

ABSTRACT
Cardiogenic shock remains a highly lethal condition. Conventional therapy including revascularization and mechanical circulatory support aims to improve cardiac output and oxygen delivery, but increasing basic and clinical observations indicate wider circulatory and cellular abnormalities, particularly at the advanced stages of shock. Progressive cardiogenic shock is associated with microcirculatory and cellular abnormalities. Cardiogenic shock is initially characterized by a failure to maintain global oxygen delivery; however, progressive cardiogenic shock is associated with the release of pro‐inflammatory cytokines, derangement of the regulation of regional blood flow, microcirculatory abnormalities, and cellular dysoxia. These abnormalities are analogous to septic shock and may not be reversed by increase in oxygen delivery, even to supranormal levels. Earlier mechanical circulatory support in cardiogenic shock may limit the development of microcirculatory and cellular abnormalities.
Introduction
Low cardiac output despite adequate or elevated filling pressure is one of the defining features of cardiogenic shock. As cardiac output is a key determinant of global oxygen delivery (DO2), cardiogenic shock can also be defined as a failure of global DO2 to meet oxygen consumption (VO2), resulting in tissue hypoperfusion. The mortality rate from cardiogenic shock in acute myocardial infarction (AMI) remains stubbornly high despite revascularization1 and hemodynamic support.2 This review will discuss the concept of DO2, the wider circulatory and cellular changes, and the therapeutic implications in cardiogenic shock.
Global Oxygen Delivery
Global DO2 is the total oxygen carried (convected) by blood to tissue and is calculated as the product of the oxygen content in arterial blood (CaO2) and the cardiac output (CO). Oxygen in blood is usually estimated from hemoglobin (Hb) concentration (Hb in g/L), the amount of Hb that has oxygen bound to it (percent saturation), and the partial pressure of oxygen (PO2) dissolved in plasma (Figure 1):
Figure 1.
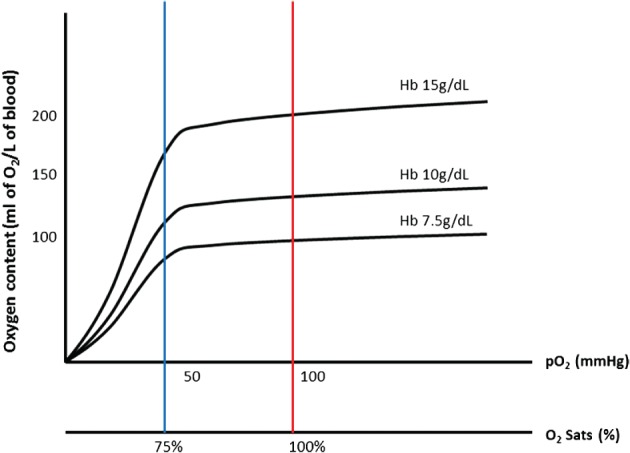
Oxygen content as a function of hemoglobin concentration and oxygenation. Abbreviations: Hb, hemoglobin; PO2, partial pressure of oxygen; sats, saturations.
This is expressed as milliliters of oxygen per liter of blood, where k 1 is the Hüfner constant and k 2 is the solubility coefficient of oxygen, which is 0.003 mL of oxygen dissolved/mm Hg/dL of plasma. Dissolved oxygen contributes little to total oxygen content due to the limited solubility. Hence, DO2 is a function of Hb concentration, oxygenation, and CO:
Oxygen Delivery in Response to Oxygen Consumption
Oxygen delivery is responsive to (patho‐)physiological changes in any of the 3 components of DO2 (Hb, oxygenation, and CO) and changes in VO2. In the case of acute hypoxia or acute anemia, CO increases to maintain normal DO2. However, there is no acute compensatory mechanism for acute reduction in CO. Acute reduction in DO2 due to a drop in CO (eg, AMI) when VO2 is unchanged is “compensated” by greater oxygen extraction (ER), resulting in a drop in mixed venous oxygen saturation (SvO2; Figure 2).
Figure 2.
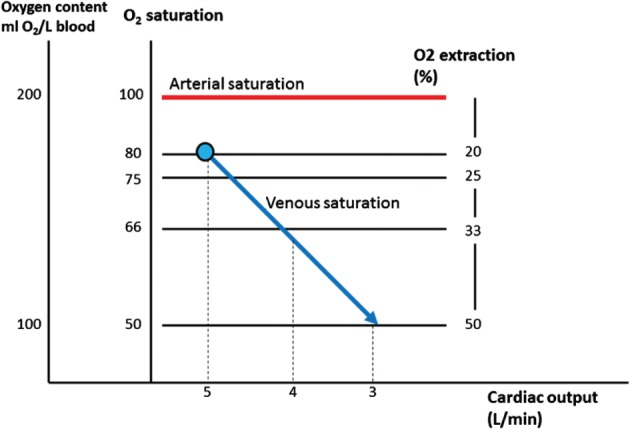
Increase in oxygen extraction with reduction in mixed venous saturation with reduction in cardiac output.
Oxygen delivery is also responsive to the changing metabolic needs and VO2, exemplified by the increase in DO2 when VO2 increases from rest to exercise (mediated almost completely by an increase of CO). The proportionate increase in DO2 maintains the ratio of delivery to consumption (DO2‐VO2 ratio) at approximately 5:1. This DO2‐VO2 ratio is high enough that cellular respiration is not supply‐dependent, and VO2 is predominantly a function of tissue oxygen demand: “consumption drives delivery.” Failure to maintain the DO2‐VO2 ratio is initially compensated by increased oxygen extraction and fall in mixed venous oxygen content. Studies have suggested VO2 becomes supply‐dependent when the DO2‐VO2 ratio falls below 2:1, producing a biphasic DO2‐VO2 relationship (Figure 3).3 This critical level of DO2‐VO2 relationship—so‐called critical DO2—corresponds to the maximal oxygen extraction.
Figure 3.
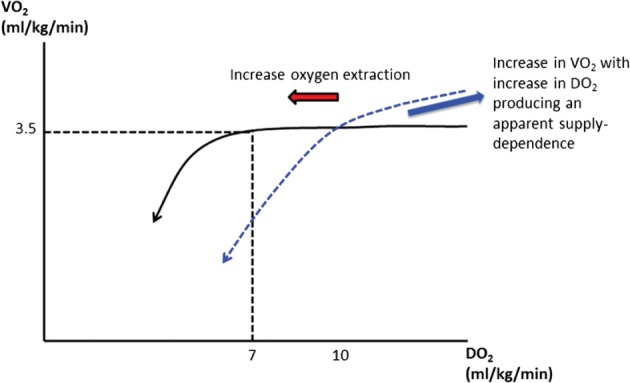
Oxygen consumption becomes supply (DO2)‐dependent when the DO2‐VO2 ratio falls below 2:1. The blue dashed line indicates higher critical DO2 threshold (eg, in splanchnic circulation) and apparent supply‐dependent increase in VO2 as oxygen debt is repaid. Abbreviations: DO2, oxygen delivery; VO2, oxygen consumption.
Below this critical level of DO2, VO2 falls in a near‐linear fashion with ensuing tissue hypoxia. The cells become almost completely reliant on inefficient anaerobic metabolism, generating adenosine triphosphate (ATP) at relatively low rates, and at the unsustainable cost of acidosis induced by unopposed ATP hydrolysis and lactic acid accumulation. “Oxygen debt” occurs because of the excessive production of lactic acid, which, in the absence of oxygen, is a metabolic end product. Restoration of DO2 before irreversible tissue death repays this oxygen debt, resulting in transient increase in VO2 as DO2 is increased (producing an apparent supply‐dependent increase in oxygen uptake), lactic acidosis is cleared, and organ dysfunction is reversed.4 Therefore, intuitively, correction of DO2 should improve survival from cardiogenic shock.
Beyond Global Oxygen Delivery in Cardiogenic Shock
Mortality in cardiogenic shock has often been attributed to the downward spiral associated with progressive pump failure and loss of CO and DO2. As such, the management of cardiogenic shock has centered on the correction and optimization of CO and DO2. However, although reduction in CO and DO2 are defining hemodynamic features of cardiogenic shock, a significant proportion of patients perish despite improvement and even normalized CO and DO2. In addition, many of these patients have succumbed to a state of low systemic vascular resistance analogous to septic shock, despite improvement in DO2.5 The latter suggests wider circulatory and cellular abnormalities with progression of cardiogenic shock.
Regional Blood Flow and Oxygen Consumption
Under normal physiological conditions, metabolic demand alters local arteriolar tone to direct the necessary increase in regional blood flow to the appropriate tissues: “demand drives supply.” The arteriolar smooth muscles actively contract in response to a range of systemic autonomic nervous, humoral, and local mediators, through a number of receptor types that are distributed with great organ‐to‐organ variability. Arteriolar smooth‐muscle relaxation is mediated principally by the synthesis and release of nitric oxide (NO) by the vascular endothelium, which, via soluble guanylate cyclase and generation of cyclic adenosine monophosphate, effects the inhibition of cross‐bridge cycling in the smooth muscle and relaxation.6 The arteriolar smooth‐muscle activity in critical organs with high metabolic activity may be subjected to additional controls, based on oxygen availability within the organ.
Many of the (auto‐)regulatory mechanisms may be lost with the development of the systemic inflammatory response syndrome (SIRS), due to inflammatory mediator–induced abnormalities of the NO system. The increased expression of inducible nitric oxide synthase (iNOS) and excess NO production induces vasodilation. The variable expression of iNOS in different organs and different vascular beds within an organ results in heterogeneous flow with abnormally high blood flow and pathological shunting in some vascular beds with high iNOS activity (if coupled with impaired oxygen utilization) with diminished response to catecholamines, and underperfusion of areas lacking iNOS. The abnormal distribution of blood flow and shunting of blood extends into the microcirculation due to endothelial damage/dysfunction and allows leukocyte adhesion and formation of microthrombi. The consequent “consumption‐perfusion mismatch” and “shunting” of blood across metabolically nonactive (or relatively less active) tissues may result in regional tissue hypoxia despite normal (or even supranormal) DO2. Hence, SvO2 may be normal/elevated in the presence of regional tissue hypoxia, and measures of global DO2 may not reflect local tissue oxygen levels during critical illness and SIRS. Indeed, hypoxia in specific organs is often the result of disordered regional distribution of blood flow both between and within the organs, and not due to inadequacy of global DO2‐VO2 matching.
Cardiogenic shock may be complicated by the development of SIRS,7 particularly with increased duration of shock (in the absence of infection).8 The level of pro‐inflammatory cytokine interleukin‐6 is elevated in cardiogenic shock and may achieve levels comparable with septic shock.9 The excessive NO production results in vasodilation that contributes to the adverse hemodynamics in cardiogenic shock, as evidenced by the increase in blood pressure with NG‐monomethyl‐L‐arginine in patients with persistent shock despite vasopressors.10 This increase in blood pressure with NG‐monomethyl‐L‐arginine was not associated with reduction mortality in a randomized trial,11 possibly due to the nonspecific inhibition of NO production or adverse effects on ventriculoarterial coupling in patients with severe cardiac dysfunction.12
This regional difference or mismatch in blood flow is compounded by regional variation in the threshold of critical oxygen delivery, with the development of SIRS.13, 14 In the critically ill patient, some parts of the body may become ischemic at higher levels of DO2, such as the gut mucosa.15 The reduction in splanchnic perfusion by endogenous and the use of exogenous vasoconstrictors, coupled with an increase in the critical DO2 threshold with SIRS,16 reduces the tolerance for reduced DO2 and increases the susceptibility to splanchnic ischemia. The failure to maintain enteral nutrition also compromises the integrity of the gut mucosa. Splanchnic ischemia and loss of gut mucosal integrity allow translocation of endotoxin and bacteria into the portal circulation, overwhelming hepatic clearance and exacerbating widespread endothelial damage and inflammatory response. Treatment aimed at maintaining or improving splanchnic perfusion may reduce the incidence of multiple organ failure and mortality,17 although this has not been examined specifically in patients with cardiogenic shock.
Microcirculatory Blood Flow
The regional regulation of blood flow extends to the microcirculation, including capillaries where oxygen diffusion to the tissues takes place. Hemorheology and capillary patency are the main determinants of capillary blood flow. Hemoglobin in the red blood cell may function as an oxygen sensor during hypoxia, enabling the red blood cells to regulate blood flow by releasing the vasodilators NO and ATP.18 The vascular endothelium, by modulating local coagulation/fibrinolysis and leukocyte migration through the release of biologically active factors such as NO, prostacyclin, adenosine, and endothelin, influences microcirculatory blood flow.19 Endothelial damage and exposure to the subendothelium facilitate leukocyte and platelet aggregation that results in microthrombosis and local tissue hypoxia.
Of note, it is red blood cell flow specifically, and not blood in general, that determines oxygen delivery, because oxygen has a low solubility in plasma. Red blood cells facilitated by the oxygen‐carrying capacity of Hb are largely responsible for the convective transport of oxygen by the blood. In the microcirculation, capillary hemodynamics can be quantified as red blood cell flux (expressed as red blood cells per second), also termed red blood cell supply rate.20 Red blood cell flux accounts for both red blood cell velocity (V, in mm/s) and capillary hematocrit or red blood cell lineal density (LD, as red blood cell/mm):
Oxygen flow in capillaries (qO2) can then be calculated from the red blood cell flux, the red blood cell saturation, and the oxygen‐carrying capacity of a single red blood cell (K = 0.0362 mL oxygen/red blood cell at 100% oxygen saturation),21 analogous to the global oxygen delivery equation:
Also analogous to the global DO2 parameters, capillary oxygen extraction ratio (ERc) can be calculated from difference in capillary oxygen flow rates at the arterial, or inflow into the capillary (qO2in) and venous, or capillary outflow (qO2out):
The alteration in microvascular geometry, capillary hemodynamics, functional capillary density, and microvascular oxygen transport in SIRS produces microvascular blood flow heterogeneity and consequent oxygen flow heterogeneity. Capillary oxygen extraction will be greater in microvascular beds with lower qO2, with ensuing hypoxia when the ERc is exhausted, which may contribute to tissue/organ injury and failure in critically ill patients.
A number of studies have documented microvascular abnormalities in patients with cardiogenic shock. First, forearm blood flow using venous plethysmography at rest and reactive hyperemia were diminished in a subgroup of patients with cardiogenic shock, indicating increased vasoconstriction and an impaired capacity for vasodilation.22 Second, the proportion of perfused small vessels (<20 µm) and perfused capillary density were lower in patients with cardiogenic shock,23 and the lower proportion of perfused vessels was associated with poorer survival. Third, diminished sublingual perfused capillary density at baseline or following treatment was associated with development of multiorgan failure and poor outcomes in patients with AMI complicated by cardiogenic shock.24 Improvement in microcirculatory abnormalities in patients with acute heart failure has been described with low‐dose nitroglycerin,25 but this has yet to be validated in clinical trials. It is not clear if these microcirculatory abnormalities can be limited by earlier correction of DO2.
Microcirculatory Oxygen Diffusion
Oxygen diffuses over relatively short distances from arterioles and capillaries in all directions based on the local PO2 gradient. Oxygen diffusion is limited by oxygen solubility (k), oxygen diffusivity (D), and the PO2 gradient (dPO2/dr). The PO2 gradient drives the net movement of oxygen from a region of high PO2 to a region of low PO2. Oxygen flux can be described by Fick's first law of diffusion:
The oxygen diffusion distance is the distance from the Hb in red blood cells to the mitochondria (Figure 4). The critical oxygen diffusion distance, which is the maximum distance that mitochondria can be away from an oxygen source without impaired function, is determined by these oxygen diffusion parameters and by capillary PO2 and tissue oxygen consumption. The intercapillary distance may be increased under hypoxic conditions, particularly “hypoxic” hypoxia (low arterial PO2) and tissue edema (increased vascular permeability or excessive fluid administration; Figure 4). Hence, in the microcirculation, it may be PO2 rather than DO2 (ie, diffusion rather than convection) that is vital for local tissue oxygenation. Avoiding tissue edema by avoiding excessive fluid loading and early treatment of congestion and arterial hypoxemia may improve tissue/cellular oxygenation.
Figure 4.
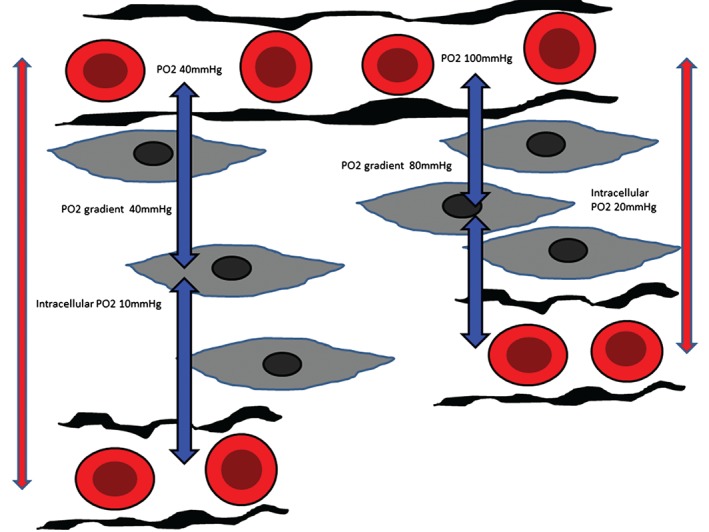
Oxygen diffusion in the microcirculation. Blue vertical arrows indicate the oxygen diffusion distance. Typical diffusion distances may range from 10 to hundreds of microns. Red vertical arrows indicate intercapillary distance. Abbreviations: PO2, partial pressure of oxygen.
Cellular Oxygen Utilization
Eukaryotic cells are generally dependent on aerobic metabolism, as mitochondrial respiration offers greater efficiency for extraction of energy from glucose than anaerobic glycolysis. Molecular oxygen readily diffuses from Hb in the capillary into the cell and the mitochondrion. In the mitochondria, molecular oxygen is the terminal electron acceptor in the electron transport chain, which is a series of protein complexes, residing in or near the inner mitochondrial membrane. The flow of electrons down this chain results in conformational changes in the protein complexes and the translocation of protons from the mitochondrial matrix to the inter membrane space. The resultant difference in both charge and proton concentration in the 2 compartments is the proton motive force that converts an intermittent supply of fuel into a constant supply of ATP.26
Inadequate perfusion from limitation in DO2 and consequent hypoxia has long been the prevailing pathophysiological model behind multiorgan dysfunction syndrome (MODS).27 Indeed, optimization of DO2 in the early period of shock when the cellular energetic machinery is still functional may ameliorate the impending cellular energetic failure and reduce the incidence/severity of organ dysfunction. However, progression of the shocked state is characterized by the failure to utilize oxygen to produce ATP due to mitochondrial failure, resulting in cytopathic hypoxia. The cells, in the face of inadequate energy due to mitochondrial dysfunction, decrease metabolic activity, which manifests clinically as multiorgan dysfunction (including sepsis‐related cardiomyopathy).28 In this regard, organ dysfunction may be an adaptive response to severe inflammatory response. Hence, impaired DO2 and the development of SIRS may instigate mitochondrial dysfunction. The latter, and not DO2, becomes the dominant pathophysiology in MODS,29 with the corollary that a strategy of increasing DO2 (even to supranormal levels) will be of little benefit.
Mitochondrial dysfunction has not been well studied in patients with cardiogenic shock. However, the evolution of the dominant pathophysiological processes corresponds well with clinical observation that shock responds better to hemodynamic resuscitation in the early stages30 and suggests that avoiding cellular dysoxia may limit the development of the syndrome of multiorgan dysfunction. The efficacy of specific intervention targeting cellular dysoxia and mitochondrial dysfunction is also likely to be dependent on the evolving pathophysiology.
Therapeutic Implications
The progression of cardiogenic shock is accompanied by the development of SIRS, vasodilation, regional circulatory abnormalities, microcirculatory abnormalities, and cellular dysoxia, a clinical phenotype that resembles septic shock (Figure 5). At these latter stages, efforts to improve blood flow to regionally hypoxic tissues by increasing DO2 (even to supranormal levels) are inefficient and even harmful.31 In addition, studies on the treatment of septic shock with the so‐called goal‐directed therapy, which is predicated on the optimization of DO2, produced little/no significant benefit32 (see Supporting Information, Table, in the online version of this article), despite encouraging results in earlier studies,33 further highlighting the limitations of medical manipulation of DO2 in the advanced stages of shock.
Figure 5.
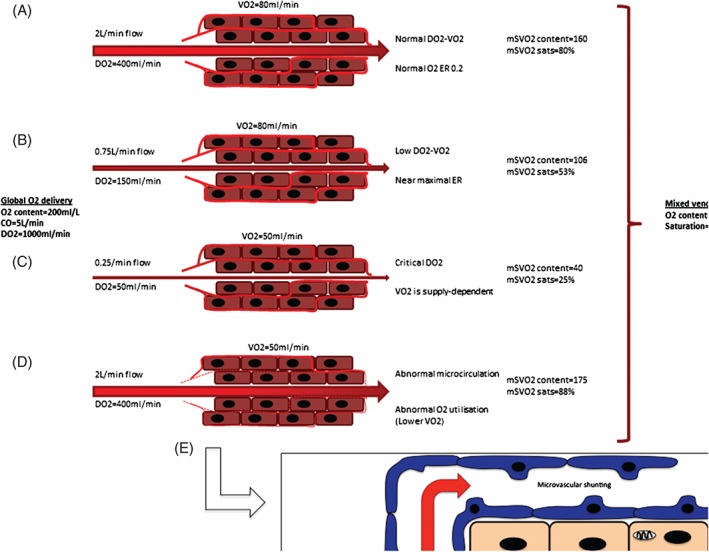
Global oxygen delivery is distributed variably to a number of vascular beds. (A) A normal oxygen delivery‐consumption (DO2‐VO2) relationship results in normal ER and normal mSVO2. (B) Reduction in blood flow (eg, vasoconstriction) reduces the DO2‐VO2 relationship, which is compensated by increased ER and reduced mSVO2. (C) Oxygen consumption becomes supply‐dependent when blood flow and DO2 drop below the critical oxygen delivery threshold. (D, E) Cellular oxygen uptake is poor even with normal blood flow in the presence of microcirculatory abnormalities and cellular cytopathic dysoxia, resulting in high mSVO2. The overall mSVO2 may remain normal despite regions of critical hypoxia. Abbreviations: CO, cardiac output; DO2, oxygen delivery; ER, oxygen extraction ratio; mSVO2, mixed venous oxygen saturation; VO2, oxygen consumption.
This pathophysiological evolution of cardiogenic shock has clinical implications:
Inotropes are routinely used to improve CO in cardiogenic shock, but at the expense of tachyarrhythmias, increased myocardial VO2, and adverse effects on regional blood flow.34 It is possible that earlier and effective mechanical circulatory support to improve DO2, while avoiding the potential toxicity associated with high‐dose catecholamines, may improve outcomes in cardiogenic shock. Acute mechanical circulatory support, including percutaneous microaxial pumps and extracorporeal life support (venoarterial membrane oxygenation), or surgical cannulation of the heart with extracorporeal ventricular assist devices, increases total blood flow and DO2 35 and has been successfully used to bridge patients with cardiogenic shock to recovery, transplantation, or an implantable left ventricular assist device.36
Impaired DO2 and tissue hypoxia is a well‐described (anaerobic) cause of high lactate levels. However, microcirculatory abnormalities may also result in anaerobic elevation in lactate levels, independent of global hemodynamic changes.37 In addition, lactate levels may be elevated in the absence of impaired global DO2 from epinephrine use or failure of pyruvate metabolism due to mitochondrial dysfunction. Therefore, lactate levels become unreliable for tissue hypoxia in critically ill patients with SIRS.38 Persistently elevated lactate may indicate microcirculatory abnormalities or cytopathic hypoxia, which may not be corrected (and even may be exacerbated) by escalation of vasopressors or inotropes and is associated with poor outcomes.39
Central or mixed venous blood oxygen saturations are widely used to guide treatment of critically ill patients. However, mixed venous blood represents the final admixture of venous blood from multiple vascular beds and may conceal regional changes in blood flow, microcirculatory abnormalities, or cytopathic hypoxia (Figure 5). Measurements of SvO2 complement blood lactate levels in guiding the management of critically ill patients; concomitant measurement of both may be more useful than the exclusive use of one or the other.
Therapeutic interventions aimed at the microcirculation and manipulation of cellular oxygen utilization (eg, the regulated induction of a hypometabolic state resembling hibernation may help the energy‐starved cell, whereas the stimulation of mitochondrial activity during the latter phases of shock may improve recovery)28 may be of benefit in advanced stages of shock.
Conclusion
Cardiogenic shock is characterized by a global reduction in DO2 due to reduction in CO, but the pathophysiology evolves with progression of shock into a hemodynamic phenotype more analogous to septic shock. Early and effective restoration of DO2 and limiting the toxicity associated with inotropes and catecholamines with mechanical circulatory support to abrogate this evolution of the shock phenotype deserves further study. Regional DO2 may be improved by exploiting the differences in receptor population and density between different vascular beds to redirect blood flow from relatively overperfused tissues to underperfused “vital” organs. Pharmacological treatment aimed at the microcirculation and manipulation of cellular oxygen utilization may also prove fruitful in the treatment of advanced cardiogenic shock.
Supporting information
Supporting TABLE 1: Multi‐centre randomised trials of early goal‐directed therapy
The authors have no funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Hochman JS, Sleeper LA, Webb JG et al; SHOCK Investigators. Early revascularization in acute myocardial infarction complicated by cardiogenic shock . Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. N Engl J Med. 1999;341:625–634. [DOI] [PubMed] [Google Scholar]
- 2. Thiele H, Zeymer U, Neumann FJ, et al; IABP‐SHOCK II Trial Investigators . Intra‐aortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med. 2012;367:1287–1296. [DOI] [PubMed] [Google Scholar]
- 3. Cain SM, Curtis SE. Experimental models of pathologic oxygen supply dependency. Crit Care Med. 1991;19:603–612. [DOI] [PubMed] [Google Scholar]
- 4. Fahey JT, Lister G. Oxygen transport in low cardiac output states. J Crit Care. 1987;2:288–305. [Google Scholar]
- 5. Lim N, Dubois MJ, De Backer D, et al. Do all nonsurvivors of cardiogenic shock die with a low cardiac index? Chest. 2003;124:1885–1891. [DOI] [PubMed] [Google Scholar]
- 6. Ignarro LJ. Biological actions and properties of endothelium‐derived nitric oxide formed and released from artery and vein. Circ Res. 1989;65:1–21. [DOI] [PubMed] [Google Scholar]
- 7. Kohsaka S, Menon V, Lowe AM, et al. Systemic inflammatory response syndrome after acute myocardial infarction complicated by cardiogenic shock. Arch Intern Med. 2005;165:1643–1650. [DOI] [PubMed] [Google Scholar]
- 8. Brunkhorst FM, Clark AL, Forycki ZF, et al. Pyrexia, procalcitonin, immune activation and survival in cardiogenic shock: the potential importance of bacterial translocation. Int J Cardiol. 1999;72:3–10. [DOI] [PubMed] [Google Scholar]
- 9. Geppert A, Steiner A, Zorn G, et al. Multiple organ failure in patients with cardiogenic shock is associated with high plasma levels of interleukin‐6. Crit Care Med. 2002;30:1987–1994. [DOI] [PubMed] [Google Scholar]
- 10. Dzavík V, Cotter G, Reynolds HR, et al; SHOCK‐2 Investigators . Effect of nitric oxide synthase inhibition on haemodynamics and outcome of patients with persistent cardiogenic shock complicating acute myocardial infarction: a phase II dose‐ranging study. Eur Heart J. 2007;28:1109–1116. [DOI] [PubMed] [Google Scholar]
- 11. Alexander JH, Reynolds HR, Stebbins AL, et al; TRIUMPH Investigators . Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: the TRIUMPH randomized controlled trial. JAMA. 2007;297:1657–1666. [DOI] [PubMed] [Google Scholar]
- 12. Nordhaug D, Steensrud T, Aghajani E, et al. Nitric oxide synthase inhibition impairs myocardial efficiency and ventriculo‐arterial matching in acute ischemic heart failure. Eur J Heart Fail. 2004;6:705–713. [DOI] [PubMed] [Google Scholar]
- 13. Schlichtig R , Klions HA, Kramer DJ, et al. Hepatic dysoxia commences during O2 supply dependence. J Appl Physiol ( 1985. ) 1992;72:1499–1505. [DOI] [PubMed] [Google Scholar]
- 14. Pinsky MR, Matuschak GM. Cardiovascular determinants of the hemodynamic response to acute endotoxemia in the dog. J Crit Care. 1986;1:18–31. [Google Scholar]
- 15. Fink MP, Kaups KL, Wang H, et al. Maintenance of superior mesenteric arterial perfusion prevents increased intestinal mucosal permeability in endotoxic pigs. Surgery. 1991;110:154–161. [PubMed] [Google Scholar]
- 16. Nelson DP, Samsel RW, Wood LD, et al. Pathological supply dependence of systemic and intestinal O2 uptake during endotoxaemia. J Appl Physiol (1985). 1988;64:2410–2419. [DOI] [PubMed] [Google Scholar]
- 17. Gutierrez G, Palizas F, Doglio G, et al. Gastric intramucosal pH as a therapeutic index of tissue oxygenation in critically ill patients. Lancet. 1992;339:195–199. [DOI] [PubMed] [Google Scholar]
- 18. Jia L, Bonaventura C, Bonaventura J, et al. S‐nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. [DOI] [PubMed] [Google Scholar]
- 19. Karimova A, Pinsky DJ. The endothelial response to oxygen deprivation: biology and clinical implications. Intensive Care Med. 2001;27:19–31. [DOI] [PubMed] [Google Scholar]
- 20. Nakajima Y, Baudry N, Duranteau J, et al. Microcirculation in intestinal villi: a comparison between hemorrhagic and endotoxin shock. Am J Respir Crit Care Med. 2001;164(8 part 1):1526–1530. [DOI] [PubMed] [Google Scholar]
- 21. Ellis CG, Bateman RM, Sharpe MD, et al. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002;282:H156–H164. [DOI] [PubMed] [Google Scholar]
- 22. Kirschenbaum LA, Astiz ME, Rackow EC, et al. Microvascular response in patients with cardiogenic shock. Crit Care Med. 2000;28:1290–1294. [DOI] [PubMed] [Google Scholar]
- 23. De Backer D, Creteur J, Dubois MJ, et al. Microvascular alterations in patients with acute severe heart failure and cardiogenic shock. Am Heart J. 2004;147:91–99. [DOI] [PubMed] [Google Scholar]
- 24. den Uil CA, Lagrand WK, van der Ent M, et al. Impaired microcirculation predicts poor outcome of patients with acute myocardial infarction complicated by cardiogenic shock. Eur Heart J. 2010;31:3032–3039. [DOI] [PubMed] [Google Scholar]
- 25. den Uil CA, Lagrand WK, Spronk PE, et al. Low‐dose nitroglycerin improves microcirculation in hospitalized patients with acute heart failure. Eur J Heart Fail. 2009;11:386–390. [DOI] [PubMed] [Google Scholar]
- 26. Rich P. Chemiosmotic coupling: the cost of living. Nature. 2003;421:583. [DOI] [PubMed] [Google Scholar]
- 27. Broder G, Weil MH. Excess lactate: an index of reversibility of shock in human patients. Science. 1964;143:1457–1459. [DOI] [PubMed] [Google Scholar]
- 28. Singer M, De Santis V, Vitale D, et al. Multiorgan failure is an adaptive, endocrine‐mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364:545–548. [DOI] [PubMed] [Google Scholar]
- 29. Fink MP. Cytopathic hypoxia: mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219–237. [DOI] [PubMed] [Google Scholar]
- 30. Gu WJ, Wang F, Bakker J, et al. The effect of goal‐directed therapy on mortality in patients with sepsis—earlier is better: a meta‐analysis of randomized controlled trials. Crit Care. 2014;18:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hayes MA, Timmins AC, Yau EH, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–1722. [DOI] [PubMed] [Google Scholar]
- 32. Angus DC, Barnato AE, Bell D, et al. A systematic review and meta‐analysis of early goal‐directed therapy for septic shock: the ARISE, ProCESS and ProMISe Investigators. Intensive Care Med. 2015;41:1549–1560. [DOI] [PubMed] [Google Scholar]
- 33. Rivers E, Nguyen B, Havstad S, et al. Early goal directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. [DOI] [PubMed] [Google Scholar]
- 34. Meier‐Hellmann A, Reinhart K, Bredle DL, et al. Epinephrine impairs splanchnic perfusion in septic shock. Crit Care Med. 1997;25:399–404. [DOI] [PubMed] [Google Scholar]
- 35. Werdan K, Gielen S, Ebelt H, et al. Mechanical circulatory support in cardiogenic shock. Eur Heart J. 2014;35:156–167. [DOI] [PubMed] [Google Scholar]
- 36. Tayara W, Starling RC, Yamani MH, et al. Improved survival after acute myocardial infarction complicated by cardiogenic shock with circulatory support and transplantation: comparing aggressive intervention with conservative treatment. J Heart Lung Transplant. 2006;25:504–509. [DOI] [PubMed] [Google Scholar]
- 37. De Backer D, Creteur J, Dubois MJ, et al. The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med. 2006;34:403–408. [DOI] [PubMed] [Google Scholar]
- 38. James JH, Luchette FA, McCarter FD, et al. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999;354:505–508. [DOI] [PubMed] [Google Scholar]
- 39. Vincent JL, Dufaye P, Berré J, et al. Serial lactate determinations during circulatory shock. Crit Care Med. 1983;11:449–451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting TABLE 1: Multi‐centre randomised trials of early goal‐directed therapy