

1. INTRODUCTION
Anti‐IgLON5 disease is a novel antibody‐mediated disorder characterized by the positive antibody to a neuronal cell adhesion protein named IgLON5 in serum and/or cerebral spinal fluid (CSF).1 Patients with anti‐IgLON5 disease developed a characteristic sleep disorder usually accompanied by some other symptoms such as bulbar symptoms, gait abnormalities, oculomotor problems, and cognitive decline.2 Since Lidia et al reported the first anti‐IgLON5 disease case in 2014,3 only a few patients have been reported worldwide include 2 patients in Chinese population.4, 5, 6 Here, we report the first anti‐IgLON5 disease patient with motor neuron disease‐like (MND‐like) phenotype.
2. CASE REPORT
The patient was a 57‐year‐old male with a chief complaint of progressive dysphagia for 2 y, limb weakness and dyspnea for 5 mo. Mild pharynx and larynx discomfort were noticed at first which then developed to mild dysphagia 2 y ago. The symptoms aggravated progressively during the following 2 y. Slowly limb weakness and rapidly progressed intrinsic hand muscle atrophy were noticed 19 mo after disease onset (Figure 1 A, B). Simultaneously, mild dyspnea developed, which appeared to be more obvious during sleep at night. The wife of the patient complained that her husband had prominent sleep abnormalities with snoring and involuntary movement (Video S1[Link]). Having developed transient unconsciousness at night, the patient was admitted to a local hospital 20 mo after disease onset. During the next four mo, recurrent respiratory failure accompanied abnormal behavior and consciousness was noticed. Mechanical ventilation was used to provide respiratory support. After ruling out the possibility of any infectious disease, immunosuppressive therapy was initiated with prednisolone 1000 mg i.v. per d for 3 d, 500 mg i.v. per d for 3 consecutive d and gradually tapered to 80 mg i.v. per d. Intravenous immunoglobulin (IVIg) (0.4 g/Kg i.v. for 5 consecutive d) was also given. The above symptoms slightly improved and spontaneous respiration was recovered. The patient was then referred to our hospital for further diagnosis and treatment. There was no family history of note.
Figure 1.
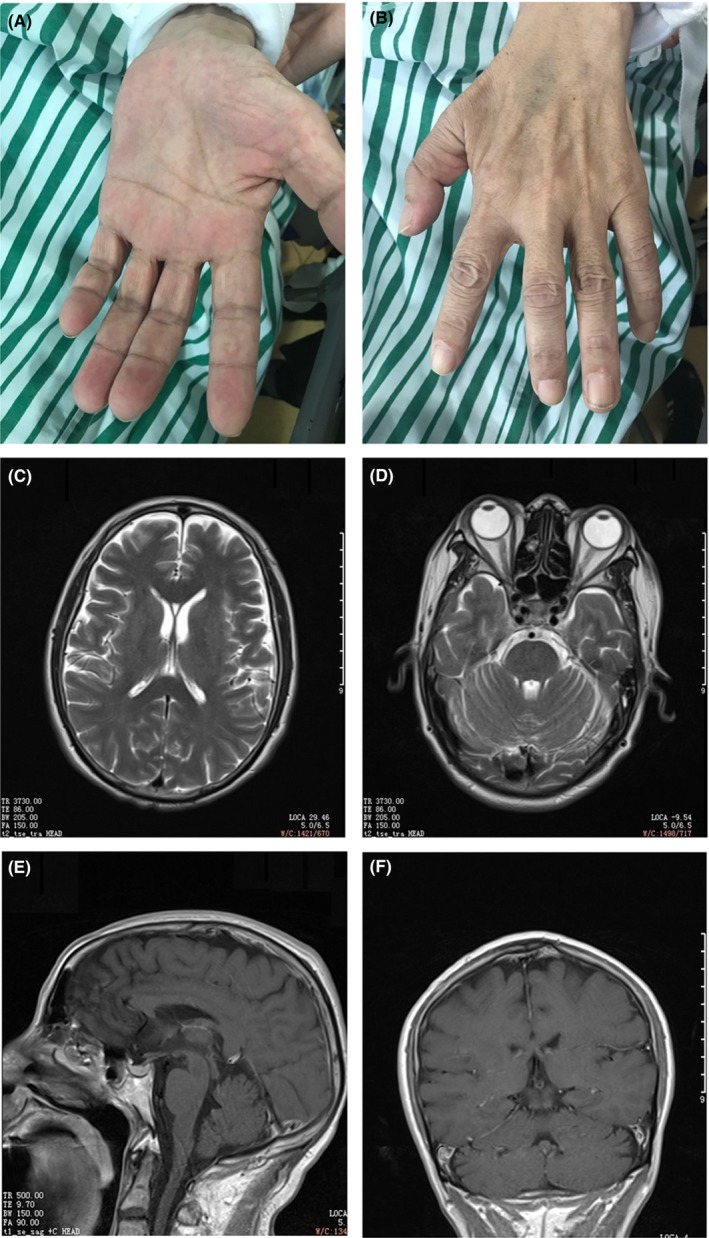
Clinical findings in the patient with anti‐IgLON5 disease. Rapidly progressed intrinsic hand muscle atrophy (A‐B); Brian MRI showed no abnormal signals nor atrophy (C‐F)
Neurological examination revealed a normal mental status and slightly slurred speech. Examinations of the cranial nerves showed bilaterally disappeared pharynx reflex. Muscle weakness involved all his extremities, with a mild decreased strength of all extremities (4/5). Atrophy of bilateral intrinsic hand muscles was observed. Hoffmann and Babinski sign was absent bilaterally. Tendon reflexes were normal in all four extremities. Sensory and cerebellar function was normal.
Routine blood test, blood biochemical test, vitamin B12, folate, C‐reactive protein (CRP), erythrocyte sedimentation rate (ESR), and thyroid function were within normal ranges. Serology for HIV, hepatitis, and syphilis were negative as well. Tumor markers examination showed mild elevation of carcinoembryonic antigen with 7.1 ng/mL (normal range < 5.0 ng/mL). Blood gas analysis showed the partial pressure of carbon dioxide was 52.9 mm Hg (normal range < 44.0 mm Hg).The lumbar puncture showed intracranial pressure was 170 mm H2O and CSF analysis showed normal protein, glucose, cell counts, and IgG synthesis rate. No abnormalities were found in brain magnetic resonance imaging (MRI) examination (Figure 1C‐F). Electromyography (EMG) test showed extensive denervation in limb muscles and thoracic paraspinal muscles (Table 1). The PSG results showed no obvious obstructive sleep apnea (OSA) events with an apnea‐hypopnea index (AHI) of 2.3/h (normal ≤ 5/h). Mean oxygen saturation was 93% with a nadir of 90%. The examination of a group of autoantibodies showed positive anti‐IgLON5 antibody in both serum (1:320) and CSF (1:1) with IFT method. (Oumeng, Hangzhou, China) thereby confirming the diagnosis. Antibodies to other cell surface or synaptic proteins including, NMDA, AMPA, GABAB, DPPX, LGI1, and Caspr2 were negative. Additionally, a high association with the HLA‐DQB1*05:01 and HLA‐DRB1*10:01 alleles was reported previously.7 Thus, human leukocyte antigen (HLA) typing was performed and both of the HLADQB1*0501 and HLA‐DRB1*1001 alleles were identified. No specific abnormalities were found in whole body FDG PET/CT scan.
Table 1.
Needle electromyography (EMG) results of the patient
| Needle EMG results | ||||||||
|---|---|---|---|---|---|---|---|---|
| Side | Muscle | Insertional activity | Spontaneous activity | Voluntary activity | ||||
| Positive sharp waves | Fibrillation | Fasciculation | Amplitude (mv) | Duration (ms) | Polyphasics (%) | |||
| Right | Anterior tibial muscle | Normal | (−) | (−) | (−) | 7.8 | 14.6 | 50 |
| Left | Medial femoral muscle | Normal | (−) | (−) | (−) | 5.3 | 15.1 | 20 |
| Right | First dorsal interosseous muscle | Extend | (+) | (+) | (+) | 4.2 | 14.1 | 50 |
| Left | Deltoid | Extend | (+) | (+) | (−) | 6.5 | 12.2 | 30 |
| Right | Sternocleidomastoid | Extend | (+) | (+) | (−) | 1.6 | 11.5 | 60 |
| Right | Rectus abdominis | Normal | (−) | (−) | (−) | 2.4 | 12.3 | 10 |
The patient's condition deteriorated 1 month after immunomodulatory therapy. The titer of anti‐IgLON5 antibody in serum was assayed again and it decreased to (1:100), indicating that there was no correlation between clinical manifestations and the titer of antibody. To date, only one study reported that a patient has good outcome after immunotherapy using IVIg followed by mycophenolate mofetil 1.5 g/d as a chronic immunotherapy.4 Unfortunately, the patient in our study suffered from suspicious tuberculosis with a positive T‐SPOT when we screened for the possible contraindication of chronic immunotherapy. The patient underwent additional plasmapheresis treatments. However, the treatment was not effective. The patient could not maintain spontaneous breathing soon and mechanical ventilation was used to provide respiratory support.
3. DISCUSSION
As a relatively novel disease, clinical spectrum of anti‐IgLON5 disease has not been fully described and is easy to be ignored if not raise awareness. According to a recent study which analyzed 22 patients with anti‐IgLON5 disease, four syndromes have been summarized as the phenotype of the disorder by the time of diagnosis include sleep disorder, bulbar syndrome, progressive supranuclear palsy (PSP‐like), and cognitive decline with or without chorea.2 Rare clinical manifestations of the disease still need to be investigated. A recent study reported that peripheral involvement may also occur in anti‐IgLON5 disease.8
To the best of our knowledge, this is the first reported patient with anti‐IgLON5 disease featured with MND‐like phenotype, including progressively dysphagia, limbs weakness with muscles atrophy, and extensive denervation in limb muscles and thoracic paraspinal muscles.9 From the perspective of neuropathology, it is not surprise that MND‐like phenotype is associated with anti‐IgLON5 disease as neuron‐specific tau deposits have been found preferentially involving the hypothalamus, the tegmental nuclei of the brainstem, the upper cervical cord as well as anterior horn of spinal cord.7 The prognosis of the disease appears to be unoptimistic as most patients did not well respond to common immunotherapies and a considerable number of reported patients died from sudden death.2, 10 The patient still could not maintain spontaneous breathing at last follow‐up time (2 months). Collectively, the results reported here expanded the known clinical features for this rare disease. In addition, anti‐IgLON5 disease should be considered in differential diagnosis of motor neuron disease.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
REFERENCES
- 1. Dale RC, Ramanathan S. Cell surface antibody‐associated neurodegeneration: The case of anti‐IgLON5 antibodies. Neurology. 2017;88(18):1688‐1690. [DOI] [PubMed] [Google Scholar]
- 2. Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti‐IgLON5 disease. Neurology. 2017;88(18):1736‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sabater L, Gaig C, Gelpi E, et al. A novel non‐rapid‐eye movement and rapid‐eye‐movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post‐mortem study. Lancet Neurol. 2014;13(6):575‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haitao R, Yingmai Y, Yan H, et al. Chorea and parkinsonism associated with autoantibodies to IgLON5 and responsive to immunotherapy. J Neuroimmunol. 2016;300:9‐10. [DOI] [PubMed] [Google Scholar]
- 5. Zhu L, Liu L, Cui S, et al. One case of anti‐IgLON5 syndrome. Chin J Neurol. 2017;10:763‐765. [Google Scholar]
- 6. Hasselbacher K, Steffen A, Wandinger KP, Bruggemann N. IgLON5 antibodies are infrequent in patients with isolated sleep apnea. Eur J Neurol. 2018;25(4):e46–e47. [DOI] [PubMed] [Google Scholar]
- 7. Gelpi E, Hoftberger R, Graus F, et al. Neuropathological criteria of anti‐IgLON5‐related tauopathy. Acta Neuropathol. 2016;132(4):531‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wenninger S. Expanding the clinical spectrum of IgLON5‐syndrome. J Neuromuscul Dis. 2017;4(4):337‐339. [DOI] [PubMed] [Google Scholar]
- 9. Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17(1):4‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Honorat JA, Komorowski L, Josephs KA, et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials