

Watch a video presentation of this article
Abbreviations
- Ach
acetylcholine
- ADMA
asymmetric dimethylarginine
- βb
beta blockers
- CBDL
common bile duct ligation
- CCl4
carbon tetrachloride
- cGMP
cyclic guanosine monophosphate
- CLD
chronic liver disease
- DDAH‐2
dimethylarginine dimethylaminohydrolase 2
- ECM
extracellular matrix
- eNOS
endothelial nitric oxide synthase
- FXR
farnesoid X receptor
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- HVPG
hepatic venous pressure gradient
- IGCI
indocyanine green clearance index
- IHVR
intrahepatic vascular resistance
- KC
Kupffer cell
- KLF2
Kruppel‐like Factor 2
- LSEC
liver sinusoidal endothelial cell
- MLCP
myosin light chain phosphatase
- Mtx
methoxamine
- NO
nitric oxide
- OCA
obeticholic acid
- PBF
portal blood flow
- PH
portal hypertension
- PP
portal pressure
- PPP
portal perfusion pressure
- ROS
reactive oxygen species
- TAA
thioacetamide
- VC
vasoconstrictors
Pathophysiology of Portal Hypertension
Portal hypertension (PH) is the most frequent and dreadful complication of chronic liver disease (CLD). The primary factor determining its development is a pathological increment in the intrahepatic vascular resistance (IHVR) to portal blood flow, which occurs early during CLD and is caused by fibrosis‐driven architectural distortion and sinusoidal microcirculatory dysfunction. Secondary to the increase in IHVR, a progressive splanchnic vasodilatation that increments portal blood flow further aggravates and perpetuates the PH syndrome1 (Fig. 1).
Figure 1.
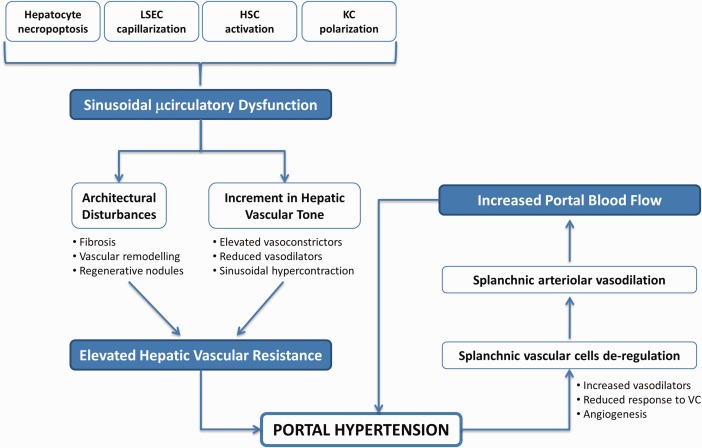
Pathophysiology of PH. Increment in the hepatic vascular resistance caused by sinusoidal microcirculatory dysfunction is the primary factor in the development of PH, which is further aggravated by an ulterior increment in the portal blood inflow. KC, Kupffer cells; VC, vasoconstrictors.
The mechanisms underlying the elevation in the IHVR directly relate to profound modifications in the phenotype of hepatic cells caused by the hepatoaggressor. It is important to note that, although the spatiotemporal sequence may differ among cell types, all liver cells become markedly deregulated because of chronic injury and negatively contribute to the increment in the IHVR, and therefore to PH development. Concretely, continuous hepatocyte necroapoptosis leads to nonregulated regeneration reactions ultimately resulting in the formation of disorganized regenerative nodules. Transdifferentiation of hepatic stellate cells (HSC) toward a myofibroblastic‐like cell (termed “activated HSC” with proliferative and hypercontractile properties) is accompanied by marked continuous extracellular matrix deposition that further contributes to the tortuosity of the liver parenchyma. In addition, loss of liver sinusoidal endothelial cells' (LSEC) healthy phenotype (a process known as “capillarization”) leads to a profound imbalance between vasodilators and vasoconstrictors and a reduction in sinusoidal porosity, which promote elevation of hepatic vascular tone and parenchymal extinction, respectively. Kupffer cells also become polarized during CLD, further contributing to both fibrosis and sinusoidal dysfunction. In addition to toxicant‐direct effects on hepatic cells, paracrine factors released from each injured cell affect neighboring cells, further exacerbating the sinusoidal dysfunction of CLD livers.2
Secondarily to the increment in IHVR, the cells of the splanchnic vascular bed modify their phenotype toward a vasodilator and proangiogenic one, trying to compensate the elevated vascular tone found within the portal system. As a consequence, portal blood flow increases, worsening the elevation in portal pressure.
New Therapeutic Targets and Pharmacological Approaches
Diminishing IHVR, splanchnic hyperemia, or both can significantly reduce the portal pressure. Until now, nonselective beta blockers, vasopressin analogues, and somatostatin analogues have demonstrated efficient reduction in portal pressure because of the correction in the splanchnic hyperemia. In contrast, safe and reliable strategies to reduce the IHVR in cirrhotic patients still represent a pending issue. Recent work developed in preclinical models of cirrhotic PH (Fig. 2) define two main therapeutic avenues with high potential to reduce the IHVR in cirrhosis and easy translation to the bedside.
Figure 2.
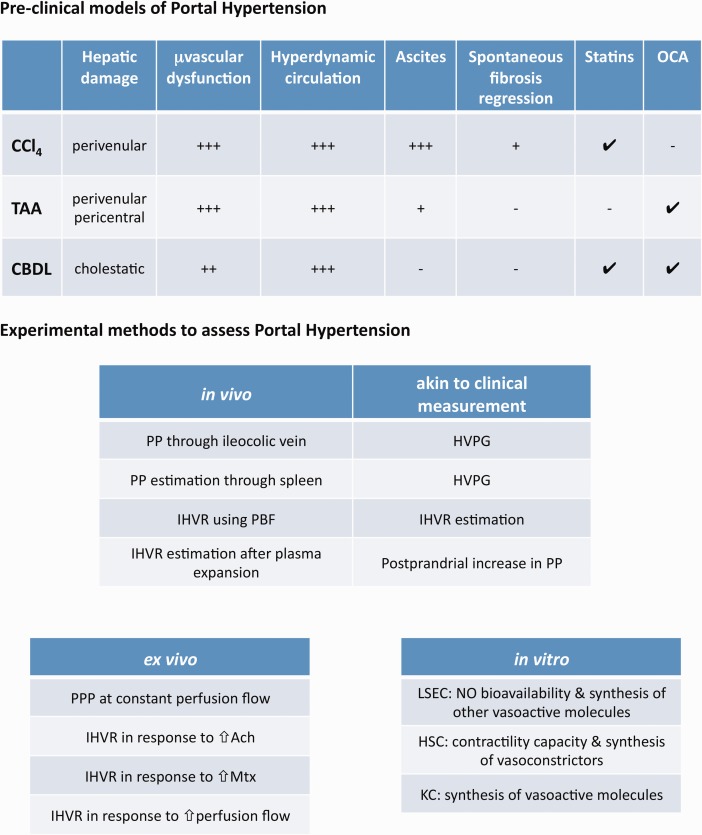
Preclinical models and methods of PH. (Top) Rodent preclinical models of cirrhotic PH. (Bottom) Most common methods to assess PH and liver microvascular dysfunction. Ach, acetylcholine; CBDL, common bile duct ligation; CCl4, carbon tetrachloride; HVPG, hepatic venous pressure gradient; KC, Kupffer cells; Mtx, methoxamine; PBF, portal blood flow; PP, portal pressure; PPP, portal perfusion pressure; TAA, thioacetamide.
The Kruppel‐like Factor 2‐Nitric Oxide Pathway: Statins
Nitric oxide (NO), a gaseous vasodilator physiologically synthesized by the endothelium, is markedly reduced in cirrhotic livers. Its low bioavailability derives from both a reduction in its synthesis and elevated scavenging by hepatic oxidative stress. Almost 15 years ago, a groundbreaking study directed by Prof. Jaime Bosch aimed at investigating whether statins, which were developed to reduce circulating cholesterol levels, but also conferred potent vasoprotection unrelated to their lipid‐lowering effects, would be able to increment intrahepatic NO levels, and therefore improve PH syndrome in cirrhotic patients.3 That study demonstrated for the first time the beneficial effects of statins in reducing portal pressure. Subsequent preclinical investigations revealed that simvastatin‐derived activation of the transcription factor Kruppel‐like Factor 2 (KLF2) was the underlying mechanism for its hepatoprotection. Indeed, administration of statins, particularly simvastatin, markedly improves the phenotype of sinusoidal cells through the induction of KLF2‐derived protective transcriptional programs (Fig. 3). As result of KLF2 induction, livers from animals with cirrhosis exhibit marked amelioration in PH syndrome that results from both regression in liver fibrosis and marked improvement in hepatic microvascular dysfunction.4
Figure 3.
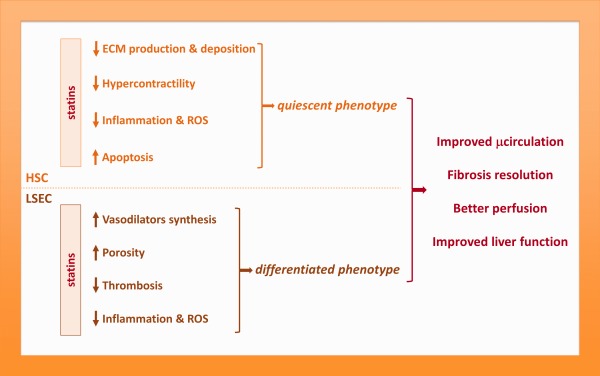
Framework of statins effects on sinusoidal cells phenotype. Statins improve the phenotype of sinusoidal cells through a variety of mechanisms, most of them depending on the transcription factor KLF2, ultimately leading to global hepatic improvement. ECM, extracellular matrix; ROS, reactive oxygen species.
Back to the bedside, in 2009 a phase II randomized controlled trial demonstrated that when administered to patients with cirrhosis and PH simvastatin was safe and promoted a moderate decrease in portal pressure, both when given alone and with nonselective beta blockers.5 The effect of simvastatin in portal pressure did not modify liver blood flow, implying a decrease in IHVR. Moreover, patients receiving simvastatin exhibited a marked improvement in liver function, suggesting an amelioration of metabolic exchange at the liver microcirculation, altogether likely reflecting decreased liver fibrosis. Those findings, together with the preclinical data, provided a strong rationale for evaluating the effects of simvastatin versus placebo in a double‐blind multicenter clinical trial performed in patients with cirrhosis who were randomized after admission because of variceal bleeding.6 The results of this recently published study show that simvastatin administration did not improve the probability of rebleeding, but was associated with a 61% reduction in the risk for death, especially in patients with moderately severe liver failure (Child‐Pugh A/B). Figure 4 summarizes the clinical studies analyzing the effects of statins on PH and cirrhosis.
Figure 4.
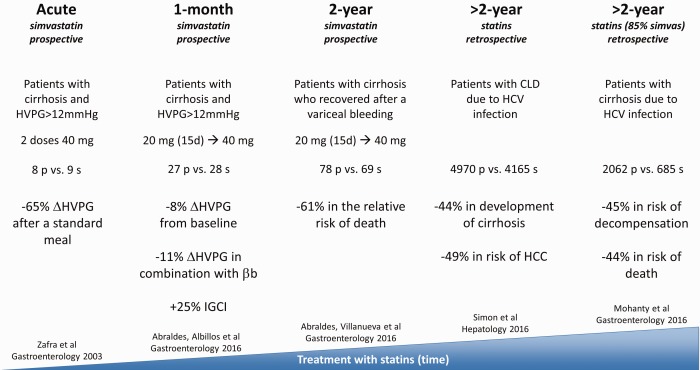
Effects of statins in patients with liver disease. Summary of five major studies analyzing the effects of statins on cirrhosis and portal hypertension. βb, beta blockers; CLD, chronic liver disease; IGCI, indocyanine green clearance index; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HVPG, hepatic venous pressure gradient.
The Farnesoid‐X‐Receptor Pathway: Obeticholic Acid
The farnesoid X receptor (FXR) is a bile‐acid response transcription factor belonging to the nuclear receptor superfamily that controls the expression of different pivotal genes involved in metabolic regulation, inflammation, and regeneration. FXR deficiency has been documented in experimental and human cirrhosis and found to be associated with complications of cirrhosis, such as spontaneous bacterial peritonitis.7, 8 The apparent cardinal role of FXR dysfunction in different disorders has led to the development of a first‐in‐class semisynthetic FXR agonist 6‐alpha‐ethyl‐chenodeoxycholic acid or obeticholic acid (OCA), which has opened the door for translational and clinical research for numerous conditions, including PH.
Short‐term treatment with OCA has been shown to reduce PH in two different cirrhotic rodent models without deleterious impact on mean arterial pressure or liver biochemistry. The basis for this beneficial effect relates to restoration of intrahepatic endothelial NO synthase activity, albeit via different mechanisms depending on the cause of experimental cirrhosis (Fig. 5).7 In humans, there is at present only one open‐label phase 2a proof‐of‐concept study in support of these translational data. Mookerjee et al.9 reported that 9 of 16 patients with alcoholic cirrhosis responded with a mean reduction of 28% in hepatic venous pressure gradient after 7 to 12 days of OCA. We are awaiting final results, as well as larger controlled confirmatory trials.
Figure 5.
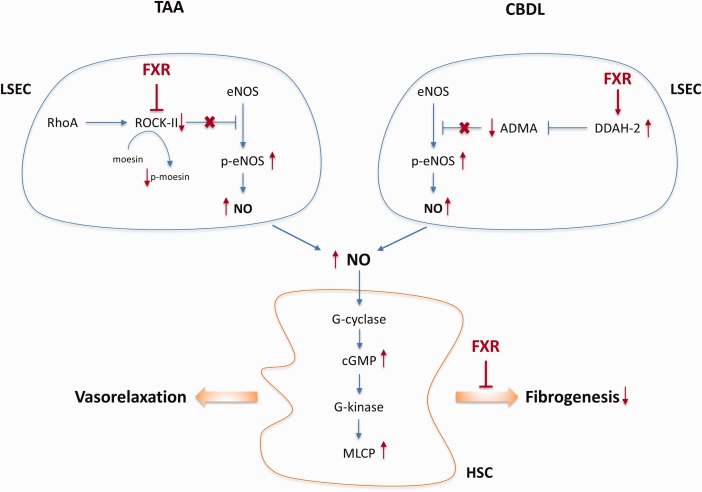
Effects of FXR agonist on PH and cirrhosis. Molecular mechanisms underlying the vasodilator and antifibrotic effects of the FXR agonist OCA in two preclinical models of cirrhosis. ADMA, asymmetric dimethylarginine; CBDL, common bile duct ligation; cGMP, cyclic guanosine monophosphate; DDAH‐2, dimethylarginine dimethylaminohydrolase 2; eNOS, endothelial nitric oxide synthase; MLCP, myosin light chain phosphatase; TAA, thioacetamide.
In addition to potential impact on intrahepatic endothelial dysfunction, long‐term administration of OCA was also shown to affect hepatic fibrosis, and thus might carry antifibrotic effects. A rat model of thioacetamide‐induced long‐term treatment with OCA was able to reduce hepatic fibrosis during ongoing injury, but also to reverse fibrosis after first obtaining cirrhosis, resulting in decreased IHVR and improved PH. The molecular basis hereof was found to be related to indirect effects on immune activation and hepatic cell turnover, associated with a marked overall decrease in nuclear factor‐κB activation and cytokines promoting fibrosis and inflammation within the liver parenchyma.10 The FLINT trial substantiated this antifibrotic effect in patients treated with OCA for nonalcoholic steatohepatitis (but without cirrhosis).11
This study was supported by grants FIS PI14/00029 and Explora BIO2014‐61377 from the Instituto de Salud Carlos III and Ministerio de Economıa y Competitividad & Funds FEDER “una manera de hacer Europa”, Spain (J.G.‐S.), and the Fund for Scientific Research–Flanders (FWO Vlaanderen), Belgium (W.L.).
Potential conflict of interest: Nothing to report.
REFERENCES
- 1. Bosch J, Goszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in the paradigm are leading to successful new treatments. J Hepatol 2015;62(1 Suppl):S121‐S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gracia‐Sancho J, Maeso‐Díaz R, Fernández‐Iglesias A, Navarro‐Zornoza M, Bosch J. New cellular and molecular targets for the treatment of portal hypertension. Hepatol Int 2015;2:183‐191. [DOI] [PubMed] [Google Scholar]
- 3. Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernández M, Garca‐Pagán JC, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 2004;126:749‐755. [DOI] [PubMed] [Google Scholar]
- 4. Marrone G, Maeso‐Díaz R, García‐Cardena G, Abraldes JG, García‐Pagán JC, Bosch J, et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut 2015;64:1434‐1443. [DOI] [PubMed] [Google Scholar]
- 5. Abraldes JG, Albillos A, Bañares R, Turnes J, González R, García‐Pagán JC, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651‐1658. [DOI] [PubMed] [Google Scholar]
- 6. Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez‐Guerra M, Genesca J, et al.; on behalf of the BLEPS Study Group . Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology 2016;150:1160‐1170. [DOI] [PubMed] [Google Scholar]
- 7. Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014;59:2286‐2298. [DOI] [PubMed] [Google Scholar]
- 8. Lutz P, Berger C, Langhans B, Grünhage F, Appenrodt B, Nattermann J, et al. A farnesoid X receptor polymorphism predisposes to spontaneous bacterial peritonitis. Dig Liver Dis 2014;46:1047‐1050. [DOI] [PubMed] [Google Scholar]
- 9. Mookerjee R, Rosselli M, Pieri G, Beecher‐Jones T, Hooshmand‐Rad R, Chouhan M, et al. Effects of the FXR agonist obeticholic acid on hepatic venous pressure gradient in alcoholic cirrhosis: a proof of concept phase 2A study. J Hepatol 2014;60(Suppl):S7‐S8. [Google Scholar]
- 10. Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Elst IV, et al. In toxic cirrhosis, the FXR agonist obeticholic acid reduces liver fibrosis indirectly via an anti‐inflammatory effect in liver sinusoidal endothelial cells and Kuppfer cells. J Hepatol 2016;64:PS011. [Google Scholar]
- 11. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; on behalf of NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. TG Simon, H Bonilla, P Yan, RT Chung, AA Butt. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: Results from ERCHIVES. Hepatology 2016;64:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. A Mohanty, JP Tate, G Garcia-Tsao. Statins are associated with a decreased risk of decompensation and death in veterans with hepatitis C-related compensated cirrhosis. Gastroenterology 2016;150:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]