

Abstract
Inclusion body myopathy (IBM) associated with Paget disease of the bone and frontotemporal dementia or IBMPFD is an autosomal dominant degenerative disorder caused by mutations in the valosin-containing protein (VCP) gene. We aim to establish a detailed clinical phenotype of VCP disease amongst 35 (28 affected individuals, 7 presymptomatic gene carriers) individuals versus 14 unaffected first-degree relatives in 14 families to establish useful biomarkers for IBMPFD and identify the most meaningful tests for monitoring disease progression in future clinical trials. Comprehensive studies included the Inclusion Body Myositis Functional Rating Scale (IBMFRS) and fatigue severity scale questionairres, strength measurements using the Manual Muscle Test with Medical Research Council (MRC) scales, hand-held dynamometry using the microFET and Biodex dynamometers, 6 minute walk test (6MWT), and pulmonary function studies. Strong correlation was observed between the IBMFRS and measurements of muscle strength with dynamometry and the other functional tests, indicating that it may be utilized in long-term follow-up assessments due to its relative simplicity. This cross-section study represents the most comprehensive evaluation of individuals with VCP disease to date and provides a useful guide for evaluating and possible monitoring of muscle weakness and pulmonary function progression in this unique cohort of individuals.
Keywords: Inclusion Body Myopathy, Paget disease, IBMPFD, VCP, IBMFRS, Dynamometry
1. Introduction
Inclusion Body Myopathy (IBM) associated with Paget’s disease of the bone (PDB) and/or frontotemporal dementia (FTD) or Multisystem Proteinopathy (OMIM 167,320) or more commonly called IBMPFD or VCP disease, was first reported by Kimonis et al. [1]. VCP disease comprises other less common phenotypes including Amyotrophic Lateral Sclerosis (ALS), Parkinson’s disease, cardiomyopathy, sensory motor axonal neuropathy, and sphincter disturbance [2–6]. IBMPFD is an underdiagnosed disease that has been reported in greater than 100 families worldwide, but is more commonly found in the United States and Europe. Case reports are increasing annually, including reports of unrelated families of Asian, South American and Australian descent [5,7,8].
IBMPFD is caused by dominantly inherited mutations in the valosin-containing protein (VCP) gene mapped to the human chromosomal region 9p13.3–12 and to date, more than 50 disease mutations have been identified in the VCP gene [1,9,10]. One of the main functions of the protein is as a chaperone for proteasomal protein degradation and autophagic dysfunction leading to protein aggregation [11–15].
IBMPFD is a heterogeneous disorder with variable penetrance [12,16–18]. Approximately 90% of affected individuals have been found to have myopathy, PDB was identified in 42%, and 30% had FTD [8] and approximately 10% of indviduals have classic ALS. Complete manifestation of all three symptoms is seen in 12% of affected individuals and isolated myopathy, PDB, or FTD seen in 30%, 5%, and 3% of affected individuals, respectively [12]. Health care providers therefore need to have a high index of suspicion in order to ensure appropriate screening and diagnostic testing to identify the VCP patient population for accurate management and prevention of complications such as PDB.
The myopathy is the most common feature associated with VCP disease (seen in 90%) and is typically clinically diagnosed as limb girdle muscular dystrophy, facioscapular muscular dystrophy, scapuloperoneal muscular dystrophy, and may coexist with ALS [19,20]. The diagnosis of VCP disease is typically made based on the presence of proximal limbgirdle myopathy, muscle biopsy findings of rimmed vacuoles, ubiquitin, and TDP-43-positive inclusions, and the presence of either PDB [1,10,18,21–23] and/or FTD [24,25] and/or ALS. Affected individuals typically die in their 50s–60s from respiratory insufficiency, cardiac failure or complications from early stage dementia, however, demise has occurred as early as the 20s [15,17,26]. Currently, there are no curative treatments for the myopathy or FTD component of the disease. Paget disease of bone is responsive to treatment with bisphosphonates [27,28] therefore early diagnosis could prevent advanced stages of PDB in at-risk individuals with VCPassociated disease. Because of the overlapping pathology of VCP disease, its study has led to dramatic progress in understanding the pathology of common related disorders in which TDP-43 is a major component of inclusions. TDP-43 characterizes VCP-associated myopathy, FTD and ALS pathology, thereby placing VCP disease in a unique category of neurodegenerative diseases termed TDP-43 proteinopathies [25,29,30]. VCP disease is increasingly being recognized in patients in recent years and often diagnosed following nextgeneration panel or exome sequencing.
Small cross-sectional studies are often the basis for future studies and clinical trials, which is especially useful in rare diseases. Previous cross-sectional studies of rare diseases examined diseases with a dearth of published information, and helped provide knowledge to physicians to diagnose these diseases. In previous studies of VCP disease, Shi et al. (2012) [7] found six new Asian families when only one had been described prior and characterized pathological features of muscles with VCP mutations, while Stojkovic et al. reported the clinical variability in French and Spanish families [31]. Pulmonary function studies have been conducted on similar neuromuscular degenerative diseases and disorders including ALS and Pompe disease. These studies established the importance of utilizing respiratory function measurements in monitoring disease evolution, as well as predicting patient survival rates [32–35]. To date, however, there have been no prior studies conducted on the respiratory functions of VCP disease. In studies of patients with Pompe disease, cross-sectional studies have characterized symptoms, profiled phenotypes and genotypes, and correlated clinical symptoms to molecular changes [36,37]. Small studies in patients with GNE myopathy have documented clinical and genetic characteristics, as well as genotype–phenotype correlations [38,39]. These studies allow for multiple aspects of the disease to be assessed and expand documented knowledge for clinical management and therapeutic interventions.
We aim to expand the clinical spectrum of VCP disease, correlate the testing parameters, and detect early features in presymptomatic mutation carriers from a cross-sectional database of 35 individuals in 14 families harboring six missense mutations.
2. Methods
2.1. Subjects
Informed consent was obtained from all the individuals for these studies. Subjects were evaluated at the ICTS (Institue of Clinical Translational Science), University of California, Irvine for a single two-day visit. All patients with VCP disease that Dr. Kimonis follows were invited to participate in the study, with inclusion criteria consisting of age of at least 18 years and a confirmed VCP gene mutation. All individuals were tested in the Kimonis laboratory, however, their mutation status was confirmed in a CLIA certified laboratory. The clinically affected group consisted of 28 individuals (17 males/11 females) and seven carriers (2 males/5 females), who carried the VCP mutation but did not show any obvious clinical evidence of VCP disease (Table 1). Presymptomatic carriers and unaffected individuals at 50% prior risk of inheriting the familial mutation underwent genetic counseling and testing; the results of the predictive testing have been reported by Surampalli et al. [40]. Fourteen (7 males/7 females) firstdegree VCP-negative relatives who served as controls were matched for age, sex and BMI as closely as possible and had no relevant clinical features. The mean ages in affected, carrier and control groups were 52 years, 43 years and 49 years, respectively. Of the affected individuals, all had myopathy, 11 had myopathy and Paget disease, and three had myopathy and early FTD (Supplemental Table 1). Three patients in this cohort had ALS and two others had Parkinson’s disease [41]. The molecular genetic testing indicated affected individuals and carriers carried one of six mutations: R155H, R155C, R155P, R159C, R159H, and G97E.
Table 1.
Demographics, Primary outcomes: Questionnaires for functional capacity in Affected, Carrier and Unaffected relatives, Muscle strength measurement: 6 min walk test.
| M/F | Affected 17/11 (61% M, 39% F) Mean (SD) |
Carriers 2/5 (29% M, 71%F) Mean (SD) |
Unaffected relatives 7/7 (50% M, 50%F) Mean (SD) |
p-value* |
|---|---|---|---|---|
| Age (year) (N = 28, 7, 14) |
52.40 (8.96) | 43.31 (7.53) | 48.79 (14.86) | 0.57, 0.13, 0.52 |
| BMI (kg/m2) (N = 24, 5, 9) |
35.13 (26.17) | 32.56 (10.95) | 29.32 (4.89) | 0.77, 0.97, 0.96 |
| IBMFRS (N = 26,6,8) (out of 40) |
27.31 (9.54) | 38.00 (2.45) | 39.88 (0.35) | 0.01, 0.02, 0.90 |
| Fatigue scale (N = 24,4,9) (out of 63) |
45.29 (12.72) | 34.50 (24.77) | 26.89 (11.86) | 0.02** |
| 6 min walk test (m) (N = 14,6,13) |
390.86 (168.64) | 458.13 (95.33) | 550.82 (98.23) | 0.02, 0.65, 0.49 |
Sample sizes (N) are listed in order of affected, carriers, and unaffected relatives.
p-values are indicated in the order of differences between affected and unaffected individuals, differences between affected individuals and carriers, and differences between carriers and unaffected individuals. p-values ≤ 0.05 indicate significant differences
Indicates difference between affected and unaffected. The small sample size of the group precluded statistical analysis of the carriers.
2.2. Questionnaires for functional capacity
2.2.1. Inclusion Body Myositis Functional Rating Scale (IBMFRS)
The IBMFRS scale based on the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) is a diseasespecific 10-point functional rating [42] that has shown highly significant correlation with online and on-site assessments, as well as correlation of telephone assessments [43,44]. The ALSFRS assesses activities of daily living [45], predicts survival time, and has good inter-rater reliability [46–48]. Studies in inclusion body myositis revealed that this scale achieved statistical significant correlations with MMT, SF36, ALS-FRS, and DXA lean body mass and handgrip dynamometry, with a mean change of −1.257 (SD 2.45) from baseline to 24 weeks [42]. The items are graded on a scale of zero (indicating the individual is unable to perform the task) to four (signifying normal performance) with a total possible score of 40. The IBMFRS has never been previously validated in VCP myopathy.
2.2.2. Fatigue severity scale (FSS)
The FSS questionnaire contains nine statements that rate the severity of fatigue symptoms. A low value (e.g. 1) indicates strong disagreement with the statement, whereas a high value (e.g. 7) indicates strong agreement. A total score of 36 or more suggests that the individual has significant fatigue. The FSS has shown good reliability and validity in individuals with spinal muscular atrophy type II, congenital myopathies [49,50] and Pompe disease in which affected individuals had significantly higher FSS scores than healthy controls ( p < 0.001) [51].
2.3. Motor function assessment
2.3.1. Manual muscle testing (MMT) using the Medical Research Council scale (MRC)
Muscle strength was scored using the manual muscle testing scale proposed by the MRC, which is a reliable and validated scale for assessing muscle weakness [52]. All individuals were evaluated clinically and neurologically for proximal muscle weakness particularly of the arms, legs, shoulder, and pelvic girdle and distal muscle groups. This scale uses the numeral grades 0–5 in different muscle groups with increasing muscle strength. Totals were calculated out of 130, measuring neck, right and left knee, elbow, hip, and wrist extension and flexion, shoulder and hip abduction, and ankle dorsiflexion and plantarflexion.
2.3.2. The 6 min walk test (6MWT)
The 6MWT is a useful measure of functional capacity, targeted at people with at least moderately severe impairment. It has been widely used for measuring the response to therapeutic interventions for pulmonary and cardiac disease and was performed according to the guidelines of the American Thoracic Society [53,54]. The 6MWT has shown reliability and concurrent validity with other clinical endpoints [55,56].
2.3.3. Isometric dynamometry
Muscle grip strength was determined using the Jamar (Jamar Technologies, Hatfield, PA) and of the other muscle groups (shoulder, elbow, grip, wrist, hip, knee, ankle) using the microFET2 dynamometer (Hoggan Scientific, LLC, Salt Lake City, UT) after standardization and calibration [57]. To avoid variability hand-held dynamometry testing was restricted to a single examiner VK [58]. The average of 3 different measurements was documented for all subjects.
2.3.4. Isokinetic dynamometry
The Biodex system 3 pro isokinetic dynamometer (Biodex Medical Systems, Inc., Shirley, New York) has been proven to show mechanical reliability for assessment of muscle strength [59]. Specific muscle groups that were tested include bilateral knee extensors and flexors. Torque was measured at angular velocities of 60 and 180 °/s (1.05 and 3.14 rad/s, respectively). These velocities were chosen because they reflect the torque that can be produced at slow (i.e., 60 °/s) and moderate (i.e., 180 °/s) velocities and, as such, provide insight regarding force-velocity properties (the most important property of skeletal muscle) of the muscles studied. Each testing session included 1 set of 4 repetitions for each muscle group at each velocity. The Biodex system controls the speed of the motion through a feedback loop and measures torque continuously throughout each set. To avoid confounding strength changes due to exercise training or greater familiarity with the test equipment, subjects were familiarized with the Biodex dynamometer prior to the first session of strength testing. Subjects were verbally encouraged to produce maximal efforts throughout each test session.
2.4. Pulmonary function studies
Pulmonary function tests (PFTs) included measurement of forced vital capacity (FVC), forced expiratory volume (FEV), forced expiratory flow (FEF), peak expiratory flow (PEF), maximal inspiratory (MIP), and maximal expiratory (MEP) mouth pressure [60,61]. Measurements were determined using the Vmax® Encore PFT system. Measurements were also reported as a percentage of the individual’s reference value, which takes age, height, sex, and ethnicity into consideration.
2.5. Statistical analysis
Comparison of the symptomatic individuals, asymptomatic carriers, and unaffected controls was performed using oneway analysis of variance (ANOVA) with the Tukey post hoc test. p-values ≤0.05 were considered significant. Pearson correlation studies describing the association between different measurement variables including the MRC scales, IBMFRS, and dynamometry were examined. Correlations were also performed between these measurements and PFTs. Correlation coefficients with magnitudes between 0.9 to 1.0 were considered to be very highly correlated, 0.7 to 0.9 were considered strong correlations or highly correlated, 0.5 to 0.7 were considered moderately correlated, and coefficients between 0.3 and 0.5 were considered low correlation. For sample sizes less than five, statistical significance was not reported. Independent sample t-tests were performed differences between affected individuals and controls for Biodex dynamometry and pulmonary function test results, as the small sample size of carriers precluded statistical analysis. Independent sample t-tests were also performed for the comparisons of gender in affected individuals. Paired sample t-tests were also performed to compare lateral differences in affected individuals. p-values for primary outcomes (IBMFRS, FSS, 6MWT) were corrected for multiple comparisons using the false discovery rate. Secondary outcomes (isometric and Biodex isokinetic dynamometry, MRC scales, PFT) were deemed exploratory and were not corrected.
3. Results
3.1. Questionnaires
The average IBMFRS score for affected individuals was 27 for presymptomatic carriers 38 and unaffected individuals was 39.9 out of a possible maximum score of 40, with the difference between the affected individuals and controls being significant ( p = 0.01) (Table 1). Affected individuals with a longer disease duration not surprisingly had decreased IBMFRS scores compared to those with a shorter duration of disease (Fig. 1). This same phenomenon was seen when comparing affected individuals at different ages, and was exhibited in carriers to a milder degree but not in unaffected individuals (Fig. 2). Affected individuals showed higher scores in the fatigue rating scale, with an average FSS score of 45 out of a possible score of 63, compared to a score of 34.5 in carriers and an average of 27 in unaffected individuals (Table 1).
Fig. 1. Comparison of affected individuals with various disease durations.
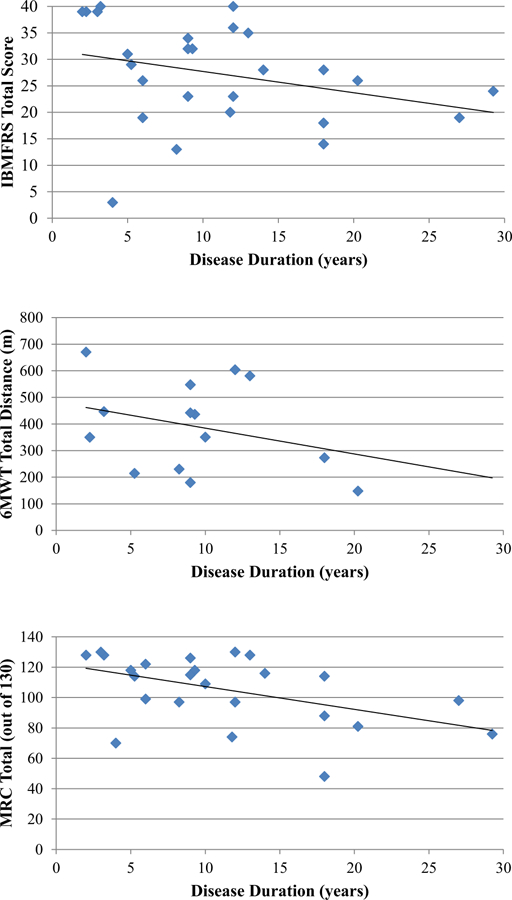
There is a declining trend in affected individuals with increased disease duration compared to more recently symptomatic individuals. However, the relationships are not strong. (a) IBMFRS score vs. Disease Duration (y = −0.4017x + 31.735, r 2 = 0.09) (b) 6MWT distance vs. Disease Duration (y = −9.7058x + 481.33, r 2 = 0.10) (c) MRC Total vs. Disease Duration (y = −1.5003x + 122.25, r 2 = 0.24).
Fig. 2. Comparison of patients of varying ages.
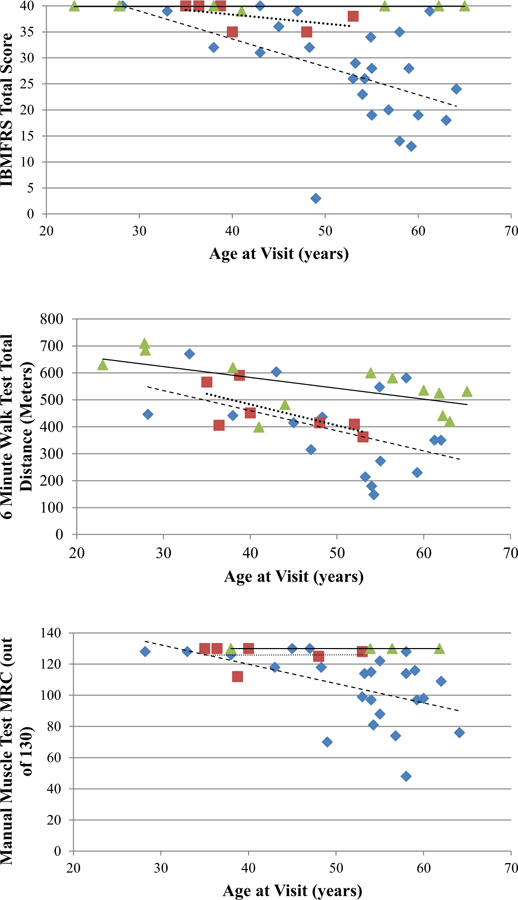
Affected are represented as diamonds, carriers as squares, and unaffected as triangles. (a) IBMFRS Score v. Age. IBMFRS declines in affected individuals with older individuals in this cohort, while unaffected stays constant. The trendline for unaffected individuals is solid (y = 0.0009x + 39.838, p-value = 0.92). The trendline for carriers is dotted (y = −0.1735x + 45.264, p-value = 0.31). The trendline for affected individuals is dashed (y = −0.5363x + 55.096, p-value = 0.01). (b) 6MWT v. Age. Decline in all groups at older age, regardless of condition. The trendline for unaffected individuals is solid (y = −4.0252x + 744.03, p-value = 0.02). The trendline for carriers is dotted (y = −7.828x + 796.11, p-value = 0.12). The trendline for affected individuals is dashed (y = −7.4643x + 758.23, p-value = 0.08). (c) Manual Muscle Test Total MRC Score v. Age. MRC score declines for older affected individuals, while unaffected stays constant. The trendline for unaffected individuals is solid (y = −0.0591x + 282.6, p-value = 0.40). The trendline for carriers is dotted (y = 0.0155x + 266.35, p-value = 0.99). The trendline for affected individuals is dashed (y = −3.4974x + 385.03, p-value = 0.01).
3.2. Muscle functional studies
3.2.1. Six-minute walk test
Affected individuals walked 390.86 ±168.64 m compared to presymptomatic carriers who walked 458.13 ±95.33m and unaffected individual who walked 550.82 ±98.23 m on average (Table 1). As with IBMFRS, affected individuals with a longer disease duration did not walk as far as individuals with a shorter duration of disease (Fig. 1). Interestingly, in all three groups, older individuals had lower 6MWT scores compared to that of younger individuals of the same condition (Fig. 2).
3.2.2. MRC scores
The MRC whole body total score for the affected individuals was 105.17 ±22.51 compared to the score in carriers (125.83 ±7.06) and unaffected individuals (130 ±0.00) out of a maximum score of 130 (Supplemental Table 2). Significant differences were found between affected and unaffected individuals of shoulder abduction and hip flexion. Evaluation of the distribution of the MRC therefore indicated proximal weakness in the majority of affected individuals and distal weakness in one individual, typical of the hip and shoulder girdle weakness previously reported in patients with VCP myopathy. Interestingly, these differences in shoulder abduction and hip flexion were also seen between carriers and unaffected individuals, indicating muscle weakness in the presymptomatic carriers. MRC showed the same trend as the IBMFRS and 6MWT when comparing scores of individuals with longer disease durations (Fig. 1). This phenomenon continued in affected individuals at different ages, but was not exhibited in carriers and unaffected individuals (Fig. 2).
3.3.3. Isometric dynamometry
All muscle groups, except the wrists, tested with the microFET dynamometry indicated a significant difference between affected and unaffected individuals. The average hand-held dynamometry results of affected individuals and presymptomatic carriers in nine muscle groups using the microFET dynamometer were expressed as a percentage of normal muscle strengths in unaffected individuals (Fig. 3). Affected individuals exhibited muscle strength at a fraction of that of unaffected individuals. For example, for knee extension, affected individuals produced a force of 38 lbs while unaffected individuals were able to produce an average force of 88 lbs and carriers produced a force of 53 lbs (Supplemental Table 3). Interestingly, the mean grip strength values using the Jamar dynamometer did not indicate significant differences between the three groups. These studies indicate that affected indviduals have proximal muscle weakness, however, the distal muscle groups strength is retained.
Fig. 3. Hand-held dynamometry in affected individuals and presymptomatic carriers.
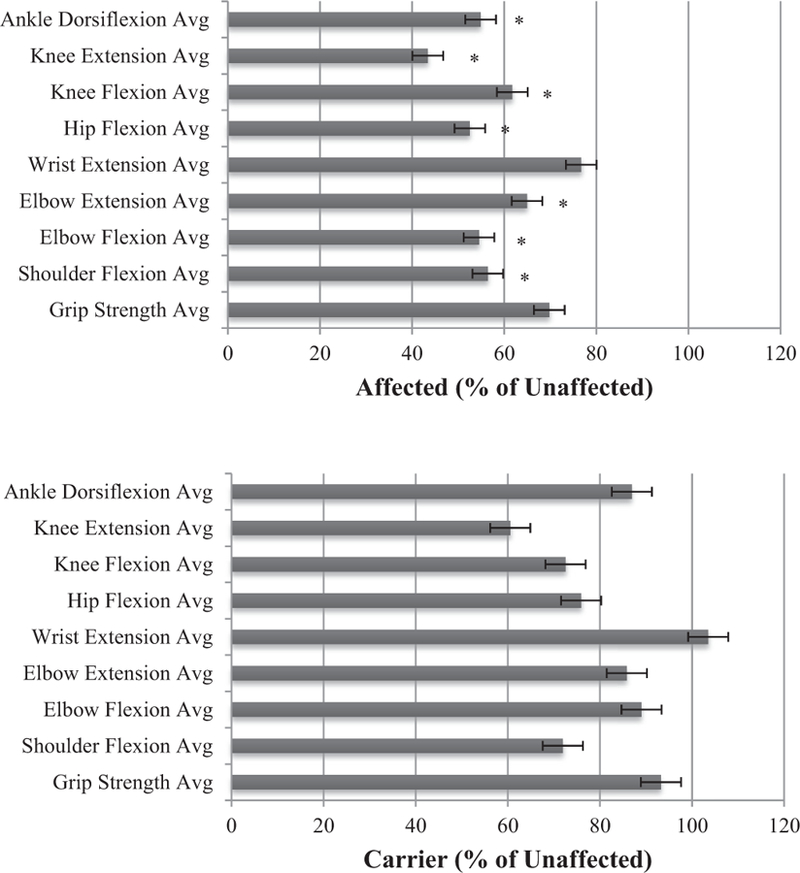
Strength in nine muscle groups expressed as percentage of normal muscle strength in unaffected individuals (top), presymptomatic carriers (bottom). ∗ denotes p-value differences ≤0.05.
3.3.4. Biodex isokinetic dynamometry
In our exploratory findings the Biodex isokinetic dynamometry measurements showed significant differences with the 60 °/s torque measurements of knee extension and flexion between affected and unaffected individuals, however, significant differences were not seen with the 180 °/s measurements (Supplemental Table 4). We believe this is mainly due to the small sample size and the wide range of individual scores, leading to large standard deviations. These results further warrant a larger study to investigate the use of handheld dynamometry versus the Biodex dynamometry for its use as an objective measure in clinical trials.
3.4. Pulmonary function tests
Values of these pulmonary function tests were lower on average in affected individuals with significant differences in mean values between affected individuals and controls for several of the tests performed. Percentages of reference volumes on average in affected individuals were 10–55% lower, indicating a decrease in respiratory function of affected individuals (Supplemental Table 5). We could not perform statistical comparisons with the carriers due to the small sample size.
3.5. Correlation analysis
There were strong significant positive correlations between IBMFRS and MRC ( r = 0.793, p < 0.0001), between IBMFRS and 6MWT ( r = 0.734, p < 0.0001) and a significant, yet low, negative correlation ( r = −0.477, p = 0.008) between IBMFRS and FSS (Supplemental Figure 1) and FSS and 6MWT ( r = −0.472, p = 0.013) (Table 2). Furthermore, IBMFRS scores were moderately or highly correlated with all hand-held dynamometry results, with significant p-values < 0.0001 for all groups (Supplemental Table 6). There was a significant correlation between MRC total and all Biodex parameters (Supplemental Table 7). IBMFRS also correlated with the right and left knee extension 180 °/s and knee flexion 60, 180 °/s Biodex measurements. Additionally, there was strong significant positive correlation between hand-held dynamometry and corresponding Biodex knee flexion and extension (Supplemental Table 8). Various pulmonary function tests including the FVC, FEV1, FEF 25–75%, PI Max, and PE Max showed significant correlation with both the IBMFRS and MRC scores (Supplemental Table 9).
Table 2.
Correlations of IBMFRS, Fatigue scale, 6 minute walk test (6MWT) and MRC scale.
| IBMFRS total Score | MRC total Whole body | Fatigue scale Total score | 6 MWT Distance | ||
|---|---|---|---|---|---|
| IBMFRS Total score | Pearson correlation | 1 | 0.793 | −0.477 | 0.734 |
| Sig. (2-tailed) | <0.0001* | 0.008* | <0.0001* | ||
| N | 46 | 30 | 30 | 26 | |
| MRC Total whole body | Pearson correlation | 0.793 | 1 | −0.250 | 0.721 |
| Sig. (2-tailed) | <0.0001* | 0.200 | <0.0001* | ||
| N | 30 | 34 | 28 | 20 | |
| Fatigue scale Total score | Pearson correlation | −0.477 | −0.250 | 1 | −0.472 |
| Sig. (2-tailed) | 0.008* | 0.200 | 0.013* | ||
| N | 30 | 28 | 30 | 27 | |
| 6 MWT | Pearson correlation | 0.734 | 0.721 | −0.472 | 1 |
| Sig. (2-tailed) | <0.0001* | <0.0001* | 0.013* | ||
| N | 26 | 20 | 27 | 33 | |
Correlation significant at the <0.01 level (2-tailed).
We analyzed the effect of dual or triple pathology in the same individual, however, we found no indication that myopathic individuals with PDB and/or FTD had more severe disease than those individuals who only had myopathy. No significant differences were found between the left and right muscle groups of affected individuals, or between the genders.
4. Discussion
This cross-sectional study represents the most comprehensive evaluation of functional status assessment, motor function assessments, and pulmonary function studies in a unique cohort of individuals with VCP disease, presymptomatic carriers and their unaffected non-carrier first-degree relatives.
As a result of our studies, we found correlations between the IBMFRS and various tests of muscle strength demonstrating concurrent validity of this questionnaire in this unique population. These results indicate that IBMFRS can be used for assessing motor function in patients with VCP disease [42] and can be used to easily monitor the progression of motor involvement in VCP disease and thus decrease study costs. The fatigue severity scale is also a useful questionnaire with affected individuals experiencing more fatigue, and is therefore useful in supplementing IBMFRS and muscle tests in future studies. Surprisingly, the carriers also scored lower than the unaffected individuals, leading us to believe carriers are beginning to experience fatigue as an early feature of the disease.
While the MRC scale has been criticized for its limited sensitivity, especially at the higher grades 4 and 5 [62], the scale appeared discriminatory in determining weakness in specific muscle groups. The strong correlations between the traditional MRC scores, hand-held dynamometry and Biodex measurements indicate that the simpler tests, MRC and hand-held dynamometry, may be helpful for the long-term follow-up assessment in these patients due to their relative simplicity and portability.
The pulmonary function tests results suggested a compromise in the lung function due to myopathic involvement of the diaphragm and other accessory muscles of respiration. Because respiratory muscle compromise is an early sign of VCP disease, low values of FVC, FEV1, MIP and MEP may have prognostic value in reflecting the state of respiratory muscle weakness [32–34]. The strong correlations between pulmonary tests and functional studies indicating respiratory muscle weakness have been reported previously [63,64].
Overall, on reviewing all the parameters in presymptomatic carriers, we found that average values ranged between affected individuals and unaffected first-degree relatives. Their scores from tests such as the FSS and 6MWT indicate that they are beginning to manifest symptoms of the myopathy. Based on the MRC scale results, the presymptomatic carriers showed muscle weakness of their shoulders and hips. A larger sample size should be studied before concluding the proper method for specifically monitoring carriers, as this study was limited by a small group of presymptomatic carriers.
Our study emphasizes that each of these simple testing methods appear to be cost-effective methods in assessing muscle function, functional impairments, and pulmonary function in VCP disease. We propose that a combination of the different tests may provide the sensitivity to measure disease severity and record changes as the disease progresses. These tests may have a role as outcome measures in future clinical trials. One of the limitations of the study is that this is a cross-sectional study derived from a single two-day visit. The patients were not consistently evaluated over time, and thus this study does not represent natural history data. However, future studies in larger group of patients and follow-up of these patients at regular intervals within a longitudinal study would provide us with information about the validity of these assessment disease parameters to monitor progression in VCP disease. Our study also hopes to increase awareness of this important disorder, which will help improve early diagnosis, understanding of the pathogenesis of VCP disease, and hopefully development of novel therapies.
Supplementary Material
Acknowledgement
We thank the patients, their families, health care providers and the numerous collaborators and researchers for their generous contribution to this work. Funding for these studies is from the National Institute of Health [Grant AR050236 (VK) ] and the UC Irvine ICTS (Institute of Clinical Translational Science), NCATS [Grant TR001414].
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.nmd.2018.06.007.
References
- [1].Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2000;2:232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Djamshidian A, Schaefer J, Haubenberger D, Stogmann E, Zimprich F, Auff E, et al. A novel mutation in the VCP gene (G157R) in a German family with inclusion-body myopathy with Paget disease of bone and frontotemporal dementia. Muscle Nerve 2009;39:389–91. [DOI] [PubMed] [Google Scholar]
- [3].Guyant-Marechal L, Laquerriere A, Duyckaerts C, Dumanchin C, Bou J, Dugny F, et al. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 2006;67:644–51. [DOI] [PubMed] [Google Scholar]
- [4].Haubenberger D, Bittner RE, Rauch-Shorny S, Zimprich F, Mannhalter C, Wagner L, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 2005;65:1304–5. [DOI] [PubMed] [Google Scholar]
- [5].Kumar KR, Needham M, Mina K, Davis M, Brewer J, Staples C, et al. Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord 2010;20:330–4. [DOI] [PubMed] [Google Scholar]
- [6].Miller TD, Jackson AP, Barresi R, Smart CM, Eugenicos M, Summers D, et al. Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): clinical features including sphincter disturbance in a large pedigree. J Neurol Neurosurg Psychiatry 2009;80:583–4. [DOI] [PubMed] [Google Scholar]
- [7].Shi Z, Hayashi YK, Mitsuhashi S, Goto K, Kaneda D, Choi YC, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501–9. [DOI] [PubMed] [Google Scholar]
- [8].Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang A, Hermann A, et al. Genotype-phenotype study in patients with VCP valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet 2018;93(1):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab 2001;74:458–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Watts GD, Thorne M, Kovach MJ, Pestronk A, Kimonis VE. Clinical and genetic heterogeneity in chromosome 9p associated hereditary inclusion body myopathy: exclusion of GNE and three other candidate genes. Neuromuscul Disord 2003;13:559–67. [DOI] [PubMed] [Google Scholar]
- [11].Ju JS, Weihl CC. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet 2010;19:R38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kimonis V, Donkervoort S, Watts G. Inclusion body myopathy associated with Paget disease of bone and/or frontotemporal dementia ‘gene genetests (wwwgenetestsorg) and University of Washington, Seattle [Review] 2011. [cited; Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301649.
- [13].Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 2011;45:522–31. [DOI] [PubMed] [Google Scholar]
- [14].Vesa J, Su H, Watts GD, Krause S, Walter MC, Martin B, et al. Valosin containing protein associated inclusion body myopathy: abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul Disord 2009;19:766–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009;19:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Waggoner B, Kovach MJ, Winkelman M, Cai D, Khardori R, Gelber D, et al. Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am J Med Genet 2002;108:187–91. [DOI] [PubMed] [Google Scholar]
- [17].Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta 2008;1782:744–8. [DOI] [PubMed] [Google Scholar]
- [18].Mehta SG, Khare M, Ramani R, Watts GD, Simon M, Osann KE, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet 2013;83:422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 2008;146A:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kimonis VE, Watts GD. Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19(Suppl 1):S44–7. [DOI] [PubMed] [Google Scholar]
- [21].Gidaro T, Modoni A, Sabatelli M, Tasca G, Broccolini A, Mirabella M. An Italian family with inclusion-body myopathy and frontotemporal dementia due to mutation in the VCP gene. Muscle Nerve 2008;37:111–14. [DOI] [PubMed] [Google Scholar]
- [22].Niwa H, Ewens CA, Tsang C, Yeung HO, Zhang X, Freemont PS. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J Biol Chem 2012;287:8561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 2008;79:1186–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 2006;65:571–81. [DOI] [PubMed] [Google Scholar]
- [25].Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 2007;66:152–7. [DOI] [PubMed] [Google Scholar]
- [26].Mehta SG, Watts GD, McGillivray B, Mumm S, Hamilton SJ, Ramdeen S, et al. Manifestations in a family with autosomal dominant bone fragility and limb-girdle myopathy. Am J Med Genet A 2006;140:322–30. [DOI] [PubMed] [Google Scholar]
- [27].Siris E. Zoledronate in the treatment of Paget’s disease. Br J Clin Pract Suppl 1996;87:19–20 discussion 22. [DOI] [PubMed] [Google Scholar]
- [28].Langston AL, Ralston SH. Management of Paget’s disease of bone. Rheumatology 2004;43:955–9. [DOI] [PubMed] [Google Scholar]
- [29].Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 2007;171:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van der Zee J, Pirici D, Van Langenhove T, Engelborghs S, Vandenberghe R, Hoffmann M, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 2009;73:626–32. [DOI] [PubMed] [Google Scholar]
- [31].Stojkovic T, Hammouda el H, Richard P, Lopez de Munain A, Ruiz–Martinez J, Camano P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord 2009;19:316–23. [DOI] [PubMed] [Google Scholar]
- [32].Ambrosino N, Confalonieri M, Crescimanno G, Vianello A, Vitacca M. The role of respiratory management of Pompe disease. Respir Med 2013;107:1124–32. [DOI] [PubMed] [Google Scholar]
- [33].Baumann F, Henderson RD, Morrison SC, Brown M, Hutchinson N, Douglas JA, et al. Use of respiratory function tests to predict survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;11:194–202. [DOI] [PubMed] [Google Scholar]
- [34].Enache I, Pistea C, Fleury M, Schaeffer M, Oswald-Mammosser M, Echaniz-Laguna A, et al. Ability of pulmonary function decline to predict death in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 2017;18(7–8):511–18. [DOI] [PubMed] [Google Scholar]
- [35].Lerario A, Bonfiglio S, Sormani M, Tettamanti A, Marktel S, Napolitano S, et al. Quantitative muscle strength assessment in duchenne muscular dystrophy: longitudinal study and correlation with functional measures. BMC Neurol 2012;12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gungor D, Schober AK, Kruijshaar ME, Plug I, Karabul N, Deschauer M, et al. Pain in adult patients with Pompe disease: a cross– sectional survey. Mol Genet Metab 2013;109:371–6. [DOI] [PubMed] [Google Scholar]
- [37].Herzog A, Hartung R, Reuser AJ, Hermanns P, Runz H, Karabul N, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J Rare Dis 2012;7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Haghighi A, Nafissi S, Qurashi A, Tan Z, Shamshiri H, Nilipour Y, et al. Genetics of GNE myopathy in the non-Jewish Persian population. Eur J Hum Genet 2016;24:243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sim JE, Park HJ, Shin HY, Nam TS, Kim SM, Choi YC. Clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy. Yonsei Med J 2013;54:578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Surampalli A, Khare M, Kubrussi G, Wencel M, Tanaja J, Donkervoort S, et al. Psychological impact of predictive genetic testing in vcp inclusion body myopathy, paget disease of bone and frontotemporal dementia. J Genet Couns 2015;24:842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chan N, Le C, Shieh P, Mozaffar T, Khare M, Bronstein J, et al. Valosin-containing protein mutation and Parkinson’s disease. Parkinsonism Relat Disord 2012;18:107–9. [DOI] [PubMed] [Google Scholar]
- [42].Jackson CE, Barohn RJ, Gronseth G, Pandya S, Herbelin L. Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity. Muscle Nerve 2008;37:473–6. [DOI] [PubMed] [Google Scholar]
- [43].Kasarskis EJ, Dempsey-Hall L, Thompson MM, Luu LC, Mendiondo M, Kryscio R. Rating the severity of ALS by caregivers over the telephone using the ALSFRS-R. Amyotroph Lateral Scler Other Motor Neuron Disord 2005;6:50–4. [DOI] [PubMed] [Google Scholar]
- [44].Maier A, Holm T, Wicks P, Steinfurth L, Linke P, Munch C, et al. Online assessment of ALS functional rating scale compares well to in-clinic evaluation: a prospective trial. Amyotroph Lateral Scler 2012;13:210–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].The Amyotrophic Lateral Sclerosis Functional Rating Scale Assessment of activities of daily living in patients with amyotrophic lateral sclerosis. The ALS CNTF treatment study (ACTS) phase I-II study group. Arch Neurol 1996;53:141–7. [PubMed] [Google Scholar]
- [46].Kaufmann P, Levy G, Montes J, Buchsbaum R, Barsdorf AI, Battista V, et al. Excellent inter-rater, intra-rater, and telephone-administered reliability of the ALSFRS-R in a multicenter clinical trial. Amyotroph Lateral Scler 2007;8:42–6. [DOI] [PubMed] [Google Scholar]
- [47].Levy G, Kaufmann P, Buchsbaum R, Montes J, Barsdorf A, Arbing R, et al. A two-stage design for a phase II clinical trial of coenzyme Q10 in ALS. Neurology 2006;66:660–3. [DOI] [PubMed] [Google Scholar]
- [48].Montes J, Levy G, Albert S, Kaufmann P, Buchsbaum R, Gordon PH, et al. Development and evaluation of a self-administered version of the ALSFRS-R. Neurology 2006;67:1294–6. [DOI] [PubMed] [Google Scholar]
- [49].Koopman FS, Brehm MA, Heerkens YF, Nollet F, Beelen A. Measuring fatigue in polio survivors: content comparison and reliability of the fatigue severity scale and the checklist individual strength. J Rehabil Med 2014;46:761–7. [DOI] [PubMed] [Google Scholar]
- [50].Rosa K, Fu M, Gilles L, Cerri K, Peeters M, Bubb J, et al. Validation of the fatigue severity scale in chronic hepatitis C. Health Qual Life Outcomes 2014;12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Werlauff U, Hojberg A, Firla-Holme R, Steffensen BF, Vissing J. Fatigue in patients with spinal muscular atrophy type II and congenital myopathies: evaluation of the fatigue severity scale. Qual Life Res 2014;23:1479–88. [DOI] [PubMed] [Google Scholar]
- [52].Compston A. Aids to the investigation of peripheral nerve injuries. Medical Research Council: Nerve Injuries Research Committee. His Majesty’s Stationery Office; 1942. pp. 48 (iii) and 74 figures and 7 diagrams; with aids to the examination of the peripheral nervous system. By Michael O’Brien for the Guarantors of Brain. Saunders Elsevier: 2010; pp. [8]64 and 94 Figures. Brain 2010;133:2838–44. [DOI] [PubMed]
- [53].Enright PL. The six-minute walk test. Respir Care 2003;48:783–5. [PubMed] [Google Scholar]
- [54].Enright PL, Sherrill DL. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med 1998;158:1384–7. [DOI] [PubMed] [Google Scholar]
- [55].van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010;362:1396–406. [DOI] [PubMed] [Google Scholar]
- [56].McDonald CM, Henricson EK, Abresch RT, Florence J, Eagle M, Gappmaier E, et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013;48:357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mentiplay BF, Perraton LG, Bower KJ, Adair B, Pua YH, Williams GP, et al. Assessment of lower limb muscle strength and power using hand-held and fixed dynamometry: a reliability and validity study. PLoS One 2015;10:e0140822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wikholm JB, Bohannon RW. Hand-held dynamometer measurements: tester strength makes a difference. J Orthop Sports Phys Ther 1991;13:191–8. [DOI] [PubMed] [Google Scholar]
- [59].Drouin JM, Valovich-mcLeod TC, Shultz SJ, Gansneder BM, Perrin DH. Reliability and validity of the Biodex system 3 pro isokinetic dynamometer velocity, torque and position measurements. Eur J Appl Physiol 2004;91:22–9. [DOI] [PubMed] [Google Scholar]
- [60].Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Rev Mal Respir 2007;24:S27–49. [Google Scholar]
- [61].Green M, Road J, Sieck GC, Similowski T. Tests of respiratory muscle strength. Am J Respir Crit Care Med 2002;166:528–47. [Google Scholar]
- [62].Bohannon RW. Manual muscle testing: does it meet the standards of an adequate screening test? Clin Rehabil 2005;19:662–7. [DOI] [PubMed] [Google Scholar]
- [63].Wang Y, Shao WB, Gao L, Lu J, Gu H, Sun LH, et al. Abnormal pulmonary function and respiratory muscle strength findings in Chinese patients with Parkinson’s disease and multiple system atrophy–comparison with normal elderly. PLoS One 2014;9:e116123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sahin G, Ulubas B, Calikoglu M, Erdogan C. Handgrip strength, pulmonary function tests, and pulmonary muscle strength in fibromyalgia syndrome: is there any relationship? South Med J 2004;97:25–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.