

Abstract
Mineralocorticoid receptor (MR) activation plays an essential role in promoting inflammation, fibrosis, and target organ damage. Currently, no studies are investigating MR antagonism in patients with type 2 diabetes mellitus (T2DM) with chronic kidney disease, at high risk for cardiovascular complications, who are otherwise not candidates for MR antagonism by virtue of heart failure. Further, there is limited information on candidate therapies that may demonstrate differential benefit from this therapy. We hypothesized that MR antagonism may provide additional protection from atherosclerosis progression in higher‐risk patients who otherwise may not be candidates for such a therapeutic approach. In this double‐blind, randomized, placebo‐controlled trial, subjects with T2DM with chronic kidney disease (≥ stage 3) will be randomized in a 1:1 manner to placebo or spironolactone (12.5 mg with eventual escalation to 25 mg daily over a 4‐week period). The co‐primary efficacy endpoint will be percentage change in total atheroma volume in thoracic aorta and left ventricular mass at 52 weeks in patients treated with spironolactone vs placebo. Secondary outcomes include 24‐hour mean systolic blood pressure, central aortic blood pressure, and insulin resistance (HOMA‐IR) at 6 weeks. A novel measure in the study will be changes in candidate miRNAs that regulate expression of NR3C2 (MR gene) as well as measuring monocyte/macrophage polarization in response to therapy with spironolactone. We envision that our strategy of simultaneously probing the effects of a drug combined with analysis of mechanisms of action and predictive response will likely provide key information with which to design event‐based trials.
Keywords: Mineralocorticoid Receptor, Atherosclerosis, Inflammation, Macrophage, Monocyte, Magnetic Resonance Imaging, miRNA
1. INTRODUCTION
Atherosclerosis is a major cause of morbidity and mortality in patients with type 2 diabetes mellitus (T2DM). Mineralocorticoid receptor (MR) activation plays an essential role in promoting inflammation, fibrosis, and target organ damage.1, 2, 3, 4 The importance of antagonizing the MR is well recognized in patients with hypertension, heart failure, and heart failure post–myocardial infarction.5, 6, 7, 8, 9 Evidence from experimental animal models and humans suggests that MR activation promotes endothelial dysfunction, vascular oxidative stress, inflammation, proliferation, migration, vasoconstriction, vascular remodeling, fibrosis, and hypertrophy.10, 11, 12, 13, 14 Emerging data links MR activation with multiple components of the metabolic syndrome and T2DM (Figure 1).15, 16 One key process by which MR activation could influence metabolic function is through its role in regulating macrophage function and inflammation.
Figure 1.
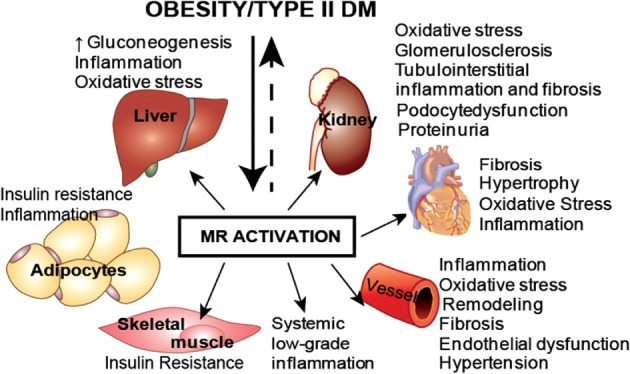
Various pathophysiologic pathways that lead to adverse outcomes as a result of MR activation. Abbreviations: DM, diabetes mellitus; MR, mineralocorticoid receptor.
In humans, MR antagonism results in anti‐inflammatory, antifibrotic, and renoprotective effects. Furthermore, recent studies have suggested a favorable effect of short‐term MR antagonism on measures of insulin resistance (IR).16 Indeed, MR activation may affect IR via: (1) impaired expression of insulin receptor; (2) reduced expression of glucose transporter type 4; (3) abnormal phosphorylation of IR substrates, leading to activation of multiple downstream kinases; (4) reduced insulin signaling in the cardiovascular and skeletal system; and (5) promoting hepatocyte oxidative stress and gluconeogenesis.17 Taken together, these data would indicate a potential role for MR antagonism as a therapeutic strategy in T2DM that may favorably affect outcomes via modulation of multiple key pathophysiologic pathways. The ability to favorably alter multiple disparate pathways by a single drug entity is an essential attribute for success of a pharmacologic entity in T2DM, given the complexity of the disease.
There is an additional need to understand response to therapies given the revolution of new knowledge in pharmacogenomics of cardiovascular disease. MicroRNAs (miRNAs) are noncoding RNAs of 18 to 25 nucleotides that regulate multigene expression and modulate biological pathways.18, 19 miRNAs regulate a multitude of targets and may influence pathways of interest involved in the pathogenesis of insulin resistance and DM, many of which could also be regulated by the MR. Additionally, recent studies seem to indicate that MR itself may be a target of multiple miRNAs. There has been a substantial interest in identifying key miRNA sequences that may predict complications in T2DM and may also provide a novel pharmacogenomic indicator of responsiveness to MR antagonism (see Supporting Information, Figure 1, in the online version of this article). In this prospective study, we will provide evidence for key specific miRNA sequences that may provide a pharmacogenomic measure of responsiveness. We envision that our strategy of simultaneously probing the effects on 2 endpoints of a drug to assess potentially concordant effects of change in atheroma burden and left ventricular mass (LVM), combined with analysis of mechanisms of action and predictive response, will likely provide key information with which to design event‐based (eg, myocardial infarction, stroke) trials.
2. METHODS
The main study phase is a multicenter, double‐blind, randomized, placebo‐controlled trial with spironolactone (http://www.ClinicalTrials.gov NCT02169089). The primary endpoints are percent change in atheroma volume (PAV) in the thoracic aorta of patients taking spironolactone vs placebo and LVM regression by cardiac magnetic resonance imaging (cMRI). The study has multiple secondary and tertiary objectives, detailed in sections below and summarized in Table 1. All MRI images will have identifying data removed for analysis, and images will be blinded to other study data and outcomes and sent to the MRI core lab for analysis (University Hospitals, Cleveland Medical Center, Core Laboratory).
Table 1.
Clinical endpoints
| Primary endpoints |
|---|
| % change in atheroma volume (PAV) in thoracic aorta |
| LV mass regression |
| Secondary endpoints |
| Central aortic SBP |
| 24‐hour mean SBP |
| Insulin resistance (HOMA‐IR) at 6 weeks |
| Tertiary measures |
| Evaluate miRNAs involved in regulating MR expression and differentially regulated miRNAs as predictors of disease progression and response |
| Monocyte inflammatory activation in spironolactone vs placebo groups at 6 weeks |
Abbreviations: HOMA‐IR, homeostatic model assessment of insulin resistance; LV, left ventricular; miRNA, microRNA; MR, mineralocorticoid receptor; SBP, systolic blood pressure.
2.1. Study population
Male and female patients age >45 years who are able to provide informed consent and meet the inclusion criteria and none of the exclusion criteria will be included in the study (Table 2). Specifically, the study will enroll T2DM patients with glycated hemoglobin (HbA1c) ≤8.0% on stable antiglycemic therapy, with normal baseline ejection fraction, who have a glomerular filtration rate (GFR) <90 mL/min/1.73 m2 and evidence of proteinuria (urine albumin/creatinine [Cr] ratio >30 mg/g or equivalent) in a urine specimen within 12 months or GFR <60 mL/min/1.73 m2 regardless of proteinuria. Patients must be on statin, angiotensin‐converting enzyme inhibitor (ACEI), and/or angiotensin II receptor blocker (ARB) therapy with no planned dose adjustments.
Table 2.
Inclusion and exclusion criteria
| Inclusion criteria |
|---|
| 1. M or F patients age >45 years and able to provide informed consent (females must be either postmenopausal for 1 year, surgically sterile, or using effective contraception. Oral contraceptives are disallowed). |
| 2. Patients with T2DM with HbA1c ≤8.0% on stable antiglycemic regimen that may include oral and/or injectable therapy (eg, GLP‐1/insulin). No new drugs are allowed. Changes in dose of glycemic regimen are allowed during the course of the trial if felt to be clinically appropriate. |
| 3. GFR <90 mL/min/1.73 m2 and evidence of proteinuria (urine albumin/Cr ratio of >30 mg/g or equivalent) in a urine specimen within 12 months, or GFR <60 mg/g regardless of proteinuria |
| 4. One of the following additional high‐risk features: |
| a. Established CVDa |
| b. Evidence of documented retinopathy or laser photocoagulation from patient |
| c. LVH on echocardiogram or 12‐lead ECG |
| d. Family history of premature CAD (first‐degree relative, male age <55 years, female age <65 years) |
| 5. Patients must be on ACEI and/or ARB therapy with no planned dose adjustments. |
| Exclusion criteria |
| 1. Uncontrolled HTN (SBP >160 mm Hg and/or DBP >95 mm Hg at visit 0 (screening) and SBP >145 mm Hg at visit 2) |
| 2. GFR (MDRD) of <30 mg/g at visit 0 (screening) |
| 3. Hyperkalemia, defined as serum K+ ≥5.1 mEq/L at visit 0 (screening) |
| 4. LDL‐C >150 mg/dL |
| 5. Plasma TG >300 mg/dL |
| 6. Contraindications to MRI (metallic implants, severe claustrophobia) |
| 7. ACS, TIA, CVA, or critical limb ischemia during the last 6 months, or coronary/peripheral revascularization within the last 3 months |
| 8. Evidence of a secondary form of HTN |
| 9. Initiation of new therapy with statins, ACEI/ARBs, antioxidants, CCBs, diuretics, or β‐blockers |
| 10. T1DM |
| 11. Known contraindication, including history of allergy, to spironolactone |
| 12. Any surgical or medical condition that might alter pharmacokinetics of drug (eg, renal transplant, liver failure, liver transplant) |
| 13. Concurrent potentially life‐threatening arrhythmia or symptomatic arrhythmia |
| 14. Significant hyponatremia, defined as Na <130 mEq/L |
| 15. History of prior malignancy, including leukemia and lymphoma (but not basal cell skin cancer, cured squamous cell cancer, and cured prostate cancer) |
| 16. History of any severe, life‐threatening disease |
| 17. Any surgical or medical conditions that place the patient at higher risk derived from his/her participation in the study or that are likely to prevent the patient from complying with study requirements |
| 18. History of drug abuse within the last 2 years, noncompliance, and unwillingness/inability to consent |
| 19. Pregnant women and nursing mothers |
| 20. NYHA class III or IV CHF |
| 21. Primary hyperaldosteronism |
Abbreviations: AAA, abdominal aortic aneurysm; ABI, ankle‐brachial index; ACEI, angiotensin‐converting enzyme inhibitor; ACS, acute coronary syndrome; ARB, angiotensin II receptor blocker; CABG, coronary artery bypass grafting; CAD, coronary artery disease; CCB, calcium channel blocker; CHF, congestive heart failure; Cr, creatinine; CT, computed tomography; CVA, cerebrovascular accident; CVD, cardiovascular disease; DBP, diastolic blood pressure; ECG, electrocardiogram; F, female; GFR, glomerular filtration rate; GLP‐1, glucagon‐like peptide‐1; HbA1c, glycated hemoglobin; HTN, hypertension; K+, potassium; LDL‐C, low‐density lipoprotein cholesterol; LVH, left ventricular hypertrophy; M, male; MDRD, Modification of Diet in Renal Disease equation; MI, myocardial infarction; MRI, magnetic resonance imaging; Na, sodium; NYHA, New York Heart Association; PCI, percutaneous coronary intervention; SBP, systolic blood pressure; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TG, triglycerides; TIA, transient ischemic attack.
1CVD is defined as a previous MI, prior catheterization (patient report alone also sufficient) documenting coronary disease, previous CABG or PCI, prior CT angiogram documenting CAD, positive stress test, prior lower extremity revascularization or stenosis or intervention, prior large‐vessel ischemic CVA (patient report alone also sufficient), abnormal ABI, evidence of carotid and/or subclavian and/or renal stenosis by imaging, AAA.
2.2. Primary endpoint
The primary endpoint in the trial, PAV, will be evaluated by MRI using a modified spin‐echo T1‐weighted sampling perfection with application optimized contrasts using different flip‐angle evolution (SPACE) sequence at 3.0T using electrocardiographically (ECG) and navigator gated acquisition as previously described, which provides a motion‐free 3‐dimensional dataset of the entire thoracic aorta, including the arch, at submillimeter resolution.20, 21 Following acquisition, pre‐ and postdata sets (before and following therapy) will be co‐registered on a workstation (Fusion; Siemens Healthcare, Inc., Erlangen, Germany) to ensure congruence of positioning. Multiplanar images perpendicular to the aorta will be generated from both datasets at the same location and evaluated using a custom‐designed plaque‐analysis tool (TeraRecon Inc., San Mateo, CA). Figure 2 demonstrates a screenshot of analysis of the thoracic aorta. For practical and clinically relevant reasons, we are specifically interested in evaluating atheroma burden, a surrogate marker for inflammation and fibrosis, rather than directly imaging these endpoints. There will be no contrast administered. Utilizing noncontrast MRI is advantageous compared with gated computed tomography imaging, which, in addition to radiation disadvantage, confers an increased risk of contrast‐induced nephropathy in the study population.
Figure 2.
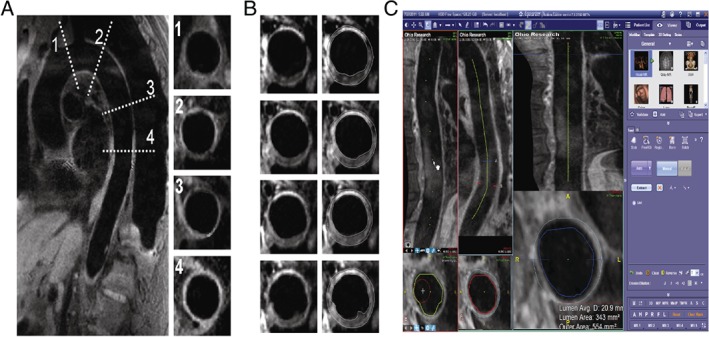
(A) ECG‐navigated gated T1‐SPACE acquisition of thoracic and abdominal aorta at 3T. Resolution 1.1 × 1.1 × 1.1 mm3; TR/TE = R‐R/21 ms; variable flip angle; NEX = 1.4; slices/slab = 56; turbo factor = 45; slice turbo factor = 1; echo train per slice = 3; echo train duration = 136; BW = 781 Hz/pixel. (B) and (C) Selected axial MPR co‐registered images pre/post treatment and analysis. Abbreviations: Avg, average; BW, bandwidth; ECG, electrocardiographically; MPR, multiplanar reconstruction; NEX, number of acquisitions; SPACE, sampling perfection with application optimized contrasts using different flip‐angle evolution; TE, echo time; TR, repetition time.
Measurements of lumen cross‐section area (LCSA) and media‐adventitia cross‐section area (MCSA) will be done. Vessel wall area measurements will be generated by subtracting the luminal area from outer border of the vessel wall for the images. Total plaque area at each time point (1, baseline; 2 post‐treatment) for each patient will be defined as the sum of the difference between MCSA and the LCSA in each slice. Derivation of absolute change (ΔTPV group) and percent change in atheroma volume (PAV or (% ΔTPV group)) will be calculated as shown below (for details, see Supporting Information, Appendix, in the online version of this article):
| (A) |
| (B) |
where Ngroup is the number of patients in the group and PAV (or % change in total atheroma volume) is the primary endpoint.
Measurement of the co–primary endpoint of LVM will be performed on a 3.0T clinical scanner (Siemens; or Philips Healthcare, Best, Netherlands). All images will be acquired during repeated breathholds and will be ECG gated using a cine steady‐state free precession sequence. Sequential short‐axis images will be acquired every 8 mm without a gap to cover the entire LV. Quantitative analysis of LVM will be performed by manually tracing the epicardial border (excluding epicardial fat) and endocardial borders (excluding papillary muscles) at end‐diastole and end‐systole for each short‐axis slice using custom software (Argus; Siemens). LVM regression will be defined as the percent difference between the LVM index (LVMI) measured at baseline and the LVMI measured at 52 weeks:
2.3. Secondary endpoints
2.3.1. Estimation of 24‐hour ambulatory BP
Ambulatory blood pressure (BP) will be measured with the Oscar 2 system (SunTech Medical, Morrisville, NC) at visit 2 (week 5, baseline) and visit 6 (week 11, midpoint). The machine will measure both ambulatory and central aortic BP at 30‐minute intervals during the daytime (6 am to 11 pm) and at 1‐hour intervals during the nighttime (11 pm to 6 am). The maximum BP measurement time will be set to 140 seconds, and the monitors will be set for a maximum pressure of 220 mm Hg.
2.3.2. Clinical SBP
The protocol for clinical systolic blood pressure (SBP) will follow the Systolic Blood Pressure Intervention Trial (SPRINT) protocol.22 Seated BP and pulse will be measured at each clinic visit after a rest period using an automated device in a quiet room in the absence of study personnel. This approach reduced potential for observer bias and decreased demand on staff in terms of training and effort in data collection.
2.3.3. Estimation of HOMA‐IR
Patients will undergo homeostatic model assessment of insulin resistance (HOMA‐IR) measurements after overnight fasting, after resting for 30 minutes after arrival, and prior to venous access and/or MRI assessment. HOMA ‐ IR = insulin (μU/mL) × glucose (mmol/L) / 22.5.23
Fasting (9‐hour) basal insulin sensitivity will be measured by the validated HOMA‐IR index. The average of 3 fasting blood samples taken 5 minutes apart will be used due to the pulsatile nature of insulin release.
2.4. Tertiary endpoints
Our preliminary data suggest miRNA control of MR expression and alternate monocyte/macrophage activation, which may represent a mechanism of regulatory control of inflammation and therapeutic response (see Supporting Information, Figure 2, in the online version of this article). We plan to measure monocyte subsets using flow cytometry and a panel of miRs that have been previously shown to be important in regulation of MR expression at baseline that may serve as indicators of pharmacogenomic efficacy of MR antagonism.24 In addition, the differential change in monocyte subpopulations and in candidate miRs following 6 weeks of therapy with MR antagonist or placebo will be evaluated.
The main study itself will have 4 subphases: screening, placebo run‐in, dose escalation, and treatment (Figure 3). The 2‐week single‐blind lead‐in period will permit assessment of patient compliance as well as determination of baseline seated BPs. Subjects with T2DM who meet all the inclusion and exclusion criteria (Table 2) will be eligible to participate in the study.
Figure 3.
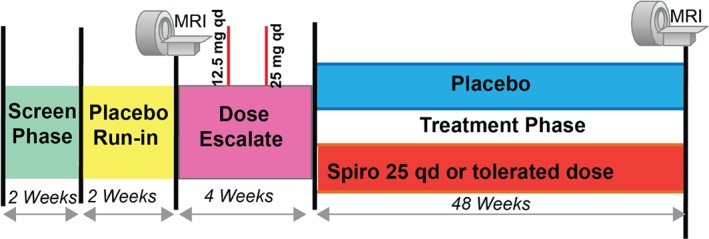
Main study phase with 4 subphases. Abbreviations: MRI, magnetic resonance imaging; qd, daily; spiro, spironolactone.
Written informed consent and Health Insurance Portability and Accountability Act (HIPAA) authorization will be obtained prior to starting any study‐related activity. An initial screening assessment will include a complete physical examination, baseline 12‐lead ECG, cardiovascular risk‐factor assessment, and diet and lifestyle questionnaire. At fixed time intervals throughout the study, SBP, body weight, medications, dietary salt intake, and an interim event log will be monitored. The eligible patients will be randomized 1:1 to placebo or spironolactone 12.5 mg on week 0. Patients will be escalated to 25 mg daily spironolactone or maximal tolerated dose over a 4‐week period with safety checks (Figure 3). Dose may be adjusted during the study due to safety concerns from laboratory results (eg, hyperkalemia) or SBP <100 mm Hg; dose adjustments will be performed by a nurse who will be unblinded to the intervention under the supervision of a nephrologist who will not be directly involved with the study.
2.4.1. Management of serum potassium
Hyperkalemia is a common side effect of renin‐angiotensin‐aldosterone blockade. It is even more common when an MR antagonist is added to renin‐angiotensin system blockade, especially in patients who may have reduced renal function. Decisions to manage elevations of hyperkalemia will be standardized using a protocol adopted in a previous trial that investigated the effects of dual ACEI and ARB therapy in patients with chronic kidney disease (CKD).25 For instance, loop diuretics will be utilized with or without sodium bicarbonate. Depending on subsequent change in serum potassium, the polymer patiromer will be added to further facilitate control of chronic hyperkalemia. This will allow consistency of treatment during the course of the clinical trial. If continued elevation in serum potassium occurs despite mitigation efforts, the patient will be withdrawn from the study and appropriate acute and/or chronic measures utilized (see Supporting Information, supplemental Table 1, in the online version of this article).
2.4.2. Management of other side effects and concomitant medications
Other common side effects include increase in blood urea nitrogen and Cr, for which the renal function will be monitored closely. While on the study protocol, changes in dose of glycemic regime are allowed, but addition of new antiglycemic agents is not permitted. All subjects should be on an ACEI and/or ARB and statin therapy with no planned dose adjustments. Oral contraceptives are prohibited during the study.
2.4.3. Safety and monitoring plan
All clinically significant events will be evaluated by an independent data‐safety monitoring committee. The safety board will meet every 3 months to review significant events (sooner if warranted). Because dietary sodium intake is an important determinant of MR responsiveness, we will ensure that all patients receive dietary survey/instructions (visit 0) on maintaining a personalized diabetic diet that is stable and is medium to low in sodium (<4 g). We will obtain 24‐hour sodium, potassium, and Cr levels (visit 1) to assess sodium intake. Patients will be stratified by sodium excretion for analysis of atherosclerosis response.
2.5. Statistical analysis
A total of 130 subjects will be enrolled. We expect that at week 6 there will be ≥135 subjects providing complete data (dropout of 10% due to hyperkalemia or other side effects). At visit 7 (week 25), which is approximately the halfway point, we expect additional loss to follow‐up, so that the number of subjects on study on each arm at 52 weeks with complete data will be ≥60/group.
2.5.1. Primary endpoint analysis
Power calculations for the primary endpoint with 62 patients/group will have 80% power to detect at least a 1.45‐fold difference in change in PAV between the 2 groups (α = 0.05; coefficient of variation [CV] = 83%). A 1.45‐fold difference would represent a 31% reduction in ΔPAV (eg, a change in ΔPAV from 2% to 1.38%). The CV was calculated using data for ΔPAV in an ongoing randomized controlled trial in n = 37 at 10 months (supplemental Table 2). The increase in PAV of 2% was based on assessment from a prior trial where the progression in a nondiabetic patient population was 1.4% in <1 year after 40 weeks of follow‐up.26 The change in total wall volume was 4%. We are performing our study in a higher‐risk patient population over a longer duration that will likely result in larger atheroma burden.
For the co–primary endpoint of LVMI, we will perform the “posteriori” analysis of LVM: If we are unable to meet statistical significance for PAV at the 0.05 level, we will then test for statistical significance for LVM at a more stringent α value of 0.025. We estimated that a sample size of 140 patients (n = 70 per study arm, assigned equally to treatment or placebo) will be required to detect a 11.6% change in LVMI on cMRI, providing 80% power, assuming a significance level of P < 0.025 (2‐sided) and a dropout of 10%. The CV of 22% was estimated by averaging the mean and SD from a double‐blinded randomized controlled trial that investigated 112 CKD patients over the course of 40 weeks; a conservative estimate, because patients in the proposed study will be followed for 52 weeks, where the treatment effects are anticipated to improve with time.27 (See Supporting Information, supplemental Table 2, in the online version of this article for the power for mean differences on reproducibility of central aortic BP and pulse wave velocity [PWV] from our own data.) These studies have shown that within‐observer and between‐observer differences in the noninvasive measurement of central systolic pressures are small with no group mean difference (intra‐ or interobserver) exceeding 4.6 mm Hg with SD of 5.4.28, 29, 30 Prior studies with eplerenone on central SBP and PWV have documented ≥10 mm Hg and 1.7 m/s change in radio‐femoral velocities, respectively, after 14 weeks of treatment.31 PWV is highly reproducible with mean differences (intra and inter) for repeat measurements not exceeding 0.30 m/s and SD of difference not exceeding 1.4.32, 33, 34, 35
2.5.2. Secondary endpoint analysis
We anticipate more than adequate power to assess changes in HOMA‐IR with this study. A previous study assessed the effect of spironolactone after 8 weeks of therapy. HOMA‐IR decreased from 4.4 ± 0.9 to 2.8 ± 0.5 in mildly insulin‐resistant patients.36 We expect that the mean response to spironolactone in the proposed study will be similar to, if not greater than, the response obtained in the previous study, as our study will involve severely insulin‐resistant subjects with T2DM.
For the functional significance of candidate miRNAs, THP‐1 experiments, n = 7 replicates per group (scrambled antagomirs/mimics), we will have 80% power to detect a 2‐fold effect of antagomirs/mimics on luciferase reporter expression (CV = 30%, α = 0.05/5 for 5 miRs). For confirmation experiment using human macrophages, n = 7 human donors will be needed for each miRNA study. An interaction contrast in an ANOVA model will be used to test the effects of overexpression of miRNAs (miRNA mimic) or antagomirs in various experiments where we will test the effect of miRs alone or in the presence of lipopolysaccharide or interleukin‐4/13. The effect of various interventions on NR3C2 gene expression and MR protein expression as well as markers of alternate activation will be tested.
3. DISCUSSION
This is the first systematic prospective study evaluating the effect of MR antagonism on change in atherosclerosis in a patient population that is at high risk for developing progressive atherosclerosis (DM and CKD) and who are otherwise not candidates for such a therapy under the current guidelines. We plan to demonstrate changes in 24‐hour mean SBP, PWV, and IR. Additionally, we will assess the link between miRNAs, macrophage activation (AΦ), and inflammation in disease progression. It is designed as a “proof‐of‐concept” study to test the utility of MR antagonism in attenuating development and progression of atherosclerosis, a surrogate of inflammation, fibrosis, and target organ damage. The inclusion of LVM, a BP‐sensitive validated endpoint that has been previously shown to be modulated by MR antagonism, together with an endpoint that responds to other stimuli (other than BP), strengthens our ability to look for discordant effects of MR antagonism on LVM and BP.
A novel aspect of the protocol is the implementation of new MRI pulse sequence design that enables motion‐free high‐resolution coverage of the entire aorta, including the arch, to evaluate longitudinal changes in PAV in patients on spironolactone vs placebo. Coverage of the entire aorta allows detection of smaller changes and inclusion of smaller numbers of patients and provides an improvement over invasive surrogate measures acquired in limited portions of the arterial bed.37, 38
Currently, there are no large trials of MR antagonism in patients with diabetic atherosclerosis and no trials investigating predictors of response. Results from our trial will provide a basis for a definitive intervention trial. However, monocyte/macrophage activation states in metabolic and cardiovascular disease are yet to be fully understood, and whether or not pharmacologic blockade of MR modifies monocyte inflammation in humans is unknown. Our novel study may elucidate an important role for MR in AΦ and will incorporate miRNA analysis to predict response, which is innovative and is based on preliminary data suggesting that several candidate miRNAs regulated MR expression and AΦ, and we may therefore shed light on the novel role and mechanism of MR blockade in reducing vascular inflammation and the burden of atherosclerosis.
An additional aspect of the clinical trial is to employ an effective mitigation strategy for managing elevations in serum potassium. This will demonstrate the clinical utility of this novel approach for the treatment of atherosclerosis. The ultimate clinical utility of adding MR antagonism to diabetic patients with heart and/or kidney disease may be limited by changes in serum potassium. The principal predictors of higher serum potassium levels in cohort studies are related to the presence of DM, higher protein intake, lower serum bicarbonate, Caucasian race, and, most important, lower estimated GFR.39, 40 In addition, the concurrent use of renin‐angiotensin system blockade and the MR antagonism will likely be an additional factor leading to hyperkalemia, which could result in cessation of the study drug. Our study will utilize novel mitigation strategies to assure the ability to maintain acceptable potassium levels despite the concomitant use of renin‐angiotensin system blockade and MR antagonism. Close attention to monitoring of serum potassium and utilization of diuretics and other clinical treatment modalities, such as the polymer patiromer, will assist in the process of mitigation.
4. CONCLUSION
As a consequence of this clinical trial, we hope to demonstrate not only the clinical utility of MR antagonism in patients with atherosclerosis, but also a successful mitigation strategy to better maintain long‐term effective treatment that may provide additional protection from atherosclerosis progression in a higher‐risk patient population that may not otherwise be candidates for such a therapeutic approach.
Conflicts of interest
The authors declare no potential conflicts of interest.
Supporting information
Appendix S1.
Figure S1. Sites for miR families conserved among vertebrates predicted by Targetscan 5.2. B. Monocyte miRs predicted to target NR3C2 from atherosclerosis derivation Cohort were initially predicted in‐silico using 12 different mIR prediction algorithms**. The presence of these miRs were then tested in a derivation cohort of normal subjects, and then evaluated in a validation cohort of patients with atherosclerosis (N=10) or controls. All patients were on statins, ACEI or ARB therapy. ↓ Symbol indicates down‐regulated in atherosclerosis versus controls. P<10−10 for most.*miRs identified by Diana, MicroInspector, Miranda, mirtarget2, mitarget, nbmirtar, pictar, pita, rna22, rnahybrid.
Figure S2. miRNAs and transcription factors such as MR regulate entire functional gene networks that through an intermediate phenotype (alternate macrophage activation) may affect atherosclerosis.
Table S1.
Table S2.
Rajagopalan S, Alaiti MA, Broadwater K, Goud A, Gaztanaga J, Connelly K, Fares A, Shirazian S, Kreatsoulas C, Farkouh M, Fink JC and Weir MR. Design of the Magnetic Resonance Imaging Evaluation of Mineralocorticoid Receptor Antagonism in Diabetic Atherosclerosis (MAGMA) Trial. Clin Cardiol. 2017;40:633–640. 10.1002/clc.22718
Funding Information National Heart, Lung, and Blood Institute, Grant/Award number: R01HL127422.
REFERENCES
- 1. Rupprecht R, Arriza JL, Spengler D, et al. Transactivation and synergistic properties of the mineralocorticoid receptor: relationship to the glucocorticoid receptor. Mol Endocrinol. 1993;7:597–603. [DOI] [PubMed] [Google Scholar]
- 2. Fuller PJ, Lim‐Tio SS, Brennan FE. Specificity in mineralocorticoid versus glucocorticoid action. Kidney Int. 2000;57:1256–1264. [DOI] [PubMed] [Google Scholar]
- 3. Farman N, Bocchi B. Mineralocorticoid selectivity: molecular and cellular aspects. Kidney Int. 2000;57:1364–1369. [DOI] [PubMed] [Google Scholar]
- 4. Funder JW, Pearce PT, Smith R, et al. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585. [DOI] [PubMed] [Google Scholar]
- 5. Pitt B, Remme W, Zannad F, et al; Eplerenone Post‐Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators . Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction [published correction appears in N Engl J Med. 2003;348:2271]. N Engl J Med . 2003;348:1309–1321. [DOI] [PubMed] [Google Scholar]
- 6. Rajagopalan S, Pitt B. Aldosterone as a target in congestive heart failure. Med Clin North Am. 2003;87:441–457. [DOI] [PubMed] [Google Scholar]
- 7. Pitt B, Zannad F, Remme WJ, et al; Randomized Aldactone Evaluation Study Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med . 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 8. Bulluck H, Fröhlich GM, Mohdnazri S, et al. Mineralocorticoid receptor antagonist pretreatment to MINIMISE reperfusion injury after ST‐elevation myocardial infarction (the MINIMISE STEMI Trial): rationale and study design. Clin Cardiol. 2015;38:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sudano I, Naegele M, Roas S, et al. Vascular effects of eplerenone in coronary artery disease with preserved ejection fraction: a double‐blind, randomized, placebo‐controlled trial. Clin Cardiol. 2016;39:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohmine T, Miwa Y, Takahashi‐Yanaga F, et al. The involvement of aldosterone in cyclic stretch‐mediated activation of NADPH oxidase in vascular smooth muscle cells. Hypertens Res. 2009;32:690–699. [DOI] [PubMed] [Google Scholar]
- 11. Miyata K, Rahman M, Shokoji T, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. 2005;16:2906–2912. [DOI] [PubMed] [Google Scholar]
- 12. Keidar S, Kaplan M, Pavlotzky E, et al. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin‐converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004;109:2213–2220. [DOI] [PubMed] [Google Scholar]
- 13. Xiao F, Puddefoot JR, Vinson GP. Aldosterone mediates angiotensin II‐stimulated rat vascular smooth muscle cell proliferation. J Endocrinol. 2000;165:533–536. [DOI] [PubMed] [Google Scholar]
- 14. Brown NJ, Nakamura S, Ma L, et al. Aldosterone modulates plasminogen activator inhibitor‐1 and glomerulosclerosis in vivo. Kidney Int. 2000;58:1219–1227. [DOI] [PubMed] [Google Scholar]
- 15. Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. [DOI] [PubMed] [Google Scholar]
- 16. Bender SB, McGraw AP, Jaffe IZ, et al. Mineralocorticoid receptor‐mediated vascular insulin resistance: an early contributor to diabetes‐related vascular disease? Diabetes. 2013;62:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamashita R, Kikuchi T, Mori Y, et al. Aldosterone stimulates gene expression of hepatic gluconeogenic enzymes through the glucocorticoid receptor in a manner independent of the protein kinase B cascade. Endocr J. 2004;51:243–251. [DOI] [PubMed] [Google Scholar]
- 18. Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mihai G, Chung YC, Merchant A, et al. T1‐weighted‐SPACE dark blood whole body magnetic resonance angiography (DB‐WBMRA): initial experience. J Magn Reson Imaging. 2010;31:502–509. [DOI] [PubMed] [Google Scholar]
- 21. Mihai G, Chung YC, Kariisa M, et al. Initial feasibility of a multi‐station high‐resolution three‐dimensional dark blood angiography protocol for the assessment of peripheral arterial disease. J Magn Reson Imaging. 2009;30:785–793. [DOI] [PubMed] [Google Scholar]
- 22. Wright JT Jr, Williamson JD, Whelton PK, et al; SPRINT Research Group . A randomized trial of intensive versus standard blood‐pressure control. N Engl J Med. 2015;373:2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jayagopal V, Kilpatrick ES, Jennings PE, et al. Biological variation of homeostasis model assessment‐derived insulin resistance in type 2 diabetes. Diabetes Care. 2002;25:2022–2025. [DOI] [PubMed] [Google Scholar]
- 24. Sõber S, Laan M, Annilo T. MicroRNAs miR‐124 and miR‐135a are potential regulators of the mineralocorticoid receptor gene (NR3C2) expression. Biochem Biophys Res Commun. 2010;391:727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON‐D Investigators . Combined angiotensin inhibition for the treatment of diabetic nephropathy [published correction appears in N Engl J Med. 2014;158:A7255]. N Engl J Med . 2013;369:1892–1903. [DOI] [PubMed] [Google Scholar]
- 26. Mihai G, Varghese J, Kampfrath T, et al. Aliskiren effect on plaque progression in established atherosclerosis using high resolution 3D MRI (ALPINE): a double‐blind placebo‐controlled trial. J Am Heart Assoc. 2013;2:e004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edwards NC, Steeds RP, Stewart, PM , et al. Effect of spironolactone on left ventricular mass and aortic stiffness in early‐stage chronic kidney disease: a randomized controlled trial. J Am Coll Cardiol. 2009;54:505–512. [DOI] [PubMed] [Google Scholar]
- 28. Asmar R, Benetos A, Topouchian J, et al. Assessment of arterial distensibility by automatic pulse wave velocity measurement: validation and clinical application studies. Hypertension. 1995;26:485–490. [DOI] [PubMed] [Google Scholar]
- 29. Crilly M, Coch C, Bruce M, et al. Repeatability of central aortic blood pressures measured non‐invasively using radial artery applanation tonometry and peripheral pulse wave analysis. Blood Press. 2007;16:262–269. [DOI] [PubMed] [Google Scholar]
- 30. Rajagopalan S, Kariisan M, Dellegrottaglie S, et al. Angiotensin receptor blockade improves vascular compliance in healthy normotensive elderly individuals: results from a randomized double‐blind placebo‐controlled trial. J Clin Hypertens (Greenwich) . 2006;8:783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. White WB, Duprez D, St Hillaire R, et al. Effects of the selective aldosterone blocker eplerenone versus the calcium antagonist amlodipine in systolic hypertension [published correction appears in Hypertension. 2003;42:e20]. Hypertension. 2003;41:1021–1026. [DOI] [PubMed] [Google Scholar]
- 32. Wilkinson IB, Fuchs SA, Jansen IM, et al. Reproducibility of pulse wave velocity and augmentation index measured by pulse wave analysis. J Hypertens . 1998;16(12 part 2):2079–2084. [DOI] [PubMed] [Google Scholar]
- 33. Oliver JJ, Webb DJ. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events. Arterioscler Thromb Vasc Biol. 2003;23:554–566. [DOI] [PubMed] [Google Scholar]
- 34. Tripkovic L, Hart K, Frost G, et al. Interindividual and intraindividual variation in pulse wave velocity measurements in a male population. Blood Press Monit. 2014;19:233–241. [DOI] [PubMed] [Google Scholar]
- 35. Safar M. Mechanical factors predicting cardiovascular risk and drug treatment of hypertension. J Hypertens. 2002;20:349–352. [DOI] [PubMed] [Google Scholar]
- 36. Polyzos SA, Kountouras J, Zafeiriadou E, et al. Effect of spironolactone and vitamin E on serum metabolic parameters and insulin resistance in patients with nonalcoholic fatty liver disease. J Renin Angiotensin Aldosterone Syst. 2011;12:498–503. [DOI] [PubMed] [Google Scholar]
- 37. Lichy MP, Wietek BM, Mugler JP 3rd, et al. Magnetic resonance imaging of the body trunk using a single‐slab, 3‐dimensional, T2‐weighted turbo‐spin‐echo sequence with high sampling efficiency (SPACE) for high spatial resolution imaging: initial clinical experiences. Invest Radiol . 2005;40:754–760. [DOI] [PubMed] [Google Scholar]
- 38. Park J, Mugler JP 3rd, Hughes T. Reduction of B1 sensitivity in selective single‐slab 3D turbo spin echo imaging with very long echo trains. Magn Reson Med. 2009;62:1060–1066. [DOI] [PubMed] [Google Scholar]
- 39. Hayes J, Kalantar‐Zadeh K, Lu JL, et al. Association of hypo‐ and hyperkalemia with disease progression and mortality in males with chronic kidney disease: the role of race. Nephron Clin Pract. 2012;120:c8–c16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kovesdy CP, Regidor DL, Mehrotra R, et al. Serum and dialysate potassium concentrations and survival in hemodialysis patients. Clin J Am Soc Nephrol. 2007;2:999–1007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Figure S1. Sites for miR families conserved among vertebrates predicted by Targetscan 5.2. B. Monocyte miRs predicted to target NR3C2 from atherosclerosis derivation Cohort were initially predicted in‐silico using 12 different mIR prediction algorithms**. The presence of these miRs were then tested in a derivation cohort of normal subjects, and then evaluated in a validation cohort of patients with atherosclerosis (N=10) or controls. All patients were on statins, ACEI or ARB therapy. ↓ Symbol indicates down‐regulated in atherosclerosis versus controls. P<10−10 for most.*miRs identified by Diana, MicroInspector, Miranda, mirtarget2, mitarget, nbmirtar, pictar, pita, rna22, rnahybrid.
Figure S2. miRNAs and transcription factors such as MR regulate entire functional gene networks that through an intermediate phenotype (alternate macrophage activation) may affect atherosclerosis.
Table S1.
Table S2.