

Summary
Hereditary Huntington's disease (HD) is characterized by cell dysfunction and death in the brain, leading to progressive cognitive, psychiatric, and motor impairments. Despite molecular and cellular descriptions of the effects of the HD mutation, no effective pharmacological treatment is yet available. In addition to well‐established alterations of glutamatergic and dopaminergic neurotransmitter systems, it is becoming clear that the GABAergic systems are also impaired in HD. GABA is the major inhibitory neurotransmitter in the brain, and GABAergic neurotransmission has been postulated to be modified in many neurological and psychiatric diseases. In addition, GABAergic neurotransmission is the target of many drugs that are in wide clinical use. Here, we summarize data demonstrating the occurrence of alterations of GABAergic markers in the brain of HD carriers as well as in rodent models of the disease. In particular, we pinpoint HD‐related changes in the expression of GABAA receptors (GABAARs). On the basis that a novel GABA pharmacology of GABAARs established with more selective drugs is emerging, we argue that clinical treatments acting specifically on GABAergic neurotransmission may be an appropriate strategy for improving symptoms linked to the HD mutation.
Keywords: basal ganglia, disease progression, inhibitory postsynaptic currents, inhibitory tonic currents, rest/activity fragmentation, synapse
1. INTRODUCTION
Huntington's disease (HD) is a hereditary neurodegenerative disease, caused by a mutation of the huntingtin protein that impairs cell functions and produces cell death. HD is associated with cognitive and psychiatric disturbances that precede chorea and other motor impairments. Several postmortem studies in humans have shown only limited signs of cell loss in the brain, despite overt clinical symptoms and the genetic confirmation of HD,1, 2, 3 suggesting that neuronal and synaptic dysfunction, rather than cell death, may underlie the early behavioral manifestations of the HD mutation.4, 5 In addition, cognitive, psychiatric, motor, electrophysiological, and neuroimaging assessments in humans also suggest that early cognitive deficits occur years prior to cell death or predicted clinical diagnosis in HD carriers, and are probably due to synaptic and cellular dysfunction.6, 7, 8
The expression of the huntingtin protein is ubiquitous, and the mutation for HD affects virtually all brain structures. However, alterations are most obvious in the striatum. This structure is the main input nucleus of the basal ganglia, a group of subcortical nuclei that are closely connected with several brain areas, including the cerebral cortex and the thalamus.9 The basal ganglia are considered to be involved in voluntary movement, memory, and cognitive functions. Medium spiny neurons (MSNs) are major constituents of the striatum and are comprised of 2, anatomically nonsegregated, subpopulations: 1 expressing mainly dopamine D1 receptors and the other expressing dopamine D2 receptors.10 The 2 subpopulations are at the origin of the direct striatonigral and indirect striatopallidal pathways, respectively. These projection neurons are also regulated by different classes of local interneurons.11, 12 Being tightly connected to the striatum via the indirect pathway, the external globus pallidus (GPe) is considered as a hub within the basal ganglia.13, 14, 15 It is important to note, furthermore, that an alteration in GPe function is linked to HD symptoms.16, 17, 18, 19, 20 The GPe also receives GABAergic collaterals from the direct pathway,21 glutamatergic projections from the subthalamic nucleus, and some dopaminergic inputs from the substantia nigra pars compacta.14 The GPe projects to virtually all basal ganglia components, including the striatum.22 Interestingly, direct connections with the cortex have been recently described.23, 24 Therefore, the GPe may play an important integrative role in coordinating neuronal activity throughout the basal ganglia with direct links to the cortex.
Studies in animal models have established that glutamate neurotransmission, including glutamate release, clearance and receptor trafficking, is altered in HD (reviewed in4, 5, 25). An early alteration of glutamatergic neurotransmission has been recently described in the subthalamic nucleus neurons of BAC transgenic and Q175 knock‐in mouse models of HD, leading to the loss of autonomous pacemaking.26 Because the striatum receives extensive excitatory glutamatergic innervation from the cerebral cortex and thalamus and is primarily affected in HD, changes in glutamatergic neurotransmission resulting in excitotoxicity and neuronal damage have been considered as a key event in HD pathogenesis. Besides the impairment of the predominant excitatory glutamate neurotransmission, inhibitory GABAergic neurotransmission, although largely neglected, is also altered in the HD‐affected brain.27, 28, 29, 30 This review focuses mainly on our current knowledge of the alteration in inhibitory neurotransmission through changes in GABAA receptor (GABAAR) subtype expression (Figure 1A). We will also consider whether current pharmacological tools targeting GABAergic transmission could be of relevance for future treatment of HD symptoms.
Figure 1.
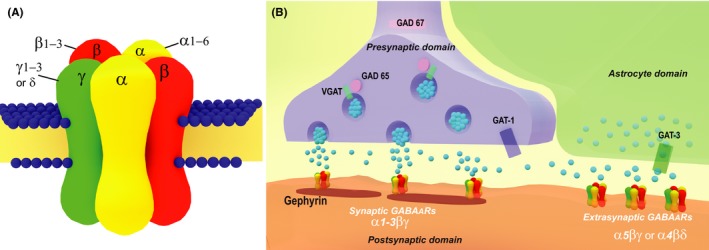
Schematic drawing of GABAA receptors (GABAARs, A) and a GABAergic synaptic terminal (B). Native GABAARs, which are heteropentameric structures containing α1, α2, or α3 with β and γ subunits, are located at the synapse and mediate a phasic inhibitory effect correlated with presynaptic action potentials. The aggregation of receptors on the postsynaptic domain depends on scaffolding proteins, including gephyrin. GABAARs containing the α5 or δ subunits are extrasynaptic with a high sensitivity to GABA and mediate tonic activity as a function of ambient transmitter level in the extracellular space. GABA levels depend on the expression of 2 isoforms of glutamate decarboxylase (GAD 67 and 65), the vesicular transporter VGAT, and the transporters GAT‐1 and GAT‐3. These GABAergic components are all subject to alteration in Huntington's disease
2. GABAergic NEUROTRANSMISSION
GABA exerts inhibitory control on many neurons in the central nervous system. The diversity in GABAergic signaling is due to several peri‐, pre‐, and postsynaptic factors (Figure 1B) that are the target of many drugs that are currently in wide clinical use.31, 32 It is also well documented that an alteration in any aspect of this system is linked to several neurological and neurodevelopmental disorders.33, 34, 35, 36, 37, 38, 39, 40 GABAARs are the main inhibitory receptors in the brain, and their heteromeric structure contributes in several ways to the physiological properties of brain GABAergic neurotransmission. GABA also acts on GABABRs which have different molecular and functional properties to those of GABAARs (eg, see 41). As data on alterations of GABAB neurotransmission in HD are sparse, they are not considered further in this review.
3. STRUCTURE AND FUNCTION OF GABAARs
Ionotropic GABAA receptors are responsible for fast and flexible postsynaptic transmission. GABA binding results in the opening of anion‐selective intrinsic channels through which primarily chloride anions flow. This in turn changes neuron excitability. Many studies have demonstrated that the subunit composition determines both the functional properties and subcellular localization of GABAARs. These receptors are heteromeric structures composed of a combination of 5 of 19 different subunits, grouped in several classes.42, 43, 44 Different subtypes of GABAARs are generated by a coassembly of the α1‐6, β1‐3, γ1‐3, δ, ε, θ, π, and ρ1‐3 subunits.45 Strong evidence supports a model in which subunit composition confers a distinctive cellular distribution, functional properties, and the specific effect of allosteric modulators like benzodiazepines or neurosteroids.36, 45, 46 In brain regions, including the striatum, synaptic neurotransmission mediating phasic inhibition is linked to GABAARs composed of α1, α2, or α3 in combination with β and γ2 subunits (Figure 1A). The substitution of an α5 by an α1‐3 or a δ by a γ2 subunit has been shown to form extrasynaptic receptors (Figure 1B) mediating tonic inhibition.47, 48 Indeed, it is now established that receptor subtypes are associated with significant physiological outcomes and specific cognitive functions.37, 49, 50 These findings have in turn led to the search for selective drugs with enhanced efficacy and fewer side effects.51
4. ALTERATION OF THE GABA SYSTEM IN THE HD BRAIN
Over the last decades, benzodiazepine or muscimol binding on GABAARs has been widely used. Postmortem analyses in the human HD brain have shown a decrease in benzodiazepine binding in the caudate nucleus or the putamen.52, 53, 54 In contrast, binding is increased in the cerebellum, frontal cortex, and the GPe.53, 54, 55, 56 Other studies have measured concentrations of GABA and found a decrease in the caudate putamen and the GPe.57, 58 More recently, positron emission tomographic (PET) imaging using [11C]flumazenil as a marker of GABAARs has been performed (reviewed in59). In patients with early HD, benzodiazepine binding levels were found to be reduced in the caudate nucleus, while no changes were observed in the putamen, thalamus, frontal cortex, or cerebellum.60, 61 Transcranial magnetic stimulation investigations have revealed significant GABA‐mediated cortical inhibitory deficits in premanifest and early symptomatic HD patients.62
The most thorough and recent investigations on GABAergic neurotransmission, including molecular and functional analyses, have been conducted on transgenic rodent models. Interestingly, quantitative autoradiography has been used to assess neurotransmitter receptor densities in several brain regions in a rat model for the HD mutation.63 These analyses showed that the expression levels of receptors of the cholinergic, dopaminergic, serotoninergic, noradrenergic, glutamatergic, and GABAergic systems are either increased, decreased, or remain unchanged. These findings in turn suggest that receptor alterations in HD are subtype selective and regionally differential. Moreover, the most recent studies on GABAergic neurotransmission in the striatum have analyzed receptor subtype alterations in identified neurons.
5. ALTERATION OF GABA NEUROTRANSMISSION IN MSNs
In the striatum of transgenic mouse or rat models of HD, no changes in benzodiazepine or GABA binding 63, 64 or a slight, albeit significant increase65 have been reported. Functional and molecular analyses of GABAergic neurotransmission in the striatum have revealed complex changes in this brain structure (Figure 2). The kinetics of evoked GABAergic currents are altered in MSNs of animal models of HD,66 associated with a reduction both in rise and in decay times leading to faster currents. Because it is well established that α1 is responsible for fast inhibitory currents,67 this kinetic alteration is probably linked to the global increase in α1 subunit expression in the MSN neuropil of HD mice.68, 69 In addition to the increased expression of α1, immunohistochemical labeling analyses of MSN cell body membranes showed an increased number of clusters containing the α2 subunit at postnatal 2 months followed by a decreased expression at 6 months in presymptomatic and symptomatic R6/1 mice, respectively.69 Among α subunits, α2 is the major component of GABAARs in MSNs. Thus, a decreased expression of the α2 subunit on MSN cell bodies of 6‐month‐old R6/1 mice69 is consistent with a decreased number of GABAergic terminals in contact with MSN somata in the zQ175 KI mouse model of HD.29 In vitro analyses have shown that cell surface receptor expression, as well as the expression of α1 and α2 subunits in MSNs, is regulated by dopamine and GABAAR activity.70, 71 Thus, changes in GABAAR subtypes might be directly linked to alterations of both dopaminergic and GABAergic neurotransmission in HD.66, 72 An alteration of GABAAR trafficking resulting in reduced mIPSCS has also been shown in striatal MSNs of the N171‐82Q mouse model.73
Figure 2.
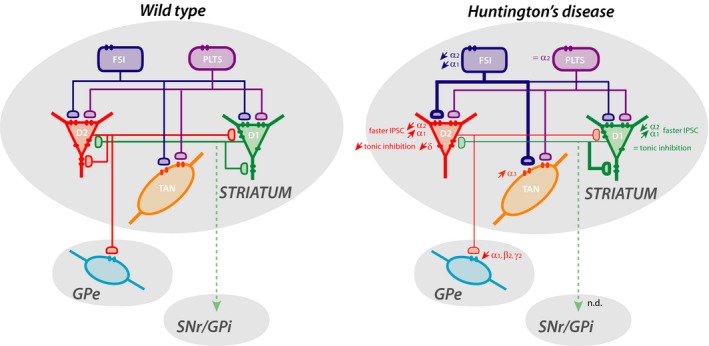
Schematic representation of striatal local circuits highlighting alterations of inhibitory currents and GABAAR subunit expression in identified neurons in mouse models of HD. The striatum includes medium‐sized output neurons expressing dopamine D1 or D2 receptors, and fast‐spiking, persistent low‐threshold spiking, or tonically active interneurons. The 2 main projection pathways to the external globus pallidus and substantia nigra pars reticulata/internal globus pallidus are also indicated. Alteration of inhibitory currents in symptomatic HD mice is indicated by thicker (increase) or thinner (decrease) lines. Alteration in subunit expression or tonic inhibition is indicated by↑, increase; ↓, decrease; or =, no change. The figure was constructed using data from30, 66, 69, 80, 101
It is also interesting to note that the α1 subunit is present in postsynaptic structures facing dopaminergic striatal synapses 74 which are believed to corelease dopamine and GABA onto MSNs.75, 76, 77 Although the functional role of this GABAergic inhibition remains elusive, it would be of interest to analyze the impact of the HD mutation on nigrostriatal synaptic transmission.
6. ALTERATION OF GABA NEUROTRANSMISSION IN STRIATAL INTERNEURONS
Other than the predominant MSN projection neurons in the striatum, several local interneurons (INs) are also GABAergic (Figure 2). It is well established that the GABAergic control of MSNs originates from different classes of INs78: fast‐spiking INs (FSI) expressing parvalbumin, persistent low‐threshold spiking INs (PLTS) expressing somatostatin or nNOS, INs expressing calretinin, as well as collaterals from MSNs themselves (Figure 2). In addition to these GABAergic neurons, the striatum also contains cholinergic INs that regulate local inhibitory circuits.79 Striatal INs are relatively spared from degeneration in HD, although there is evidence for dysfunctions.66, 80, 81, 82, 83, 84
In the brain, GABAergic INs are involved in the coordination and regulation of network functions, and many pathological conditions are linked to their alteration.47, 78, 85, 86 In the striatum, it has been suggested that the functional role of inhibition from fast‐spiking PV cells might be in shaping striatal output conveyed in both direct and indirect pathways.87 This feed‐forward inhibition from fast‐spiking PV INs to MSNs is altered in HD.66 We found a decreased expression of the α1 subunit in striatal PV interneuron cell bodies at postnatal 2 and 6 months, while the expression of α2 is increased at 2 months and decreased at 6 months.69 Although a comprehensive study of the molecular, pharmacological, and functional properties of GABAergic conductances in these cell types is still lacking, the increased expression of the α2 subunit and decreased expression of α1 in mutant mice predict that GABAergic currents in PV cells from 2‐month‐old R6/1 should have lower decay times compared to their WT counterparts.67 Interestingly, following a striatal‐dependent cognitive task, we found an alteration in activation of interneurons that express parvalbumin in the dorsomedial striatum at both presymptomatic and early symptomatic ages, thereby confirming a severe and early impairment of these INs in HD mice.84
Cholinergic INs also play a major role in striatal physiology.83 It has been shown that in human and animal models, there is no, or a limited, loss of these INs, whereas the level of vesicular acetylcholine transporters and choline acetyltransferase is decreased.81, 88, 89 In symptomatic R6/1 or R6/2 mice, a decrease in acetylcholinesterase expression and acetylcholine levels has also been reported.69, 90 With immunohistochemical labeling, we found an increased expression of the α3 GABAAR subunit in cholinergic INs.69 The α3 subunit is the main α subunit expressed in cholinergic striatal INs,91 which is likely to represent the major GABAAR subtype in these neurons. In R6/2 mice, striatal cholinergic INs receive more GABAergic inhibitory postsynaptic currents compared to their WT counterparts.80 Together these data suggest that an increased number of α3‐containing postsynaptic GABAARs are involved in the increased inhibition of striatal cholinergic INs in HD (Figure 2) and underlie the decreased level of acetylcholine.
7. ALTERATION OF TONIC INHIBITORY NEUROTRANSMISSION
Both δ‐ and α5‐GABAARs are responsible for generating tonic inhibitory conductances in the brain (Figure 1), which is recognized as a key factor in controlling local networks.35, 92 These 2 GABAAR subtypes are developmentally regulated in MSNs.48 Tonic inhibition is decreased in MSNs in mouse models of HD.66, 93 In addition, a decrease in striatal δ subunit mRNA expression has been reported in different HD mouse models as well as in human patients,69, 94, 95 suggesting that a reduction in δ subunit expression plays a major role in the tonic inhibition decrease. It is also of note that it has been shown93 that an alteration in tonic GABA currents in HD might be also due to a reduced release of GABA from surrounding astrocyte processes (Figure 1B). However, the role played by astrocytes in the regulation of GABA homeostasis remains poorly understood, and further studies should be conducted to tackle this important question. A striking finding in a recent study from our laboratory was that α5 and δ subunit expression in the striatum is increased and decreased, respectively.69 In addition to MSNs, the expression of both α5 and δ subunits has been identified in striatal INs.48, 66, 93, 96 Because tonic inhibition of interneurons may also be modified in many movement or psychiatric disorders,47, 78 it would be of interest to identify the neuron types whose specific α5 or δ subunit expression is modified in HD.
Taken together, analyses in the striatum of HD brains show that GABAergic neurotransmission undergoes complex changes leading to alterations in synaptic and extrasynaptic GABAergic functions. The change in GABAAR subunit composition would likely influence the pharmacological and gating properties of the GABAARs of interneurons and MSNs and thus alter inhibitory neurotransmission in the striatum of the HD brain. Interestingly, the change in synaptic GABAA receptor subunit expression in HD follows the opposite trend in experimental models of Parkinson's disease,97 suggesting the occurrence of mirror alterations in GABAergic synaptic neurotransmission in hyperkinetic and hypokinetic disorders, respectively.
8. ALTERATION OF GABA NEUROTRANSMISSION IN THE GPe
The GPe receives many sources of GABA. In addition to massive GABAergic input from the striatal indirect pathway,98 local GPe collaterals99 and bridging collaterals from the striatal direct pathway21 control GPe neuronal excitability. In humans, the GPe is overactive in HD,19 and GPe deep brain stimulation has been shown to alleviate motor and cognitive dysfunctions in both human patients and a rat model of HD.16, 17, 18, 20 Ex vivo, the firing pattern of GPe neurons is altered in R6/2 mice where blockade of GABAARs facilitates bursting activity.100 It is then reasonable to anticipate that the HD mutation has an impact on GABAergic neurotransmission in this brain structure.
In R6/1 mice, we showed a decreased expression of α1, β2, and γ2 subunits (Figure 2) in addition to a decreased expression of the vesicular GABA transporter (VGAT) involved in the synaptic release of GABA as well as gephyrin and neuroligin 2, both of which are involved in inhibitory synapse formation.101 We also found a decrease in the frequency of mIPSCs (Figure 2) supported by a reduced number of synapses, whereas the amplitude of mIPSCs was not altered, suggesting that the number of receptors in individual synapses is not decreased. Interestingly, the modification of mIPSC kinetics in 2‐month‐old R6/1 mice in the absence of a change in the short‐term facilitation of striatopallidal synapses suggested postsynaptic alterations in GABAAR subunit composition.102
In addition to α1, β2, and γ2 subunits, α2, α3, and γ1 are also expressed in the GPe.103, 104 The GPe also contains a number of distinct neuronal subtypes and projections with different physiological functions.13, 14 It would thus be of interest to investigate the localization of the different subunits expressed in GPe neurons and correlate functional property alterations with specific GABAAR subtypes. It is of note that data on the GPe from R6/1 and HdhQ111 mouse models 101 are at odds with immunohistolabeling from the postmortem human GPe.105 This may highlight one limitation of rodent models where a limited neuronal death is observed.106 It also suggests that rodent transgenic models display an alteration occurring in humans, before any dramatic cell damage occurs in the brain. The hyperactivity of the GPe in animal models and human HD 20, 107 may be the consequence of a decrease in GABAergic neurotransmission and GABAAR synapses. The development of PET analyses in the future should allow monitoring the evolution of GABA binding, well before the overt onset of the disease.
9. POTENTIAL THERAPEUTIC AVENUES
Although GABAA neurotransmission is clearly altered in HD, drugs, whether already approved or under clinical trials, are currently being employed to target other neuromodulatory systems aiming to improve motor dysfunction in HD.5 Only limited treatment evaluations have been conducted,108, 109, 110, 111 and to our knowledge, no drug therapies that target the alteration in GABAergic neurotransmission in HD are currently available (reviewed in112). A new benzodiazepine pharmacology acting on GABAARs and comprising selective hypnotics, nonsedative anxiolytics, and cognition enhancement is emerging.32 This suggests potential therapeutic avenues for nonmotor symptoms including anxiety, sleep alteration, cognitive dysfunctions, or psychiatric disorders linked to the HD mutation that are present long before the appearance of overt motor symptoms (reviewed in113). In addition, an altered tonic inhibitory conductance as found in HD (see above) is also the therapeutic target for the treatment of several other diseases.114, 115
Sleep disturbances are believed to contribute to HD symptoms,116, 117, 118 and recent findings suggest that the GABA system may be a target for preventing sleep alteration and reduce the cognitive and psychiatric symptoms of this neurodegenerative disease.119, 120, 121 In transgenic mouse models, it has been shown recently that treatment with zolpidem, an hypnotic benzodiazepine acting preferentially on α1‐containing GABAARs, corrects EEG abnormalities.119 In addition, the regulation of sleep/wake activity with a benzodiazepine treatment has been found to improve cognitive function and apathy.121 Interestingly, consistent with the well‐established alteration of rest/activity in HD,101, 116, 122, 123, 124 a role for the basal ganglia in sleep/wake regulation has been highlighted (reviewed in125), and lesions of the GPe in rats have a profound effect on sleep/wakefulness fragmentation.126 A link between the striatopallidal pathway and the regulation of sleep/wake behavior has also been shown very recently.127 Therefore, it is tempting to speculate that an early alteration in GABAergic neurotransmission in the striatum or GPe and an early modification in rest/activity are not independent phenomena.
Based on analyses with point‐mutated mice of a benzodiazepine binding site, it was possible to attribute specific behavioral responses following benzodiazepine treatments to specific GABAARs comprising either α1, α2, α3, or α5 subunits, thereby providing the opportunity to selectively modulate brain areas or neuronal networks (review in128). In HD, changes in GABAAR subtype expression are specific not only to brain area53, 63, 69, 101, 129 but also to neuronal cell type.66, 69 The fact that the expression of GABAAR subunits in the different striatal neuron classes is differentially altered suggests that subtype‐specific benzodiazepine drugs may act on particular symptoms linked to HD disorders.
Besides the altered expression of GABAARs containing α1‐3 subunits essentially present in the postsynaptic domain, the expression of extrasynaptically localized α5‐ and δ‐containing GABAARs is also altered in human HD carriers and animal models.69, 94, 95, 129 These extrasynaptic receptors are key factors in the control of local networks35 and are potential therapeutic targets for synthetic compounds or endogenous neuroactive steroids.34, 130, 131, 132 On the basis of an increased expression of α5 in the striatum of R6/1 mice,69 it would be relevant to test whether an α5‐specific antagonist could slow disease progression or symptoms in HD, as is the case in mouse models of Down syndrome.32, 115 Our preliminary data point to the validity of such a possibility because an alleviation of HD symptoms was observed following an acute treatment with an inverse α5 agonist (M. Garret, M.C. Potier, Y.H. Cho, unpublished observations). Although a clinical trial with 1 synthetic agonist of δ‐containing GABAARs failed to improve symptoms of HD patients,111 it would be of interest to test the effect of several available compounds on animal models during the progression of HD.114 Interestingly, striatal tonic GABAA currents mediated by δ‐containing GABAARs have neuroprotective effects against excitotoxicity133 and might be a relevant target to slow disease evolution.
10. CONCLUSION
GABAergic neurotransmission is altered early in HD and is likely to precede the appearance of overt symptoms. Significantly, studies on animal models suggest that such alterations depend on the particular brain structure, neuronal cell type, and the stage of the disease. It remains to study these changes during aging in HD carriers using new technologies such as PET imaging59 with a view to determine whether the GABA system could be a therapeutic target for preventing sleep alteration and reducing the cognitive and psychiatric symptoms of this neurodegenerative disease.32, 118 Although HD‐related changes have been mostly studied in the basal ganglia, it would be of further interest to assess whether GABAergic neurotransmission is also altered in other areas of the HD‐affected brain. Because deficits in the ontogeny of GABAergic neurotransmission may impact on adult brain functions,134 it would also be relevant to assess whether synapses involving GABAARs are altered during development in HD carriers.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Garret M, Du Z, Chazalon M, Cho YH, Baufreton J. Alteration of GABAergic neurotransmission in Huntington's disease. CNS Neurosci Ther. 2018;24:292–300. 10.1111/cns.12826
REFERENCES
- 1. Caramins M, Halliday G, McCusker E, Trent RJ. Genetically confirmed clinical Huntington's disease with no observable cell loss. J Neurol Neurosurg Psychiatry. 2003;74:968‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizuno H, Shibayama H, Tanaka F, et al. An autopsy case with clinically and molecular genetically diagnosed Huntington's disease with only minimal non‐specific neuropathological findings. Clin Neuropathol. 2000;19:94‐103. [PubMed] [Google Scholar]
- 3. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559‐577. [DOI] [PubMed] [Google Scholar]
- 4. Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy? Trends Mol Med. 2003;9:414‐420. [DOI] [PubMed] [Google Scholar]
- 5. Tyebji S, Hannan AJ. Synaptopathic mechanisms of neurodegeneration and dementia: insights from Huntington's disease. Prog Neurobiol. 2017;153:18‐45. [DOI] [PubMed] [Google Scholar]
- 6. Schippling S, Schneider SA, Bhatia KP, et al. Abnormal motor cortex excitability in preclinical and very early Huntington's disease. Biol Psychiat. 2009;65:959‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Orth M, Schippling S, Schneider SA, et al. Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J Neurol Neurosurg Psychiatry. 2010;81:267‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict‐HD study. J Neurol Neurosurg Psychiatry. 2008;79:874‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tepper JM, Abercrombie ED, Bolam JP. Basal ganglia macrocircuits. Prog Brain Res. 2007;160:3‐7. [DOI] [PubMed] [Google Scholar]
- 10. Bertran‐Gonzalez J, Herve D, Girault JA, Valjent E. What is the degree of segregation between Striatonigral and Striatopallidal projections? Front Neuroanat. 2010;4:136 10.3389/fnana.2010.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tepper JM, Tecuapetla F, Koos T, Ibanez‐Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150 10.3389/fnana.2010.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abdi A, Mallet N, Mohamed FY, et al. Prototypic and arkypallidal neurons in the dopamine‐intact external globus pallidus. J Neurosci. 2015;35:6667‐6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gittis AH, Berke JD, Bevan MD, et al. New roles for the external globus pallidus in Basal Ganglia circuits and behavior. J Neurosci. 2014;34:15178‐15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hegeman DJ, Hong ES, Hernandez VM, Chan CS. The external globus pallidus: progress and perspectives. Eur J Neurosci. 2016;43:1239‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beste C, Muckschel M, Elben S, et al. Behavioral and neurophysiological evidence for the enhancement of cognitive control under dorsal pallidal deep brain stimulation in Huntington's disease. Brain Struct Funct. 2015;220:2441‐2448. [DOI] [PubMed] [Google Scholar]
- 17. Ligot N, Krystkowiak P, Simonin C, et al. External globus pallidus stimulation modulates brain connectivity in Huntington's disease. J Cereb Blood Flow Metab. 2011;31:41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagel SJ, Machado AG, Gale JT, Lobel DA, Pandya M. Preserving cortico‐striatal function: deep brain stimulation in Huntington's disease. Front Syst Neurosci. 2015;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reiner A. Can lesions of GPe correct HD deficits? Exp Neurol. 2004;186:1‐5. [DOI] [PubMed] [Google Scholar]
- 20. Temel Y, Cao C, Vlamings R, et al. Motor and cognitive improvement by deep brain stimulation in a transgenic rat model of Huntington's disease. Neurosci Lett. 2006;406:138‐141. [DOI] [PubMed] [Google Scholar]
- 21. Cazorla M, de Carvalho FD, Chohan MO, et al. Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron. 2014;81:153‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mallet N, Micklem BR, Henny P, et al. Dichotomous organization of the external globus pallidus. Neuron. 2012;74:1075‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen MC, Ferrari L, Sacchet MD, et al. Identification of a direct GABAergic pallidocortical pathway in rodents. Eur J Neuorsci. 2015;41:748‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saunders A, Oldenburg IA, Berezovskii VK, et al. A direct GABAergic output from the basal ganglia to frontal cortex. Nature. 2015;521:85‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raymond LA. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem Biophys Res Comm. 2017;483:1051‐1062. [DOI] [PubMed] [Google Scholar]
- 26. Atherton JF, McIver EL, Mullen MR, Wokosin DL, Surmeier DJ, Bevan MD. Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington's disease. eLife. 2016;5:e21616 10.7554/eLife.21616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummings DM, Cepeda C, Levine MS. Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington's disease. ASN Neuro. 2010;2:e00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington's disease: time‐dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rothe T, Deliano M, Wojtowicz AM, et al. Pathological gamma oscillations, impaired dopamine release, synapse loss and reduced dynamic range of unitary glutamatergic synaptic transmission in the striatum of hypokinetic Q175 Huntington mice. Neuroscience. 2015;311:519‐538. [DOI] [PubMed] [Google Scholar]
- 30. Andre VM, Fisher YE, Levine MS. Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington's disease. Front Syst Neurosci. 2011;5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Foster AC, Kemp JA. Glutamate‐ and GABA‐based CNS therapeutics. Curr Opin Pharmacol. 2006;6:7‐17. [DOI] [PubMed] [Google Scholar]
- 32. Mohler H. The rise of a new GABA pharmacology. Neuropharmacology. 2011;60:1042‐1049. [DOI] [PubMed] [Google Scholar]
- 33. Braat S, Kooy RF. The GABA receptor as a therapeutic target for neurodevelopmental disorders. Neuron. 2015;86:1119‐1130. [DOI] [PubMed] [Google Scholar]
- 34. Braudeau J, Delatour B, Duchon A, et al. Specific targeting of the GABA‐A receptor alpha5 subtype by a selective inverse agonist restores cognitive deficits in down syndrome mice. J Psychopharmacol. 2011;25:1030‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brickley SG, Mody I. Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking‐mediated plasticity of inhibitory synapses. Neuron. 2011;70:385‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rudolph U, Mohler H. GABA receptor subtypes: therapeutic potential in down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol. 2013;54:483‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim YS, Yoon BE. Altered GABAergic signaling in brain disease at various stages of life. Exp Neurobiol. 2017;26:122‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martino D, Stamelou M, Bhatia KP. The differential diagnosis of Huntington's disease‐like syndromes: ‘red flags’ for the clinician. J Neurol Neurosurg Psychiatry. 2013;84:650‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fritschy JM. Epilepsy, E/I balance and GABA(A) receptor plasticity. Front Mol Neurosci. 2008;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park A, Hoffman K, Keller A. Roles of GABAA and GABAB receptors in regulating thalamic activity by the zona incerta: a computational study. J Neurophysiol. 2014;112:2580‐2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bonnert TP, McKernan RM, Farrar S, et al. theta, a novel gamma‐aminobutyric acid type A receptor subunit. Proc Natl Acad Sci U S A. 1999;96:9891‐9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma‐aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schofield PR, Darlison MG, Fujita N, et al. Sequence and functional expression of the GABA A receptor shows a ligand‐gated receptor super‐family. Nature. 1987;328:221‐227. [DOI] [PubMed] [Google Scholar]
- 45. Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chua HC, Chebib M. GABAA receptors and the diversity in their structure and pharmacology. Adv Pharmacol. 2017;79:1‐34. [DOI] [PubMed] [Google Scholar]
- 47. Ferando I, Mody I. Interneuronal GABAA receptors inside and outside of synapses. Curr Opin Neurobiol. 2014;26:57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luo R, Partridge JG, Vicini S. Distinct roles of synaptic and extrasynaptic GABAAreceptors in striatal inhibition dynamics. Front Neural Circuits. 2013;7:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific gamma‐aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796‐800. [DOI] [PubMed] [Google Scholar]
- 50. McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587‐592. [DOI] [PubMed] [Google Scholar]
- 51. Jucaite A, Cselenyi Z, Lappalainen J, et al. GABAA receptor occupancy by subtype selective GABAAalpha2,3 modulators: PET studies in humans. Psychopharmacology. 2017;234:707‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mohler H, Okada T. The benzodiazepine receptor in normal and pathological human brain. Br J Psychiatry. 1978;133:261‐268. [DOI] [PubMed] [Google Scholar]
- 53. Reisine TD, Wastek GJ, Speth RC, Bird ED, Yamamura HI. Alterations in the benzodiazepine receptor of Huntington's diseased human brain. Brain Res. 1979;165:183‐187. [DOI] [PubMed] [Google Scholar]
- 54. Faull RL, Waldvogel HJ, Nicholson LF, Synek BJ. The distribution of GABAA‐benzodiazepine receptors in the basal ganglia in Huntington's disease and in the quinolinic acid‐lesioned rat. Prog Brain Res. 1993;99:105‐123. [DOI] [PubMed] [Google Scholar]
- 55. Trifiletti RR, Snowman AM, Whitehouse PJ, Marcus KA, Snyder SH. Huntington's disease: increased number and altered regulation of benzodiazepine receptor complexes in frontal cerebral cortex. Neurology. 1987;37:916‐922. [DOI] [PubMed] [Google Scholar]
- 56. Walker FO, Young AB, Penney JB, Dovorini‐Zis K, Shoulson I. Benzodiazepine and GABA receptors in early Huntington's disease. Neurology. 1984;34:1237‐1240. [DOI] [PubMed] [Google Scholar]
- 57. Storey E, Beal MF. Neurochemical substrates of rigidity and chorea in Huntington's disease. Brain 1993;116(Pt 5):1201‐1222. [DOI] [PubMed] [Google Scholar]
- 58. Perry TL, Hansen S, Kloster M. Huntington's chorea. Deficiency of gamma‐aminobutyric acid in brain. N Engl J Med. 1973;288:337‐342. [DOI] [PubMed] [Google Scholar]
- 59. Pagano G, Niccolini F, Politis M. Current status of PET imaging in Huntington's disease. Eur J Nucl Med Mol Imaging. 2016;43:1171‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holthoff VA, Koeppe RA, Frey KA, et al. Positron emission tomography measures of benzodiazepine receptors in Huntington's disease. Ann Neurol. 1993;34:76‐81. [DOI] [PubMed] [Google Scholar]
- 61. Kunig G, Leenders KL, Sanchez‐Pernaute R, et al. Benzodiazepine receptor binding in Huntington's disease: [11C]flumazenil uptake measured using positron emission tomography. Ann Neurol. 2000;47:644‐648. [PubMed] [Google Scholar]
- 62. Philpott AL, Cummins TD, Bailey NW, Churchyard A, Fitzgerald PB, Georgiou‐Karistianis N. Cortical inhibitory deficits in premanifest and early Huntington's disease. Behav Brain Res. 2016;296:311‐317. [DOI] [PubMed] [Google Scholar]
- 63. Bauer A, Zilles K, Matusch A, Holzmann C, Riess O, von Horsten S. Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington's disease mutation. J Neurochem. 2005;94:639‐650. [DOI] [PubMed] [Google Scholar]
- 64. Dowie MJ, Bradshaw HB, Howard ML, et al. Altered CB1 receptor and endocannabinoid levels precede motor symptom onset in a transgenic mouse model of Huntington's disease. Neuroscience. 2009;163:456‐465. [DOI] [PubMed] [Google Scholar]
- 65. Kennedy L, Shelbourne PF, Dewar D. Alterations in dopamine and benzodiazepine receptor binding precede overt neuronal pathology in mice modelling early Huntington disease pathogenesis. Brain Res. 2005;1039:14‐21. [DOI] [PubMed] [Google Scholar]
- 66. Cepeda C, Galvan L, Holley SM, et al. Multiple sources of striatal inhibition are differentially affected in Huntington's disease mouse models. J Neurosci. 2013;33:7393‐7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vicini S, Ferguson C, Prybylowski K, Kralic J, Morrow AL, Homanics GE. GABA(A) receptor alpha1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;21:3009‐3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cepeda C, Starling AJ, Wu N, et al. Increased GABAergic function in mouse models of Huntington's disease: reversal by BDNF. J Neurosci Res. 2004;78:855‐867. [DOI] [PubMed] [Google Scholar]
- 69. Du Z, Tertrais M, Courtand G, et al. Differential alteration in expression of striatal GABAAR subunits in mouse models of Huntington's disease. Front Mol Neurosci. 2017;10:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arama J, Abitbol K, Goffin D, et al. GABAA receptor activity shapes the formation of inhibitory synapses between developing medium spiny neurons. Front Cell Neurosci. 2015;9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goffin D, Ali AB, Rampersaud N, et al. Dopamine‐dependent tuning of striatal inhibitory synaptogenesis. J Neurosci. 2010;30:2935‐2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Francelle L, Galvan L, Brouillet E. Possible involvement of self‐defense mechanisms in the preferential vulnerability of the striatum in Huntington's disease. Front Cell Neurosci. 2014;8:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yuen EY, Wei J, Zhong P, Yan Z. Disrupted GABAAR trafficking and synaptic inhibition in a mouse model of Huntington's disease. Neurobiol Dis. 2012;46:497‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Uchigashima M, Ohtsuka T, Kobayashi K, Watanabe M. Dopamine synapse is a neuroligin‐2‐mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc Natl Acad Sci USA. 2016;113:4206‐4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron. 2014;82:63‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non‐canonical release of GABA. Nature. 2012;490:262‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tritsch NX, Oh WJ, Gu C, Sabatini BL. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. ELife. 2014; 3: e01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35:557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. English DF, Ibanez‐Sandoval O, Stark E, et al. GABAergic circuits mediate the reinforcement‐related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Holley SM, Joshi PR, Parievsky A, et al. Enhanced GABAergic inputs contribute to functional alterations of cholinergic interneurons in the R6/2 mouse model of Huntington's disease. ENeuro. 2015;2(1): e0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Massouh M, Wallman MJ, Pourcher E, Parent A. The fate of the large striatal interneurons expressing calretinin in Huntington's disease. Neurosci Res. 2008;62:216‐224. [DOI] [PubMed] [Google Scholar]
- 82. Picconi B, Passino E, Sgobio C, et al. Plastic and behavioral abnormalities in experimental Huntington's disease: a crucial role for cholinergic interneurons. Neurobiol Dis. 2006;22:143‐152. [DOI] [PubMed] [Google Scholar]
- 83. Pisani A, Bernardi G, Ding J, Surmeier DJ. Re‐emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545‐553. [DOI] [PubMed] [Google Scholar]
- 84. Cabanas M, Bassil F, Mons N, Garret M, Cho YH. Changes in striatal activity and functional connectivity in a mouse model of Huntington's disease. PLoS ONE. 2017;12(9):e0184580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 86. Klaus A, Planert H, Hjorth JJ, Berke JD, Silberberg G, Kotaleski JH. Striatal fast‐spiking interneurons: from firing patterns to postsynaptic impact. Front Syst Neurosci. 2011;5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Planert H, Szydlowski SN, Hjorth JJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast‐spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499‐3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Smith R, Chung H, Rundquist S, et al. Cholinergic neuronal defect without cell loss in Huntington's disease. Hum Mol Genet. 2006;15:3119‐3131. [DOI] [PubMed] [Google Scholar]
- 89. Suzuki M, Desmond TJ, Albin RL, Frey KA. Vesicular neurotransmitter transporters in Huntington's disease: initial observations and comparison with traditional synaptic markers. Synapse. 2001;41:329‐336. [DOI] [PubMed] [Google Scholar]
- 90. Farrar AM, Callahan JW, Abercrombie ED. Reduced striatal acetylcholine efflux in the R6/2 mouse model of Huntington's disease: an examination of the role of altered inhibitory and excitatory mechanisms. Exp Neurol. 2011;232:119‐125. [DOI] [PubMed] [Google Scholar]
- 91. Waldvogel HJ, Kubota Y, Fritschy J, Mohler H, Faull RL. Regional and cellular localisation of GABA(A) receptor subunits in the human basal ganglia: an autoradiographic and immunohistochemical study. J Comp Neurol. 1999;415:313‐340. [DOI] [PubMed] [Google Scholar]
- 92. Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262‐269. [DOI] [PubMed] [Google Scholar]
- 93. Wojtowicz AM, Dvorzhak A, Semtner M, Grantyn R. Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT‐3. Front Neural Circuits. 2013;7:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kuhn A, Goldstein DR, Hodges A, et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild‐type huntingtin dosage. Hum Mol Genet. 2007;16:1845‐1861. [DOI] [PubMed] [Google Scholar]
- 95. Seredenina T, Luthi‐Carter R. What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis. 2012;45:83‐98. [DOI] [PubMed] [Google Scholar]
- 96. Schwarzer C, Berresheim U, Pirker S, et al. Distribution of the major gamma‐aminobutyric acid(A) receptor subunits in the basal ganglia and associated limbic brain areas of the adult rat. J Comp Neurol. 2001;433:526‐549. [DOI] [PubMed] [Google Scholar]
- 97. Glajch KE, Kelver DA, Hegeman DJ, et al. Npas1 + pallidal neurons target striatal projection neurons. J Neurosci. 2016;36:5472‐5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kita H. Parvalbumin‐immunopositive neurons in rat globus pallidus: a light and electron microscopic study. Brain Res. 1994;657:31‐41. [DOI] [PubMed] [Google Scholar]
- 99. Sadek AR, Magill PJ, Bolam JP. A single‐cell analysis of intrinsic connectivity in the rat globus pallidus. J Neurosci. 2007;27:6352‐6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Akopian G, Barry J, Cepeda C, Levine MS. Altered membrane properties and firing patterns of external globus pallidus neurons in the R6/2 mouse model of Huntington's disease. J Neurosci Res. 2016;94:1400‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Du Z, Chazalon M, Bestaven E, et al. Early GABAergic transmission defects in the external globus pallidus and rest/activity rhythm alteration in a mouse model of Huntington's disease. Neuroscience. 2016;329:363‐379. [DOI] [PubMed] [Google Scholar]
- 102. Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569‐575. [DOI] [PubMed] [Google Scholar]
- 103. Gross A, Sims RE, Swinny JD, Sieghart W, Bolam JP, Stanford IM. Differential localization of GABA(A) receptor subunits in relation to rat striatopallidal and pallidopallidal synapses. Eur J Neuorsci. 2011;33:868‐878. [DOI] [PubMed] [Google Scholar]
- 104. Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013;236:345‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Allen KL, Waldvogel HJ, Glass M, Faull RL. Cannabinoid (CB(1)), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington's disease. J Chem Neuroanat. 2009;37:266‐281. [DOI] [PubMed] [Google Scholar]
- 106. Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013;14:708‐721. [DOI] [PubMed] [Google Scholar]
- 107. Starr PA, Kang GA, Heath S, Shimamoto S, Turner RS. Pallidal neuronal discharge in Huntington's disease: support for selective loss of striatal cells originating the indirect pathway. Exp Neurol. 2008;211:227‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Peiris JB, Boralessa H, Lionel ND. Clonazepam in the treatment of choreiform activity. Med J Aust. 1976;1:225‐227. [PubMed] [Google Scholar]
- 109. Stewart JT. Treatment of Huntington's disease with clonazepam. South Med J. 1988;81:102. [DOI] [PubMed] [Google Scholar]
- 110. Farrell DF, Hofmann WW. A quantitative evaluation of the effect of diazepam in Huntington's chorea. Arch Phys Med Rehabil. 1968;49:586‐591. [PubMed] [Google Scholar]
- 111. Foster NL, Chase TN, Denaro A, Hare TA, Tamminga CA. THIP treatment of Huntington's disease. Neurology. 1983;33:637‐639. [DOI] [PubMed] [Google Scholar]
- 112. Mason SL, Barker RA. Emerging drug therapies in Huntington's disease. Expert Opin Emerg Drugs. 2009;14:273‐297. [DOI] [PubMed] [Google Scholar]
- 113. Cardoso F. Nonmotor symptoms in Huntington disease. Int Rev Neurobiol. 2017;134:1397‐1408. [DOI] [PubMed] [Google Scholar]
- 114. Whissell PD, Lecker I, Wang DS, Yu J, Orser BA. Altered expression of deltaGABAA receptors in health and disease. Neuropharmacology. 2015;88:24‐35. [DOI] [PubMed] [Google Scholar]
- 115. De San Martin JZ, Delabar JM, Bacci A, Potier MC. GABAergic over‐inhibition, a promising hypothesis for cognitive deficits in down syndrome. Free Radic Biol Med. 2017;114:33‐39. [DOI] [PubMed] [Google Scholar]
- 116. Lebreton F, Cayzac S, Pietropaolo S, Jeantet Y, Cho YH. Sleep physiology alterations precede plethoric phenotypic changes in R6/1 Huntington's disease mice. PLoS ONE. 2015;10:e0126972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Morton AJ. Circadian and sleep disorder in Huntington's disease. Exp Neurol. 2013;243:34‐44. [DOI] [PubMed] [Google Scholar]
- 118. Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 2010;11:589‐599. [DOI] [PubMed] [Google Scholar]
- 119. Kantor S, Varga J, Morton AJ. A single dose of hypnotic corrects sleep and EEG abnormalities in symptomatic Huntington's disease mice. Neuropharmacology. 2016;105(Pt 7):298‐307. [DOI] [PubMed] [Google Scholar]
- 120. Pallier PN, Maywood ES, Zheng Z, et al. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington's disease. J Neurosci. 2007;27:7869‐7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pallier PN, Morton AJ. Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington's disease. Brain Res. 2009;1279:90‐98. [DOI] [PubMed] [Google Scholar]
- 122. Goodman AO, Rogers L, Pilsworth S, et al. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington's disease. Curr Neurol Neurosci Rep. 2011;11:211‐217. [DOI] [PubMed] [Google Scholar]
- 123. Kantor S, Szabo L, Varga J, Cuesta M, Morton AJ. Progressive sleep and electroencephalogram changes in mice carrying the Huntington's disease mutation. Brain 2013;136(Pt 7):2147‐2158. [DOI] [PubMed] [Google Scholar]
- 124. Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep‐wake cycle and circadian timing in Huntington's disease. J Neurosci. 2005;25:157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lazarus M, Chen JF, Urade Y, Huang ZL. Role of the basal ganglia in the control of sleep and wakefulness. Curr Opin Neurobiol. 2013;23:780‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Qiu MH, Vetrivelan R, Fuller PM, Lu J. Basal ganglia control of sleep‐wake behavior and cortical activation. Eur J Neurosci. 2010;31:499‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yuan XS, Wang L, Dong H, et al. Striatal adenosine A2A receptor neurons control active‐period sleep via parvalbumin neurons in external globus pallidus. ELife. 2017;6:e29055 10.7554/eLife.29055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mohler H. The legacy of the benzodiazepine receptor: from flumazenil to enhancing cognition in down syndrome and social interaction in autism. Adv Pharmacol. 2015;72:1‐36. [DOI] [PubMed] [Google Scholar]
- 129. Hsu YT, Chang YG, Chang CP, et al. Altered behavioral responses to gamma‐aminobutyric acid pharmacological agents in a mouse model of Huntington's disease. Mov Disord. 2017;11:1600‐1609. [DOI] [PubMed] [Google Scholar]
- 130. Sarkar J, Wakefield S, MacKenzie G, Moss SJ, Maguire J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid‐sensitive GABAA receptors. J Neurosci. 2011;31:18198‐18210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang JM, Singh C, Liu L, et al. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:6498‐6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Holm MM, Nieto‐Gonzalez JL, Vardya I, et al. Hippocampal GABAergic dysfunction in a rat chronic mild stress model of depression. Hippocampus. 2011;21:422‐433. [DOI] [PubMed] [Google Scholar]
- 133. Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience. 2010;167:644‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Grantyn R, Henneberger C, Juttner R, Meier JC, Kirischuk S. Functional hallmarks of GABAergic synapse maturation and the diverse roles of neurotrophins. Front Cell Neurosci. 2011;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]