytokine expression, or local glia activation, causing neurotoxicity to the central nerve system.9, 12 Prostaglandin E2 (PGE2) is one of the most popular products of inflammation reaction. It is biosynthesized from Arachidonic acid (AA) by sequential actions of rate‐limiting cyclooxygenase (COX) enzymes and prostaglandin E synthases.13, 14 It is currently believed that PGE2 plays a crucial role in transferring the information received from circulating immune factors to brain parenchymal cells.15, 16, 17 A substantial amount of evidence suggests that PGE2 is upregulated in the brain and can impair memory in response to a wide variety of adverse stimuli, including injury/trauma, infection, neurodegeneration, and severe psychological stress.18, 19, 20
Preclinical studies have indicated that an increased level of PGE2 in rodent hippocampus after surgery and those selective COX‐2 inhibitors, such as meloxicam and parecoxib, ameliorate postoperative cognitive decline.21, 22 This is in concordance with clinical data that advocate that surgical trauma could upregulate PGE2 in the central nervous system and parecoxib decreases POCD incidence.23, 24 However, in the central nervous system, there exist 4 distinct PGE2 receptors (EP1‐4),25 and the exact mechanism of PGE2 meditated development of PCOD need further exploration.
In this study, we investigated PGE2 downstream pathway in the development of POCD based on an animal model of laparotomy26, 27, 28 and identified that PGE2‐EP3 pathway contributes most other than EP1, EP2, and EP4.
2. MATERIALS AND METHODS
2.1. Animals
Experimental protocols were approved by Institutional Animal Care and Use Committee of Huazhong University of Science and Technology and met guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. A total number of 116 male C57BL/6J aged mice (16‐month old) were purchased from the Experimental Animal Center of Tongji Medical College, Huazhong University of Science and Technology. Five mice were housed per cage in a temperature‐controlled holding room (22 ± 1°C) on a 12 hour light/dark cycle and given food and water ad libitum. The experimental protocol is presented in Figure S1A‐C.
2.2. Surgery
Postoperative cognitive dysfunction is a frequent complication occurring in geriatric patients after abdominal surgery. A well‐studied laparotomy model was used in this study with minor modifications26 and briefly described as following. Mice were maintained at a constant depth of anesthesia on the heated pad in a supine position with 1.5% isoflurane in 100% oxygen via a conical mask. The skin of the operation area was sanitized, and a length of 1.5 cm vertical incision, below the lower right rib, was made. The surgeon vigorously manipulated the viscera and musculature using cotton swabs moistened in saline. The intestine was then exteriorized and rubbed between the surgeon's thumb and forefinger for 30 seconds. Hemorrhage in the intestinal wall or lesions of the gut vessels should be avoided. The intestines were then placed back. The peritoneum, muscular wall, and the skin were closed with sutures, respectively. The surgical procedure lasted approximately 25 minutes. Mice were then placed on a heated pad and allowed to recover from anesthesia. For sham group, mice were anesthetized, shaved, and cleaned as described above and maintained at a constant depth of isoflurane anesthesia for the same amount of time as the surgical counterpart.
2.3. Cannulation and intracerebroventricular interventions
Mice were anesthetized with sodium pentobarbital (60 mg/kg, ip) and positioned in a stereotaxic frame (RWD Life Science Co., Ltd, Shenzhen, China). Ophthalmic ointment was applied to prevent corneal desiccation. A small craniotomy was performed with a hand‐held dental drill above the left lateral ventricle (AP‐0.4 mm, ML‐1 mm, DV‐1.6 mm) according to the mouse brain atlas of Paxinos and Watson, as previously described.29 A 26‐gauge stainless steel cannula (RWD Life Science Co., Ltd, Shenzhen, China) was then fixed to the skull with a layer of cyanoacrylate glue followed by dental cement. Mice were given a period of 1 week for postoperative recovery. L‐798,106 (an antagonist of EP3 receptors; Sigma, St.Louis, MO, USA) was first dissolved in dimethyl sulfoxide (DMSO) and then diluted in artificial cerebrospinal fluid (ACSF) to make a 1% final DMSO concentration. The effective dose of 10 nmol in 2 μL was delivered once daily for 7 days, as shown in Figure S1B, according to the previous studies.30 For the vehicle controls, the same amount of 1% DMSO in ACSF (2 μL) was used.
2.4. Virus injection
Stereotaxic surgeries were performed as described above. Double small burr holes were drilled above the target area. The lentivirus was delivered using a 10 μL Hamilton microsyringe attached with a 33‐gauge metal needle, into both left and right dorsal hippocampus (AP‐2.0 mm, ML±1.5 mm, DV‐1.5 mm). The target mRNA sequence was as follows: GGTCACTGGCTTCGTGGGCAA. Lentivirus that targeted a nonspecific sequence (TTCTCCGAACGTGTCACGT) was constructed as the controls. All of them were packaged from Shanghai GeneChem Co., Ltd. To suppress EP3 expression, mice were bilaterally injected with EP3 short‐hairpin RNA (shRNA) lentivirus (2 × 108 TU/mL) at a volume of 1 UL on each side. Equal amount of scrambled sequence lentivirus was delivered as control. The injection volume and flow rate (1 UL at 0.1 UL/min) were controlled by an injection pump, and the needle was maintained in place for an additional 5 minutes before slowly retracting it. After the injection, mice were given 1‐week period before commencing the experiment to allow sufficient infection in the hippocampus and to allow recovery.
2.5. Open field test
All of the behavioral experiments were conducted between 8:00 am to 6:00 pm in the light phase and recorded by an experimenter who was blinded to the treatment. The exploratory locomotor activities of the mice were evaluated using the open field test 6 days after the laparotomy. Each mouse was placed in the center of a white opaque plastic chamber (50 × 50 × 40 cm) and allowed to freely explore for 5 minutes. Activity in the center and periphery of the field was measured using an automated video‐tracking system (AVTAS v3.3; AniLab Software and Instruments Co., Ltd., Ningbo, China). Between each test, the surface of the arena was cleaned with 75% alcohol to avoid the presence of olfactory cues.
2.6. Fear conditioning test
The fear conditioning paradigm was conducted in standard operant chambers (AniLab Software & Instruments Co., Ltd., Ningbo, China) to study memory performance as previously described.31 Mice were habituated to handling in 5 minutes epochs every day, for 2 days prior to training. Two hours after the open field test, mice were placed in the conditioning chamber. During training, each mouse was typically allowed to freely explore the chamber for 120 seconds. Thereafter, the auditory tone (3.6 kHz, 70 dB) was presented for 20 seconds, and an electric foot shock (2 seconds, 0.5 mA) was given to the mice during the last 2 seconds of the sound. Each of these sequences was presented 3 times, separated by 60 seconds. Following the final foot shock, the mice were left undisturbed in the chambers for an additional 60 seconds. Twenty‐four hours after the training session, contextual fear conditioning test was assessed by placing the mouse in the original context for 5 minutes. Two hours later, auditory‐cued fear conditioning test was evaluated by placing the mouse in a novel chamber. Freezing was measured for 2.5 minutes in this new cage, and then the training tone was sounded for 2.5 minutes, during which conditioned freezing was measured. Freezing behavior was defined as the absence of all visible movement except for respiration, and it was recorded and expressed as the percentage of the observation period using an automated video‐tracking system. The apparatus was cleaned with 75% alcohol between each testing session to avoid the interference of olfactory cues. The experimental paradigm is illustrated in Figure S1D‐F.
2.7. Quantitative real‐time polymerase chain reaction (qRT‐PCR)
Total RNA was extracted from hippocampus by use of Trizol reagent (Aidlab, China) according to the protocol of the manufacturer. RNA samples were quantified by use of a spectrophotometer (Eppendorf, Germany) and then synthesized to cDNA using reverse transcription. Quantitative real‐time PCR protocol was performed on the ABI7900 real‐time detection system (Illumina, USA) by use of SYBR Green Master Mix (TAKARA, Japan) to detect amplification. The PCR reaction conditions were carried out in accordance with the manufacturer's protocol: Incubation was set at 50°C for 2 minutes and then at 95°C for 10 minutes, followed by 40 cycles at 95°C for 30 seconds and 60°C for 30 seconds. The sequences of the specific primers for qRT‐PCR were designed based on the previously reported sequence of mouse genes (EP1, EP2, EP3, EP4, and GAPDH) by biotechnology company (Qingke, China) and detailed in Table S1^. For standardization, the housekeeping gene GAPDH was used as an internal control. Relative changes in gene expression were calculated by the use of the comparative (2−ΔΔCT) method.
^[Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH” in the sentence.]
2.8. Enzyme‐linked immunosorbent assay (ELISA)
Hippocampus samples were homogenized and then centrifuged at 10 000 g for 15 minutes at 4°C. The supernatants were collected, and levels of PGE2 were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA). The procedures were performed according to the manufacturer's instructions, and the concentration of PGE2 was presented as pg/100 μg of total hippocampal protein.
2.9. Western blot
Hippocampal samples were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer containing freshly added phosphatase and protease inhibitors. Protein concentrations were determined using the BCA Protein Assay Kit (Boster, Wuhan, China). Equal amounts of total protein (30‐50 μg/lane) were separated by SDS‐PAGE gels and subsequently transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) by electroblotting. The membrane was blocked in 5% skim milk in TBST (0.1%) for 2 hours at room temperature and then incubated with primary antibody overnight at 4°C. Antibodies used include rabbit anti‐EP1 antibody (1:200, #101740, Cayman Chemical, Ann Arbor, MI); rabbit anti‐EP2 antibody (1:200, #101750, Cayman Chemical, Ann Arbor, MI); rabbit anti‐EP3 antibody (1:200, #101760, Cayman Chemical, Ann Arbor, MI); rabbit anti‐EP4antibody (1:200, #101775, Cayman Chemical, Ann Arbor, MI); rabbit anti‐cAMP response element‐binding protein (CREB) antibody (1:1000, #9197, Cell Signaling Technology, MA, USA); rabbit anti‐pCREB antibody (1:1000, #9198, Cell Signaling Technology, MA, USA); rabbit anti‐brain‐derived neurotrophic factor (BDNF) antibody (1:1000, #ab108319, Abcam, Cambridge, UK); rabbit anti‐activity‐regulated cytoskeletal‐associated protein (Arc) antibody (1:1000, #ab118929, Abcam, Cambridge, UK); mouse anti‐GAPDH antibody (1:500, #BM1623, Boster, Wuhan, China). After incubation with horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit and goat anti‐mouse secondary antibodies (1:2000, Boster, Wuhan, China) for 2 hours at room temperature, bands were visualized using an enhanced chemiluminescence (ECL) detection reagents (Thermo Scientific, Rockford, IL, USA) and exposed to an imaging film. The mean intensities of selected areas and the areas of these images were calculated using Image J software and normalized to values of GAPDH.
[Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH” in the sentence.]
2.10. Hematoxylin‐Eosin (HE) staining
The cannula position was examined by HE staining. Mice were anesthetized with pentobarbital sodium (60 mg/kg, ip) and transcardially perfused with phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. The brains were removed, postfixed in 4% PFA and then dehydrated in 30% sucrose in PBS overnight at 4°C. Coronal 5 μm‐thick brain sections were cut with a Leica cryostat (CM1900, Wiesbaden, Germany). Every section underwent 8 minutes immersion in Harris's hematoxylin solution. Then, they were differentiated in 1% acid alcohol for 30 seconds. After rinsing using 95% alcohol, brain sections were counterstained in eosin‐phloxine solution for 1 minutes and dehydrated in graded series of ethanol. After being cleared in xylene solutions, these brain slices were mounted on coverslips. Images were examined under bright field illumination using microscope (DM2500, Leica, Mannheim, Germany).
2.11. Immunohistochemistry staining and image analysis
The mice brains were collected as described above. Coronal 30 μm‐thick brain sections were serially cut with a Leica cryostat. These brain sections underwent three 10‐minutes washes in PBS and permeabilized using PBST (1% Triton X‐100 in PBS) for 40 minutes. After washing, brain slices were incubated with blocking solution (5% bovine serum albumin) for 2 hours. For double‐labeling immunofluorescence, brain sections were incubated with a mixture of rabbit anti‐EP3 antibody (1:100, #101760, Cayman Chemical, Ann Arbor, MI), and specific biomarkers as mouse anti‐GFAP antibody (1:400, #MAB360, Merck Millipore, Darmstadt, Germany), goat anti‐Iba‐1 antibody (1:200, #ab5076, Abcam, Cambridge, UK), and mouse anti‐NeuN antibody (1:200, #MAB377, Merck Millipore, Darmstadt, Germany), respectively, for 48 hours at 4°C. The next day, slices were washed with PBS 3 times and incubated with a mixture of Alexa Fluor 594‐ and 488‐conjugated secondary antibodies for 2 hours at room temperature followed by counterstaining with DAPI (Invitrogen). Thereafter, these brain slices were mounted on coverslips. Fluorescent images were captured by a scanning confocal microscope (C2, Nikon, Japan).
2.12. Statistical analyses
Statistical analyses were conducted using SPSS software (IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp.). Unpaired Student's t‐test was used for comparisons between 2 groups. For multigroup comparison, two‐way analysis of variance (ANOVA) was used, followed by Bonferroni post hoc tests. Surgery and time, or surgery and drug, were considered as 2 independent factors. Data in this study are represented as mean ± standard error of the mean (SEM). Statistical significance was set at P < 0.05.
3. RESULTS
3.1. Laparotomy‐induced impairment of contextual fear memory and alterations hippocampal PGE2 concentration and EP1‐4 mRNA levels in aged mice
Locomotor activities and anxiety performance were tested using an open field test at 6 days after surgery. No significant differences were found in the path length, velocity, and percent of time spent in the center of the field between sham and laparotomy mice (Figure 1A‐C). Fear conditioning paradigm was used to assess long‐term memory performance, and there were also no significant differences in pretone and tone freezing time between 2 groups (Figure 1D). This demonstrates an intact cued fear memory in response to tone, which is hippocampus independent.32, 33 However, the freezing time to context was significantly lower in the laparotomy group than that of the sham group (25.81% ± 3.12% vs 52.97% ± 3.72%; t = 5.588, P < 0.001; Figure 1E), as analyzed with unpaired Student's t‐test. Together, these experiments suggest that the laparotomy affected only the hippocampus‐dependent task in aged mice.
Figure 1.
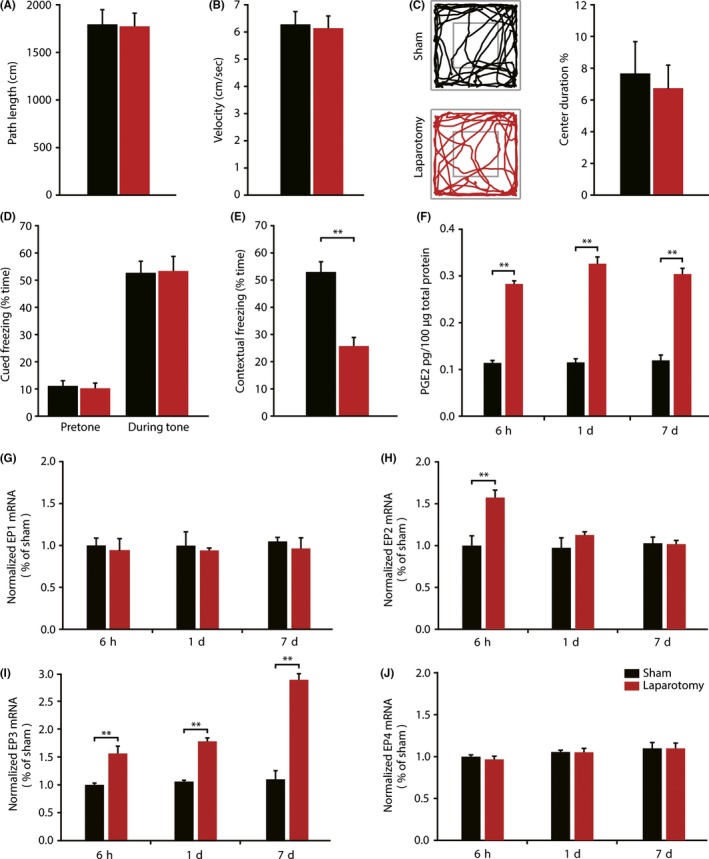
Laparotomy induced impairment of contextual fear memory and alterations of hippocampal PGE2 concentration and EP1‐4 mRNA Levels. (A‐C) In the open field test, sham and laparotomy mice had similar path length, velocity, and percent of time spent in the center of the field. Representative exploration traces are presented. (n = 10) (D‐E) There were no significant differences in pretone and tone freezing time in the auditory‐cued fear conditioning test between 2 groups. When exposed to context, mice undergoing laparotomy demonstrated a significant reduction in freezing time compared with sham mice. (n = 10) (F) Laparotomy induced a marked upregulation of PGE2 expression at 6 h, 1 day, and 7 day postsurgery, measured by ELISA. (n = 3) (G‐J) Quantitative real‐time PCR results showed that laparotomy‐induced prolonged elevation of EP3 mRNA levels at 6 h, 1 day, and 7 days, but not EP1 and EP4. EP2 mRNA levels only increased at 6 h after surgery but not at 1 day and 7 days.(n = 3) Data presented as mean ± SEM. **P < 0.01
Time course of changes in the hippocampal PGE2 content after surgery was assessed by ELISA. A two‐way ANOVA produced a significant effect of surgery (F (2,12) = 507.036; P < 0.001) but not of time (F (2,12) = 2.404; P = 0.132), and there was no interaction (F (2,12) = 2.150; P = 0.159; Figure 1F). Laparotomy‐induced significant increases of PGE2 expression at 6 hours (P < 0.001) and constant increase at 1 day (P < 0.001) and 7 days (P < 0.001) after surgery, compared to mice receiving the same anesthesia without surgery. Sham group mice produced no significant changes of PGE2 at any time point, indicating that anesthesia alone does not interfere with PGE2 synthesis.
Then, we examined the hippocampal transcription level of EP1‐4 after laparotomy at 6 hours, 1 day, and 7 days by qRT‐PCR. There was a statistically significant interaction between the effects of surgery type and time course on EP2 mRNA levels (F (2,12) = 6.086; P = 0.015; Figure 1H). Laparotomy induced a rapid and transient increase in EP2 mRNA levels at 6 hours (P < 0.001) and back to levels comparable with sham group at 1 d postsurgery (P = 0.230). The effect of surgery (F (2,12) = 162.298; P < 0.001), time (F (2,12) = 29.217; P < 0.001), and surgery × time interaction (F (2,12) = 22.865; P < 0.001) on EP3 levels was compared by two‐way ANOVA. Post hoc tests confirmed that the expression of EP3 mRNA in the hippocampus was substantially increased at all‐time points (6 hours: P = 0.002; 1 day: P < 0.001;7 day: P < 0.001; Figure 1I). However, the levels of EP1 and EP4 mRNA were not affected at any time point (Figure 1G, J). These data indicate that laparotomy exclusively induced persistent elevation of EP3 mRNA levels.
3.2. Laparotomy elevated EP3 protein expression and cell‐type specificity of EP3 in mice hippocampus
The protein expression of all 4 PGE2 receptors was further detected by Western blot analysis at 7 days postsurgery. The hippocampal protein levels of EP3 were robustly increased at 7 days postsurgery (t = −7.252, P = 0.002; Figure 2C) compared with sham group, which is consistent with qRT‐PCR results. However, there were no significant differences in the expression of any other PGE2 receptors, when compared with sham group (Figure 2A, B, D). Therefore, we next focused on EP3 receptor. Double immunofluorescence staining showed that EP3 positive cells in the hippocampal CA1 region coexpressed the neuron marker NeuN (Figure 2E) but did not colocalize with the astrocyte marker GFAP or the microglia marker Iba‐1 at 7 day postsurgery.
Figure 2.
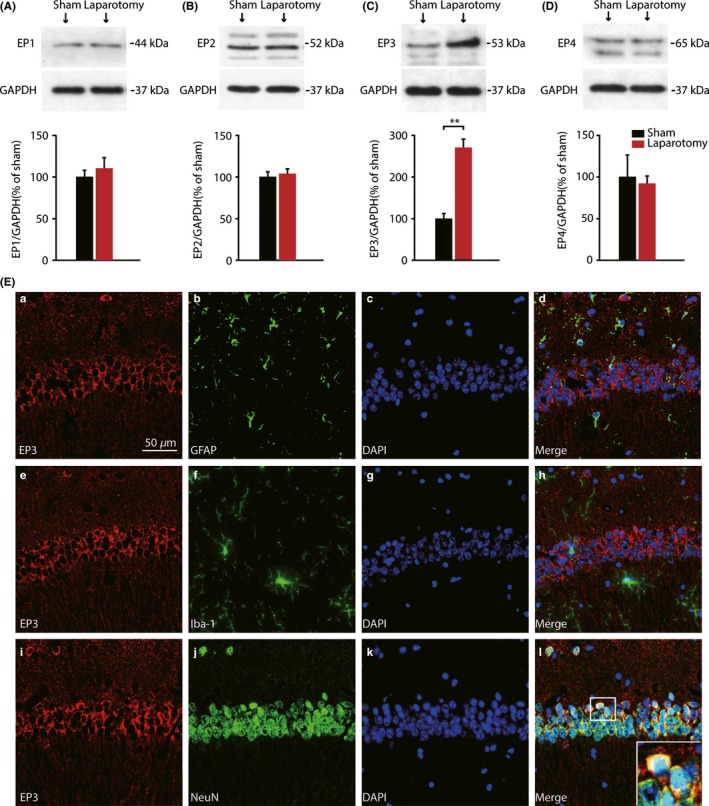
Protein levels of EP1‐4 in the hippocampus of postsurgery mice and cellular localization of EP3 expression. (A‐D) Western blot analysis demonstrated that laparotomy exclusively elevated hippocampal EP3 receptors expression at 7 days postsurgery, but not EP1, EP2, and EP3. (n = 3) (E) Immunofluorescence double staining showed that anti‐EP3 receptor staining colocalized with the neuron marker NeuN (i‐l), but not astrocyte marker GFAP (a‐d), nor microglia marker Iba‐1 (e‐h) in the CA1 of the hippocampus 7 days after laparotomy. Nuclei were stained with DAPI. (Scale bars: 50 μm). Data presented as mean ± SEM. **P < 0.01 [Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH” in figure 2.]
3.3. Pharmacological blocking EP3 receptor by L‐798,106 improved hippocampus‐dependent memory performance after laparotomy
To investigate the role of hippocampal EP3 receptors in the progression of POCD, the EP3 receptors antagonist L‐798,106 or vehicle was injected into the lateral ventricle of the mice. HE staining was used to validate the cannula position (Figure 3F) 7 days after ICV cannulation. No significant differences were found in the path length, velocity, and percent of time spent in the center of the field among 4 groups (Figure 3A‐C). In the auditory‐cued fear conditioning test, there were no significant differences in pretone and tone freezing time among 4 groups (Figure 3D). In the contextual fear conditioning test, there was an interaction effect (F (1,36) = 12.019, P = 0.001; Figure 3E). Post hoc analysis showed that the laparotomy mice treated with vehicle demonstrated a significant reduction in contextual freezing time compared with sham mice treated with vehicle (P < 0.001), which was prevented by the administration of L‐798,106 (P < 0.001). Thus, we can conclude that EP3 receptors regulate mechanisms that are essential for hippocampus‐dependent cognitive impairments induced by laparotomy.
Figure 3.
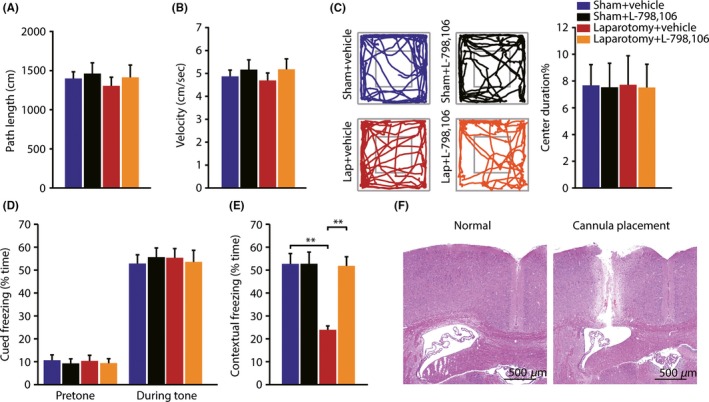
Pharmacological blocking EP3 receptor by L798,106 improved contextual fear memory in mice after laparotomy. (A‐C) In the open field test, no significant differences were found in path length, velocity, and the percent of time spent in the center of the field, among 4 groups. Representative exploration traces are presented. (n = 10) (D‐E) In the auditory‐cued fear conditioning test, there were no significant differences in pretone and tone freezing time among 4 groups. In the contextual fear conditioning test, the decline in freezing time to context was completely blocked by the administration of L‐798,106. (n = 10) (F) The cannula position was examined by HE staining, and needle track was located at the expected position (Scale bars: 500 μm). Data presented as mean ± SEM. **P < 0.01
3.4. Pharmacological blocking EP3 receptor by L798,106 rescued the expression of synaptic plasticity‐related proteins in mice hippocampus
To gain insight into molecular pathways which are coupled to the PGE2‐EP3 signaling cascades in surgery‐induced memory deficits, we examined the expression of some representative molecules which have been found to be closely related to hippocampal synaptic plasticity and memory formation, including CREB, Arc, and BDNF.34, 35, 36 Quantitative analysis of Western blot results showed that there was an interaction effect (pCREB: F(1,8) = 7.214, P = 0.028; Arc: F(1,8) = 17.811, P = 0.003; BDNF: F(1,8) = 10.414 P = 0.012). Post hoc tests revealed that there was a marked reduction in pCREB phosphorylation at Ser133 (P < 0.001, Figure 4B) in laparotomy + vehicle group, compared with sham + vehicle group, without a change in expression of total CREB. Additionally, the expression of Arc and BDNF, which were CREB‐dependent, also significantly decreased in laparotomy + vehicle group, compared with sham + vehicle group (Arc: P = 0.001; BDNF: P = 0.011, Figure 4C, D). But the decreased expression of pCREB, Arc, and BDNF could have been completely rescued by L‐798,106 administration (pCREB: P = 0.002; Arc: P = 0.005; BDNF: P < 0.001).
Figure 4.
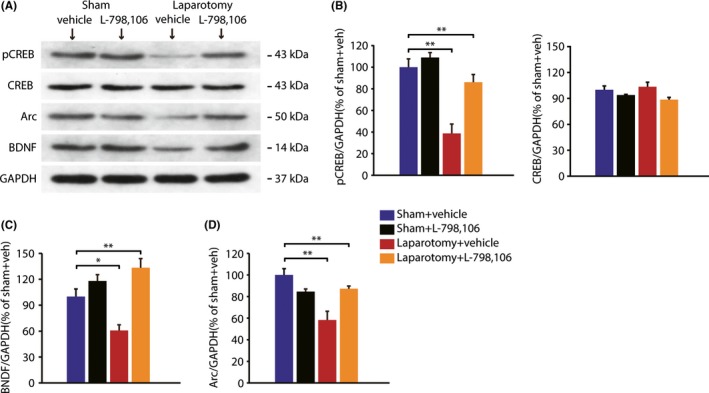
Pharmacological blocking EP3 receptor by L798,106 rescued the expression of synaptic plasticity‐related protein in mice hippocampus after laparotomy. (A) The visualization of protein bands of pCREB, CREB, Arc, and BDNF. (B‐D) Quantitative analysis showed that there was a marked reduction in pCREB Ser133, Arc, and BDNF, in laparotomy + vehicle group, compared with sham + vehicle group, which were completely blocked by L‐798,106.(n = 3) Data presented as mean ± SEM. *P < 0.05, **P < 0.01 [Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH”; and “BNDF” was changed to “BDNF”in figure 4.]
3.5. Efficient and specific knockdown expression of hippocampal EP3 receptors using a lentiviral vector
To further determine the role of hippocampal EP3 receptors, we constructed the recombinant lentivirus containing the sequence for a short‐hairpin RNA specific for EP3 (LV‐shEP3). Lentivirus encoding a scrambled short‐hairpin RNA sequence (LV‐shSCR) was chosen as the negative control. The lentivirus (1 UL/site) was microinjected into the CA1 region (Figure 5A). The brain tissues were analyzed 7 days after laparotomy. As traced by enhanced green fluorescent protein (Figure 5B), lentivirus was found to result in CA1‐specific expression, which was previously established to play a critical role in the fear conditioning memory test.37, 38 The protein levels of EP1‐4 were quantified by Western blot. Quantitative analysis showed that EP3 protein levels were remarkably reduced by LV‐shEP3, in comparison with the negative control lentivirus (t = 7.393, P = 0.002, Figure 5E). However, the protein levels of EP1, EP2, and EP4 remained unchanged (Figure 5C, D, F).
Figure 5.
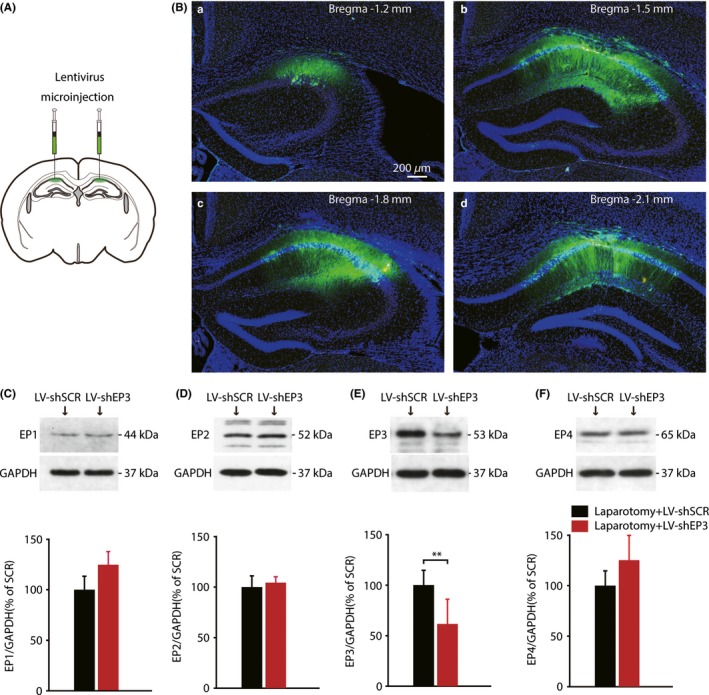
Efficient and specific knockdown expression of hippocampal EP3 protein based on a viral vector. (A) Bilateral in vivo microinjection of lentivirus into the CA1 region. (B) Schematic coronal sections from mice showed the extent of viral transfection, which was determined by enhanced green fluorescent protein (EGFP). The anteroposterior stereotaxic coordinates for the sections are shown (in mm). (Scale bars: 200 μm) (C‐F) Western blot analysis showed that the LV‐shEP3 vector specifically reduced hippocampal EP3 levels; whereas the protein levels of other PGE2 receptor remained unchanged. (n = 3) Data presented as mean ± SEM. **P < 0.01 [Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH” in figure 5.]
3.6. Selective knockdown of EP3 receptors ameliorated the detrimental effects of laparotomy
Next, we investigated the effect of downregulation of EP3 expression in the development of POCD. There were no significant differences in the path length, velocity, or percent of time spent in the center of the field between 2 groups (Figure 6A‐C). In the fear conditioning test, no significant differences were found in the pretone and tone freezing time between 2 groups (Figure 6D); however, contextual freezing time was significantly increased in laparotomy mice injected with LV‐shEP3, compared with mice injected with LV‐shSCR (t = −6.342, P < 0.001; Figure 6E). Western blot analysis also showed a significant increase of pCREB, Arc, and BDNF expression in hippocampus injected with LV‐shEP3, compared with the negative control lentivirus (pCREB: t = −19.749, P < 0.001, Arc: t = −8.603, P = 0.001;BDNF: t = −3.709, P = 0.021, Figure 6 G‐I). These results demonstrate that selective knockdown of EP3 receptors in hippocampal CA1 could ameliorate the detrimental effects of surgery.
Figure 6.
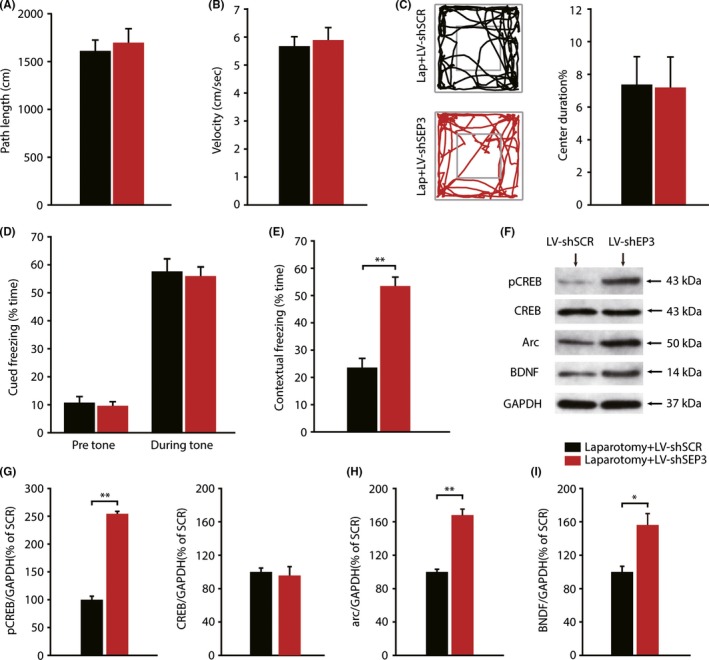
Specific knockdown of EP3 receptors ameliorated the cognitive function in mice after laparotomy. (A‐C) The open field test was comparable between the 2 group. Representative exploration traces are presented. (n = 10) (D‐E) There were no significant differences in pretone and tone freezing time between the 2 groups. Contextual freezing time was significantly increased by knocking down of EP3 expression. (n = 10) (F) The visualization of protein bands of pCREB, CREB, Arc, and BDNF. (G‐I) Quantitative analysis showed that hippocampal expression of pCREB, Arc, and BDNF in LV‐shEP3 injection mice was significantly higher than that in negative control lentivirus injection mice.(n = 3) Data presented as mean ± SEM. *P < 0.05, **P < 0.01 [Correction added on 03 April 2018, after first online publication: The word “GADPH” was changed to “GAPDH”; and “BNDF” was changed to “BDNF” in figure 6.]
4. DISCUSSION
POCD affects a significant number of patients, especially geriatrics,3, 11 resulting in prolonged hospitalization and delayed recovery from illness,39 but the underlying neurobiological basis has remained elusive. In the present study, we have identified PGE2‐EP3 signaling pathway in mediating development of POCD following laparotomy in aged mice. Pharmacological and genetic suppression of EP3 reversed surgery‐induced memory deficits. The mechanism could be at least partially due to rescuing the expression of synaptic plasticity‐related molecular impairments, including pCREB, Arc, and BDNF (Figure S2).
The present research was based on an established animal model using isoflurane anesthesia and exploratory laparotomy.26, 27 Our data showed that surgery trauma could induce contextual fear memory impairment in aged mice which was hippocampus‐dependent, while leaving an intact cued fear memory, consistent with previous reports.28, 40 Accumulating evidence revealed that surgical trauma‐induced systemic inflammation could lead to neuroinflammation, featured as elevated levels of proinflammatory molecules including cytokines and prostaglandins in the central nervous system.23, 40 During systemic inflammation, prostaglandins released from perivascular macrophages and brain endothelial cells can diffuse directly across the BBB and into the brain parenchyma, due to their hydrophilic characteristic.16, 41 In the current study, laparotomy produced a prolonged elevation of PGE2 levels up to 7 days following surgery, which was similar but somewhat different from a previous study by Peng et al.22 We infer that the differences in animal species and surgical model may explain the discrepancy in experiment results.
Both preclinical and clinical studies suggest that selective COX‐2 inhibitors such as meloxicam and parecoxib, ameliorate postoperative cognitive decline.21, 22, 24 Moreover, selective COX‐2 inhibitor has been shown to improve memory function by downregulation of PGE2 levels in Alzheimer's diseases (AD) which have similar clinical symptoms of POCD, and these beneficial effects on memory do not depend on lowered levels of cytokines such as IL‐1 and TNF‐α.42 However, chronic use of COX‐2 inhibitors also results in adverse cardiovascular side effects, which could impose tighter restrictions on the future use of COX‐2 inhibitors,43 particularly for the elderly with POCD or AD. Consequently, future studies should aim at local and specific targets rather than the nonselective block of the entire COX‐2 signaling cascade. In the central nervous system, PGE2 can bind 4 subtypes of EP receptors, designated EP1‐4, but each evokes cellular response via distinct signaling cascades.
Of all 4 subtypes of EP receptors, EP3 is most abundant in the brain and has the highest affinity for PGE2.44, 45 EP3 signaling in response to PGE2 is primarily coupled to Gi protein, which reduces cAMP formation, and therefore termed the “inhibitory” receptor.20, 46 Previous studies have confirmed PGE2‐EP3 signaling axis in modulating multiple forms of brain disorders, including ischemic and hemorrhagic stroke, and Alzheimer's diseases, using EP3 knockout mice.47, 48, 49 In this study, we found that it is the EP3 receptor that dramatically upregulated at both transcription and translation levels after mice received laparotomy, especially in neurons, but not other EP receptors (EP1, EP2, and EP4), which may imply the direct influence of EP3 receptors on neuronal function. Although the EP2 mRNA level increased at 6 hours after surgery, it failed to do so at 1 day and 7 days. Furthermore, in protein level, translation level of EP2 receptor did not change either. So here we concluded that the hippocampal PEG2 mediated the development of hippocampus‐dependent memory deficient after surgery mainly through EP3 receptor pathway. Furthermore, blocking EP3 receptor by ICV administration of EP3 specific receptor antagonist or downregulating the expression of EP3 receptor in hippocampus, the laparotomy‐induced hippocampus‐dependent memory deficient could be rescued, which also strongly supports that PEG2‐EP3 signal pathway contributes to the development of POCD.
In our study, we also observed that CREB, Arc, and BDNF dramatically decreased after laparotomy in mice. That means the synaptic plasticity mechanism is possibly involved in the development of POCD. Further studies showed that the decreased levels of pCREB, Arc, and BDNF could be rescued by L‐798,106 (highly selective EP3 receptors antagonist) treatment. Additionally, EP3 shRNA lentivirus was used to further confirm this hypothesis. A possible mechanism is that EP3 induced a decrease in intracellular calcium and cAMP levels, and downregulated phosphorylation of CREB together with its downstream products Arc and BDNF, through the inhibition of Ras/MAPK of PKA pathways.20, 50 Molecules, such as CREB, BDNF, and Arc, participate in the formation of long‐term potentiation (LTP),51, 52, 53 which is most widely considered an electrophysiological phenomenon in synaptic plasticity that underlies learning and memory. According to electrophysiological studies on acute brain slices, EP3 receptors are critical downstream mediators of PGE2‐induced LTP impairment.54, 55 All these facts combine to support a strong association of EP3 receptors in the progression of POCD.
There are several limitations in the current study. First, although our data showed laparotomy‐induced hippocampus‐dependent memory deficit involved in the progression of POCD, other regions of the brain including prefrontal, insular, and thalamus, which were implicated in the cognitive functions,56 may not be excluded in the neurobiology of POCD. Further studies on these brain regions and the paralleling of the behavior paradigm should be evaluated. In addition, the function of 1 brain area cannot be isolated. Given that the hippocampus receives numerous afferent fibers from the entorhinal cortex (EC), thalamus, and amygdala,57, 58, 59 whether fluctuations in hippocampal function in POCD attributes to the adjacent brain regions still remains unknown. Recent advances in optogenetic approaches offer us a new tool for identifying the brain regions and the underlying behaviors.60 Further studies on the specific neural circuits involved in the pathogenesis of POCD are urgently needed.
In summary, our findings indicate that surgical trauma disrupts PGE2‐EP3 signaling, by reducing expression levels of hippocampal synaptic plasticity‐related proteins, which in turn compromise hippocampus‐dependent learning and memory. Unraveling molecular mechanisms responsible for surgery‐induced cognitive dysfunction may stimulate the development of prophylactic and therapeutic management for POCD. Our research may provide novel insights into mechanisms of POCD, and result in the identification of EP3 receptors that could be targeted as promising treatment strategies.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This research was financed by a grant from the National Natural Science Foundation of China (No.81571053, No.81371250 to Y.T., No.81670240 to H.X and No. 81771191 to F.G.).
Xiao J‐Y, Xiong B‐R, Zhang W, et al. PGE2‐EP3 signaling exacerbates hippocampus‐dependent cognitive impairment after laparotomy by reducing expression levels of hippocampal synaptic plasticity‐related proteins in aged mice. CNS Neurosci Ther. 2018;24:917–929. 10.1111/cns.12832
Contributor Information
Xue‐Bi Tian, Email: tianxuebi@gmail.com.
Yu‐Ke Tian, Email: yktian@tjh.tjmu.edu.cn.
REFERENCES
- 1. Newman S, Stygall J, Hirani S, Shaefi S, Maze M. Postoperative cognitive dysfunction after noncardiac surgery: a systematic review. Anesthesiology. 2007;106:572‐590. [DOI] [PubMed] [Google Scholar]
- 2. Deiner S, Silverstein JH. Postoperative delirium and cognitive dysfunction. Br J Anaesth. 2009;103(Suppl 1):i41‐i46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinmetz J, Christensen KB, Lund T, Lohse N, Rasmussen LS, ISPOCD Group . Long‐term consequences of postoperative cognitive dysfunction. Anesthesiology. 2009;110: 548‐555. [DOI] [PubMed] [Google Scholar]
- 4. Monk TG, Weldon BC, Garvan CW. Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology. 2008;108:18‐30. [DOI] [PubMed] [Google Scholar]
- 5. Moller JT, Cluitmans P, Rasmussen LS. Long‐term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post‐Operative Cognitive Dysfunction. Lancet. 1998;351:857‐861. [DOI] [PubMed] [Google Scholar]
- 6. Androsova G, Krause R, Winterer G, Schneider R. Biomarkers of postoperative delirium and cognitive dysfunction. Front Aging Neurosci. 2015;7:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kapila AK, Watts HR, Wang T, Ma D. The impact of surgery and anesthesia on post‐operative cognitive decline and Alzheimer's disease development: biomarkers and preventive strategies. J Alzheimers Dis. 2014;41:1‐13. [DOI] [PubMed] [Google Scholar]
- 8. Terrando N, Eriksson LI, Ryu JK. Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol. 2011;70:986‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hovens IB, Schoemaker RG, van der Zee EA, Absalom AR, Heineman E, van Leeuwen BL. Postoperative cognitive dysfunction: Involvement of neuroinflammation and neuronal functioning. Brain Behav Immun. 2014;38:202‐210. [DOI] [PubMed] [Google Scholar]
- 10. Skvarc DR, Berk M, Byrne LK. Post‐operative cognitive dysfunction: an exploration of the inflammatory hypothesis and novel therapies. Neurosci Biobehav Rev. 2017;84:116‐133. [DOI] [PubMed] [Google Scholar]
- 11. Terrando N, Brzezinski M, Degos V. Perioperative cognitive decline in the aging population. Mayo Clin Proc. 2011;86:885‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wan Y, Xu J, Ma D, Zeng Y, Cibelli M, Maze M. Postoperative impairment of cognitive function in rats: a possible role for cytokine‐mediated inflammation in the hippocampus. Anesthesiology. 2007;106:436‐443. [DOI] [PubMed] [Google Scholar]
- 13. Markovic T, Jakopin Z, Dolenc MS, Mlinaric‐Rascan I. Structural features of subtype‐selective EP receptor modulators. Drug Discov Today. 2017;22:57‐71. [DOI] [PubMed] [Google Scholar]
- 14. Rael E. Unraveling the complexity of leukotriene and prostaglandin inflammatory signaling. J Allergy Clin Immunol. 2016;137:299‐300. [DOI] [PubMed] [Google Scholar]
- 15. Engblom D, Ek M, Saha S, Ericsson‐Dahlstrand A, Jakobsson PJ, Blomqvist A. Prostaglandins as inflammatory messengers across the blood‐brain barrier. J Mol Med (Berl). 2002;80:5‐15. [DOI] [PubMed] [Google Scholar]
- 16. Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson‐Dahlstrand A. Inflammatory response: pathway across the blood‐brain barrier. Nature. 2001;410:430‐431. [DOI] [PubMed] [Google Scholar]
- 17. Quan N. Immune‐to‐brain signaling: how important are the blood‐brain barrier‐independent pathways? Mol Neurobiol. 2008;37:142‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181‐213. [DOI] [PubMed] [Google Scholar]
- 19. Furuyashiki T, Narumiya S. Stress responses: the contribution of prostaglandin E(2) and its receptors. Nat Rev Endocrinol. 2011;7:163‐175. [DOI] [PubMed] [Google Scholar]
- 20. Hein AM, O'Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamer AR, Galoyan SM, Haile M. Meloxicam improves object recognition memory and modulates glial activation after splenectomy in mice. Eur J Anaesthesiol. 2012;29:332‐337. [DOI] [PubMed] [Google Scholar]
- 22. Peng M, Wang YL, Wang FF, Chen C, Wang CY. The cyclooxygenase‐2 inhibitor parecoxib inhibits surgery‐induced proinflammatory cytokine expression in the hippocampus in aged rats. J Surg Res. 2012;178:e1‐e8. [DOI] [PubMed] [Google Scholar]
- 23. Buvanendran A, Kroin JS, Berger RA. Upregulation of prostaglandin E2 and interleukins in the central nervous system and peripheral tissue during and after surgery in humans. Anesthesiology. 2006;104:403‐410. [DOI] [PubMed] [Google Scholar]
- 24. Zhu YZ, Yao R, Zhang Z, Xu H, Wang LW. Parecoxib prevents early postoperative cognitive dysfunction in elderly patients undergoing total knee arthroplasty: a double‐blind, randomized clinical consort study. Medicine (Baltimore). 2016;95:e4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661‐690. [DOI] [PubMed] [Google Scholar]
- 26. Barrientos RM, Hein AM, Frank MG, Watkins LR, Maier SF. Intracisternal interleukin‐1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J Neurosci. 2012;32:14641‐14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu LL, Luo D, Zhang H. Nox‐2‐mediated phenotype loss of hippocampal parvalbumin interneurons might contribute to postoperative cognitive decline in aging mice. Front Aging Neurosci. 2016;8:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qiu LL, Ji MH, Zhang H. NADPH oxidase 2‐derived reactive oxygen species in the hippocampus might contribute to microglial activation in postoperative cognitive dysfunction in aged mice. Brain Behav Immun. 2016;51:109‐118. [DOI] [PubMed] [Google Scholar]
- 29. Wang XD, Su YA, Wagner KV. Nectin‐3 links CRHR1 signaling to stress‐induced memory deficits and spine loss. Nat Neurosci. 2013;16:706‐713. [DOI] [PubMed] [Google Scholar]
- 30. Zhang ZH, Yu Y, Wei SG, Nakamura Y, Nakamura K, Felder RB. EP(3) receptors mediate PGE(2)‐induced hypothalamic paraventricular nucleus excitation and sympathetic activation. Am J Physiol Heart Circ Physiol. 2011;301:H1559‐H1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang DS, Zurek AA, Lecker I. Memory deficits induced by inflammation are regulated by alpha5‐subunit‐containing GABAA receptors. Cell Rep. 2012;2:488‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844‐852. [DOI] [PubMed] [Google Scholar]
- 33. Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207‐234. [DOI] [PubMed] [Google Scholar]
- 34. Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533‐535. [DOI] [PubMed] [Google Scholar]
- 36. Viola H, Furman M, Izquierdo LA. Phosphorylated cAMP response element‐binding protein as a molecular marker of memory processing in rat hippocampus: effect of novelty. J Neurosci. 2000;20:RC112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee SH, Choi JH, Lee N. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253‐1256. [DOI] [PubMed] [Google Scholar]
- 38. Seidenbecher T, Laxmi TR, Stork O, Pape HC. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science. 2003;301:846‐850. [DOI] [PubMed] [Google Scholar]
- 39. Hudetz JA, Iqbal Z, Gandhi SD. Postoperative cognitive dysfunction in older patients with a history of alcohol abuse. Anesthesiology. 2007;106:423‐430. [DOI] [PubMed] [Google Scholar]
- 40. Fidalgo AR. Experimental insights into age‐exacerbated cognitive dysfunction after peripheral surgery. Aging Cell. 2013;12:523‐524. [DOI] [PubMed] [Google Scholar]
- 41. Poon DC, Ho YS, Chiu K, Wong HL, Chang RC. Sickness: From the focus on cytokines, prostaglandins, and complement factors to the perspectives of neurons. Neurosci Biobehav Rev. 2015;57:30‐45. [DOI] [PubMed] [Google Scholar]
- 42. Kotilinek LA, Westerman MA, Wang Q. Cyclooxygenase‐2 inhibition improves amyloid‐beta‐mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ganesh T. Prostanoid receptor EP2 as a therapeutic target. J Med Chem. 2014;57:4454‐4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu P, Genc A, Zhang X, Zhang J, Bazan NG, Chen C. Heterogeneous expression and regulation of hippocampal prostaglandin E2 receptors. J Neurosci Res. 2005;81:817‐826. [DOI] [PubMed] [Google Scholar]
- 45. Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Comp Neurol. 2000;421:543‐569. [DOI] [PubMed] [Google Scholar]
- 46. Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;12:425‐434. [DOI] [PubMed] [Google Scholar]
- 47. Leclerc JL, Lampert AS, Diller MA, Dore S. PGE2‐EP3 signaling exacerbates intracerebral hemorrhage outcomes in 24‐mo‐old mice. Am J Physiol Heart Circ Physiol. 2016;310:H1725‐H1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Han X, Lan X, Li Q. Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin‐induced brain injury. J Cereb Blood Flow Metab. 2016;36:1059‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi J, Wang Q, Johansson JU. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:788‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hein AM, Stutzman DL, Bland ST. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin‐1 beta injections into the dorsal hippocampus. Neuroscience. 2007;150:754‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alberi L, Liu S, Wang Y. Activity‐induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron. 2011;69:437‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mamiya N, Fukushima H, Suzuki A. Brain region‐specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pittenger C, Huang YY, Paletzki RF. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus‐dependent spatial memory. Neuron. 2002;34:447‐462. [DOI] [PubMed] [Google Scholar]
- 54. Maingret V, Barthet G, Deforges S, Jiang N, Mulle C, Amedee T. PGE2‐EP3 signaling pathway impairs hippocampal presynaptic long‐term plasticity in a mouse model of Alzheimer's disease. Neurobiol Aging. 2017;50:13‐24. [DOI] [PubMed] [Google Scholar]
- 55. Akaneya Y, Tsumoto T. Bidirectional trafficking of prostaglandin E2 receptors involved in long‐term potentiation in visual cortex. J Neurosci. 2006;26:10209‐10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hovens IB, Schoemaker RG, van der Zee EA, Heineman E, Izaks GJ, van Leeuwen BL. Thinking through postoperative cognitive dysfunction: how to bridge the gap between clinical and pre‐clinical perspectives. Brain Behav Immun. 2012;26:1169‐1179. [DOI] [PubMed] [Google Scholar]
- 57. Yang X, Yao C, Tian T. A novel mechanism of memory loss in Alzheimer's disease mice via the degeneration of entorhinal‐CA1 synapses. Mol Psychiatry. 2016;23:199‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Y, Wang ZH, Jin S. Opposite monosynaptic scaling of BLP‐vCA1 inputs governs hopefulness‐ and helplessness‐modulated spatial learning and memory. Nat Commun. 2016;7:11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lisman JE, Pi HJ, Zhang Y, Otmakhova NA. A thalamo‐hippocampal‐ventral tegmental area loop may produce the positive feedback that underlies the psychotic break in schizophrenia. Biol Psychiatry. 2010;68:17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Adamantidis A, Arber S, Bains JS. Optogenetics: 10 years after ChR2 in neurons–views from the community. Nat Neurosci. 2015;18:1202‐1212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials