

Abstract
Statins are the accepted standard for lowering low‐density lipoprotein cholesterol (LDL‐C). However, 5% to 10% of statin‐treated patients report intolerance, mostly due to muscle‐related adverse effects. Challenges exist to objective identification of statin‐intolerant patients. Evolocumab is a monoclonal antibody that binds proprotein convertase subtilisin/kexin type 9 (PCSK9), resulting in marked LDL‐C reduction. We report the design of Goal Achievement After Utilizing an Anti‐PCSK9 Antibody in Statin‐Intolerant Subjects 3 (GAUSS‐3), a phase 3, multicenter, randomized, double‐blind, ezetimibe‐controlled study to compare effectiveness of 24 weeks of evolocumab 420 mg monthly vs ezetimibe 10 mg daily in hypercholesterolemic patients unable to tolerate an effective statin dose. The study incorporates a novel atorvastatin‐controlled, double‐blind, crossover phase to objectively identify statin intolerance. Eligible patients had LDL‐C above the National Cholesterol Education Project Adult Treatment Panel III target level for the appropriate coronary heart disease risk category and were unable to tolerate ≥3 statins or 2 statins (one of which was atorvastatin ≤10 mg/d) or had a history of marked creatine kinase elevation accompanied by muscle symptoms while on 1 statin. This trial has 2 co‐primary endpoints: mean percent change from baseline in LDL‐C at weeks 22 and 24 and percent change from baseline in LDL‐C at week 24. Key secondary efficacy endpoints include change from baseline in LDL‐C, percent of patients attaining LDL‐C <70 mg/dL (1.81 mmol/L), and percent change from baseline in total cholesterol, non–high‐density lipoprotein cholesterol, and apolipoprotein B. Recruitment of 511 patients was completed on November 28, 2014.
Introduction
Clinicians who treat lipid disorders consider the intolerance of patients to effective doses of HMG‐CoA reductase inhibitors (statins) one of the most vexing problems in clinical practice. Patients reporting statin intolerance typically describe a variety of muscle symptoms, including muscle pain or weakness, when treated with a statin, and often report relief from the symptoms when the drug is withdrawn or the dose decreased. More severe forms are associated with marked elevation in the concentration of creatine kinase (CK), which may, in rare cases, result in rhabdomyolysis. However, CK elevations are only observed in a small subset of patients who report muscle symptoms. The reported incidence of statin intolerance in observational studies varies widely, but ranges from 5% to 10% of patients.1, 2, 3 The precise definition of this syndrome has been elusive, in part because of the lack of established biomarkers that identify statin intolerance.4, 5 Accordingly, the diagnosis of this disorder is typically subjective and is usually based upon patient‐reported symptoms rather than objective criteria. Regardless of severity or definition, many at‐risk patients stop taking their statins as prescribed, which worsens the impact of hypercholesterolemia‐associated morbidity on public health.3, 5, 6, 7
The vague nature of the complaints, lack of consistent diagnostic biomarkers, and incidence of similar symptoms in placebo‐treated patients has resulted in skepticism within the health care and regulatory communities about the true incidence of statin intolerance. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors represent a promising approach to lowering low‐density lipoprotein cholesterol (LDL‐C) in patients who experience intolerable muscle‐related adverse effects during statin therapy. These new drugs, alirocumab and evolocumab, which were approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) in 2015, are highly effective in reducing LDL‐C. Currently available data demonstrate few untoward muscle‐related adverse effects in patients administered PCSK9 inhibitors.8, 9 In this setting, we designed a rigorous randomized controlled trial to identify patients with reproducible statin‐induced symptoms to compare the effectiveness of 2 therapies: ezetimibe or evolocumab.
Methods
Study Design
The Goal Achievement After Utilizing an Anti‐PCSK9 Antibody in Statin Intolerant Subjects 3 (GAUSS‐3) trial is a phase 3, multicenter, randomized, double‐blind, ezetimibe‐controlled, parallel‐group study of the PCSK9 inhibitor evolocumab in hypercholesterolemic patients unable to tolerate an effective dose of a statin (http://www.clinicaltrials.gov NCT01984424). The study incorporates a novel double‐blind, 2‐period, placebo‐controlled crossover design to identify patients with objectively documented statin intolerance prior to randomization to evolocumab or ezetimibe (Figure).
Prior to randomization, patients undergo a 4‐week washout phase, during which ezetimibe and statins are withdrawn. This study has 3 active parts: Part A is a double‐blind, 2‐period, placebo‐controlled, 24‐week crossover phase in which patients with a history of statin intolerance are randomly assigned in a 1:1 ratio to either atorvastatin 20 mg daily or matching placebo for the first 10 weeks (Period 1), then undergo a 2‐week washout period, with subsequent crossover to the alternate therapy for a second 10‐week period (Period 2). Patients who experience intolerable muscle‐related symptoms during either period do not need to complete the full 10 weeks of exposure. Patients who experience muscle‐related symptoms which, in the clinical judgment of the investigator, would lead them to stop the study treatment (or if the symptoms are deemed intolerable by the patient) enter a 2‐week washout period prior to entering Period 2 or Part B of the study.
Part B is a 24‐week double‐blind, double‐dummy, randomized active‐controlled comparison of subcutaneously (SC) injected evolocumab 420 mg monthly with oral ezetimibe 10 mg daily with a 2:1 ratio of evolocumab to ezetimibe. Two categories of patients can enter Part B of the study: (1) patients from Part A who experience muscle‐related symptoms while taking atorvastatin and do not experience these symptoms while taking placebo; and (2) any patient with a documented history of CK elevation >10× the upper limit of normal accompanied by muscle symptoms while on statin therapy with resolution of both CK elevation and muscle symptoms upon discontinuation of statin therapy. These study procedures are designed to ensure that only patients with rigorously documented statin‐induced muscle symptoms enter Part B of the study.
Part C is a 2‐year open‐label extension phase, during which all patients who complete Part B receive evolocumab to evaluate the long‐term safety and efficacy in patients with objectively documented statin intolerance. (http://www.clinicaltrials.gov NCT01854918.)
Figure 1.
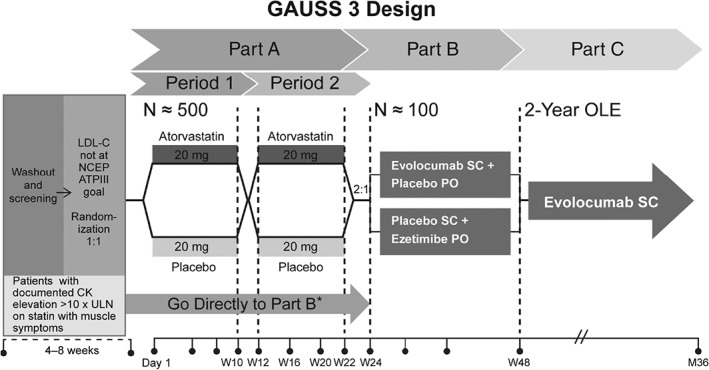
Study design for GAUSS‐3. Abbreviations: ATP III, Adult Treatment Panel III; CK, creatine kinase; GAUSS‐3, Goal Achievement After Utilizing an Anti‐PCSK9 Antibody in Statin‐Intolerant Subjects 3; NCEP, National Cholesterol Education Project; OLE, open‐label extension; PO, oral; SC, subcutaneous; ULN, upper limit of normal. *Patients are randomized 2:1 to the Part B treatment arms.
Study Objectives and Endpoints
The primary objective of GAUSS‐3 is to evaluate the effect of 24 weeks of evolocumab administered monthly compared with ezetimibe on percent change from baseline in LDL‐C in hypercholesterolemic patients unable to tolerate an effective dose of a statin due to muscle‐related side effects (MRSE) as confirmed by statin rechallenge. Secondary objectives of the study include evaluation of the safety and tolerability of monthly SC evolocumab compared with ezetimibe; change from baseline in LDL‐C; percent change from baseline in total cholesterol, non–high‐density lipoprotein cholesterol (non–HDL‐C), apolipoprotein B (ApoB), total cholesterol/HDL‐C ratio, ApoB/apolipoprotein A1 (ApoA1) ratio, lipoprotein(a) [Lp(a)], triglycerides (TG), HDL‐C, and very low‐density lipoprotein cholesterol (VLDL‐C); and percent of patients attaining LDL‐C <70 mg/dL (1.81 mmol/L). An exploratory objective of the study is to evaluate the incidence of MRSE during a double‐blind, placebo‐controlled, crossover atorvastatin rechallenge. The primary hypothesis of GAUSS‐3 is that evolocumab 420 mg monthly will be well tolerated and result in greater reduction of LDL‐C than ezetimibe in hypercholesterolemic patients unable to tolerate an effective dose of a statin.
GAUSS‐3 has 2 co‐primary endpoints: mean percent change from baseline in LDL‐C at Weeks 22 and 24 of Part B and percent change from baseline in LDL‐C at Week 24 of Part B. Tier 1 co‐secondary efficacy endpoints of the means at Weeks 22 and 24 and at Week 24 of Part B include change from baseline in LDL‐C; LDL‐C response (LDL‐C <70 mg/dL [1.81 mmol/L]); and percent change from baseline in total cholesterol, non–HDL‐C, ApoB, total cholesterol/HDL‐C ratio, and ApoB/ApoA1 ratio. Tier 2 co‐secondary efficacy endpoints of the means at Weeks 22 and 24 and at Week 24 of Part B are percent change from baseline in Lp(a), TG, HDL‐C, and VLDL‐C.
Inclusion and Exclusion Criteria
The major inclusion criteria are summarized in Table 1. Briefly, they include an LDL‐C above the target level specified in the National Cholesterol Education Project (NCEP) Adult Treatment Panel III (ATP III) for the appropriate coronary heart disease risk category and an inability to tolerate ≥3 statins (one of which must be at the lowest‐approved starting average daily dose) or an inability to tolerate 2 statins (one of which must be atorvastatin at an average daily dose of ≤10 mg). The key exclusion criteria are summarized in Table 2.
Table 1.
Main Inclusion Criteria
| Male or Female age ≥18 to ≤80 years at signing of informed consent |
|---|
| Patient not at LDL‐C goal by NCEP ATP III risk category and the following LDL‐C levels by central laboratory at screening: |
| Fasting LDL‐C ≥100 mg/dL (2.59 mmol/L) for patients with diagnosed CHD or CHD risk equivalent, or |
| Fasting LDL‐C ≥130 mg/dL (3.37 mmol/L) for patients without diagnosed CHD or risk equivalent and ≥2 risk factors, or |
| Fasting LDL‐C ≥160 mg/dL (4.14 mmol/L) for patients without diagnosed CHD or risk equivalent and with ≥1 risk factors, or |
| Fasting LDL‐C ≥190 mg/dL (4.9 mmol/L) for patients without diagnosed CHD or risk equivalent and with no risk factors |
| Patient must have a history of statin intolerance as evidenced by the following: |
| Unable to tolerate atorvastatin at an average daily dose of 10 mg and unable to tolerate any other statin at any dose due to skeletal muscle‐related symptoms (eg, pain, aches, weakness, or cramping) |
| OR |
| Unable to tolerate ≥3 statins: 1 statin at the lowest starting average daily dose (rosuvastatin 5 mg, simvastatin 10 mg, pravastatin 40 mg, lovastatin 20 mg, fluvastatin 40 mg, or pitavastatin 2 mg) and any other 2 statins at any dose, due to skeletal muscle‐related symptoms (eg, pain, aches, weakness, or cramping) |
| OR |
| A documented history of CK elevation >10× ULN accompanied by muscle symptoms while on statin therapy and documented resolution of both CK elevation and muscle symptoms upon discontinuation of statin therapy |
| AND |
| Symptoms resolved or improved when statin dose was decreased or discontinued |
| Lipid‐lowering therapy has been stable prior to LDL‐C screening for ≥4 weeks if currently on a bile‐acid sequestering resin and/or stanol; if patient is on statin or ezetimibe at start of screening, statin or ezetimibe must be discontinued for ≥4 weeks before LDL‐C screening |
| Fasting TG ≤400 mg/dL (4.52 mmol/L) by central laboratory at screening |
Abbreviations: CHD, coronary heart disease; CK, creatine kinase; F, female; LDL‐C, low‐density lipoprotein cholesterol; M, male; NCEP ATP III, National Cholesterol Education Project Adult Treatment Panel III; TG, triglycerides; ULN, upper limit of normal.
Table 2.
Major Exclusion Criteria
| History of hemorrhagic stroke |
|---|
| Personal or family history of hereditary muscular disorders |
| NYHA class III or IV HF, or last known LVEF <30% |
| Uncontrolled serious cardiac arrhythmia in the past 3 months prior to randomization |
| MI, UA, PCI, CABG, or stroke within 3 months prior to randomization |
| Planned cardiac surgery or revascularization |
| Type 1 DM, poorly controlled type 2 DM (HbA1c >8.5%), newly diagnosed type 2 DM within 6 months of randomization |
| Uncontrolled HTN defined as sitting SBP >160 mm Hg or DBP >100 mm Hg |
| Subject who has taken, in the last 4 weeks, red yeast rice, >200 mg/d niacin, or prescription lipid‐regulating drugs (eg, fibrates and derivatives, statins, or ezetimibe) other than bile‐acid sequestering resin or stanols and stanol esters |
| Patient who has taken a CETP inhibitor in the last 12 months prior to LDL‐C screening, such as anacetrapib, dalcetrapib, or evacetrapib |
| Treatment in the last 3 months prior to LDL‐C screening with any of the following drugs: systemic cyclosporine, systemic corticosteroids |
| Uncontrolled hypothyroidism or hyperthyroidism |
| eGFR <30 mL/min/1.73 m2 at screening |
| Active liver disease or hepatic dysfunction, defined as AST or ALT >3× ULN |
| Known active infection or major hematologic, renal, metabolic, GI, or endocrine dysfunction in the judgment of the investigator |
| Diagnosis of DVT or PE within 3 months prior to randomization |
| Unreliability as a study participant based on the investigator's (or designee's) knowledge of the patient (eg, alcohol or other drug) |
| Currently enrolled in another investigational device or drug study, or <30 days since ending another investigational device or drug study(s), or receiving other investigational agent(s) |
| Female patient who has either (1) not used ≥1 highly effective method of contraception for ≥1 month prior to screening or (2) is not willing to use such a method during treatment and for an additional 15 weeks after the end of treatment, unless the patient is sterilized or postmenopausal |
| Patient who is pregnant or breastfeeding, or planning to become pregnant during treatment and/or within weeks after the end of treatment |
| Patient who has previously received evolocumab or any other investigational therapy to inhibit PCSK9 |
| Malignancy except nonmelanoma skin cancers, cervical carcinoma, or breast DCIS within the last 5 years |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CABG, coronary artery bypass grafting; CETP, cholesteryl ester transfer protein; DBP, diastolic blood pressure; DCIS, ductal carcinoma in situ; DM, diabetes mellitus; DVT, deep vein thrombosis; eGFR, estimated glomerular filtration rate; GI, gastrointestinal; HbA1c, glycated hemoglobin; HF, heart failure; HTN, hypertension; LDL‐C, low‐density lipoprotein cholesterol; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NYHA, New York Heart Association; PCI, percutaneous coronary intervention; PCSK9, proprotein convertase subtilisin/kexin type 9; PE, pulmonary embolism; SBP, systolic blood pressure; UA, unstable angina; ULN, upper limit of normal.
Treatments and Key Procedures
At screening, subjects were given placebo SC injections within 5 to 10 days of the screening LDL‐C evaluation via 3 prefilled 1‐mL autoinjector pens prior to randomization in Part A. In Part A, patients were assigned to 20 mg atorvastatin or matching placebo taken orally daily for 10 weeks or until considered intolerant. After a 2‐week washout, patients switch therapies for an additional 10 weeks. In Part B, the patients were randomized 2:1 to either 420 mg evolocumab SC monthly and placebo orally daily or placebo SC monthly and daily 10 mg ezetimibe. Injections were administered via 3 prefilled 1‐mL autoinjector pens containing either 140 mg/mL of evolocumab or placebo. In Part C, all patients will receive open‐label evolocumab 420 mg SC monthly.
Randomization in Part B was stratified by screening LDL‐C level (<180 mg/dL [4.66 mmol/L] vs ≥180 mg/dL) at study baseline. The following describes procedures during Part B: (1) blinded investigational product (IP), evolocumab or placebo, was administered at the study site using a prefilled autoinjector pen; (2) central laboratory results of the lipid panel, ApoA1, ApoB, Lp(a), and high‐sensitivity C‐reactive protein were blinded for the duration of the study; (3) investigators were not permitted to perform nonprotocol lipid testing until ≥12 weeks after the last blinded IP administration (investigators were informed if TG are >1000 mg/dL (11.3 mmol/L) to enable appropriate management); (4) the last dose of blinded SC IP was given at Week 20 of Part B for all patients.
Patients were encouraged to complete all planned visits regardless of their adherence to IP administration (intention‐to‐treat approach). In countries where permitted, patients were invited to consent to pharmacogenetic analyses. The study included collection of biomarker samples where approved by the independent ethics committee, applicable regulatory, and other authorities. The study included adjudication of deaths and major cardiovascular (CV) events by an independent Clinical Events Committee with oversight by an independent Data Monitoring Committee. An independent external biostatistical group provided analyses for the Data Monitoring Committee.
Statistical Analysis
The number of patients needed in Part B assumed the smallest treatment effect was approximately 25% with a common SD of 20% based on evolocumab phase 2 results. It was anticipated that the treatment effect would be attenuated due to: (1) ∼ 15% of randomized subjects stopping IP early; (2) ∼ 5% of randomized subjects ending study participation early; (3) 2% of randomized subjects not receiving any IP. After accounting for treatment attenuation, the sample size provides approximately 98% power for each co‐primary endpoint.
The number of patients needed for Part A was calculated to achieve a planned sample size of 100 patients in Part B. In the double‐blind, placebo‐controlled statin rechallenge phase, the estimated rate of MRSE by blinded statin rechallenge was 20%. Thus, the expected number of patients to be enrolled in Part A was 500. To ensure that the target number of patients was achieved, enrollment in Part A continued until the Part B target sample size was met. All patients who completed Part B are eligible to participate in Part C. It was estimated that approximately 80% to 100% of patients in Part B would continue to Part C. The 20% estimate of MRSE was selected based on a previous study with a similar crossover design examining the efficacy of co‐enzyme Q10 with simvastatin in statin‐intolerant patients.10 This prior single‐center study showed a 34% incidence of MRSE by blinded statin rechallenge. We selected a more conservative 20% estimate for our global multicenter study to ensure that sufficient patients were enrolled to adequately test the principal hypotheses.
Efficacy and safety analyses will be performed on all randomized patients in Part B who received ≥1 dose of IP in Part B. Multiplicity adjustments will be applied for primary analyses of co‐primary and co‐secondary endpoints to control the overall family‐wise error rate at 0.05.
The long‐term efficacy and safety analysis will be descriptive and will include all enrolled patients in Part C of the study who received ≥1 dose of IP in Part C. Events of death, myocardial infarction, hospitalization for unstable angina, coronary revascularization, stroke, transient ischemic attack, and hospitalization for HF will be adjudicated by an independent Clinical Events Committee. Patient incidence of exploratory endpoint events will be summarized for each treatment group.
Analyses of Co‐Primary and Co‐Secondary Endpoints
To assess the co‐primary endpoints of the mean percent change in LDL‐C from baseline at Weeks 22 and 24 of Part B and the percent change from baseline at Week 24 of Part B, a repeated measures linear effects model will be used to compare the efficacy of evolocumab with ezetimibe. The repeated measures model will include terms for treatment group, stratification factor, scheduled visit, and the interaction of treatment with scheduled visit. Missing values will not be imputed when the repeated measures linear effects model is used. The statistical model for the co‐secondary efficacy endpoints will be similar to the co‐primary endpoints; however, LDL‐C response will be analyzed using the Cochran‐Mantel‐Haenszel test adjusted by the stratification factors.
Subgroup Analyses
The following baseline covariates were prespecified for subgroup or covariate analyses: age: <65 years or ≥65 years, sex, race, baseline LDL‐C < median or ≥ median, family history of premature coronary heart disease, and PCSK9 level < baseline median or ≥ baseline median. Because of the small size of the study, the protocol did not emphasize such analyses.
Safety Analyses
Adverse events (AEs) will be coded using the current version of the Medical Dictionary for Regulatory Activities (MedDRA). Patient incidence of treatment emergent AEs, serious AEs, and AEs leading to discontinuation of IP will be tabulated by randomized treatment group. Measurements of laboratory parameters and vital signs will be summarized over time. The incidence and percentages of patients who develop anti‐evolocumab antibodies (binding and neutralizing) at any time will also be tabulated. Data for patients with 2 consecutive values <25 mg/dL (0.65 mmol/L) are provided to the Data Monitoring Committee for their review.
Baseline Demographics
The GAUSS‐3 study completed enrollment of 511 patients on November 28, 2014, of whom 492 entered Part A and 19 bypassed Part A and directly entered Part B. A majority of randomized patients had high CV risk and had failed ≥3 statins (Table 3). Median baseline LDL‐C in Part A was 198 mg/dL (5 mmol/L). The male‐to‐female ratio was 1:1 and 94% of patients were white.
Table 3.
Part A Baseline Demographics and Laboratory Values
| All Randomized, N = 511 | |
|---|---|
| Sex | |
| F | 254 (49.7) |
| M | 257 (50.3) |
| Age, y | 62.0 (54.0–68.0) |
| Race | |
| White | 483 (94.5) |
| Other | 28 (5.5) |
| BMI, kg/m2 | 28.5 (24.9–30.9) |
| CAD | 170 (33.3) |
| Cerebrovascular disease or PAD | 115 (22.5) |
| CV risk factors | |
| Current cigarette use | 54 (10.6) |
| Type 2 DM | 64 (12.5) |
| HTN | 283 (55.4) |
| Family history of premature CHD | 196 (38.4) |
| Low HDL‐C | 189 (37.0) |
| Patients with ≥2 risk factors | 242 (47.4) |
| NCEP risk category | |
| High | 316 (61.8) |
| Moderately high | 55 (10.8) |
| Moderate | 76 (14.9) |
| Lower | 64 (12.5) |
| History of intolerance to statins, per patient | |
| 1 statin | 8 (1.6) |
| 2 statins | 102 (20.0) |
| 3 statins | 217 (42.5) |
| ≥4 statins | 184 (36.0) |
| Worst muscle‐related side‐effect | |
| Myalgia | 412 (80.6) |
| Myositis | 85 (16.6) |
| Rhabdomyolysis | 14 (2.7) |
| Baseline laboratory values | |
| TC, mg/dL | 286.5 (254.5–334.0) |
| LDL‐C, mg/dL | 197.5 (170.3–243.0) |
| HDL‐C, mg/dL | 48.0 (40.0–60.0) |
| TG, mg/dL | 168.5 (122.0–231.0) |
| Lipoprotein(a), nmol/L | 32.0 (15.0–146.0) |
| hsCRP, mg/L | 1.55 (0.83–3.41) |
Abbreviations: BMI, body mass index; CAD, coronary artery disease; CHD, coronary heart disease; CV, cardiovascular; DM, diabetes mellitus; HDL‐C, high‐density lipoprotein cholesterol; hsCRP, high‐sensitivity C‐reactive protein; HTN, hypertension; F, female; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; M, male; NCEP, National Cholesterol Education Program; PAD, peripheral arterial disease; TC, total cholesterol; TG, triglycerides.
Data are presented as n (%) or median (IQR).
Discussion
Patient‐reported statin intolerance, predominantly due to statin‐associated muscle symptoms, is a relatively common and difficult to manage condition that affects millions of patients worldwide. The Prediction of Muscular Risk in Observational Conditions (PRIMO) study reported muscle symptoms in 10.5% of 7924 patients treated with statins.1 Other observational studies suggest an incidence approaching 20% of statin‐treated patients in routine clinical practice.3 In a recent retrospective cohort study involving >100000 patients, 17% had ≥1 statin‐related event documented, of which 27% (5% of the total study population) referred to myalgia or myopathy.3 In the context of strong evidence from randomized controlled trials of the benefits of lowering of LDL‐C in both the primary and secondary prevention settings, the inability to treat patients with statin intolerance undermines public‐health efforts to reduce the burden of cardiovascular disease. Accordingly, development of alternative treatment strategies for adequate lipid management in statin‐intolerant patients represents an important unmet medical need.
Current approaches to management of patients with muscle‐related statin intolerance have many limitations. Most clinicians first try a series of alternative statins, which is successful in some patients, although recurrent symptoms occur in many others. Some authorities recommend administration of small doses of a high efficacy statin 1× to 3× weekly with gradual titration to reach the maximally tolerated dosage.11 This strategy has proven useful, although many patients are unable to reach a dosage that is both sufficient to reduce LDL‐C to desirable levels and well tolerated. Ezetimibe is also commonly prescribed in statin‐intolerant patients, but the LDL‐C–lowering effect of this agent is modest (about 16%–20%) and the effect on CV outcomes relatively small. The only controlled CV outcome trial using ezetimibe demonstrated a 6.4% reduction in CV events when added to a statin after 7 years of treatment, which was driven by the reduction in nonfatal events.12
The identification of patients with MRSE has been challenging due to the subjective nature of symptoms. Most patients do not exhibit either myositis or overt rhabdomyolysis, both of which are characterized by a substantial elevation of CK. Furthermore, successful statin rechallenge in some patients with previously reported statin‐related muscle symptoms has led to skepticism about the true incidence of statin intolerance and concerns about overdiagnosis of muscle‐related statin intolerance. As a consequence, no pharmacological therapy has received approval from the US Food and Drug Administration to lower LDL‐C in patients unable to tolerate adequate dosages of statins.
The development of PCSK9 inhibitors represents a potentially important addition to the therapeutic armamentarium for management of statin‐intolerant patients. These agents reduce LDL‐C by ≥50% when administered at doses of 140 mg every 2 weeks or 420 mg every 4 weeks (evolocumab) or 150 mg every 2 weeks (alirocumab).13, 14 Preliminary studies have shown a low incidence of myalgia or myositis, including trials conducted in patients with a history of statin intolerance.8, 9 In the GAUSS trial, in patients unable to tolerate ≥1 statin, evolocumab demonstrated robust reductions in LDL‐C and was well tolerated, with a low incidence of muscle‐related adverse effects.9 A second study, GAUSS‐2, which included ∼20% of patients with myositis or rhabdomyolysis, compared evolocumab with ezetimibe in patients intolerant to ≥2 statins and demonstrated a >50% reduction from baseline in LDL‐C with no differences in muscle‐related adverse effects compared with ezetimibe.8 Similar findings have been reported with another PCSK9 inhibitor.15
Despite the promise of these agents, regulatory approval in the United States for the treatment of statin‐intolerant patients will require the highest‐quality evidence of efficacy in a population with demonstrated statin intolerance. GAUSS‐3 was designed to supplement the prior GAUSS trials by providing additional stringent evidence regarding the potential role of evolocumab in patients with demonstrated statin intolerance. In GAUSS‐3, screening for enrollment into Part A required the strong evidence of statin intolerance, including either the inability to tolerate an entry dosage of atorvastatin (10 mg daily) and another statin at any dose or failure to tolerate 3 statins with ≥1 administered at the lowest approved average daily dose. For randomization into Part B, enrollment included only those patients who experienced muscle‐related symptoms on atorvastatin but not placebo or patients with a documented history of CK elevation >10× upper limit of normal accompanied by muscle symptoms while on statin therapy with resolution of both upon discontinuation of statin therapy. Accordingly, GAUSS‐3 will provide a high degree of confidence that enrolled patients are truly unable to tolerate effective statin therapy. The study will also provide insights into the incidence of statin intolerance after a blinded statin rechallenge. Although not the primary endpoints, the percentage and characteristics of patients who complete Part A will provide valuable objective data regarding the incidence of statin intolerance during blinded rechallenge in patients who report prior MRSE.
Conclusion
The enrollment criteria for GAUSS‐3 selected a patient population with a particularly high unmet clinical need that met an objective definition of statin‐intolerance. The median LDL‐C is approximately 200 mg/dL, a level considered unacceptably high, even in primary prevention. Approximately half of the enrolled patients have coronary, cerebrovascular, or peripheral arterial disease, and >50% of patients fell into the highest‐risk category defined by the NCEP ATP III guidelines. The need to substantially reduce LDL‐C in such patients is self‐evident. The results of GAUSS‐3 will inform clinicians about the relative efficacy of evolocumab compared with ezetimibe in such patients and will provide both clinicians and the regulatory community with a better understanding of the potential role of PCSK9 inhibition in the management of these challenging patients.
GAUSS‐3 Investigators
David Sullivan, Royal Prince Alfred Hospital, Australia; Sam Lehman, Adelaide Medical Research, Australia; Karam Kostner, Dr Heart Pty Ltd, Australia; Andrew Don Wauchope, Hamilton Health Sciences Hamilton General Hospital, Canada; Michael Hartleib, Kawartha Cardiology Clinical Trials, Canada; Alexis Baass, Institut de recherches cliniques de Montréal, Canada; Jean Bergeron, Clinique des Maladies Lipidiques de Quebec Incorporated, Canada; Tisha Joy, London Health Sciences Centre, University Hospital, Canada; Richard Ceska, Vseobecna fakultni nemocnice v Praze, Czech Republic; Vera Adamkova, Institut klinicke a experimentalni mediciny, Czech Republic; Vladimir Blaha, Fakultni nemocnice Hradec Kralove, Czech Republic; Jorgen Jeppesen, Glostrup Hospital, Denmark; Henrik Kjaerulf Jensen, Aarhus Universitets Hospital, Denmark; Eric Bruckert, Hôpital Pitié‐Salpêtrière, France; Michel Krempf, Centre Hospitalier Universitaire de Nantes, Hôpital Nord Laennec, France; Chantal Bully, Groupe Hospitalier Mutualiste, France; Peter Bosiljanoff, Herz‐Gefäβ‐Zentrum Nymphenburg am Klinikum Dritter Orden, Germany; Ioanna Gouni‐Berthold, Universitätsklinikum Köln, Germany; Elisabeth Steinhagen‐Thiessen, Charité–Universitätsmedizin Berlin, Charité Campus Virchow‐Klinikum, Germany; Paolo Pintus, Azienda Ospedaliera Brotzu, Italy; Claudio Borghi, Azienda Ospedaliero Universitaria di Bologna Policlinico S Orsola Malpighi, Italy; Tiziana Sampietro, Fondazione Toscana Gabriele Monasterio, Stabilimento Ospedaliero di Pisa, Italy; Giovanni Battista Vigna, Azienda Ospedaliero Universitaria di Ferrara Nuovo Ospedale S Anna, Italy; Elmo Mannarino, Ospedale Santa Maria della Misericordia Università degli Studi di Perugia, Italy; Claudio Pozzi, Azienda Ospedaliera istituti Clinici di Perfezionamento Ospedale E Bassini, Italy; Erik Stroes, Academisch Medisch Centrum, Netherlands; Anho Liem, Sint Franciscus Gasthuis, Netherlands; Rudolf Van Leendert, Albert Schweitzer Ziekenhuis Locatie Zwijndrecht, Netherlands; Russell Scott, Lipids and Diabetes Research Group, New Zealand; Thorbjorn Kjaernli, Medi 3 Klinikk, Norway; Gisle Langslet, Oslo Universitetssykehus/Rikshopitalet, Norway; Andrew Jacovides, Midrand Medical Centre, South Africa; Eric Klug, Sunninghill Hospital, South Africa; Dirk Blom, Lipid Laboratory, South Africa; David Preiss, Glasgow Royal Infirmary, United Kingdom; Dermot Neely, Royal Victoria Infirmary, United Kingdom; Charlotte Dawson, Queen Elizabeth Hospital, United Kingdom; Michael Miller, University of Maryland Medical Center, United States; Robert Rosenson, Mount Sinai Icahn School of Medicine, United States; Kevin McCullum, York Hospital, United States; Christie Ballantyne, Baylor College of Medicine, United States; Michael Rocco, Cleveland Clinic Heart and Vascular Institute, United States; Prediman Shah, Cedars Sinai Medical Center, United States; Norman Lepor, Westside Medical Associates of Los Angeles, United States; Paul Rosenblit, Diabetes Lipid Management and Research Center, United States; Gregory Pokrywka, IRC Clinics Inc., United States; Michael Blazing, Duke Health Center at Southpoint, United States; Peter Toth, Community General Hospital Main Clinic, United States; Patrick Moriarty, University of Kansas Medical Center, United States; Melvyn Rubenfire, University of Michigan Health System, Dominos Farms, United States; Pamela Morris, Medical University of South Carolina, United States; Arshed Quyyumi, Emory University Hospital, United States; Stephen Kopecky, Mayo Clinic Rochester, United States.
Data Monitoring Committee Members
Charles H. Hennekens, MD, DrPH; Chair, Felicita Andreotti, MD, PhD; Colin Baigent, FFPH; W. Virgil Brown, MD; Barry R. Davis, MD, PhD; John Newcomer, MD; Sarah K. Wood, MD.
Clinical Events Committee Members
Stephen D. Wiviott, MD, Chair; Cheryl Lowe, Director; Eric Awtry, MD; Clifford J. Berger, MD; Kevin Croce, MD; Akshay Desai, MD; Eli Gelfand, MD; Carolyn Ho, MD; David E. Leeman, MD; Mark S. Link, MD; Andrew D. Norden, MD; Ashvin Pande, MD; Natalia Rost, MD; Frederick Ruberg, MD; Scott Silverman, MD; Aneesh Singhal, MD; Joseph Vita, MD (deceased).
Acknowledgments
The authors thank Meera Kodukulla, PhD, CMPP of Amgen Inc. and Laura Evans, PharmD, on behalf of Amgen Inc., for editorial support.
Dr. Nissen reports that the Cleveland Clinic Center for Clinical Research receives funding to perform clinical trials from AstraZeneca, Amgen, Inc., Cerenis, Eli Lilly, Pfizer, The Medicines Company, Novartis, Takeda, Orexigen, and Eli Lilly. Dr. Nissen is involved in these clinical trials but receives no personal remuneration for his participation. Dr. Nissen consults for many pharmaceutical companies, but he requires them to donate all honoraria or consulting fees directly to charity so that he receives neither income nor a tax deduction. Dr. Rosenson has participated on advisory boards for Akcea, Amgen, Inc., AstraZeneca, CVS Caremark, GSK, Regeneron, and Sanofi. Dr. Rosenson has received institutional research grants from Amgen, Inc., Catabasis, and Sanofi; honoraria from Kowa; and royalties from UpToDate, Inc. Dr. Stroes reports receiving (nonsubstantial) lecturing fees from Aegerion, Amgen, Inc., Merck, Novartis, Regeneron, and Sanofi. Dr. Sattar has consulted for Amgen, Inc., Sanofi, AstraZeneca, and Eli Lilly. Dr. Preiss has consulted for Sanofi during previous employment. Dr. Mancini reports consulting fees (modest) from Amgen, Inc., and Sanofi; honoraria (modest) from Amgen, Inc., Sanofi, Regeneron, Lilly, AstraZeneca, Roche, and Merck; and research grants from Amgen, Inc. and Merck. Dr. Ballantyne has received research grants and consulting fees from Amgen, Inc., Abbott, Amarin, Genentech, Merck, Novartis, Pfizer, Roche, Sanofi, and Regeneron; consulting fees from Cerenis, Aegerion, Esperion, Kowa, Genzyme, and Arena; and research grants from Eli Lilly and GSK. Dr. Catapano has received lecturing fees from Amgen, Inc., Abbott, Genentech, Merck, Pfizer, Roche, Sanofi, and Regeneron; consulting fees from Aegerion, Kowa, Genzyme, and Merck; and research grants from Merck, Pfizer, and Sanofi. Dr. Gouni‐Berthold has received lecturing and consulting fees from Amgen, Inc., AstraZeneca, Sanofi, Genzyme, Eli Lilly, Chiesi, and GSK. Dr. Stein has received consulting fees from Amgen, Inc., BMS/Adnexus Therapeutics, Genentech, Regeneron, and Sanofi related to PCSK9 inhibitors and his institution has received research funding related to PCSK9 clinical trials from Amgen, Inc., Alnylam, BMS/Adnexus Therapeutics, Genentech, Sanofi, and Regeneron. Dr. Thompson has received research support from Genomas, Roche, Sanofi, Regeneron, Esperion, Amarin, and Pfizer; has served as a consultant for Amgen, Inc., Regeneron, Merck, Esperion, and Sanofi; has received speaker honoraria from Merck, AstraZeneca, Regeneron, Sanofi, and Amgen, Inc.; owns stock in AbbVie, Abbott Labs, CVS, General Electric, Johnson & Johnson, Medtronic, and JA Willey; and has provided expert legal testimony on exercise‐related cardiac events and statin myopathy. Drs. Dent‐Acosta, Xue, Scott, Somaratne, and Wasserman are employees and stockholders of Amgen, Inc.
The study was funded by Amgen, Inc.
The authors have no other funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high‐dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–414. [DOI] [PubMed] [Google Scholar]
- 2. Thompson PD, Clarkson P, Karas RH. Statin‐associated myopathy. JAMA. 2003;289:1681–1690. [DOI] [PubMed] [Google Scholar]
- 3. Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stroes ES, Thompson PD, Corsini A, et al. Statin‐associated muscle symptoms: impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36:1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenson RS, Baker SK, Jacobson TA, Kopecky SL, Parker BA. An assessment by the Statin Muscle Safety Task Force: 2014 update. J Clin Lipidol. 2014;8:S58–71. [DOI] [PubMed] [Google Scholar]
- 6. Avorn J, Monette J, Lacour A, et al. Persistence of use of lipid‐lowering medications: a cross‐national study. JAMA. 1998;279:1458–1462. [DOI] [PubMed] [Google Scholar]
- 7. Ellis JJ, Erickson SR, Stevenson JG, et al. Suboptimal statin adherence and discontinuation in primary and secondary prevention populations. J Gen Intern Med. 2004;19:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stroes E, Colquhoun D, Sullivan D, et al; GAUSS‐2 Investigators . Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–2548. [DOI] [PubMed] [Google Scholar]
- 9. Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low‐density lipoprotein cholesterol levels in statin‐intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–2506. [DOI] [PubMed] [Google Scholar]
- 10. Taylor BA, Lorson L, White CM, et al. A randomized trial of coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis. 2015;238:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every‐other‐day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother. 2008;42:341–346. [DOI] [PubMed] [Google Scholar]
- 12. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE‐IT Investigators . Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. [DOI] [PubMed] [Google Scholar]
- 13. Blom DJ, Hala T, Bolognese M, et al; DESCARTES Investigators . A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–1819. [DOI] [PubMed] [Google Scholar]
- 14. Robinson JG, Farnier M, Krempf M, et al; ODYSSEY LONG TERM Investigators . Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. [DOI] [PubMed] [Google Scholar]
- 15. Moriarty PM, Thompson PD, Cannon CP, et al; ODYSSEY ALTERNATIVE Investigators . Efficacy and safety of alirocumab vs ezetimibe in statin‐intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9:758–769. [DOI] [PubMed] [Google Scholar]