

Watch a video presentation of this article
Watch the interview with the author
Abbreviations
- GABA
gamma‐hydroxybutyric acid
- GS
glutamine synthetase
- HE
hepatic encephalopathy
- LPS
lipopolysaccharide
- MHE
minimal hepatic encephalopathy
- NH3
ammonia
- NH4
ammonium
- NHS
National Health Service
- NIHR
National Institute for Health Research
- NMDA
N‐Methyl‐D‐aspartic acid
- OHE
overt hepatic encephalopathy
Introduction
Hepatic encephalopathy (HE) represents a diverse spectrum of complex neuropsychiatric disturbance resulting from liver disease and its concomitant metabolic and immunological derangements. It is characterized by deficits in cognitive, psychiatric, and motor function and can range in severity from minimal (or covert) hepatic encephalopathy (MHE) to overt hepatic encephalopathy (OHE), coma, and death. Patients with MHE demonstrate neuropsychological alterations including disrupted sleep–wake cycle, personality changes, impairment of attention, cognitive and memory dysfunction, and changes in motor coordination. These can progress through to higher grades of OHE, including lethargy, stupor, coma, and death; these are more pronounced in patients with acute liver failure. HE is a frequent and debilitating manifestation of decompensated liver disease affecting patients and their careers alike. The scope of this review is to outline the current understanding and ongoing research into the pathophysiological mechanisms underlying this complex neuropsychiatric condition.
Ammonia
Ammonia has long been regarded as the key metabolic factor underpinning the development of HE since the original description of the “meat intoxication syndrome” in portacaval‐shunted dogs at the end of the 19th century. In the presence of liver failure, decreased utilization of ammonia as a substrate in the hepatic urea cycle (the major mammalian ammonia detoxification pathway) and portosystemic shunting lead to the accumulation of ammonia in the systemic circulation, which readily crosses the blood–brain barrier. Cerebral ammonia detoxification occurs via glutamine synthetase, exclusively expressed in cerebral astrocytes, in the formation of glutamine, which is an important precursor of the main excitatory and inhibitory neurotransmitters: glutamate and gamma‐hydroxybutyric acid (GABA), respectively. Astrocytic glutamine accumulation exerts an osmotic effect resulting in swelling and cytotoxic edema, which leads to increased brain water on magnetic resonance imaging and worsening HE (Fig. 1).
Figure 1.
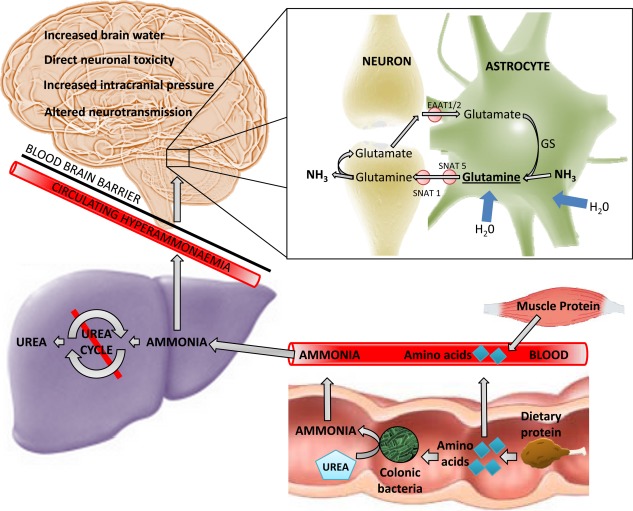
The role of ammonia in the development of hepatic encephalopathy. Decreased hepatic urea‐cycle metabolism in the context of liver cirrhosis and/or portosystemic shunting leads to the accumulation of ammonia (NH3), a product of protein catabolism, in the systemic circulation. Ammonia readily crosses the blood–brain barrier and is metabolized in a cerebral detoxification pathway by GS, with the formation of glutamine occurring exclusively in cerebral astrocytes. Accumulation of glutamine exerts an osmotic effect, with the influx of water leading to astrocytic swelling. Glutamine is shuttled via transporters (SNAT 5/SNAT 1) to presynaptic neurones and converted to GABA or glutamate before release into the inhibitory or excitatory synaptic cleft, respectively, and subsequently scavenged by astrocytic reuptake transporters (EAAT1/2).
Hyperammonemia and subsequent glutamine accumulation induce changes in cerebral neurotransmission. Acute hyperammonemia leads to excessive glutamate‐induced N‐Methyl‐D‐aspartic acid (NMDA)‐receptor activation, which can cause neuronal death. Blockade of the NMDA receptor has been shown to be protective in this context.1 Also noted is an increase in “GABAergic tone,” with an observed clinical response to flumazenil, a GABAA receptor antagonist. Rabbits exposed to benzodiazepines and rabbits with acute liver failure demonstrate similar visual‐evoked potentials.2
Inflammation
However, circulating hyperammonemia does not explain all of the pathophysiological processes underpinning HE. Arterial ammonia concentrations correlate poorly with clinical presentations of HE in cirrhosis, and increasingly recognized is the importance of the synergistic role between hyperammonemia and inflammation/infection in the development of HE. Progressive encephalopathy was noted to occur in patients with higher systemic inflammatory response scores in acute liver failure3; and infection was shown to exacerbate neurocognitive dysfunction following an ammonia load in patients with cirrhosis, which resolved following antibiotic therapy.4 Bile‐duct–ligated cirrhotic rats progressed to precoma stages of encephalopathy following intraperitoneal administration of lipopolysaccharide, a gram‐negative cell wall peptide.5 A proinflammatory cytokine plasma milieu verus arterial ammonia or the severity of liver disease has been shown to independently correlate with the presence and severity of HE.6 Potential pathophysiological mechanisms explaining the susceptibility to developing HE during episodes of infection include cerebral hyperemia with increased brain ammonia delivery, astroglial oxidative stress with microglial activation and neuronal dysfunction, and innate immune system dysfunction arising from systemic inflammation and circulating endotoxemia.7 The maintenance of a high index of suspicion of infection underlying clinical presentations of HE and expedient treatment remains pivotal in its management.
The Intestinal Microbiome and Hepatic Encephalopathy
Changes in the gut microbiota of patients with liver cirrhosis are considered central to bacterial translocation, endotoxemia, and systemic inflammation that can result in the development of HE. Liu et al. demonstrated that cirrhotic patients with MHE had significant fecal overgrowth of Escheria Coli and Staphylococcus Spp. and went on to show that treatment with synbiotics reduced blood ammonia and endotoxemia and improved MHE scores.8 More recently, the advent of culture‐independent techniques in quantitating and speciating bacterial colonic flora has sparked a burgeoning interest in the role of the intestinal microbiota in HE.
Overgrowth of pathogenic bacteria in the gut microbiome relative to autochthonous (commensal) bacteria correlates with worsening liver disease severity, with studies demonstrating that the change in composition of the microbiota is associated with higher model for end‐stage liver disease scores.9 Furthermore, significant changes in enteric microbiota have been described between patients with MHE and OHE, with the latter group demonstrating a higher dysbiosis ratio10 (Fig. 2).
Figure 2.
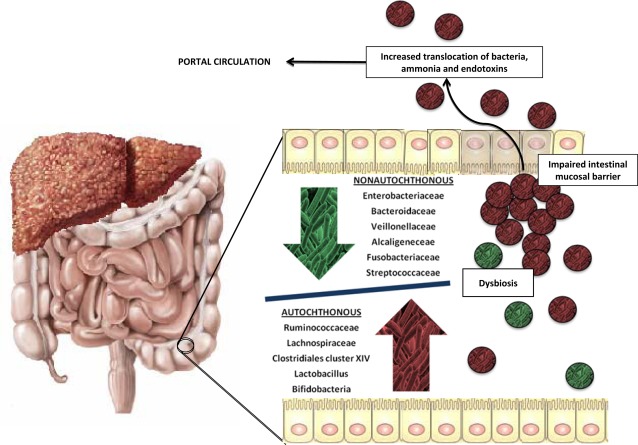
Changes in microbiota with worsening liver disease severity and cognitive performance. Comparing patients with differing severity of liver cirrhosis and comparing cirrhotic patients with and without the presence of OHE reveals an observed difference in the intestinal bacterial composition, with a relative paucity of autochthonous floral bacteria that have pathological species overrepresented, creating a dysbiosis. This changes gut ammonia metabolism and contributes to colonic inflammation, portal circulation endotoxemia, increased inflammatory burden in the presence of portal hypertensive enteropathy, and impaired intestinal mucosal barrier. Manipulation of the intestinal microbiome represents an attractive therapeutic target in the management of cirrhotic patients with HE.
Lactulose is a nonabsorbable disaccharide that has a pleiotropic role in the treatment of HE and has long formed the cornerstone of empirical HE treatment. It has been shown to change the colonic pH favoring retention of ammonium salts in the bowel lumen and exerts a prebiotic effect favoring the colonization of Lactobacillus and Bifidobacterium; it also reduces intestinal transit time and may therein reduce ammonia absorption. Similarly, probiotics tilt the intestinal microbiota toward nonurease‐producing organisms, decrease luminal production of ammonia, and have been shown to be effective in the prevention of HE in cirrhosis.11 Rifaximin‐α is a nonabsorbable antibiotic that maintains remission from OHE with a reduction in hospitalizations due to HE over a 6‐month period.12 Interestingly, it was not shown to alter the relative abundance of pathogenic bacteria; instead, it may be exerting its therapeutic effect by reducing circulating endotoxemia and manipulating bacterial function and virulence.13
Conclusion
HE remains one of the major challenges and morbidities facing patients with decompensated liver cirrhosis. Even in its subclinical presentation, it exerts a profound influence on patient quality of life and functional capability and confers a damning prognosis. Recurrent encephalopathy is in itself an extended criterion for consideration of liver transplantation. Increasing understanding of the interplay between the liver, the intestinal microbiome, and the innate immune system allows for the development of exciting new technologies and treatments to include in the clinician's armory for tackling this neurophysiological manifestation of decompensated liver disease (Fig. 3). A low threshold of suspicion and early specialist review, complex neuropsychological evaluation, and identification and treatment of precipitants are required in the approach to a patient with liver cirrhosis and altered mentation.
Figure 3.
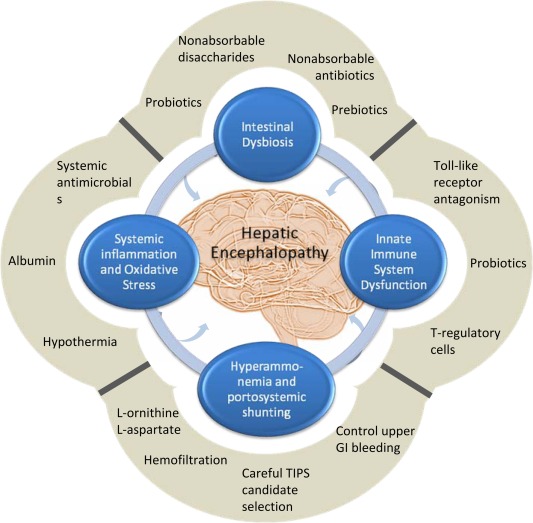
Overlapping pathophysiological mechanisms underpinning the development of hepatic encephalopathy and revealing targets for therapeutic intervention. No single pathophysiological pathway explains in full the development of HE; and it is increasingly recognized that hyperammonemia, systemic inflammation, and intestinal dysbiosis act in a synergistic cascade that terminates in the development of HE. Equally, no single treatment is completely effective at ensuring resolution and preventing the recurrence of HE. Recognizing the multifaceted pathophysiological process driving the development of HE has allowed for innovative therapeutic targeting. The utilization of a multipronged treatment strategy is central to management of this condition.
Acknowledgment
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
This study was supported by grants by the Medical Research Council Centre for Transplantation, King's College London, UK (MRC grant no. MR/J006742/1) and the NIHR Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London.
Potential conflict of interest: Nothing to report.
References
- 1. Kosenko E, Kaminski Y, Lopata O, Muravyov N, Felipo V. Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Radic Biol Med 1999;26:1369‐1374. [DOI] [PubMed] [Google Scholar]
- 2. Sergeeva OA. GABAergic transmission in hepatic encephalopathy. Arch Biochem Biophys 2013;536:122‐130. [DOI] [PubMed] [Google Scholar]
- 3. Rolando N, Davalos M, Wendon J, Philpott‐Howard J, Willams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000;32:734‐739. [DOI] [PubMed] [Google Scholar]
- 4. Shawcross DL, Davies NA, Williams R, Jalan R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol 2004;40:247‐254. [DOI] [PubMed] [Google Scholar]
- 5. Wright G, Davies NA, Shawcross DL, Hodges SJ, Zwingmann C, Brooks HF, et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007;45:1517‐1526. [DOI] [PubMed] [Google Scholar]
- 6. Shawcross DL, Wright G, Olde Damink SW, Jalan R. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab Brain Dis 2007;22:125‐138. [DOI] [PubMed] [Google Scholar]
- 7. Coltart I, Tranah TH, Shawcross DL. Inflammation and hepatic encephalopathy. Arch Biochem Biophys 2013;536:189‐196. [DOI] [PubMed] [Google Scholar]
- 8. Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Rioran SM. Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004;39:1441‐1449. [DOI] [PubMed] [Google Scholar]
- 9. Chen Y, Yang F, Wang B, Chen Y, Lei D, Wang Y, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011;54:562‐572. [DOI] [PubMed] [Google Scholar]
- 10. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 2014;60:940‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lunia MK, Sharma BC, Sharma P, Sachdeva S, Srivastava S. Probiotics prevent hepatic encephalopathy in patients with cirrhosis: a randomized controlled trial. Clin Gastroenterol Hepatol 2014;12:1003‐1008.e1. [DOI] [PubMed] [Google Scholar]
- 12. Bass NM, Mullen KD, Sanyal A, Poordad F, Neff G, Leevy CB, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med 2010;362:1071‐1081. [DOI] [PubMed] [Google Scholar]
- 13. Bajaj JS, Heuman M, Sanyal AJ, Hylemon PB, Sterling RK, Stravitz R, et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS One 2013;8:e60042. [DOI] [PMC free article] [PubMed] [Google Scholar]