

Abstract
Background
Frequent exacerbations of allergic asthma leads to airway remodeling and a decline in pulmonary function, producing morbidity. Cat dander is an aeroallergen associated with asthma risk.
Objective
We sought to elucidate the mechanism of cat dander-induced inflammation-remodeling.
Methods
We identified remodeling in mucosal samples from allergic asthma by Q-RT-PCR. We developed a model of aeroallergen-induced experimental asthma by repetitive cat dander extract exposure. We measured airway inflammation using immunofluorescence, leukocyte recruitment and Q-RT-PCR. Airway remodeling was measured using histology, collagen content, myofibroblast numbers and selected reaction monitoring. Inducible NFκB-BRD4 interaction was measured by proximity ligation assay in situ.
Results
Enhanced mesenchymal signatures are observed in bronchial biopsies from patients with allergic asthma. Cat dander induces innate inflammation via NFκB signaling, followed by producing a pro-fibrogenic, mesenchymal transition (EMT) in primary human small airway epithelial cells. The IκB kinase (IKK)-NFκB signaling pathway is required for mucosal inflammation-coupled airway remodeling and myofibroblast expansion in the mouse model of aeroallergen exposure. Cat dander induces NFκB/RelA to complex with and activate BRD4, resulting in modifying the chromatin environment of inflammatory and fibrogenic genes through its atypical histone acetyltransferase (HAT) activity. A novel, small-molecule BRD4 inhibitor (ZL0454) disrupts BRD4 binding to the NFκB-RNA Pol II complex and inhibits its HAT activity. ZL0454 prevents EMT, myofibroblast expansion, IgE sensitization, and fibrosis in airways of naïve mice exposed to cat dander.
Conclusions:
NFκB-inducible BRD4 activity mediates cat dander-induced inflammation and remodeling. Therapeutic modulation of the NFκB-BRD4 pathway affects allergen-induced inflammation, epithelial cell-state changes, ECM production and expansion of the subepithelial myofibroblast population.
Keywords: Bromodomain-containing protein 4 (BRD4), epigenetics, myofibroblast, airway remodeling, aeroallergen, nuclear factor-κB (NFκB), histone acetyltransferase activity
Capsule Summary:
Allergen-induced exacerbations are associated with remodeling and loss of lung function. Repetitive exposure to cat dander activates TLR-NFκB signaling. Persistent activation results in epigenetic reprogramming, mesenchymal transition and myofibroblast expansion through the BRD4 histone acetyltransferase.
Graphical Abstract
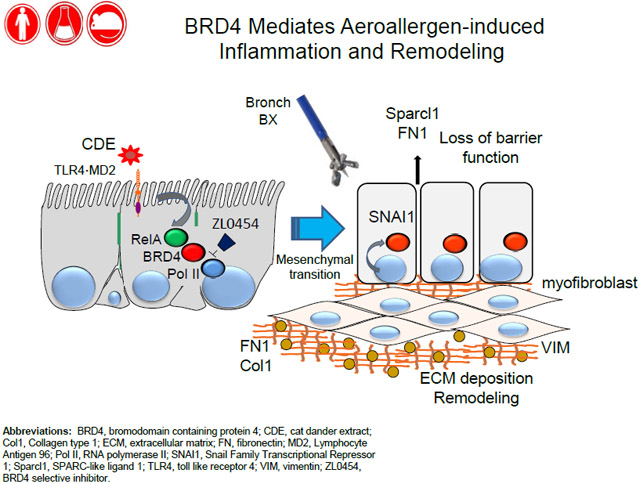
INTRODUCTION
Allergen-induced airway remodeling1 functionally impacts the quality of life for over 300 million patients with asthma2-4. Proteolytic enzymes and NADPH-oxidase activities intrinsic to aeroallergens 5, 6 trigger toll-like receptors (TLRs) and protease-activated receptors (PARs) 7-9, resulting in innate inflammation. This innate pathway disrupts the mucosal barrier function and activates dendritic cell recruitment, leading to enhanced antigen penetration, Th2 polarization and IgE production 10. Consequently allergic sensitization produces a stable submucosal population of Th2 lymphocytes producing fibrogenic cytokines leading to epithelial cell state changes, barrier dysfunction, goblet cell metaplasia, and mucosal thickening. The mechanisms how allergen-induced innate signaling causes mucosal remodeling are incompletely understood.
Cat dander is one of the most prevalent indoor house aeroallergens. In large scale epidemiological studies, cat dander sensitization is associated with asthma in ~ 29% of individuals11 and is found in household dust at levels far above those necessary to induce an IgE response 12. In unsensitized airways, acute cat dander exposure is a potent innate inflammatory stimulus, activating the epithelial TLR4-MD2 complex, a TLR4 signaling pathway independent of that triggered by LPS 5, 6. Downstream, the IKK-NFκB arm of the innate pathway is activated, resulting in oxidative DNA damage, CXCL2 expression, and neutrophil recruitment 13. The mechanisms how these acute inflammatory responses disrupt epithelial barrier leading to airway remodeling are not fully understood.
Recently, we have identified a molecular link between epithelial TLR3 stimulation, NFκB activation, and airway remodeling 14-16. In response to dsRNA, TLR3 activates RelA translocation and production of intracellular ROS 17. Intracellular ROS activate Ribosomal S6 kinases to phosphorylate liberated RelA on Ser 276. Phospho-276 RelA is rapidly acetylated on Lys residues by p300/CBP and binds BRD4 through bromodomain (BD)-acetylated Lys interactions 18, 19. Through site-specific DNA binding, RelA recruits BRD4 to the promoters of mesenchymal genes, where it phosphorylates Ser 2 of the carboxyl terminus of RNA Pol II. Phospho Ser 2 licenses RNA Pol II to produce full-length mRNA transcripts 17, 20. In addition, we recently found that RelA binding induces the atypical histone acetyltransferase (HAT) activity of BRD4, acetylating histone H3 on Lys (K) 122, a modification that destabilizes nucleosomes, enhancing transcription through compacted G-C rich gene bodies 21, 22. These studies demonstrated the central role of NFκB-BRD4 in remodeling inflammatory and fibrogenic genes controlling a coordinate- phenotypic transition to undergo type II epithelial mesenchymal transition (EMT) 14, 17, 23. The mechanisms how NFκB mediates allergen-induced inflammation and airway remodeling downstream of TLR4 are unknown, however.
In bronchial specimens of patients with allergic asthma and atopy, we detect gene expression signatures of EMT and mesenchymal growth factors, a finding that provides direct evidence of enhanced mucosal fibrogenic reprogramming. Because cat dander exposures are repeated chronically over time, we established a mouse model to study its mechanism. We found that repetitive mucosal cat dander extract (rCDE) exposures induces acute inflammation, followed by epithelial mesenchymal transition (EMT), interstitial fibrosis, and expansion of the pro-fibrogenic myofibroblast population mediated through the IKK-NFκB/RelA pathway. rCDE induces RelA to complex and activate the BRD4 HAT. A novel potent highly selective BRD4 small molecule inhibitor recently developed by us 24 prevents acute inflammation and chronic EMT, mucous metaplasia, myofibroblast expansion, and fibrosis. These data validate BRD4 as a therapeutic target to prevent airway remodeling and allergic sensitization.
METHODS
Further details can be found in the Methods section in this article’s Online Repository at www.jaci.org.
Ethical statement
Human subjects were part of clinicaltrials.gov registration NCT02264691. Animal experiments were performed according to the NIH Guide for Care and Use of Experimental Animals and approved by the University of Texas Medical Branch (UTMB) Animal Care and Use Committee (No. 1312058A).
Repetitive CDE (rCDE) challenges
18 week old male wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). CDE (Stallergenes/Greer laboratories, 20 μg/dose) was administered to naïve C57BL6/J mice every other day via the intranasal route for a total of 15 administrations (n=5 mice/group). In the IKK inhibitor experiments, mice were either pretreated with vehicle or the selective IKK inhibitor [BMS345541, 10 mg/kg via the intraperitoneal (ip) route]. In the BRD4 inhibitor experiments, mice were either pretreated with vehicle or the selective BRD4 inhibitor (ZL0454, 10 mg/kg via ip). Mice were sacrificed 12 d later after the last CDE challenge to allow resolution of the acute inflammation.
Histological assessment of inflammation and fibrosis
Formalin-fixed lungs were embedded in paraffin, sectioned at a 4 μm thickness, and stained with hematoxylin and eosin or Masson's trichrome. Microscopy was performed on a NIKON Eclipse Ti System 16, 17. Pulmonary fibrosis was graded using a modified Ashcroft scoring method 16, 17. To determine aggregate histopathology score, the entire left and right lungs were longitudinally sectioned. Each lung was scored separately (score range, 0 to 9) and scores combined (total score range, 0 to 18) 16, 17.
Statistical analysis
One-way ANOVA was performed when looking for time differences followed by Tukey's post-hoc test to determine significance. P<0.05 was considered significant.
RESULTS
Activation of the EMT gene program in human asthmatic mucosa
Although epithelial stress response and damage are characteristic findings in human asthma 25, the role of EMT in allergic disease is not known 26-28. Seven normal and 7 atopic asthmatics with positive allergen skin tests were randomly selected from a subset of a prospective observational study (See Table E1 available in this article’s Online Repository at www.jaci.org.) and assayed for mesenchymal gene expression by Q-RT-PCR. Relative to controls, we observed a significant 4.1-fold increase in the EMT core regulators, SNAI1 and ZEB1, in mucosa from allergic asthmatics (Fig 1). In addition, a 4-fold increase in the ECM gene, fibronectin (FN1), and 5-fold increase in the myofibroblast activation marker, CD274/Programmed Death Ligand (PD-L1) was observed (Fig 1). Collectively, these data suggest the activation of the mesenchymal remodeling program in mucosa of allergic asthmatics.
Fig 1. EMT signature in mucosal samples in human severe asthma.

Q-RT-PCR for bronchial mucosal biopsies from normal or mild-moderate asthma for EMT regulators SNAI1, ZEB1, and FN1, or myofibroblast activation marker, PD-L1 mRNA expression. Shown as fold-change mRNA abundance normalized to PPIA (cyclophilin A). **, p<0.01, compared to healthy human samples (n=5 normals; 7 AAs), t-test.
Tonic CDE exposure induces epithelial mesenchymal transition
Because dander exposure is chronic in households with cats, we initially studied the effect of tonic CDE on model human small airway epithelial cells. Tert-hSAECs are telomerase-immortalized Scgb1a1/CC10- expressing cells that maintain expression of differentiated cytokeratins 29 and exhibit overlapping genomic and proteomic signatures with those of terminally differentiated primary cells 21, 29, 30. Knowing that CDE activates NFκB6, a transcription factor that mediates TGFβ-and TLR3-induced mesenchymal transition 15, 16, we examined the effect of tonic CDE activation of TLR4 on mesenchymal transition. Tonic CDE stimulation induced hSAECs to assume an elongated shape with enhanced formation of filamentous (F) actin throughout the cytoplasm and nucleus (see Fig E1, A, in this articles Online Repository available at www.jaci.org ), suggesting acquisition of front-rear polarity and stress fiber accumulation, chacteristic factors of mesenchymal transition15, 16.
The EMT program silences epithelial markers and activates mesenchymal genes, a process mediated by the SNAI1 and RelA transcription factors 15, 28, 31. We observed that tonic CDE exposure induced RelA and SNAI1 expression with SNAI1 monotonically increasing to an apparent plateau of 12-fold (see Fig E1, B, in this articles Online Repository available at www.jaci.org). Additionally, CDE induced expression of the mesenchymal intermediate protein, vimentin (VIM) and the ECM remodeling proteins FN1, collagen (COL1A), and matrix metalloproteinase 9 (MMP9) (see Fig E1, C, in this articles Online Repository available at www.jaci.org). In addition to enhanced expression of IL6 and TGFβ mesenchymal growth factors, CDE also induced NOX4 mRNA, an NADPH oxidase responsible for ROS stress and DNA damage response 32-34.
NFκB/RelA is required for CDE-induced mesenchymal reprogramming of airway epithelial cells
We next determined the role of NFκB signaling in mediating the CDE-induced mesenchymal cellular program, using Tert-hSAECs stably expressing doxycycline (Dox)-inducible RelA shRNA. Dox stimulation reduced RelA mRNA by >90% relative to that of non-Dox treated cells indicating highly effective silencing (WT, Fig 2, A).
Fig 2. NFκB/RelA mediates CDE-induced mesenchymal transition of airway epithelial cells.

A, RelA shRNA-expressing hSAECs treated with/without 2 μg/ml doxycycline (Dox), 5d. Afterwards, cells were treated with CDE (20 μg/mL) for 0 or 15 d prior to analysis by Q-RT-PCR. Fold changes in indicated mRNAs are shown. *p< 0.01 compared to control shRNA, #, p<0.01 compared to without CDE, n=3, t-test. B, Western immunoblots. Whole cell extracts from the same experiment were fractionated by SDS-PAGE and subjected to western immunoblot with indicated Abs, β-actin was probed as loading controls. Similar results were found in experiments repeated three times independently. C, Immunofluorescent confocal microscopy (IFCM) assays of WT and RelA-shRNA hSAECs. Cells were stained with either Alexa Fluor 568-conjugated phalloidin (upper panel, red color), or primary antibodies to VIM, SNAI1, and CDH1 Abs followed by secondary detection using Alexa 488-(green, for VIM and CDH1) or 568-(red, SNAI1) conjugated goat anti-rabbit IgG. Nuclei were counterstained with DAPI (blue). Images were acquired at 63X magnification. Right, quantifications of fluorescence intensities shown as fold changes compared to control hSAECs. * p<0.01, n = 5, t-test. FI: relative total fluorescence intensity. D, IFCM assays of total RelA, phospho-Ser276 RelA, H3K122 Ac, and phospho-Ser 2 CTD Pol II (pPol II). Secondary detection was Alexa 488-(green color, for RelA and H3K122ac) or 568-(red color, for p276 RelA and pPol II) conjugated goat anti-rabbit IgG. At the right are quantifications (X ± SD) of total fluorescence intensities, * p<0.01, n = 5, t-test.
The significant downregulation of CDH1 mRNA produced by tonic CDE stimulation was blocked in the RelA-depleted cells (Fig 2, A). Conversely, the 15-fold induction of SNAI1 mRNA produced by CDE stimulation in WT hSAECs was reduced to less than 3-fold in the RelA-depleted cells. Also, RelA was similarly required for the 7-fold upregulation of VIM because RelA silencing reduced VIM mRNA abundance to that of unstimulated controls (Fig 2, A). Similarly, the CDE-mediated increase of FN1 and COL1A mRNAs were significantly attenuated in the RelA-depleted cells.
The induction of VIM and SNAI1 as well as inhibition of CDH1 proteins by CDE in wild type Tert-hSAECs were confirmed in Western immunoblots (Fig 2, B). A similar inhibition of VIM and SNAI1 was also observed in the RelA−/− Tert-hSAECs by Western (Fig 2, B). RelA−/− had a less dramatic effect on CDH1 downregulation in a manner similar to its effects on CDH1 mRNA expression (c.f. Figs 2, A and B). CDE induced F-actin formation, VIM and SNAI1 expression in Tert-hSAECs in immunofluorescence confocal microscopy (IFCM; Fig 2, C). These data demonstrate that NFκB/RelA is required for CDE-induced mesenchymal reprogramming.
RelA mediates CDE-induced BRD4 HAT and phospho-Pol II kinase activities.
Because of the central role of RelA binding in activation of the atypical BRD4 HAT and/or phospho-Pol II activity, we asked whether BRD4 was activated in a RelA-dependent manner in response to CDE. We found that CDE stimulation induced a uniform translocation of the cytoplasmic RelA into the nucleus (Fig 2, D); the nuclear translocated form of RelA was serine 276 phosphorylated (Fig 2, D). The specificity of RelA and phospho-Ser 276 RelA staining was confirmed in the RelA shRNA expressing hSAECs (Fig 2, D). Strikingly, CDE stimulation also induced the global accumulation of nuclear H3K122 Ac marks; this induction was RelA dependent (Fig 2, D). Similar findings were observed for the phospho-Ser 2 CTD RNA Pol II (Fig 2, D).
Repetitive CDE (rCDE) exposures induce an NFκB-dependent fibrotic program in vivo
To determine whether repetitive CDE exposures induce airway remodeling through the IKK-NFκB pathway, unsensitized (naïve) C57BL/6 mice were subjected to repetitive CDE exposures in the absence or presence of the selective IKK inhibitor (IKKi), BMS-345541 (BMS) 35 (see Fig E2, in this articles Online Repository available at www.jaci.org).
rCDE induced a 3.8-fold increase in the numbers of total leukocytes in the BALF, whose numbers were significantly reduced in the IKKi-treated mice (Fig 3, A). Induction of CDE-specific circulating IgE was also observed, indicating sensitization occurred during the exposure. This sensitization was completely blocked by the IKKi treatment (Fig 3, B).
Fig 3. Repetitive CDE (rCDE) exposure induces airway remodeling in mice.

C57BL6/J mice were pretreated with and without IKK inhibitor BMS345541 and given repetitive intranasal challenges of CDE. A, Total cells and macrophages count in the bronchoalveolar lavage fluid (BALF), expressed as total number of cells X 103/ml (left) and macrophages X 103/ml (right). **, p<0.01, n=5 mice per group. B, CDE specific serum IgE levels were quantitated. **, p<0.01, n=5, t-test. C, Masson Trichrome staining of lung sections from mice in the absence (left panel) or presence of CDE (middle), or those treated with rCDE and IKK inhibitor BMS345541 (right panel). The images were taken at magnifications of 10X and 40X respectively. D, Modified Ashcroft scoring for treatment groups. *, p<0.05, compared to without CDE; #, p<0.05, compared to CDE alone, t-test. E, Quantification of hydroxyproline. Left, hydroxyproline levels in BALF. Right, hydroxyproline content in total lung tissue. *, p<0.05, compared to without CDE; #, p<0.05 compared to CDE alone, t-test. F, PAS staining (pink) by treatment groups. At right is quantification of accumulated mucin in airway epithelial cells. *, p<0.05, compared to without CDE; #, p<0.05 compared to CDE alone, n=5 mice per group. G, IFCM for eosinophil Major Basic Protein (MBP, green color) with DAPI counterstain.
The rCDE treated lungs showed marked hypercellularity, epithelial hyperplasia, and subepithelial collagen deposition around medium sized airways, within the intersititium, and surrounding the blood vessels (Fig 3, C). Increased staining of the subepithelial fibroblast/smooth muscle cell layer was also observed. IKKi treatment reduced the fibrosis and hypercellularity, although not completely in the alveoli. A nearly 9-fold increase in the modified Ashcroft score observed in rCDE treated mice was significantly reduced in IKKi treatment group (Fig 3, D). Additionally, rCDE stimulation produced 2-fold increase in hydroxyproline concentration in BALF and lung tissue that was normalized in the IKKi treatment group (Fig 3, E). We also observed that rCDE produced a marked increase in PAS staining throughout the airway and that this effect was also blocked by the IKKi treatment (Fig 3, F). Despite the presence of IgE in the response to rCDE exposure, few eosinophils were observed in BALF or lung tissue as determined by Major Basic Protein staining in IFCM (Fig 3, G). This is in contrast to the induction of eosinophils seen in an acute CDE exposure (Fig 3, G). We conclude that chronic NFκB-mediated inflammation plays a central role in rCDE exposure-induced airway fibrosis, mucous metaplasia, and remodeling.
rCDE activates the IKK-NFκB pathway and mesenchymal transition in the airway mucosa
rCDE induced a 9-fold increase in phospho-IKKα/β abundance in the airway epithelial layer, an induction reduced to control values by the IKKi treatment, indicating its therapeutic effect (see Fig E3, A in this articles Online Repository available at www.jaci.org). We also observed that rCDE exposure produced a 14-fold increase in mucosal phospho-Ser 276 RelA formation; this induction was also blocked by IKKi treatment (see Fig E3, A in this articles Online Repository available at www.jaci.org).
A similar induction of mouse (m) RelA, α-smooth muscle actin (αSMA), SNAI1, COL1A, FN1, VIM, CXCL1/KC, and IL6 genes was observed in total lung RNA (see Fig E3, B in this articles Online Repository available at www.jaci.org ). This induction pattern of pro-fibrotic mesenchymal gene expression signatures matches those of cultured hSAECs in response to tonic CDE stimulation. The induction of all of these genes were inhibited by IKKi treatment.
We observed that rCDE exposure induced a 12-fold increase in SNAI1 expression, most intensely in the epithelium and interstitial myofibroblasts around medium- and small-sized airways (Figs 4, A and B). Similarly, the mesenchymal intercellular contractile protein, VIM, was upregulated by 18-fold as well as the ECM proteins COL1A and FN1 (Figs 4 A, and B). Expression of these mesenchymal and ECM proteins were completely blocked in the IKKi treatment arm (Fig 4 A, and B). These data suggested that rCDE triggered the mesenchymal program in the mucosa. To confirm this finding, we measured the presence of extracellular mesenchymal proteins in the BALF using stable isotope dilution selected reaction monitoring (SRM) in a triple quadrupole MS 36. We observed a 4-fold increase in BALF mFN1 and 3.5-fold increase in Sparc-ligand 1 in response to rCDE that was reduced to untreated levels by the IKKi inhibitor (Fig. 4, C). Collectively, these data indicate that the rCDE induces mesenchymal transition in an NFκB-dependent manner.
Fig 4. rCDE induces mucosal EMT in an IKK-NFκB-dependent manner.

A, IFCM of lung sections from control (PBS), CDE, or BMS+CDE treated mice. Sections were stained for the EMT markers SNAI1, VIM, COL1A, and FN1, Images are acquired at 63X magnification. B, quantitation of relative changes in fluorescence intensity for each treatment group. *, p<0.05, compared to without CDE; #, p<0.05 compared to CDE alone, n=5, t-test. C, Stable isotope dilution-Selected Reaction Monitoring (SID-SRM) of extracellular matrix proteins, mFN1 and mSparcl1 in BALF. Shown are mean ± SD of native to stable isotope standards (SIS) for n=5 animals in two technical replicates. *, p<0.05 compared to control; #, p<0.01 compared to CDE treatment only, t-test. D. IFCM of the BRD4 activation marker H3K122-Ac. Left panel, quantitation of relative changes in fluorescence intensity of H3K122-actin. *, p<0.05 compared to control; #, p<0.01 compared to CDE treatment only, t-test.
rCDE triggers atypical BRD4 HAT activity in vivo
Because of the significant activation of NFκB in the airway mucosa, and BRD4’s dependence on NFκB activation in vitro (Fig 2), we examined whether rCDE also stimulated the atypical BRD4 HAT activity in vivo. rCDE exposure induced a 17-fold increase in H3K122 Ac mucosal staining over that of PBS controls (Fig 4, D). H3K122 Ac is blocked to control levels by the IKKi (Fig. 4, D).
Myofibroblast expansion is IKK-NFκB pathway-dependent
The finding that rCDE produces expansion of the subepithelial fibroblast layer and induced expression of IL6, TGFβ, and αSMA mRNAs prompted us to study the effects of rCDE on the myofibroblast population. We observed that rCDE induced a population of αSMA+/Col1A+ myofibroblasts37 in the subepithelial layer of small airways, a distribution consistent with the expanded fibroblast observed earlier (Fig 5,A; c.f. Fig 3,C). An average of 34.6 costaining cells/high powered field (hpf) were seen in the rCDE treated animals vs 0 for the PBS treatment. Expansion of this population was blocked by the IKKi with only 2.6 αSMA+/Col1A+ myofibroblasts been found in rCDE mice pretreated with IKKi (Fig 5,B, p<0.01), suggesting mesenchymal transition and myofibroblast expansion were dependent on allergen-induced epithelial NFκB signaling.
Fig 5. rCDE induces subepithelial myofibroblast expansion.

Confocal immunofluorescence microscopy of lung tissues in control (PBS), CDE, or BMS+CDE treated mice stained with rabbit anti-alpha smooth muscle actin (αSMA, green color) and mouse anti-COL1 (red color) and counterstained with DAPI (blue color). Merged images shown as top, 63X. Experiments were independently repeated twice with 5 animals in each treated group. Total 10 fields of each treatment were examined by 2 investigators who were blind to the treatment groups (n=10, *, p<0.01, t-test).
Functional role of BRD4 in CDE-mediated mesenchymal transition of airway mucosa
We next tested the requirement of BRD4 HAT on rCDE-induced inflammation-remodeling. For this purpose, a highly selective small molecular BRD4 antagonist was synthesized targeting the BRD4 bromodomain domain (BD) with nanomolar binding affinities and submicromolar potency. Our earlier studies demonstrated that ZL0454 disrupts BRD4 binding to Pol II and histones in cellulo, releasing it from chromatin into the soluble fraction of the nucleoplasm24.
The requirement for BRD4 on the CDE-induced mesenchymal transition was validated in hSAECs. Here, the CDE-induced inhibition of CDH1 was blocked by ZL0454 treatment (see Fig E4, A in this articles Online Repository available at www.jaci.org ). Similarly the upregulation of SNAI1, FN1, VIM, COL1A, and IL6 mRNAs were all significantly blocked by ZL0454 treatment (see Fig E4, B-D in this articles Online Repository available at www.jaci.org). Consistently, ZL0454 prevented the inducible formation of actin stress fibers, H3K122 Ac, SNAI1, and VIM expression (see Fig E4, E in this articles Online Repository available at www.jaci.org ).
In vivo, we observed that ZL0454 blocked acute CDE-induced BALF neutrophilia, observed within 24 h of treatment (see Fig E4, F in this articles Online Repository available at www.jaci.org ). Based on this finding and our earlier studies that ZL0454 is well tolerated over 3 months of administration without apparent toxicity24, indicated that ZL0454 could be used as a probe for BRD4 actions in vivo.
rCDE-induced airway fibrotic program is mediated by BRD4
In our standard rCDE model, we observed that ZL0454 treatment reduced collagen formation and hypercellularity surrounding the bronchioles and alveolar spaces (Fig 6, A). The striking Ashcroft Score of 9 produced by rCDE was reduced to 1.5 by concomitant ZL0454 treatment (Fig 6, B). Similarly, the 4-fold increase in hydroxyproline content in the lung (Fig 6, C) and 2.2-fold increase in BALF (Fig 6, C) were both reduced by ZL0454 treatment. Importantly, ZL0454 also reduced the pan-epithelial mucous metaplasia in PAS staining (Figs 6, D and E).
Fig 6. BRD4 inhibitor blocks rCDE-induced airway remodeling.

C57BL/6 mice were subjected to 15 treatments with PBS (in), rCDE (20 μg/dose, in), ZL0454 (10 mg/kg body weight, ip) or rCDE + ZL0454 for total 30 d and lungs were harvested 12 d after the last CDE challenge. A, Masson-Trichrome staining taken at 10X, 20X, and 40X magnification. B, Modified Ashcroft Score by treatment group. *, p<0.05, compared to without CDE; #, p<0.05, compared to CDE alone, t-test. C, Upper, hydroxyproline level in BALF. Lower, hydroxyproline content in lung tissue. *, P<0.05, compared to without CDE; #, p<0.05 compared to CDE alone. D, PAS staining (pink) showing mucin production. E, Quantification of cellular mucin. *, p<0.05, compared to without CDE; #, p<0.05 compared to CDE alone, n=5. F, SID-SRM of mFN1 and mSparcl1 in BALF. Shown are mean ± SD of native to stable isotope standards (SIS) for n=5 animals in two technical replicates. *, p<0.05 compared to control; #, p<0.01 compared to CDE treatment only, t-test.
BRD4 HAT is required for rCDE-induced EMT
The striking induction of mucosal H3K122 Ac accumulation produced by rCDE was inhibited by ZL0454 (Fig 7, A), indicating that ZL0454 administration effectively inhibited BRD4 HAT activity in vivo. In the same tissues, we observed that the rCDE-induced expression of αSMA, COL1A, FN1, VIM, and MMP9 mRNAs were also inhibited by ZL0454, indicating BRD4-dependence (Fig 7, B). Similarly, the mucosal induction of SNAI1, FN1, and VIM observed in immunofluorescence microscopy (Fig 7, C) and by Western blot (see Fig E5 in this articles Online Repository available at www.jaci.org) were also inhibited by ZL0454 treatment.
Fig 7. BRD4 inhibitor blocks mucosal mesenchymal transition in vivo.

A, Confocal immunofluorescence microscopy of H3K122 Ac (red color) in mouse lungs treated with PBS, rCDE, ZL0454 or rCDE + ZL0454 respectively. Lung sections were counterstained in DAPI (blue color). X63 magnification. At the right is quantifications of relative fluorescence intensity, * p<0.01, n = 5. B, Q-RT-PCR for mRNA expression of mesenchymal and ECM genes from total RNA of mouse lungs treated with PBS, rCDE, ZL0454 or rCDE + ZL0454. * p<0.01, n = 5, t-test. C, Confocal immunofluoresence microscopy for SNAI1 (green color), FN1 (red color), and VIM (green color) in mouse lungs treated with PBS, rCDE, ZL0454 or rCDE + ZL0454. Lung sections were counterstained in DAPI (blue color). X63 magnification. At right are quantitation of relative fluorescence intensities of SNAI, FN1, and VIM. *, p<0.01, compared to CDE alone, n=5. D, PLA assay of RelA-BRD4 molecular interactions in lung sections from PBS, rCDE, ZL0454 or rCDE + ZL0454- treated mice. Foci of interactions are amplified as red foci; sections are counterstained with DAPI (blue color). X63 magnification. At the right is quantification of PLA assay. *, p<0.01, compared to CDE alone, n=5, t-test.
Activated RelA binds to BRD418, 21, an interaction mediated by the BRD4 BD domain, a target of ZL0454 24. To measure this molecular interaction in the airway mucosa, we applied a proximity ligation assay (PLA), an assay that detects atomic-distance interactions between two molecules 38. After heterotypic Ab staining, oligonucleotide ligation and PCR amplification, RelA-BRD4 interactions appear as fluorescent foci in situ21. We observed that rCDE increased RelA·BRD4 binding was increased by 7-fold; this interaction was disrupted by ZL0454 treatment (Fig 7, D).
rCDE induced myofibroblast expansion is BRD4-dependent
Finally, we found that activation of the subepithelial αSMA+/COL1+ co-staining myofibroblast cells by rCDE was also blocked by ZL0454 treatment (Fig 8). An average of 36.8 costaining cells/hpf were seen in the rCDE treated animals vs 0 for the PBS treatment and only 2.2 αSMA+/Col1A+ myofibroblasts been found in rCDE mice pretreated with ZL0454 (p<0.01), These data indicate that the allergen induced mucosal NFκB-BRD4 signaling affects airway inflammation-remodeling through its direct effects on epithelial cell state, growth factor and ECM production, indirectly producing expansion of subepithelial myofibroblasts.
Fig 8. BRD4 mediates allergen-induced myofibroblast transition.

Confocal immunofluorescence microscopy of lung sections in PBS, rCDE, ZL0454 or rCDE + ZL0454- treated mice stained with both primary antibodies of rabbit anti-alpha smooth muscle actin (αSMA, green color) and mouse anti-COL1 Abs (red color) and counterstained with DAPI (blue color). Merged images shown at top. X 63 magnification. Experiments were independently repeated twice with 5 animals in each treated group. Total 10 fields of each treatment were examined by 2 investigators who were blind to the treatment groups (n=10, *, p<0.01, t-test).
DISCUSSION
We observe here that patients with allergic asthma have evidence of mucosal remodeling and activation of fibrogenic/remodeling programs. Cat dander is a potent aero-allergen whose ubiquitous exposure has been associated with significant rates of asthma and allergy. Using this disease-relevant model, we demonstrate that rCDE signaling is sufficient to induce fibrogenic growth factor expression and produce mesenchymal cell state change in human airway epithelial cells. Although previous work has found that mucosal NFκB mediates airway remodeling to house dust mice and OVA exposures 39, 40, the mechanisms how NFκB produces remodeling is unknown. Here we demonstrate that CDE induces NFκB/RelA to associate with BRD4; this epigenetic regulator mediates innate inflammation and downstream epithelial cell-state transition, ECM production, and myofibroblast generation. Our small molecule inhibitor studies validate BRD4 as a therapeutic target whose inhibition reduces mucosal sensitization, restoration of barrier dysfunction, and prevention of structural remodeling.
Mesenchymal transition in allergen-induced epithelial damage
The normal airway epithelial surface forms a semi-impermeable barrier 10, whose barrier function is universally disrupted in asthma 25. Our work extends the mechanisms for sensitization and disruption of barrier function using an animal allergen that activates epithelial cells through binding and activating TLR4, triggering NFκB-ROS signaling. This is a common pathway shared with grass and tree allergens 13. Epithelial barrier disruption is suggested by the loss of epithelial cadherin (CDH1), histological disruption of the epithelial surface and the leakage of mesenchymal extracellular matrix proteins, FN1 and Sparcl1 appearing in the BALF after rCDE exposure. The mechanisms how aeroallergens disrupt epithelial barrier function are not fully understood. Our studies suggest that that chronic cat dander exposure downregulates CDH1, a major protein in the maintenance of aherens junctions 41, 42, via the IKKi-NFκB pathway. CDH1 downregulation produces mesenchymal transition, and may be one of the mechanisms how CDE induces changes in the epithelial barrier function. Although barrier function was not directly measured here, the presence of FN1 and Sparcl1 extracellular matrix proteins in the BALF suggests disruption of the epithelial tight junction. We propose that disruption of the epithelial barrier through mesenchymal transition enables allergens to penetrate the mucosa to interact with effector immune cells, further enhancing atopy. Despite the ubiquitous exposure to cat dander, and its effects on the mucosal barrier, not all exposures result in sensitization. Sensitization requires subsequent interactions with antigen processing cells linked to Th2 lymphocytic activity. CDE induced epithelial barrier disruption is permissive, but not sufficient for sensitization.
Mucosal inflammation is linked to mesenchymal transition via NFκB
Our earlier studies have demonstrated that CDE is a potent activator of the TLR4-ROS-NFκB pathway6. Although acutely NFκB activation produces chemokine secretion and neutrophilic inflammation, persistent activation triggers reprogramming of fibrogenic genes and the core transcriptional regulators of epithelial cell state transition 17,45. Here, activated NFκB repositions BRD4 to the promoters of COL1, FN1, SNAI, ZEB1 and other genes to trigger expression through regulated transcriptional elongation involving its atypical HAT activity17. Through this epigenetic reprogramming mechanism, persistent NFκB activation from a variety of TLR agonists induces mesenchymal cell state transition and ECM remodeling associated with airway fibrosis. Our PLA results demonstrate that NFκB dynamically forms a complex with BRD4 in the mucosa of aeroallergen validating this pathway in a complex in vivo model.
Mesenchymal transition involves a series of cell-state changes43 driven by master transcription factors and BRD4-mediated reprogramming 31 resulting in the expression of core mesenchymal regulatory factors, including SNAI1 and ZEB1, and silencing epithelial differentiation markers, such as CDH1 44. A consistent finding from our poly(I:C)24, 45, 46, and CDE challenges in the mouse model are that these aeroallergens induce mucosal changes consistent with epithelial de-differentiation and mesenchymal transition. Our identification of the mesenchymal signatures in human mucosal biopsies are concordant with the findings of others47, 48 that enhanced TGFβ signaling and mesenchymal transition is found in the airways of allergic asthmatics 28, 49. Although our data is unequivocal that the airway epithelium is at least in a partial EMT state, we recognize the controversy as whether mesenchymal transitioned epithelial cells contribute to the myofibroblast population 27. Currently pulmonary myofibroblasts are thought to originate from resident mesenchymal cells, epithelial and endothelial cells (EMT/EndMT) undergoing mesenchymal transition, perivascular fibroblasts (pericytes) as well as circulating bone marrow stem cells (“fibrocytes”) 50-52. More work will be needed to resolve the origin of the myofibroblast populations in response to TLR signaling from aeroallergens.
rCDE induces expansion of the myofibroblast population
αSMA+/COL1+- coexpressing myofibroblasts are a secretory phenotype of lung stromal mesenchymal cells that are major producers of ECM proteins and matrix metalloproteinases that contribute to lamina reticularis expansion 53, 54, an early and consistent finding in asthma 55, 56. Expanded myofibroblast populations have been observed in acute asthma, fatal severe asthma 57, refractory asthma 58, and OVA-sensitized mice 59. Mesenchymal cell state changes are associated with secretion of paracrine growth factors that expand and sustain the subepithelial myofibroblast population. Our data indicate that repetitive allergen exposures activate the epithelial expression of IL6, a growth factor that coordinates myofibroblast expansion. IL6 triggers α-SMA expression, autocrine TGFβ stimulation and extracellular matrix production in fibroblasts 60. Our work indicates that IL6 is a target of BRD4 epigenetic reprogramming. Its upregulation provides insight into the paracrine mechanism how the transitioned epithelium sustains the subepithelial myofibroblast population.
BRD4 therapeutics in NFκB-dependent allergic disease
Our findings here that rCDE exposure enhances RelA·BRD4 binding required for EMT suggests that this pathway is a common mediator of structural remodeling by growth factors and allergens. Here we extend this mechanism to show that RelA association activates both the RNA Pol II kinase-and atypical HAT activities of BRD4. Moreover, our data provides direct evidence in vivo that BRD4 kinase and HAT activity are inflammation-inducible and RelA-dependent.
Currently, no commercially available small molecule inhibitor is highly BRD4-selective 61. Our recent development and validation of ZL0454 as a highly specific BRD4 inhibitor will advance the field by providing a useful probe for the testing the role of BRD4 in pathophysiological conditions in vivo 24. The mechanism how BD-targeted inhibitors disrupt BRD4-dependent transcription is at multiple steps. Our finding that RelA recruitment and Pol II phosphorylation are disrupted by BD-directed small molecule inhibitors indicates a central role of the BRD4 BD in coordinating formation of a stable pre-initiation complex 24, 62. Similarly, BRD4 BD inhibitors displace BRD4 from high affinity chromatin binding sites 24, 63 and disrupt the multiple protein-protein interactions necessary for BRD4 function, including those with Pol II and RelA 24, 64. Our studies are designed as proof-of-principal that BRD4 plays a central role in the complex mucosal response to aero-allergen challenge. Further studies will need to be conducted using BRD4 inhibitors in a therapeutic mode to advance these compounds to the treatment of human allergic disease.
Our work has important implications for the mechanisms of chronic CDE exposures in humans, informing a viable strategy to prevent structural airway remodeling in chronic allergic airway disease. We demonstrate that repetitive allergen exposure produces innate inflammation, epithelial barrier disruption, cell-state transition, mucous metaplasia, expansion of the myofibroblast population, and consequent fibrosis. Importantly, our validation of the activation of the NFκB-BRD4 pathway and its therapeutic effect to block airway remodeling and sensitization is a therapeutically viable approaches to modify pathological responses to aeroallergen exposures.
Supplementary Material
Key messages:
1. Cat dander is a ubiquitious aeroallergen that activates mucosal TLR4-NFκB signaling producing innate inflammation.
2. Repetitive cat dander exposure repositions the atypical histone acetyltransferase, BRD4, to reprogram fibrogenic genes whose expression result in cell state transition, ECM remodeling and myofibroblast expansion.
3. Small molecule inhibitors of BRD4 disrupt aeroallergen-induced Pol II binding, inflammation, ECM deposition and myofibroblast expansion.
Acknowledgments
Funding support. Supported, in part, by NIH grants NIAID AI062885 (ARB), UL1TR001439 (ARB), NSF grant DMS-1361411/DMS-1361318 (ARB), UTMB Technology Commercialization Program and Sanofi Innovation Awards (iAwards) (ARB, JZ, BT, ER). Core laboratory support was provided by the UTMB Histopathology Core and Optical Imaging Core.
Abbreviations:
- BRD4
bromodomain-containing protein 4
- CDE
cat dander extract
- ECM
extracellular matrix
- EMT
epithelial mesenchymal transition
- H3K122Ac
acetylated Histone H3 on Lys 122
- HAT
histone acetyltransferase activity
- IKK
IκB kinase
- NFκB
nuclear factor-κB
- PLA
proximity ligation assay
- SRM
selected reaction monitoring
Footnotes
Disclosure of potential conflict of interests: Drs. Brasier, Zhou, and Tian have disclosed intellectual property on the BRD4 inhibitors, but receive no license or royalty support.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J Allergy Clin Immunol 2008; 121:560–70; quiz 71-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawankar R Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organ J 2014; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahl R, Andersen PS, Chivato T, Valovirta E, de Monchy J. National prevalence of respiratory allergic disorders. Respir Med 2004; 98:398–403. [DOI] [PubMed] [Google Scholar]
- 4.Prakash Ys, Halayko AJ, Gosens R, Panettieri RA Jr., Camoretti-Mercado B, Penn RB, et al. An Official American Thoracic Society Research Statement: Current Challenges Facing Research and Therapeutic Advances in Airway Remodeling. Am J Respir Crit Care Med 2017; 195:e4–e19. [DOI] [PubMed] [Google Scholar]
- 5.Hosoki K, Aguilera-Aguirre L, Brasier AR, Kurosky A, Boldogh I, Sur S. Facilitation of Allergic Sensitization and Allergic Airway Inflammation by Pollen-Induced Innate Neutrophil Recruitment. Am J Respir Cell Mol Biol 2016; 54:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hosoki K, Boldogh I, Aguilera-Aguirre L, Sun Q, Itazawa T, Hazra T, et al. Myeloid differentiation protein 2 facilitates pollen- and cat dander-induced innate and allergic airway inflammation. J Allergy Clin Immunol 2016; 137:1506–13 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, et al. Mucosal allergic sensitization to cockroach allergens is dependent on proteinase activity and proteinase-activated receptor-2 activation. J Immunol 2011; 186:3164–72. [DOI] [PubMed] [Google Scholar]
- 8.Kale SL, Agrawal K, Gaur SN, Arora N. Cockroach protease allergen induces allergic airway inflammation via epithelial cell activation. Sci Rep 2017; 7:42341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol 2002; 169:5904–11. [DOI] [PubMed] [Google Scholar]
- 10.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol 2008; 8:193–204. [DOI] [PubMed] [Google Scholar]
- 11.Salo PM, Arbes SJ Jr., Jaramillo R, Calatroni A, Weir CH, Sever ML, et al. Prevalence of allergic sensitization in the United States: results from the National Health and Nutrition Examination Survey (NHANES) 2005-2006. J Allergy Clin Immunol 2014; 134:350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly LA, Erwin EA, Platts-Mills TA. The indoor air and asthma: the role of cat allergens. Curr Opin Pulm Med 2012; 18:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosoki K, Redding D, Itazawa T, Chakraborty A, Tapryal N, Qian S, et al. Innate mechanism of pollen- and cat dander-induced oxidative stress and DNA damage in the airways. J Allergy Clin Immunol 2017. [DOI] [PubMed] [Google Scholar]
- 14.Kalita M, Tian B, Gao B, Choudhary S, Wood TG, Carmical JR, et al. Systems Approaches to Modeling Chronic Mucosal Inflammation. BioMed Research International 2013; 2013:505864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian B, Li X, Kalita M, Widen SG, Yang J, Bhavnani SK, et al. Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 2015; 16:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian B, Patrikeev I, Ochoa L, Vargas G, Belanger KK, Litvinov J, et al. NFkappaB Mediates Mesenchymal Transition, Remodeling and Pulmonary Fibrosis in Response to Chronic Inflammation by Viral RNA Patterns. Am J Respir Cell Mol Biol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian B, Zhao Y, Sun H, Zhang Y, Yang J, Brasier AR. BRD4 Mediates NFkB-dependent Epithelial-Mesenchymal Transition and Pulmonary Fibrosis via Transcriptional Elongation. The American Journal of Physiology -Lung Cellular and Molecular Physiology 2016; 311:L1183–L201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brasier aR, Tian B, Jamaluddin M, Kalita MK, Garofalo RP, Lu M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J Virol 2011; 85:11752–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang B, Yang XD, Zhou MM, Ozato K, Chen Lf. Brd4 Coactivates Transcriptional Activation of NF-{kappa}B via Specific Binding to Acetylated RelA. Molecular and Cellular Biology 2009; 29:1375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian B, Zhao Y, Kalita M, Edeh CB, Paessler S, Casola A, et al. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J Virol 2013; 87:7075–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian B, Yang J, Zhao Y, Ivanciuc T, Sun H, Garofalo RP, et al. Bromodomain Containing 4 (BRD4) Couples NFκB/RelA With Airway Inflammation And The IRF-RIG-I Amplification Loop In Respiratory Syncytial Virus Infection Journal of Virology 2017; 91:doi: 10.1128/JVI.00007-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol 2016; 23:540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Tian B, Sadygov RG, Zhang Y, Brasier AR. Integrative proteomic analysis reveals reprograming tumor necrosis factor signaling in epithelial mesenchymal transition. J Proteomics 2016; 148:126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian B, Liu Z, Yang J, Sun H, Zhao Y, Wakamiya M, et al. Selective Molecular Antagonists of the Bronchiolar Epithelial NF&B-Bromodomain-Containing Protein 4 (BRD4) Pathway in Viral-induced Airway Inflammation. Cell Reports 2018; 23:1138–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med 2012; 18:684–92. [DOI] [PubMed] [Google Scholar]
- 26.Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc 2004; 1:93–8. [DOI] [PubMed] [Google Scholar]
- 27.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A 2011; 108:E1475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ijaz T, Pazdrak K, Kalita M, Konig R, Choudhary S, Tian B, et al. Systems Biology Approaches To Understanding Epithelial Mesenchymal Transition (EMT) In Mucosal Remodeling And Signaling In Asthma. World Allergy Organization Journal 2014; 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Jamaluddin M, Zhang Y, Sun H, Ivanciuc T, Garofalo RP, et al. Systematic Analysis of Cell-Type Differences in the Epithelial Secretome Reveals Insights into the Pathogenesis of Respiratory Syncytial Virus-Induced Lower Respiratory Tract Infections. J Immunol 2017; 198:3345–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 2004; 64:9027–34. [DOI] [PubMed] [Google Scholar]
- 31.Chang H, Liu Y, Xue M, Liu H, Du S, Zhang L, et al. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res 2016; 44:2514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010; 65:733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiraga R, Kato M, Miyagawa S, Kamata T. Nox4-derived ROS Signaling Contributes to TGF-β-induced Epithelial-mesenchymal Transition in Pancreatic Cancer Cells. Anticancer Research 2013; 33:4431–8. [PubMed] [Google Scholar]
- 34.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 2010; 30:3553–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, et al. BMS-345541 Is a Highly Selective Inhibitor of IκB Kinase That Binds at an Allosteric Site of the Enzyme and Blocks NF-κB-dependent Transcription in Mice. Journal of Biological Chemistry 2003; 278:1450–6. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Brasier AR. Applications Of Selected Reaction Monitoring (SRM)-Mass Spectrometry (MS) For Quantitative Measurement Of Signaling Pathways. Methods 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ijaz T, Jamaluddin M, Zhao Y, Zhang Y, Finnerty CC, Jay J, et al. Coordinate activities of BRD4 and CDK9 in the transcriptional elongation complex are required for TGFb-induced Nox4 expression and myofibroblast transdifferentiation Cell Death Differ 2017; ;8(2)::e2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammond M, Nong RY, Ericsson O, Pardali K, Landegren U. Profiling Cellular Protein Complexes by Proximity Ligation with Dual Tag Microarray Readout. PLoS ONE 2012; 7:e40405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ather JL, Hodgkins SR, Janssen-Heininger YMW, Poynter ME. Airway Epithelial NF-κB Activation Promotes Allergic Sensitization to an Innocuous Inhaled Antigen. American Journal of Respiratory Cell and Molecular Biology 2011; 44:631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tully JE, Hoffman SM, Lahue KG, Nolin JD, Anathy V, Lundblad LKA, et al. Epithelial NF-κB Orchestrates House Dust Mite–Induced Airway Inflammation, Hyperresponsiveness, and Fibrotic Remodeling. The Journal of Immunology 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 2007; 26:711–24. [DOI] [PubMed] [Google Scholar]
- 42.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2:84–9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, et al. TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal 2014; 7:ra91. [DOI] [PubMed] [Google Scholar]
- 44.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian B, Patrikeev I, Ochoa L, Vargas G, Belanger KK, Litvinov J, et al. NF-kappaB Mediates Mesenchymal Transition, Remodeling, and Pulmonary Fibrosis in Response to Chronic Inflammation by Viral RNA Patterns. Am J Respir Cell Mol Biol 2017; 56:506–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Tian B, Chen H, Wang P, Brasier AR, Zhou J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. European Journal of Medicinal Chemistry 2018; 151:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol 2012; 12:53–9. [DOI] [PubMed] [Google Scholar]
- 48.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med 2009; 180:122–33. [DOI] [PubMed] [Google Scholar]
- 49.Sagara H, Okada T, Okumura K, Ogawa H, Ra C, Fukuda T, et al. Activation of TGF-beta/Smad2 signaling is associated with airway remodeling in asthma. J Allergy Clin Immunol 2002; 110:249–54. [DOI] [PubMed] [Google Scholar]
- 50.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. The Journal of Clinical Investigation; 122:2756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 2006; 103:13180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 2004; 114:438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol 2011; 128:451–62; quiz 63-4. [DOI] [PubMed] [Google Scholar]
- 54.Brewster CEP, Howarth PH, Djukanovic R, Wilson J, Holgate ST, Roche WR. Myofibroblasts and Subepithelial Fibrosis in Bronchial Asthma. American Journal of Respiratory Cell and Molecular Biology 1990; 3:507–11. [DOI] [PubMed] [Google Scholar]
- 55.Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax 2005; 60:389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J 2010; 17:e85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boser SR, Mauad T, de Araújo-Paulino BB, Mitchell I, Shrestha G, Chiu A, et al. Myofibroblasts are increased in the lung parenchyma in asthma. PLoS ONE 2017; 12:e0182378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carroll NG, Perry S, Karkhanis A, Harji S, Butt J, James AL, et al. The airway longitudinal elastic fiber network and mucosal folding in patients with asthma. Am J Respir Crit Care Med 2000; 161:244–8. [DOI] [PubMed] [Google Scholar]
- 59.Bentley JK, Popova AP, Bozyk PD, Linn MJ, Baek AE, Lei J, et al. Ovalbumin sensitization and challenge increases the number of lung cells possessing a mesenchymal stromal cell phenotype. Respiratory Research 2010; 11:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J Invest Dermatol 2013; 133:1212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Wang P, Chen H, Wold E, Tian B, Brasier AR, et al. Drug Discovery Targeting Bromodomain-Containing Protein 4 (BRD4). Journal of Medical Chemistry 2017; 60:4533–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A 2012; 109:6927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 2014; 56:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Sun H, Zhang J, Brasier AR, Zhao Y. Quantitative Assessment of the Effects of Trypsin Digestion Methods on Affinity Purification-Mass Spectrometry-based Protein-Protein Interaction Analysis. J Proteome Res 2017; 16:3068–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.