

Summary
Aims
Fingolimod is a sphingosine‐1‐phosphate (S1P) receptor modulator approved for the treatment of the relapsing form of multiple sclerosis (MS). It prevents the egress of lymphocyte subpopulations from lymphoid tissues into the circulation. Here, we explored the broad effects of fingolimod on gene expression in different immune cell subsets.
Methods
Utilizing 150 high‐resolution microarrays from Affymetrix, we obtained the transcriptome profiles of 5 cell populations, which were separated from the peripheral blood of MS patients prior to and following oral administration of fingolimod.
Results
After 3 months of treatment, significant transcriptome shifts were seen in CD4+ and CD8+ cells, which is mainly attributable to the selective homing of naive T cells and central memory T cells. Although the number of B cells was greatly reduced in the blood of fingolimod‐treated MS patients, the analysis of differential expression in CD19+ cells identified only a small set of 42 genes, which indicated a slightly higher frequency of transitional B cells. The transcriptome signatures of CD14+ monocytes and CD56+ natural killer cells were not affected.
Conclusion
Our study corroborates changes in the composition of circulating immune cells in response to fingolimod and delineates the respective implications at the RNA level. Our data may be valuable for comparing the effects of novel S1P receptor modulating agents, which may be a therapeutic option for patients with secondary progressive MS as well.
Keywords: CD19+ B cells, fingolimod therapy, peripheral blood, relapsing‐remitting multiple sclerosis, transcriptome microarray analysis
Key points.
CD4+ and CD8+ cell populations demonstrated huge shifts in the transcriptome during fingolimod therapy, reflecting the selective sequestration of smaller subpopulations in lymphoid tissues (eg, naive T cells and central memory T cells)
CD19+ cells, although reduced in number in the blood circulation by more than 80%, showed only marginal gene expression changes, indicating that fingolimod similarly affects the trafficking of the different B cell subpopulations
CD14+ monocytes and CD56+ natural killer cells showed fairly stable cell counts and transcriptome profiles in the course of treatment, with no evidence of significant gene regulatory effects independent of the sphingosine‐1‐phosphate pathway
1. INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory and demyelinating disease of the central nervous system (CNS), which is driven by autoreactive immune cells.1, 2 Distinct B cell and T cell subpopulations play different roles in the initiation and progression of this clinically heterogeneous disease. Activated cytotoxic T cells provoke inflammation and neurodegeneration in the CNS by targeting autoantigenic structures.3 T helper cells and memory B cells are mediating the destructive processes by presenting antigens and secreting proinflammatory cytokines to costimulate immune responses.4 The intrathecal formation of antibody‐producing plasma cells and plasmablasts is another hallmark of the pathophysiology of MS.5
Fingolimod is an oral drug approved for the treatment of relapsing‐remitting MS (RRMS).6 As a sphingosine‐1‐phosphate (S1P) receptor modulator, it affects the migration of lymphocytes out of secondary lymphatic tissues.7, 8 During fingolimod therapy, certain immune cell subpopulations are sequestered in lymph nodes, because their S1P receptors are internalized and degraded. In consequence, there are less autoreactive cells circulating in the blood and hence less cells are able to migrate into the CNS. Immune cells expressing lymph node homing receptors such as CCR7 and SELL are preferentially trapped. Continued administration of fingolimod thus leads to a shift of cell populations in the peripheral blood. After 3 months of therapy, there is a significant decrease in the proportions of CD4+ T cells (from 46% to 6% of lymphocytes) and CD19+ B cells (from 11% to 6%) and a moderate decrease of CD8+ T cells (from 20% to 15%),9 while frequencies of CD14+ monocytes and CD56+ natural killer (NK) cells are relatively increased.10, 11 However, although the altered trafficking of cell subsets has been well studied, the effects of fingolimod on gene expression and cellular signaling are still not fully established.
We examined the transcriptome of lymphocytes and monocytes before and during S1P receptor modulator therapy to obtain further insights on the effects on molecular signatures thought to be related to the pathophysiology of MS. To identify differentially expressed genes (DEG) in immune cells separated from the blood of treated RRMS patients, we measured all human protein‐coding and noncoding transcripts at the exon level. The data allowed to analyze to which extent transcriptome changes correlate with the fingolimod‐induced shift of circulating cell populations, to investigate gene regulatory programs potentially caused by downstream S1P signaling, and to define molecular markers for monitoring therapeutic responses.
2. METHODS
Details about study design and analysis methods were published previously.12, 13 Briefly, a large dataset comprising 150 high‐resolution microarrays was obtained for specific cell populations from blood samples of MS patients. The patients were aged between 26 and 46 and diagnosed with RRMS according to the revised McDonald criteria.14 Disease duration was, on average, 8 years. All patients were previously treated with either glatiramer acetate or interferon‐beta, and none of them received a S1P receptor modulator before. In the 12 months prior to the study, each patient experienced 1‐3 relapses. For this reason, the therapy was switched to fingolimod (standard dose of 0.5 mg orally once daily), following the treatment guidelines and recommendations for routine medical care of the German Society of Neurology. As part of our research on MS, the study was approved by the local ethics committee of the University of Rostock and conducted in accordance with the ethical principles of the Declaration of Helsinki. All patients gave prior written informed consent to participate in this study.
Blood samples (~20 mL) were always taken immediately before the start of fingolimod therapy (baseline) as well as after 24 hours (before the second dose of fingolimod) and after 3 months. Five different cell subsets were magnetically labeled based on the cell surface markers CD4, CD8, CD14, CD19, and CD56 using Whole Blood MicroBeads from Miltenyi Biotec (Bergisch Gladbach, Germany). Afterward, the positively selected cell fractions were collected and then counted under a conventional microscope. Total RNA was isolated from the cells using the mirVana Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA). For each cell population, samples of ten MS patients were prepared to analyze the gene expression in the course of therapy. In doing so, the 3 longitudinal samples for each patient and cell subset were handled at the same day by the same laboratory technician. In total, 150 high‐coverage human transcriptome arrays (HTA) 2.0 from Affymetrix (Santa Clara, CA, USA) were prepared according to the manufacturer's instructions. HTA 2.0 microarrays contain over 6 million 25mer oligonucleotide probes for measuring the transcripts of all coding and noncoding genes. They are designed with 10 probes per exon fragment and 4 probes per exon‐exon splice junction.15 The probes were summarized in the Expression Console 1.3.1 software (Affymetrix) to 70523 gene‐level probe sets and 914585 exon‐level probe sets. Quality control and data normalization by the robust multiarray average algorithm16 including a log2 transformation were performed as described elsewhere.12, 13 The processing of the microarray data was performed separately for each cell population. The complete raw and processed data are publicly available together with the clinico‐demographic data of the patients in the Gene Expression Omnibus (GEO) database (SuperSeries GSE73174).
The Transcriptome Analysis Console (TAC) software version 1.0 (Affymetrix) was used to explore the effects of fingolimod on the transcriptome in the 5 different cell populations. For each gene‐level probe set, the signal intensities of the 24‐hour samples and the 3‐month samples, respectively, were compared with those of the pretreatment samples. DEG were determined by filtering probe sets with Student's two‐tailed unpaired t test P‐value <0.001 and fold change (FC) >1.5 or <−1.5. Probe sets not mapping to genes (n = 2995; hybridization controls, etc.) were discarded. The FC was calculated by TAC in linear scale, with negative FC indicating reduced gene expression. Accordingly, a statistically significant increase of more than 50% or decrease of more than 33% in the transcript levels distinguished DEG in response to the therapy. False discovery rate (FDR) correction17 was applied to adjust the P‐values for multiple testing.
For DEG in CD19+ B cells, a gene regulatory network (GRN) model was constructed on the basis of the microarray data and information on predicted transcription factor (TF) binding sites (TFBS) in the promoter region of the genes. For this purpose, TFBS‐integrating least angle regression (TILAR) was employed as described elsewhere.18, 19 We used transcription start sites from the GeneCards database version 3.0420 and evolutionarily conserved TFBS from the tfbsConsSites track of the UCSC database build hg19.21 TFBS‐integrating least angle regression (TILAR) was applied with backward stepwise selection on TF‐gene interactions and no additional prior knowledge on gene‐TF interactions (δ = 1). All other steps of the GRN inference (eg, prevention of overfitting) were performed with the default configuration.
3. RESULTS
Moderate differences (up to ~20%) in the numbers of cells that were separated from the individual blood samples were noted 24 hours after the first administration of fingolimod. After 3 months of therapy, however, the mean cell counts of sorted CD4‐, CD8‐, and CD19‐expressing cells were significantly reduced by ~60%‐90% compared to baseline (Table 1). The numbers of circulating CD14+ and CD56+ cells showed strong interindividual differences but, on average, remained relatively stable even after continued drug administration (Figure 1A).
Table 1.
Overview of fingolimod‐induced cellular and transcriptome shifts for different blood cell populations
| Cell population | 24 h after the first dose | After 3 mo of therapy | ||
|---|---|---|---|---|
| Cell count change (%) | DEG (up/down) | Cell count change (%) | DEG (up/down) | |
| CD4+ | −21.1 | 1 (1/0) | −92.4 | 6489 (2574/3915) |
| CD8+ | 18.7 | 0 (0/0) | −59.5 | 861 (690/171) |
| CD14+ | 15.8 | 0 (0/0) | −5.3 | 1 (1/0) |
| CD19+ | −14.2 | 0 (0/0) | −87.0 | 42 (41/1) |
| CD56+ | 15.5 | 0 (0/0) | −15.7 | 0 (0/0) |
Shown are the effects of fingolimod therapy on average absolute counts of cells sorted from the peripheral blood of relapsing‐remitting MS patients (see also Figure 1A). In comparison with baseline, significant changes in the number of circulating cells were only observed after 3 mo of treatment for CD4+ cells (Student's t test P‐value = 0.00002), CD8+ cells (P = 0.027), and CD19+ cells (P = 0.00006). Moreover, the number of differentially expressed genes (DEG, t test P‐values <0.001 and fold changes >1.5 or <−1.5) is indicated for each cell population and each time point during therapy (see also Figure 1D). There was basically no increase or decrease in expression 24 h after the first dose of fingolimod. In contrast, after 3 mo of treatment, huge transcriptome alterations were seen in CD4+ cells and CD8+ cells. For CD19+ B cells, there were 42 DEG despite a strong reduction in the respective cell count of almost 90%.
Figure 1.
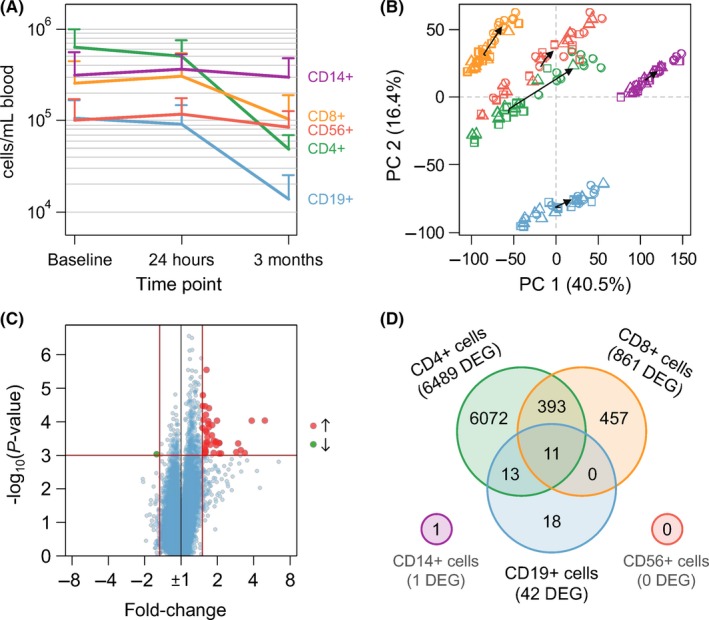
Analysis of cellular and molecular effects of fingolimod therapy in the blood of RRMS patients. Microarray‐based transcriptome profiling was performed for monocytes and lymphocyte subpopulations, which are represented in the figure by different colors (CD4+ cells in green, CD8+ cells in orange, CD14+ cells in purple, CD19+ cells in blue, and CD56+ cells in red). A, Concentration of cells (mean plus standard deviation) for each of the 5 separated cell populations and each of the 3 study time points. After 3 mo of fingolimod therapy, the numbers of sorted CD4+ cells, CD8+ cells as well as CD19+ cells were significantly reduced (Student's t test P‐values <0.05). B, Principal component (PC) analysis of the complete dataset comprising 150 microarrays (10 patients × 3 time points × 5 cell populations). The samples were plotted in the first two dimensions explaining most of the variation in the normalized and mean‐centered gene‐level probe set signal intensities (n = 70 523 per array). Dark arrows connect the centers of each time point (depicted by rectangles for baseline, triangles for “24 h,” and circles for “3 mo”). C, Volcano plot of gene expression changes in CD19+ cells within 3 mo after initiation of fingolimod therapy. Red and green dots display probe sets for gene transcripts expressed at significantly higher or lower levels, respectively (n = 42). The red vertical and horizontal lines indicate the fold change and P‐value filtering criteria. D, Venn diagram summarizing for each cell population the sets and their intersections of differentially expressed genes (DEG) after 3 mo of fingolimod therapy relative to pretreatment levels. Eleven DEG were shared among CD4+ cells, CD8+ cells, and CD19+ cells
The transcriptome analysis of the 5 immune cell populations revealed only a single DEG 24 hours after treatment initiation (Table 1). At this time point, solely the signal intensities of the probe set “TC10001500.hg.1” (no official gene symbol available) were significantly increased in the CD4+ cell subset. The lack of early gene regulatory effects reflects that the first dose of fingolimod does not have a lasting effect on the frequencies of blood cell populations and also indicates that S1P receptor downstream signaling does not elicit a discernible transcriptional response in these cells.
Significant alterations in the transcriptome profiles were seen after 3 months of fingolimod therapy (Table 1). The treatment effect was also well visible in a principal component analysis of the whole dataset comprising the signals of 70523 gene‐level probe sets for all 150 HTA 2.0 microarrays, which were aggregated after data normalization (Figure 1B). The greatest change in gene expression was observed in the CD4+ cell subpopulation with in total 6489 DEG (890 of those even had a FC >2.0 or <−2.0). For CD8+ cells and CD19+ cells, 861 genes and 42 genes, respectively, were found to be expressed at significantly higher or lower levels relative to pretreatment levels. On the other hand, there were no significant gene expression dynamics in CD56+ cells and CD14+ cells. For the latter, only 1 probe set (“TC03002386.hg.1,” no official gene symbol) fulfilled our filtering criteria (FC >1.5 or <−1.5 and t test P‐value <0.001).
The huge transcriptome shifts in CD4+ cells and CD8+ cells clearly display the selective homing of T cell subpopulations to lymphoid tissues in response to sustained fingolimod therapy.12, 13 On the contrary, the gene expression changes in CD19+ B cells were quite subtle considering the fact that their cell count was also remarkably reduced in the peripheral blood by almost 90% after 3 months (Table 1). As the purity of isolated B cells was high before and during therapy (Figure S1), this indicates that fingolimod similarly affects the trafficking of the different B cell subpopulations. Overall, 41 genes showed an up to 5‐fold increase in expression, while just 1 transcript (probe set “TC15000493.hg.1”) was expressed at lower levels in the CD19+ cell subset during therapy (Figure 1C). Five of these genes remained significant at the 5% FDR level (Table 2). Predictions of TFBS revealed 5 TF to be associated with the DEG at the significance level α = 0.05. This information was used as template for modeling the underlying GRN using TILAR.18, 19 The resulting network linked 27 of the DEG by 15 TF‐gene interactions and 42 gene‐TF interactions (Figure S2). MEF2A appeared as the most highly connected TF in the network.
Table 2.
Genes with significant changes in expression in B cells from MS patients receiving fingolimod therapy
| No | Probe set | Chromosome location | FC | P‐value | Probes | Gene symbol | 2 sets | 3 sets |
|---|---|---|---|---|---|---|---|---|
| 1 | TC01001498.hg.1 | chr1(+):171810621‐172387606 | 2.14 | 0.000883 | 305 | DNM3 | ||
| 2 | TC01001619.hg.1 | chr1(+):192127587‐192154945 | 4.95 | 0.000092 | 105 | RGS18 | ✓ | |
| 3 | TC02001958.hg.1 | chr2(−):70476123‐70476193 | 2.08 | 0.000431 | 30 | (tRNA Gly) | ✓ | |
| 4 | TC02004620.hg.1 | chr2(−):145143042‐145143557 | 1.66 | 0.000287 | 30 | (ZEB2) | ✓ | ✓ |
| 5 | TC02004622.hg.1 | chr2(−):145251832‐145254445 | 1.70 | 0.000039a | 30 | (ZEB2) | ✓ | ✓ |
| 6 | TC02004623.hg.1 | chr2(−):145268952‐145277958 | 1.63 | 0.000003a | 30 | (ZEB2) | ✓ | ✓ |
| 7 | TC04002285.hg.1 | chr4(+):156902136‐156938403 | 3.13 | 0.000705 | 50 | — | ||
| 8 | TC05002969.hg.1 | chr5(−):39383148‐39393457 | 3.37 | 0.000854 | 70 | (DAB2) | ✓ | ✓ |
| 9 | TC05000368.hg.1 | chr5(+):75699074‐76003957 | 1.62 | 0.000409 | 629 | IQGAP2 | ✓ | ✓ |
| 10 | TC05001839.hg.1 | chr5(−):138700366‐138705406 | 1.68 | 0.000117 | 19 | (SLC23A1) | ✓ | |
| 11 | TC06002615.hg.1 | chr6(+):21597661‐21598850 | 1.87 | 0.000369 | 15 | (SOX4) | ✓ | |
| 12 | TC06004064.hg.1 | chr6(+):27777842‐27779078 | 3.85 | 0.000093 | 20 | HIST1H3A‐J | ||
| 13 | TC06000759.hg.1 | chr6(+):80451636‐80451669 | 1.96 | 0.000503 | 10 | RNY4 | ✓ | |
| 14 | TC07000495.hg.1 | chr7(+):77166415‐77269388 | 1.57 | 0.000616 | 476 | PTPN12 | ✓ | ✓ |
| 15 | TC08001286.hg.1 | chr8(−):67474410‐67525529 | 1.60 | 0.000701 | 242 | MYBL1 | ✓ | ✓ |
| 16 | TC08002364.hg.1 | chr8(−):67476954‐67525175 | 1.80 | 0.000253 | 160 | (MYBL1) | ✓ | ✓ |
| 17 | TC08000531.hg.1 | chr8(+):86376081‐86393722 | 1.93 | 0.000411 | 154 | CA2 | ||
| 18 | TC08002454.hg.1 | chr8(−):102193086‐102195404 | 1.65 | 0.000410 | 30 | (ZNF706) | ||
| 19 | TC08002460.hg.1 | chr8(−):104395306‐104396082 | 1.81 | 0.000126 | 30 | — | ✓ | ✓ |
| 20 | TC09001206.hg.1 | chr9(−):74517371‐74517466 | 1.61 | 0.000062 | 30 | (Y RNA) | ✓ | |
| 21 | TC09001401.hg.1 | chr9(−):100689073‐100707138 | 1.99 | 0.000093 | 90 | HEMGN | ||
| 22 | TC09001610.hg.1 | chr9(−):130594914‐130595022 | 1.60 | 0.000821 | 30 | (Y RNA) | ||
| 23 | TC10001072.hg.1 | chr10(−):17256238‐17271983 | 1.63 | 0.000206 | 60 | VIM‐AS1 | ✓ | |
| 24 | TC10002487.hg.1 | chr10(−):17256245‐17271983 | 1.70 | 0.000146 | 90 | (VIM‐AS1) | ✓ | ✓ |
| 25 | TC10001111.hg.1 | chr10(−):25270908‐25351208 | 2.07 | 0.000889 | 120 | ENKUR | ||
| 26 | TC10002540.hg.1 | chr10(−):31109147‐31110214 | 2.97 | 0.000446 | 27 | — | ||
| 27 | TC10000448.hg.1 | chr10(+):73980510‐73980610 | 1.59 | 0.000035a | 30 | (Y RNA) | ||
| 28 | TC11002664.hg.1 | chr11(+):48191599‐48192391 | 1.53 | 0.000087 | 24 | (PTPRJ) | ✓ | |
| 29 | TC11002092.hg.1 | chr11(−):74971166‐75062875 | 1.58 | 0.000889 | 197 | ARRB1 | ✓ | |
| 30 | TC12000011.hg.1 | chr12(+):890299‐890424 | 1.61 | 0.000185 | 30 | (WNK1) | ✓ | ✓ |
| 31 | TC12002795.hg.1 | chr12(−):24332887‐24333863 | 1.87 | 0.000852 | 30 | (SOX5) | ||
| 32 | TC12000297.hg.1 | chr12(+):32552463‐32798984 | 1.51 | 0.000034a | 454 | FGD4 | ✓ | |
| 33 | TC12000977.hg.1 | chr12(+):123252646‐123252747 | 1.75 | 0.000486 | 30 | (Y RNA) | ||
| 34 | TC13001561.hg.1 | chr13(−):67787977‐67789866 | 1.68 | 0.000096 | 30 | (PCDH9) | ||
| 35 | TC15001162.hg.1 | chr15(−):31248451‐31248553 | 1.60 | 0.000778 | 30 | (MTMR10) | ✓ | |
| 36 | TC15000493.hg.1 | chr15(+):62533117‐62533147 | −1.59 | 0.000930 | 7 | — | ||
| 37 | TC16001729.hg.1 | chr16(−):223162‐223620 | 1.95 | 0.000390 | 30 | — | ||
| 38 | TC16000008.hg.1 | chr16(+):226679‐227521 | 2.87 | 0.000807 | 70 | HBA1‐2 | ||
| 39 | TC16000732.hg.1 | chr16(−):686736‐686806 | 2.08 | 0.000431 | 30 | (tRNA Gly) | ✓ | |
| 40 | TC17000978.hg.1 | chr17(−):1519486‐1519589 | 1.51 | 0.000154 | 30 | (SLC43A2) | ✓ | |
| 41 | TC17002022.hg.1 | chr17(+):5016654‐5018299 | 1.61 | 0.000503 | 30 | — | ||
| 42 | TC19002594.hg.1 | chr19(−):57804451‐57805837 | 1.52 | 0.000016a | 30 | — |
In total, 42 genes were filtered as differentially expressed with t test P‐values <0.001 and fold changes (FC) >1.5 or <−1.5 in CD19+ B cells when comparing the data at baseline with the data at 3 mo of treatment. The table provides the Affymetrix microarray gene‐level probe set identifiers, the chromosome positions of the genes (GRCh37/hg19 genome assembly) in sorted order, FC, raw t test P‐values, the number of 25mer oligonucleotide probes for each probe set as well as official gene symbols if available. Symbols in brackets (n = 21) indicate that the respective probe set interrogates only a part of a protein‐coding gene or a noncoding RNA class. We marked the genes that were also expressed at significantly higher transcript levels in CD4+ cells (“2 sets,” n = 24) or in both CD4+ cells and CD8+ cells (“3 sets,” n = 11) as depicted in Figure 1D.
Significant at false discovery rate of 5%.
A comparative analysis of the sets of DEG at the 3‐month time point showed marked overlaps. Almost half of the DEG of CD8+ cells (46.9%) were also modulated in expression in CD4+ cells, and 11 genes were consistently upregulated in CD4+, CD8+, and CD19+ cells (Figure 1D). Three of these 11 shared DEG encode the proteins IQGAP2, MYBL1, and PTPN12 (Figure 2), and the other ones constitute gene fragments and less well‐characterized noncoding transcripts. A subset of 18 genes was significantly differentially expressed in CD19+ B cells only.
Figure 2.
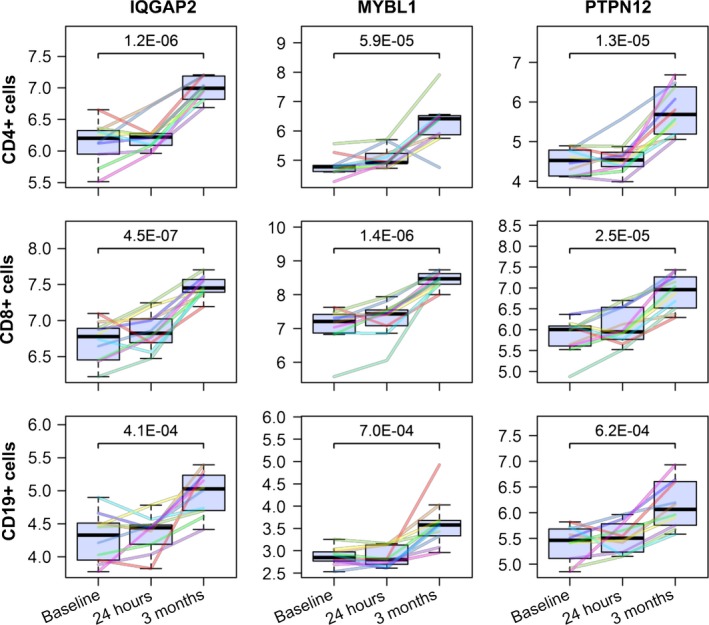
Gene expression dynamics of IQGAP2, MYBL1, and PTPN12 in the course of fingolimod therapy. Matrix of boxplots visualizing the mRNA expression of 3 selected genes (in columns) in 3 distinct immune cell populations (in rows). The cells were separated from the peripheral blood of relapsing‐remitting MS patients based on cell surface markers such as CD4 and CD8 for T cells and CD19 for B cells. Pretreatment expression values as well as levels after 24 h and 3 mo of fingolimod therapy are presented in log2 scale. Differently colored lines refer to each of the 10 individual patients per cell population. The P‐values above the brackets were calculated by Student's t test. In all 3 cell populations shown, IQGAP2, MYBL1, and PTPN12 were consistently expressed at significantly higher transcript levels in response to continued administration of fingolimod in comparison with baseline (P‐values <0.001 and fold changes >1.5)
4. DISCUSSION
We obtained a huge dataset of 150 high‐resolution HTA 2.0 microarrays to profile the transcriptome of 5 different types of circulating immune cells before and during fingolimod therapy. Alterations in gene expression were not yet observed after the first dose but could be ascertained in CD4+, CD8+, and CD19+ cells (but not in CD14+ and CD56+ cells) after continued drug exposure. This goes along with a significant reduction in the number of T and B cells and, consequently, a relative increase in the frequencies of CD14+ monocytes and CD56+ NK cells in the peripheral blood of treated patients. These changes in the composition of lymphocytes, as a result of the prevention of their egress from lymphoid tissues, have already been well established in the literature.9, 10, 11, 22 Preferential lymph node homing of naive T cells and central memory T cells, but not effector memory T cells, correlates with hundreds of DEG that we identified in the CD4+ and CD8+ cell subsets in response to fingolimod treatment. Additional gene regulatory effects independent of S1P receptor binding, however, could not be evidenced in these cells in our study. The transcriptome shift in T cells and its impact on biological processes and S1P‐related pathways are presented in detail in our previous publications.12, 13 In the following, we will focus on the results for the subset of B cells, which recently gained increasing attention in MS research.23, 24
Other studies have shown that the absolute numbers of naive B cells, memory B cells, and plasmablasts are significantly reduced in the peripheral blood during fingolimod treatment.25, 26, 27 As all these subpopulations are affected, only 42 DEG passed our filtering criteria when comparing the gene expression levels in separated CD19+ cells after 3 months of therapy vs baseline, although total B cell counts were decreased by 87%. Some of the DEG such as arrestin beta‐1 (ARRB1), hemoglobin alpha 1/2 (HBA1‐2), SRY‐box 4 (SOX4), and genes of the histone H3 family (HIST1H3A‐J) are known to be expressed at higher levels in immature B cells compared to mature B cells.28, 29, 30, 31 Their elevated expression thus likely indicates a proportionally higher frequency of circulating transitional B cells in fingolimod‐treated MS patients, which would also explain the emergence of TF families promoting early B cell development in the GRN (such as MEF232), even though classical cell surface markers such as CD24 and CD38 were not identified as DEG. Consistent with this, a relative (but not absolute) increase of newly produced immature B cells during therapy has been demonstrated by flow cytometry immunophenotyping.10, 27, 33, 34 Interestingly, lower baseline percentages of transitional B cells in the peripheral blood might predict clinical response to fingolimod,34 but this still has to be confirmed in larger patient cohorts.
The probe sets for the protein‐coding genes IQGAP2, PTPN12, and MYBL1 were consistently filtered in the transcriptome analyses of circulating CD4+, CD8+, and CD19+ cells. This implicates that some of the 42 DEG that were identified in the CD19+ cell population after 3 months of fingolimod therapy exert various functions not only in B cells but also in other immune and nonimmune cells. IQGAP2 is a broadly expressed calmodulin binding protein regulating cytoskeletal dynamics.35 PTPN12, on the other hand, is a protein tyrosine phosphatase that was shown to suppress antigen‐receptor signaling in B cells and T cells.36 The related family member PTPRJ also plays a role in lymphocyte signaling, particularly in B cell activation and B cell development.36, 37 Notably, the filtered probe set for PTPRJ (“TC11002664.hg.1”) matches only the 3′ untranslated region of the last exon, whereas the designated probe set for the full‐length transcript (“TC11000412.hg.1”) narrowly missed the significance threshold (t test P‐value = 0.004). MYBL1 is a strong transcriptional activator, which is implicated in Burkitt's lymphoma,38 a disease associated with Epstein‐Barr virus (EBV).39 Three probe sets measured increased RNA levels of gene fragments from ZEB2, a transcriptional repressor that contributes to maintenance of EBV latency by inhibiting lytic reactivation.40 Latent EBV infection of B cells is one of the strongest environmental risk factors for MS.1 However, the possible relevance of MYBL1 and ZEB2 in MS remains to be investigated. Another differentially expressed gene in B cells during fingolimod therapy was ARRB1. An earlier study demonstrated the presence of antibodies reactive with ARRB1 in sera from patients with MS.41 Moreover, the first intron of ARRB1 harbors the precursor sequence for the microRNA hsa‐miR‐326, which is dysregulated in peripheral blood cells of MS patients.42 DNM3 is also host gene of a microRNA, namely hsa‐miR‐3120. DNM3 is preferentially expressed in the brain and as a GTP‐binding protein involved in vesicular transport,43 while hsa‐miR‐3120 regulates heat shock cognate protein 70 and vesicle uncoating.44 Furthermore, some of the genes, for example, ARRB1, FGD4, IQGAP2, and RGS18, are known to act as regulators of GTPases.45
Among the DEG in CD19+ cells, there were also several noncoding transcripts, which deserve further research. For instance, Ro‐associated Y4 (RNY4) is affiliated with the Y RNA class, a group of small RNA acting as licensing factors for chromosomal DNA replication through interactions with chromatin and initiation proteins.46 In addition to RNY4, we filtered 4 probe sets corresponding to paralogous Y RNA sequences in intronic regions. Moreover, 2 copies of glycine transfer RNA were found with elevated levels of expression in the B cells from patients treated with fingolimod for 3 months in comparison with baseline levels.
The strengths of our study are that we analyzed the gene expression signatures of distinct blood cell populations longitudinally in a well‐characterized cohort of RRMS patients.12, 13 Moreover, we used 150 high‐resolution HTA 2.0 microarrays15 to obtain very accurate and comprehensive snapshots of the cellular transcriptomes. As limitations, our study does not give insights into RNA expression changes after the 3‐month time point, and it was not designed to detect prognostic biomarkers of the long‐term individual clinical response to fingolimod therapy. Subsequent studies thus may further evaluate the identified DEG in larger cohorts using focused approaches such as real‐time PCR. On the other hand, a more exhaustive characterization of immune cells from treated patients is feasible as massively parallel sequencing technologies emerged that allow to measure RNA profiles at single‐cell resolution.47, 48 Other possible extensions of this study include the analysis of alternative splicing events,49 protein levels, and other cell subpopulations. A recent study demonstrated that fingolimod also stimulates gene expression in neurons, thereby affecting axonal growth and regeneration,50 but these effects so far have not been examined in detail at the transcriptome level.
To conclude, fingolimod selectively alters the trafficking of immune cells and, consequently, changes the gene expression profiles of CD4+, CD8+, and CD19+ cells circulating in the peripheral blood. Genes induced in B cells of MS patients receiving fingolimod therapy for 3 months comprised signaling molecules and transcriptional regulators but not cell surface markers and cytokines. However, in comparison with T cells, the transcriptome shift in B cells was quite subtle. This reflects that the B cell subpopulations are sequestered to similar extents in lymphoid tissues. Basically, all treatments for MS modulate B cell subsets.51, 52 Thus, further research on their roles in the pathogenesis of MS is of critical importance. Finally, our data may also be useful to compare molecular effects of fingolimod with other emerging S1P receptor modulators being examined in clinical trials of MS and other brain‐related diseases.53
DISCLOSURE
MH received speaking fees and travel funds from Bayer HealthCare, Biogen, Novartis, and Teva. AW received speaking fees and travel funds from Bayer HealthCare, Genzyme, Merck Serono, and Novartis. UKZ received research support as well as speaking fees and travel funds from Almirall, Bayer HealthCare, Biogen, Merck Serono, Novartis, Sanofi, and Teva. ICA, DK, LR, JF, AR, BF, NB, IS, KF, HJT, and SM declare that they have no competing interests.
Supporting information
ACKNOWLEDGMENT
We thank Christa Tiffert for coordinating patient care, Nele Retzlaff for clinical documentation, and Ildikó Tóth and Annegret Zettl for laboratory assistance. The microarray experiments were partly funded by Novartis. The funder had no role in the study design, data collection, and analysis, decision to publish or preparation of the manuscript.
Angerer IC, Hecker M, Koczan D, et al. Transcriptome profiling of peripheral blood immune cell populations in multiple sclerosis patients before and during treatment with a sphingosine‐1‐phosphate receptor modulator. CNS Neurosci Ther. 2018;24:193–201. 10.1111/cns.12793
The first two authors contributed equally to this work.
REFERENCES
- 1. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545‐558. [DOI] [PubMed] [Google Scholar]
- 2. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017;133:13‐24. [DOI] [PubMed] [Google Scholar]
- 3. Denic A, Wootla B, Rodriguez M. CD8(+) T cells in multiple sclerosis. Expert Opin Ther Targets. 2013;17:1053‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15:185‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Büdingen HC, Palanichamy A, Lehmann‐Horn K, Michel BA, Zamvil SS. Update on the autoimmune pathology of multiple sclerosis: B‐cells as disease‐drivers and therapeutic targets. Eur Neurol. 2015;73:238‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wingerchuk DM, Weinshenker BG. Disease modifying therapies for relapsing multiple sclerosis. BMJ. 2016;354:i3518. [DOI] [PubMed] [Google Scholar]
- 7. Mehling M, Johnson TA, Antel J, Kappos L, Bar‐Or A. Clinical immunology of the sphingosine 1‐phosphate receptor modulator fingolimod (FTY720) in multiple sclerosis. Neurology. 2011;76:S20‐S27. [DOI] [PubMed] [Google Scholar]
- 8. Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883‐897. [DOI] [PubMed] [Google Scholar]
- 9. Muls N, Dang HA, Sindic CJ, van Pesch V. Fingolimod increases CD39‐expressing regulatory T cells in multiple sclerosis patients. PLoS ONE. 2014;9:e113025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blumenfeld S, Staun‐Ram E, Miller A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL‐10 and TGFβ in patients with Multiple Sclerosis. J Autoimmun. 2016;70:40‐51. [DOI] [PubMed] [Google Scholar]
- 11. Johnson TA, Evans BL, Durafourt BA, et al. Reduction of the peripheral blood CD56(bright) NK lymphocyte subset in FTY720‐treated multiple sclerosis patients. J Immunol. 2011;187:570‐579. [DOI] [PubMed] [Google Scholar]
- 12. Friess J, Hecker M, Roch L, et al. Fingolimod alters the transcriptome profile of circulating CD4+ cells in multiple sclerosis. Sci Rep. 2017;7:42087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roch L, Hecker M, Friess J, et al. High‐resolution expression profiling of peripheral blood cd8+ cells in patients with multiple sclerosis displays fingolimod‐induced immune cell redistribution. Mol Neurobiol. 2017;54:5511‐5525. [DOI] [PubMed] [Google Scholar]
- 14. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu W, Seok J, Mindrinos MN, et al. Human transcriptome array for high‐throughput clinical studies. Proc Natl Acad Sci U S A. 2011;108:3707‐3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249‐264. [DOI] [PubMed] [Google Scholar]
- 17. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289‐300. [Google Scholar]
- 18. Hecker M, Goertsches RH, Engelmann R, Thiesen HJ, Guthke R. Integrative modeling of transcriptional regulation in response to antirheumatic therapy. BMC Bioinformatics. 2009;10:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hecker M, Goertsches RH, Fatum C, et al. Network analysis of transcriptional regulation in response to intramuscular interferon‐β‐1a multiple sclerosis treatment. Pharmacogenomics J. 2012;12:134‐146. [DOI] [PubMed] [Google Scholar]
- 20. Safran M, Dalah I, Alexander J, et al. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010;2010:baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tyner C, Barber GP, Casper J, et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017;45:D626‐D634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kowarik MC, Pellkofer HL, Cepok S, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology. 2011;76:1214‐1221. [DOI] [PubMed] [Google Scholar]
- 23. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar‐Or A. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS‐compartmentalized inflammation. Front Immunol. 2015;6:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bittner S, Ruck T, Wiendl H, Grauer OM, Meuth SG. Targeting B cells in relapsing‐remitting multiple sclerosis: from pathophysiology to optimal clinical management. Ther Adv Neurol Disord. 2017;10:51‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamura M, Matsuoka T, Chihara N, et al. Differential effects of fingolimod on B‐cell populations in multiple sclerosis. Mult Scler. 2014;20:1371‐1380. [DOI] [PubMed] [Google Scholar]
- 26. Claes N, Dhaeze T, Fraussen J, et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12‐month follow‐up study. PLoS ONE. 2014;9:e111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyazaki Y, Niino M, Fukazawa T, et al. Suppressed pro‐inflammatory properties of circulating B cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of B‐cell subpopulations. Clin Immunol. 2014;151:127‐135. [DOI] [PubMed] [Google Scholar]
- 28. Hoffmann R, Seidl T, Neeb M, Rolink A, Melchers F. Changes in gene expression profiles in developing B cells of murine bone marrow. Genome Res. 2002;12:98‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Novershtern N, Subramanian A, Lawton LN, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haddad R, Guardiola P, Izac B, et al. Molecular characterization of early human T/NK and B‐lymphoid progenitor cells in umbilical cord blood. Blood. 2004;104:3918‐3926. [DOI] [PubMed] [Google Scholar]
- 31. Mallampati S, Sun B, Lu Y, et al. Integrated genetic approaches identify the molecular mechanisms of Sox4 in early B‐cell development: intricate roles for RAG1/2 and CK1ε. Blood. 2014;123:4064‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herglotz J, Unrau L, Hauschildt F, et al. Essential control of early B‐cell development by Mef2 transcription factors. Blood. 2016;127:572‐581. [DOI] [PubMed] [Google Scholar]
- 33. Chiarini M, Sottini A, Bertoli D, et al. Newly produced T and B lymphocytes and T‐cell receptor repertoire diversity are reduced in peripheral blood of fingolimod‐treated multiple sclerosis patients. Mult Scler. 2015;21:726‐734. [DOI] [PubMed] [Google Scholar]
- 34. Teniente‐Serra A, Hervás JV, Quirant‐Sánchez B, et al. Baseline differences in minor lymphocyte subpopulations may predict response to fingolimod in relapsing‐remitting multiple sclerosis patients. CNS Neurosci Ther. 2016;22:584‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Atcheson E, Hamilton E, Pathmanathan S, Greer B, Harriott P, Timson DJ. IQ‐motif selectivity in human IQGAP2 and IQGAP3: binding of calmodulin and myosin essential light chain. Biosci Rep. 2011;31:371‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13:439‐447. [DOI] [PubMed] [Google Scholar]
- 37. Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Sci Signal. 2010;3:rs1. [DOI] [PubMed] [Google Scholar]
- 38. Golay J, Facchinetti V, Ying G, Introna M. The A‐myb transcription factor in neoplastic and normal B cells. Leuk Lymphoma. 1997;26:271‐279. [DOI] [PubMed] [Google Scholar]
- 39. Rowe M, Fitzsimmons L, Bell AI. Epstein‐Barr virus and Burkitt lymphoma. Chin J Cancer. 2014;33:609‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ellis AL, Wang Z, Yu X, Mertz JE. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein‐Barr virus latent‐lytic switch in a cell‐type‐specific manner. J Virol. 2010;84:6139‐6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ohguro H, Chiba S, Igarashi Y, Matsumoto H, Akino T, Palczewski K. Beta‐arrestin and arrestin are recognized by autoantibodies in sera from multiple sclerosis patients. Proc Natl Acad Sci U S A. 1993;90:3241‐3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Angerstein C, Hecker M, Paap BK, et al. Integration of MicroRNA databases to study MicroRNAs associated with multiple sclerosis. Mol Neurobiol. 2012;45:520‐535. [DOI] [PubMed] [Google Scholar]
- 43. Raimondi A, Ferguson SM, Lou X, et al. Overlapping role of dynamin isoforms in synaptic vesicle endocytosis. Neuron. 2011;70:1100‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott H, Howarth J, Lee YB, et al. MiR‐3120 is a mirror microRNA that targets heat shock cognate protein 70 and auxilin messenger RNAs and regulates clathrin vesicle uncoating. J Biol Chem. 2012;287:14726‐14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thiriet M. Guanosine triphosphatases and their regulators In: Intracellular Signaling Mediators in the Circulatory and Ventilatory Systems. New York, NY: Springer; 2013:465‐646. [Google Scholar]
- 46. Zhang AT, Langley AR, Christov CP, et al. Dynamic interaction of Y RNAs with chromatin and initiation proteins during human DNA replication. J Cell Sci. 2011;124:2058‐2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stubbington MJT, Rozenblatt‐Rosen O, Regev A, Teichmann SA. Single‐cell transcriptomics to explore the immune system in health and disease. Science. 2017;358:58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Romero JP, Muniategui A, De Miguel FJ, et al. EventPointer: an effective identification of alternative splicing events using junction arrays. BMC Genom. 2016;17:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anastasiadou S, Knöll B. The multiple sclerosis drug fingolimod (FTY720) stimulates neuronal gene expression, axonal growth and regeneration. Exp Neurol. 2016;279:243‐260. [DOI] [PubMed] [Google Scholar]
- 51. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine. 2017;16:41‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dubey D, Forsthuber T, Flanagan EP, Pittock SJ, Stüve O. B‐cell‐targeted therapies in relapsing forms of MS. Neurol Neuroimmunol Neuroinflamm. 2017;4:e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O'Sullivan S, Dev KK. Sphingosine‐1‐phosphate receptor therapies: advances in clinical trials for CNS‐related diseases. Neuropharmacology. 2017;113:597‐607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials