

Abstract
Background
Since the 1950s antipsychotic medication has been extensively used to treat people with chronic mental illnesses such as schizophrenia. These drugs, however, have also been associated with a wide range of adverse effects, including movement disorders such as tardive dyskinesia (TD) – a problem often seen as repetitive involuntary movements around the mouth and face. Various strategies have been examined to reduce a person's cumulative exposure to antipsychotics. These strategies include dose reduction, intermittent dosing strategies such as drug holidays, and antipsychotic cessation.
Objectives
To determine whether a reduction or cessation of antipsychotic drugs is associated with a reduction in TD for people with schizophrenia (or other chronic mental illnesses) who have existing TD. Our secondary objective was to determine whether the use of specific antipsychotics for similar groups of people could be a treatment for TD that was already established.
Search methods
We updated previous searches of Cochrane Schizophrenia's study‐based Register of Trials including the registers of clinical trials (16 July 2015 and 26 April 2017). We searched references of all identified studies for further trial citations. We also contacted authors of trials for additional information.
Selection criteria
We included reports if they assessed people with schizophrenia or other chronic mental illnesses who had established antipsychotic‐induced TD, and had been randomly allocated to (a) antipsychotic maintenance versus antipsychotic cessation (placebo or no intervention), (b) antipsychotic maintenance versus antipsychotic reduction (including intermittent strategies), (c) specific antipsychotics for the treatment of TD versus placebo or no intervention, and (d) specific antipsychotics versus other antipsychotics or versus any other drugs for the treatment of TD.
Data collection and analysis
We independently extracted data from these trials and estimated risk ratios (RR) or mean differences (MD), with 95% confidence intervals (CI). We assumed that people who dropped out had no improvement.
Main results
We included 13 RCTs with 711 participants; eight of these studies were newly included in this 2017 update. One trial is ongoing.
There was low‐quality evidence of a clear difference on no clinically important improvement in TD favouring switch to risperidone compared with antipsychotic cessation (with placebo) (1 RCT, 42 people, RR 0.45 CI 0.23 to 0.89, low‐quality evidence). Because evidence was of very low quality for antipsychotic dose reduction versus antipsychotic maintenance (2 RCTs, 17 people, RR 0.42 95% CI 0.17 to 1.04, very low‐quality evidence), and for switch to a new antipsychotic versus switch to another new antipsychotic (5 comparisons, 5 RCTs, 140 people, no meta‐analysis, effects for all comparisons equivocal), we are uncertain about these effects. There was low‐quality evidence of a significant difference on extrapyramidal symptoms: use of antiparkinsonism medication favouring switch to quetiapine compared with switch to haloperidol (1 RCT, 45 people, RR 0.45 CI 0.21 to 0.96, low‐quality evidence). There was no evidence of a difference for switch to risperidone or haloperidol compared with antipsychotic cessation (with placebo) (RR 1 RCT, 48 people, RR 2.08 95% CI 0.74 to 5.86, low‐quality evidence) and switch to risperidone compared with switch to haloperidol (RR 1 RCT, 37 people, RR 0.68 95% CI 0.34 to 1.35, very low‐quality evidence).
Trials also reported on secondary outcomes such as other TD symptom outcomes, other adverse events outcomes, mental state, and leaving the study early, but the quality of the evidence for all these outcomes was very low due mainly to small sample sizes, very wide 95% CIs, and risk of bias. No trials reported on social confidence, social inclusion, social networks, or personalised quality of life, outcomes that we designated as being important to patients.
Authors' conclusions
Limited data from small studies using antipsychotic reduction or specific antipsychotic drugs as treatments for TD did not provide any convincing evidence of the value of these approaches. There is a need for larger trials of a longer duration to fully investigate this area.
Plain language summary
Antipsychotic‐reduction and/or cessation and antipsychotics as specific treatments for tardive dyskinesia
Review question
To determine whether stopping or reducing antipsychotic drugs helps in the reduction of tardive dyskinesia for people with schizophrenia. To examine whether specific antipsychotic drugs could be a treatment for tardive dyskinesia.
Background
People with schizophrenia often hear voices and see things (hallucinations) and have strange beliefs (delusions). The main treatment for schizophrenia is antipsychotic drugs. However, these drugs can have debilitating side effects. Tardive dyskinesia is an involuntary movement that causes the face, mouth, tongue, and jaw to convulse, spasm, and grimace. It is caused by long‐term use or high doses of antipsychotic drugs, is difficult to treat, and can be incurable. Various strategies have been proposed to reduce a person’s exposure to antipsychotic drugs. These include lowering the dose of medication, intermittent ‘drug holidays’, and stopping taking antipsychotic medication altogether.
Study characteristics
The review includes 13 trials with a total of 711 people with schizophrenia and other psychiatric diagnoses.
Key results
Due to the poor quality, small size, and limited data from the 13 studies, there is limited evidence. It is not known if strategies such as dose reduction, ‘drug holidays’, and stopping medication are helpful in the treatment of tardive dyskinesia. There is limited evidence on specific antipsychotic drugs in the treatment of tardive dyskinesia.
Quality of the evidence
Evidence is poor, small scale, and of short duration. There is a need for larger trials of a longer duration in order to fully investigate this area.
This plain language summary was adapted by the review authors from a summary originally written by Ben Gray, Senior Peer Researcher, McPin Foundation (http://mcpin.org/).
Summary of findings
Background
Description of the condition
Since the 1950s antipsychotic (neuroleptic) medication has been extensively used to treat people with chronic mental illnesses such as schizophrenia. These drugs can effectively control symptoms such as abnormal perceptions (hallucinations), disordered thinking, and fixed false beliefs (delusions). In addition, maintenance therapy with antipsychotic medication is associated with reduced risk of relapse (Schooler 1993). Antipsychotic medication, however, has also been associated with a wide range of adverse effects, including movement disorders. The appearance of these disorders can be extremely disfiguring, compounds stigma, and is associated with poor compliance to antipsychotic treatment (Barnes 1993; Tarsy 2011).
Tardive dyskinesia (TD) is one such iatrogenic movement disorder and it is characterised by abnormal repetitive and involuntary movements. The clinical features include: tongue protrusion, side‐to‐side or rotatory movement of the jaw, lip smacking, puckering and pursing, and rapid eye blinking (Casey 1999). In some people rapid movements of the arms, legs, and trunk may also occur. TD is a chronic condition of insidious onset, the severity of which fluctuates spontaneously (APA 1992). Although the most frequent cause of TD is the use of antipsychotic medication, it is striking that dose reduction can lead to a temporary exacerbation in symptoms. Conversely, increasing the dose is often associated with a temporary remission (Cavallaro 1993; Smith 1980).
The exact mechanisms of the pathophysiology of TD are unknown. Antipsychotic drugs block certain chemical receptor sites in the brain ‐ one of these is specific for dopamine (Casey 1994). One hypothesis explaining the cause of antipsychotic‐induced TD is that chronic blockade of dopamine receptors in specific cells of the brain (neurones from the nigrostriatum) causes an overgrowth of these receptors (Casey 1994). However, there is some suggestion that the chronic use of antipsychotics may also cause an abnormal production of highly active atoms and chemical groups (cytotoxic free radicals), which may damage specific cells in the brain. This, in turn, could be responsible for the appearance of TD (Cadet 1989). This theory is supported by the persistent nature of the syndrome, once established.
Studies on the natural history of TD have reported widely variable remission rates (1% to 62%) depending on patient age, psychiatric diagnosis, course of the psychiatric disorder, and duration of therapy (Bergen 1989; Fernandez 2001; Glazer 1990). TD occurs in more than 20% of people that use first‐generation antipsychotic drugs (FGAs) continually for longer than three months (Tarsy 2011). Every year 4% to 5% of adults and 25% to 30% of elderly people who continually use these drugs begin to show signs of TD (APA 1992; Correll 2004). Advancing age is a risk factor for both TD's prevalence and severity, with those who are under 60 years of age being three times more likely to spontaneously remit (Smith 1980).
When the second‐generation antipsychotic drugs were introduced in the 1990s many hoped that they would not cause TD (Miller 2007; Rosenheck 2007). Although the risk of developing TD with second‐generation antipsychotic drugs does seem to be reduced, TD risks have not been eliminated (Miller 2007; Tarsy 2011). There is even some evidence to indicate that rates of TD do not differ at all between first‐ and second‐generation anti‐psychotic drugs (Leucht 2009; Rosenheck 2007; Woods 2010). The large, definitive, US randomised trial of antipsychotic treatments for schizophrenia (CATIE), with a four‐year period of follow‐up, obtained an incidence rate of TD of around 17% (Miller 2008). Due to widespread use of second‐generation antipsychotic drugs, increased off‐label use, and an ageing population, the frequency of TD is likely to be higher than thought (Cloud 2014; Maher 2012) and increasing. The problem will be considerably greater for people in countries where use of newer drugs is less prevalent (Ballesteros 2000; Martins 2011).
Description of the intervention
Various strategies have been examined in order to reduce a person's cumulative exposure to antipsychotics. These strategies include dose reduction, intermittent dosing strategies, such as drug holidays, and antipsychotic cessation. The prevention and treatment of TD provided much of the impetus for these studies. While antipsychotic reduction or cessation, or both would seem to be a logical first step in the management of antipsychotic‐induced TD, this is not always possible in the clinical setting because of the overriding need to manage current psychotic symptoms or to reduce the risk of relapse, or both. In this review we undertook a comprehensive study of the impact of antipsychotic dose reduction and/or cessation strategies for those who were already presenting with TD.
In the search for ways to manage antipsychotic‐induced TD, certain antipsychotic medications have themselves been proposed as specific treatments for the condition. The usual rationale for such trials relates to variations in receptor‐blocking profile that distinguishes the compound of interest from antipsychotics in general.
How the intervention might work
Although the pathophysiology of TD is not entirely clear, one of the possible underlying mechanisms is believed to be hypersensitivity of postsynaptic dopamine receptors (Margolese 2005). The risk appears to increase with higher cumulative exposure to antipsychotics, especially those with stronger D2 dopamine receptor blockade. Newer antipsychotic medications, which cause less dopamine D2 blockade, have been shown to cause less TD (Correll 2004). The primary intervention of interest (reduction/cessation of antipsychotics), is likely to help by reducing the cumulative exposure to antipsychotics. The other intervention (specific antipsychotics) is likely to work as a result of the reduction in the levels of dopamine D2 receptor blockade, a characteristic property of many of the newer antipsychotic medications.
Why it is important to do this review
TD can result in considerable social and physical disability (Barnes 1993) and symptoms are often irreversible (Bergen 1989; Fernandez 2001; Gerlach 1988; Glazer 1990). Additionally, TD is frequently associated with lower quality of life (Ascher‐Svanum 2008) and a greater mortality rate (Chong 2009). Several antipsychotic medications have been produced in recent decades that claim to cause less or no TD (Lieberman 1996). These claims may or may not be true, and certainly evidence does suggest that thoughtful use of older generation drugs is not associated with more TD than newer treatments (Chouinard 2008). However, in a global context, it is likely that the less expensive and more familiar drugs ‐ such as chlorpromazine or haloperidol ‐ will continue to be the mainstay of treatment of people with schizophrenia (WHO Essential List 2010). Use of drugs such as these is associated with emergence of TD and, therefore, this condition will remain a problem for years to come.
Given the high incidence and prevalence of TD among people taking antipsychotic medication, the need for prevention or treatment is clear. Unfortunately, there has been sparse evidence to guide clinicians (NICE 2014; Taylor 2009). Although many treatments have been tested, no one intervention has been shown clearly to be effective.
This review is one in a series of Cochrane Reviews (see Table 12) evaluating treatments for antipsychotic‐induced TD, and is an update of a Cochrane Review first published in 1998 (McGrath 1998), and previously updated in 2000 (McGrath 2000) and in 2006 (Soares‐Weiser 2006).
1. Other Cochrane Reviews in this series.
| Interventions | Current reference (updates underway) |
| Anticholinergic medication | Soares‐Weiser 1997; Soares 2000 |
| Benzodiazepines | Bhoopathi 2006 |
| Calcium channel blockers | Essali 2011 |
| Cholinergic medication | Tammenmaa 2002 |
| Gamma‐aminobutyric acid agonists | Alabed 2011 |
| Miscellaneous treatments | Soares‐Weiser 2003 |
| Neuroleptic reduction and/or cessation and neuroleptics | This review |
| Non‐neuroleptic catecholaminergic drugs | El‐Sayeh 2006 |
| Vitamin E | Soares‐Weiser 2011 |
Objectives
To determine whether a reduction or cessation of antipsychotic drugs is associated with a reduction in TD for people with schizophrenia (or other chronic mental illnesses) who have existing TD. Our secondary objective was to determine whether the use of specific antipsychotics for similar groups of people could be a treatment for already established TD.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. Had there been trials that were described as double‐blind, but that did not mention whether the study was randomised, we would have included them in a sensitivity analysis. If there had been no substantive difference within primary outcomes (see 'Types of outcome measures') when these studies were added, then we would have included them in the final analysis. If there had been a substantive difference, we would have used only clearly randomised trials and described the results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people with schizophrenia or any other serious mental illness, diagnosed by any criteria, irrespective of gender, age, or nationality who required the use of antipsychotics for more than three months and who developed TD (diagnosed by any criteria) during antipsychotic treatment, and for whom the dose of antipsychotic medication had been stable for one month or more. We made a post hoc change to include studies that did not require antipsychotic medication to have been stable for one month prior to randomisation. We felt it important to include these studies as they provide additional important information. However, we planned to analyse these separately, so that they would not change existing outcome data.
Types of interventions
Reduction or cessation of the dose of antipsychotic drugs compared with the continuation of standard dose of the same compound. For the purposes of this review, we divided these trials into those that aimed to reduce the total dosage of antipsychotic medication, for example reduced‐dose and intermittent‐dosage schedule studies, and those that ceased antipsychotics (sometimes after variable periods of dose reduction).
Specific antipsychotic drugs proposed to have TD‐lessening qualities compared with placebo or no intervention. We made a decision post hoc to broaden this criteria to also include antipsychotic versus antipsychotic and antipsychotic versus other drugs for the treatment of TD.
Types of outcome measures
We have defined clinical efficacy as an improvement in the symptoms of TD of more than 50%, on any scale. We grouped outcomes into short term (less than six weeks), medium term (between six weeks and six months) and long term (more than six months).
Primary outcomes
1. Tardive dyskinesia (TD) symptoms
No clinically important improvement in the symptoms of individuals, defined as more than 50% improvement on any TD scale ‐ any time period.
2. Adverse effects
No clinically significant extrapyramidal adverse effects ‐ any time period.
Secondary outcomes
1. Tardive dyskinesia (TD) symptoms
1.1 Any improvement in the symptoms of individuals on any TD scale, as opposed to no improvement 1.2 Deterioration in the symptoms of individuals, defined as any deleterious change on any TD scale 1.3 Average change in severity of TD during the trial period 1.4 Average difference in severity of TD at the end of the trial
2. General mental state changes
2.1 The number of people per treatment group who were defined as relapsed (according to any definition) 2.2 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale 2.3 Average difference in severity of psychiatric symptoms at the end of the trial
3. Acceptability of the treatment
3.1 Acceptability of the intervention to the participant group as measured by numbers of people leaving the study early (dropping out) during the trial.
4. Adverse effects
4.1 The number of people per treatment group who had any adverse effect (other than deterioration of symptoms of TD or relapse). 4.2 Use of any antiparkinsonism drugs 4.3 Average score/change in extrapyramidal adverse effects 4.4 Acute dystonia
5. Other adverse effects, general and specific
6. Hospital and service utilisation outcomes
6.1 Hospital admission 6.2 Average change in days in hospital 6.3 Improvement in hospital status (for example: change from formal to informal admission status, use of seclusion, level of observation)
7. Economic outcomes
7.1 Average change in total cost of medical and mental health care 7.2 Total indirect and direct costs
8. Social confidence, social inclusion, social networks, or personalised quality‐of‐life measures
8.1. No significant change in social confidence, social inclusion, social networks, or personalised quality‐of‐life measures 8.2 Average score/change in social confidence, social inclusion, social networks, or personalised quality‐of‐life measures
9. Behaviour
9.1 Clinically significant agitation 9.2 Use of adjunctive medication for sedation 9.3 Aggression to self or others
10. Cognitive state
10.1 No clinically important change 10.2 No change, general and specific
In Effects of interventions we reported on all TD symptom outcomes grouped together and all adverse effects outomes grouped together, whether or not they were primary or secondary, and we indicated primary outcomes with a '*'.
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADEpro to export data from this review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from the included studies in the comparison, the magnitude of effects of interventions examined, and the sum of available data on all outcomes rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table:
1. Tardive dyskinesia 1.1 Improved to a clinically important extent 1.2 Deteriorated
2. Mental state
3. Adverse effect 3.1 Any adverse event 3.2 Adverse effects: no clinically significant extrapyramidal adverse effects
4. Acceptability of treatment 4.1 Leaving the study early
5. Social confidence, social inclusion, social networks, or personalised quality of‐life‐measures 5.1 No significant change in social confidence, social inclusion, social networks, or personalised quality‐of‐life measures for either recipients of care or caregivers
We used this summary to guide our conclusions.
Personalised quality‐of‐life is an outcome designated important to patients. We wished to add perspectives from people’s personal experience with TD to the research agenda. We planned a consultation with service users, where the previously published version of a review in the Cochrane TD series (Soares‐Weiser 2011; Table 12) and a lay overview of that review gave the foundation for the discussions. The session was planned to provide time to reflect on current research on TD and to consider gaps in knowledge. The report is published in the Health Technology Assessment (HTA) report for the UK National Institute of Health Research (Bergman 2017). We have added one figure showing one service user's expression of frustration concerning this neglected area of research (Figure 1). Informed by the results of the consultation, for this review, we have included outcomes of the consultation in the 'Summary of findings' tables.
1.

Message from one of the participants of the public and patient involvement consultation of service user perspectives on tardive dyskinesia research
Search methods for identification of studies
Electronic searches
The 2017 review update was carried out in parallel with updating eight other Cochrane TD Reviews and the search covered all nine reviews; see Table 12 for details.
1. Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On July 16, 2015 and April 26, 2017, the Information Specialist searched the register using the following string:
*Tardive Dyskinesia* in Healthcare Condition Field of Study
In a study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017).
This register is compiled by systematic searches of major resources (AMED, BIOSIS, CINAHL, ClinicalTrials.Gov, Embase, MEDLINE, PsycINFO, PubMed, WHO ICTRP) and their monthly updates, ProQuest Dissertations and Theses A&I and its quarterly update, Chinese databases (CBM, CNKI, and Wanfang) and their annual updates, hand‐searches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
For the 2017 update, reviewers RA and AG (see Acknowledgements) inspected all abstracts of studies identified as above and identified potentially relevant reports. We resolved disagreement by discussion, or where there was still doubt, we acquired the full article for further inspection. We acquired the full articles of relevant reports/abstracts meeting initial criteria for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). RA and AG were not blinded to the names of the authors, institutions or journal of publication. Where difficulties or disputes arose, we asked review author HB for help and where it was impossible to decide or if adequate information was not available to make a decision, we added these studies to those awaiting assessment and contacted the authors of the papers for clarification.
Data extraction and management
1. Extraction
For the 2017 update, reviewers RA and HB independently extracted data from all included studies. Again, we discussed any disagreement and documented decisions. We extracted data presented only in graphs and figures whenever possible, but we included data only if two review authors independently had the same result. We attempted to contact study authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multi‐centre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
For the 2017 update we extracted data online in Covidence. Extracted data are available here with a link to the original source PDF for each item.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument had not been written or modified by one of the trialists for that particular trial. Ideally the measuring instrument should either have been (a) a self‐report or (b) completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, and we noted in Description of studies if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to primarily use endpoint data, and only to use change data if the former were not available. We combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences throughout (Higgins 2011a).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant data before inclusion.
Please note, we entered data from studies of at least 200 participants in the analysis because skewed data pose less of a problem in large studies. We also entered all relevant change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
For endpoint data from studies with fewer than 200 participants:
(a) for scales starting from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. If this value was lower than 1, it strongly suggested a skew and we excluded these data. If this ratio was higher than one but below 2, there was suggestion of skew. We entered these data and tested whether their inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2 we included these data, because skew was less likely (Altman 1996; Higgins 2011a).
(b) for scales starting from a positive value (such as the Positive and Negative Syndrome Scale (PANSS, Kay 1986), which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
2.5 Common measure
Where relevant, to facilitate comparison between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week, or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we converted continuous outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this can be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the study authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for reduction or cessation of antipsychotic. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'not un‐improved') we presented data where the left of the line indicated an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
RA (see Acknowledgements) and HB independently assessed risk of bias within the included studies by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011b). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data, and selective reporting.
If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. If non‐concurrence occurred, we reported this.
We noted the level of risk of bias in the text of the review and in Figure 2; Figure 3 and 'Summary of findings' tables.
2.
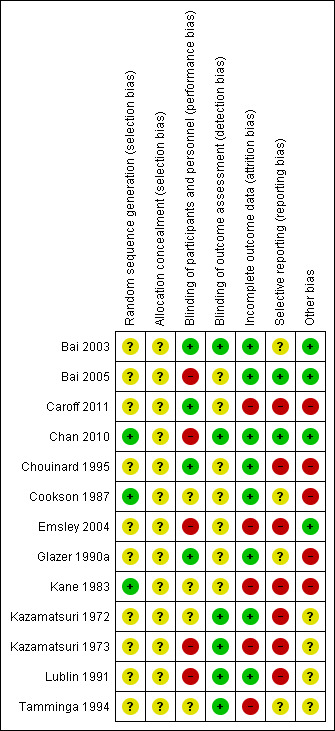
Risk of bias summary: review authors' judgements about each risk of bias item for each included study
3.
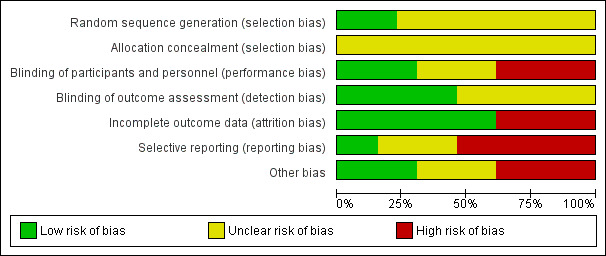
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios, as odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster‐randomised trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, study authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow, and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If any of the included trials had randomised participants by clusters, and where clustering was not accounted for in primary studies, we would have presented such data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster‐randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC) (design effect=1+(m‐1)*ICC) (Donner 2002). If the ICC was not reported we would assume it was 0.1 (Ukoumunne 1999).
If cluster studies were appropriately analysed, taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. The carry‐over effect occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary we simply added and combined within the two‐by‐two table. If data were continuous we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We did not use data where the additional treatment arms were not relevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We decided that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' tables by down‐rating quality. We also downgraded quality within the 'Summary of findings' tables should loss be 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50%, and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). We assumed that all those leaving the study early had seen no improvement in TD symptoms. We undertook a sensitivity analysis, testing how prone the primary outcomes were to change by comparing data only from people who had completed the study to that point to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
We reported and used data where attrition for a continuous outcome was between 0% and 50%, and data only from people who had completed the study to that point were reported.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the study authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and confidence intervals available for group means, and either P value or 't' value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011c). When only the standard error (SE) is reported, standard deviations (SDs) are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011a; Higgins 2011c) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges, or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which was based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently, methods such as multiple imputation or mixed effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, we preferred to use the more sophisticated approaches (e.g. MMRM or multiple‐imputation) and only presented completer analyses if some kind of ITT data were not available at all. Moreover, we addressed this issue in the item "incomplete outcome data" of the risk of bias tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise and discussed in the text if they arose.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise and discussed in the text if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 statistic method alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 statistic depends on (a) magnitude and direction of effects and (b) strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2 statistic). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic can be interpreted as evidence of substantial levels of heterogeneity (section 9.5.2 Cochrane Handbook for Systematic Reviews of InterventionsDeeks 2011). We explored and discussed in the text potential reasons for substantial levels of heterogeneity (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were ten or fewer studies, or where all studies were of similar sizes. In future versions of this review, if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We anticipated one subgroup analysis to test the hypothesis that the use of antipsychotic reduction, cessation or specific antipsychotics is most effective for those with early‐onset TD (less than five years). We hoped to present data for this subgroup for the primary outcomes.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of antipsychotic reduction/cessation or specific antipsychotics for people with TD in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage, and with similar problems.
2. Investigation of heterogeneity
We reported that inconsistency was high. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successively removed studies outside of the company of the rest to see if homogeneity was restored. For this review we decided that, should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If not, we did not pool such data and discussed issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply discussed. We did not undertake sensitivity analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
If trials were described in some way as to imply randomisation we undertook a sensitivity analysis for the primary outcomes. We included these studies in the analyses, and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, we used relevant data from these studies.
2. Assumptions for lost binary data
Where we had to make assumptions regarding people lost to follow‐up (see Dealing with missing data) we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
Where we had to make assumptions regarding missing SD data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with completer data only. We undertook a sensitivity analysis, testing how prone results were to change when completer data only were compared with the imputed data using the above assumption. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that we judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding, and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, we included data from these trials in the analysis
4. Imputed values
Had cluster‐randomised trials been included, we would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect.
If we found substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately
5. Fixed‐effect and random‐effects models
We synthesised data using a fixed‐effect model; however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether this altered the significance of the results.
Results
Description of studies
Please see Characteristics of included studies, Characteristics of excluded studies and Characteristics of ongoing studies.
Results of the search
The 2015 and 2017 searches for the 2017 review update also covered updates for the other eight Cochrane Reviews in the TD series, see Table 12.
The 2015 search retrieved 704 references for 344 studies, see Figure 4 for study flow diagram (Moher 2009). We also screened all references of the previously published review, 99 of which were not covered in the updated search. We identified 18 new potentially relevant studies for this review for which we conducted full‐text screenings. Five of these studies are newly included in this review (Bai 2005; Caroff 2011; Chan 2010; Chouinard 1995; Kazamatsuri 1973). A study previously awaiting classification was included as several more publications were identified in the new search (Bai 2003). Two studies were previously excluded because there was no placebo group, but as head‐to‐head comparisons of antipsychotic drugs for the treatment of TD has been added to the inclusion criteria in this review, these studies were subsequently included (Lublin 1991; Tamminga 1994). We also added 13 new excluded studies, and three studies previously awaiting classification were also excluded (Barnes 2002; Cai 1988; Zeng 1994).
4.
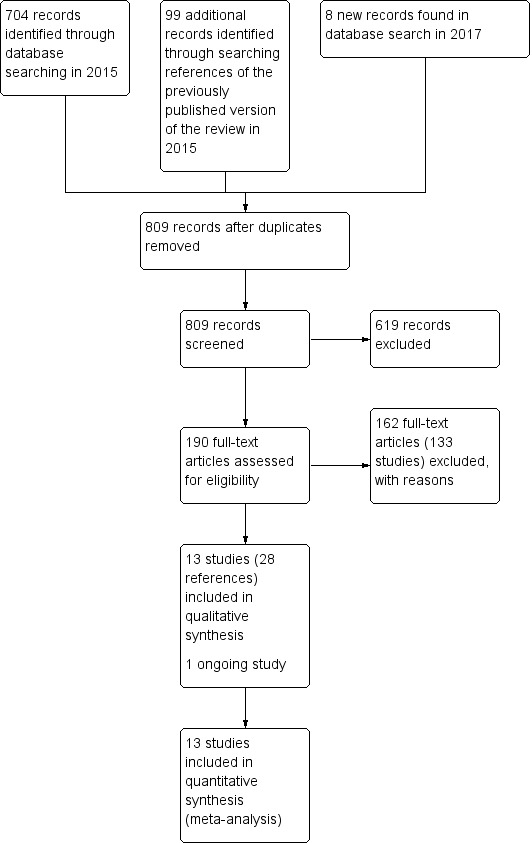
Study flow diagram for 2015 and 2017 searches for this review
The 2017 search found 8 records (5 studies). Editorial base of Cochrane Schizophrenia screened these records and no new studies were relevant to this review. They could be relevant to the other reviews in this series of TD reviews (see Table 12), and have been put into awaiting assessment of the Miscellaneous treatments review Soares‐Weiser 2006 and the benzodiazepines review Bhoopathi 2006.
Thirteen studies are now included in this review (Tamminga 1994; Cookson 1987; Kane 1983; Kazamatsuri 1973; Glazer 1990a; Bai 2005; Caroff 2011; Chan 2010; Emsley 2004; Chouinard 1995; Bai 2003; Kazamatsuri 1972; Lublin 1991).
Included studies
Overall the review now includes 13 studies with 711 participants published between 1972 and 2011.
1. Methods
Most studies stated that they were randomised and double‐blind. Two studies reported being only single‐blind (Bai 2005; Chan 2010). For further details please see sections below on allocation and blinding.
2. Design
All included studies presented a parallel longitudinal design. One of the 13 studies used a cross‐over design with two periods (Lublin 1991). We had considered this as likely when embarking on the review and have used only the data from before the first cross‐over for the reasons outlined above (Unit of analysis issues).
3. Duration
We included three short‐term studies reporting data at two to four weeks (Glazer 1990a; Kazamatsuri 1972; Lublin 1991). Five studies reported data at medium‐term, 8 to 24 weeks (Bai 2003; Bai 2005; Chan 2010; Chouinard 1995; Kazamatsuri 1973), and there were five long‐term studies reporting data at 44 weeks to 18 months (Caroff 2011; Cookson 1987; Emsley 2004; Kane 1983; Tamminga 1994).
4. Participants
Participants, now totaling 711 people, were mostly men with aged 50 to 60 years with diagnoses of various chronic psychiatric disorders, but mainly schizophrenia. All had antipsychotic‐induced TD diagnosed using Schooler and Kane’s research diagnostic criteria, except Lublin 1991, which did not report any criteria for the diagnosis of TD. The number of participants ranged from 8 to 200 (median 32).
5. Setting
Most trials were conducted in hospital. The studies themselves were from around the world, with six conducted in the USA (Caroff 2011; Glazer 1990a; Kane 1983; Kazamatsuri 1972; Kazamatsuri 1973; Tamminga 1994), three in Taiwan (Bai 2003; Bai 2005; Chan 2010), and one each in South Africa (Emsley 2004), Canada (Chouinard 1995), Denmark/Finland (Lublin 1991), and the UK (Cookson 1987).
6. Interventions
6.1 Antipsychotic reduction
6.1.1 cis(z)‐flupenthixol decanoate
Cookson 1987 used a reduction of 50% of the standard dose of cis(z)‐flupenthixol decanoate.
6.1.2 Fluphenazine decanoate
Kane 1983 compared a low dose of fluphenazine decanoate (1.25 mg to 5 mg for two weeks) to the standard dose (12.5 mg to 50 mg for two weeks).
6.2 Specific antipsychotics
6.2.1 Clozapine
Tamminga 1994 used clozapine in a mean (± SD) dose of 293.8 mg ± 171.9 mg a day for 12 months.
6.2.2 Risperidone
Four studies used risperidone in a dose ranging from 1.5 mg a day to 16 mg a day (Bai 2003; Caroff 2011; Chan 2010; Chouinard 1995).
6.2.3 Olanzapine
Three studies used olanzapine in a dose ranging from 7.5 mg a day to 12.6 mg a day (Caroff 2011; Chan 2010), Bai 2005 did not report the dose.
6.2.4 Amisulpride
Bai 2005 used amisulpride but did not report the dose.
6.2.5 Quetiapine
Two studies used quetiapine in a dose ranging from 100 mg a day to 400 mg a day (Caroff 2011; Emsley 2004).
6.2.6 Ziprasidone
Caroff 2011 used ziprasidone in a flexible dose of 40 mg.
6.2.7 Haloperidol
Seven studies used haloperidol in doses ranging from 2 mg a day to 34 mg a day (Chouinard 1995; Emsley 2004; Glazer 1990a; Kazamatsuri 1972; Kazamatsuri 1973; Lublin 1991; Tamminga 1994).
6.2.8 Molindone
Glazer 1990a used molindone in a dose from 75 mg to 145 mg.
6.2.9 Thiopropazate
Kazamatsuri 1972 used thiopropazate in a dose of 10 mg a day to 80 mg a day.
6.2.10 Zuclopenthixol
Lublin 1991 used zuclopenthixol in a dose from 16.5 mg a day to 26.6 mg a day.
6.3 Other drugs
6.3.1 Tetrabenazine
Kazamatsuri 1973 used tetrabenazine in a dose of 50 mg twice a day.
7. Outcomes
7.1 General
The included studies presented outcomes in graphs, inexact P values of differences, or a statement of significant or non‐significant difference. This made it impossible to acquire raw data for synthesis. We were unable to extract some continuous outcomes due to missing number of participants or missing means, standard deviations, or standard errors. All included studies used the LOCF strategy for the ITT analysis of dichotomous data.
7.2 Scales used to measure the TD symptoms
We have shown details of the scales that provided usable data below. We have provided reasons for exclusions of data under 'Outcomes' in the Characteristics of included studies tables.
7.2.1 Abnormal Involuntary Movement Scale (AIMS)
The AIMS (Guy 1970) is a 12‐item scale consisting of a standardised examination followed by questions rating orofacial, extremity, and trunk movements, as well as three global measurements. Each of these 10 items can be scored from 0 (none) to 4 (severe). Two additional items assess dental status. The AIMS ranges from 0 to 40, with higher scores indicating greater severity.
7.2.2 Extrapyramidal symptom rating scale (ESRS)
The ESRS was developed to assess four types of drug‐induced movement disorders (DIMD): parkinsonism, akathisia, dystonia, and TD (Chouinard 2005). The score for TD, ranging from 0 to 42, is based on the sum of all seven items in the TD objective examination.
7.2.3 St. Hans Rating Scale for extrapyramidal syndromes (SHRS)
The SHRS is a multidimensional rating scale for the evaluation of antipsychotic‐induced hyperkinesia, parkinsonism, akathisia, and dystonia (Gerlach 1993). Each item is rated from 0 (not present) to 6 (present to an extreme degree). This gives a total score from 0 to 48 for hyperkinesia and parkinsonism.
7.3 Scales used to measure adverse events related to antipsychotic medication
7.3.1 Simpson‐Angus Scale (SAS)
The SAS (Simpson 1970) is a 10‐item scale, with a scoring system of 0 to 4 for each item, that measures drug‐induced parkinsonism, a short‐term drug‐induced movement disorder. A low score indicates low levels of parkinsonism.
7.3.2 Barnes Akathisia Scale (BAS)
The BAS (Barnes 1989) is a 12‐item scale consisting of a standardised examination followed by questions rating orofacial, extremity, and trunk movements, as well as three global measurements. Each of these 10 items can be scored from 0 (none) to 4 (severe). Two additional items assess dental status. The BAS ranges from 0 to 40, with higher scores indicating greater severity.
7.3.3 UKU‐Side Effect Rating Scale
The UKU was developed to provide a comprehensive side effect rating scale with well‐defined and operationalised items to assess the side effects of psychopharmacological medications (Lingjaerde 1987). The scoring sheet includes 48 items with higher scores indicating more side effects.
7.4 Scales used to measure mental state and behaviour
7.4.1 Brief Psychiatric Rating Scale (BPRS)
The BPRS is an 18‐item scale measuring positive symptoms, general psychopathology and affective symptoms (Overall 1962). The original scale has 16 items, although a revised 18‐item scale is commonly used. Total scores can range from 0 to 126. Each item is rated on a seven‐point scale, with high scores indicating more severe symptoms.
7.4.2 Positive and Negative Syndrome Scale (PANSS)
The PANSS is a medical scale used for measuring symptom severity of people with schizophrenia (Kay 1986). The individual is rated from 1 to 7 on 30 different symptoms based on the interview as well as reports of family members or primary care hospital workers.
7.4.3 Clinical Global Impression
The CGI is a three‐item scale commonly used in studies on schizophrenia to enable clinicians to quantify severity of illness and overall clinical improvement (Guy 1976). The items are: severity of illness, global improvement, and efficacy index. A seven‐point scoring system is usually employed with low scores indicating decreased severity or greater recovery, or both.
Excluded studies
There are 133 excluded studies (163 references); the majority (n = 74) were excluded because they were not randomised. Thirty‐nine studies included participants with schizophrenia or other mental disorders who did not have TD. One study did not provide separate data from the included minority with TD; we contacted study authors who confirmed that these data were not available. Five studies investigated interventions that were not relevant for this review. Seven studies were cross‐over studies that did not provide data from the phase before crossing over to the next treatment. We contacted authors for three of these studies: author of Lal 1974 confirmed that no separate data were available, and we received no reply from authors of Lieberman 1988 or NDSG 1986, which were also excluded as they were published over 20 years ago and we assumed it very unlikely to receive a reply with data so many years later. We did not find up‐to‐date contact details for authors of four of the cross‐over studies (Bateman 1979; Delwaide 1979; Schwartz 1990; Singer 1971), and decided to also exclude these as they were published 25 to 45 years ago and we assumed it very unlikely to receive a reply so many years later. Seven studies did not provide any usable data. Authors of five of these studies (Herz 1991; Johnson 1987; Quinn 1984; Spohn 1988; Spohn 1993) confirmed that no further data were available. Authors of one of these studies did not reply (Andia 1998) and we could not find up‐to‐date contact details for authors of a final study (Borison 1987). We excluded these two studies as they were published 15 to 25 years ago and we assumed it very unlikely to receive a reply with data so many years later.
Studies awaiting classification
There are currently no studies awaiting assessment for inclusion in this review.
Ongoing studies
There is one ongoing study (N0546099389), which compares quetiapine and risperidone. This study was excluded in the previous review, but we have decided to move it to the ongoing studies section. It is a record from a trial registry, and at the time of preparing this update we were unable to locate author contact details or any more information about the study.
Risk of bias in included studies
Please refer to Figure 2 and Figure 3 for graphical overviews of the risk of bias in the included studies.
Allocation
While all 13 included studies stated that they randomised participants, we considered only three studies to be at low risk of selection bias. Two of these studies explicitly described how they generated the randomisation sequence (Chan 2010; Kane 1983), whereas one described block randomisation with stratification, and we assumed low risk of bias (Cookson 1987). None of the studies described how they concealed allocation and we rated all of them at unclear risk of selection bias.
Blinding
Eight studies stated that they were conducted on a double‐blind basis, but none tested the blindness of raters, clinicians and trial participants. Only four studies (Bai 2003; Caroff 2011; Chouinard 1995; Glazer 1990a) explicitly described how they blinded participants and personnel, and we rated them at low risk of performance bias. Five studies did not mention blinding of participants or personnel, or that the study was double‐blind (Bai 2005; Chan 2010; Emsley 2004; Kazamatsuri 1973; Lublin 1991); these studies were at high risk of performance bias. The remaining four studies were at unclear risk of performance bias; they stated that the study was double‐blind but did not describe further details of blinding.
Six studies had blinded raters and were at low risk of detection bias (Bai 2003; Chan 2010; Kazamatsuri 1972; Kazamatsuri 1973; Lublin 1991; Tamminga 1994). The remaining seven studies were at unclear risk of detection bias.
Incomplete outcome data
Eight studies were at low risk of attrition bias; they either had no dropouts (Cookson 1987) or low dropout rate and reported on dropouts adequately (Bai 2003; Bai 2005; Chan 2010; Chouinard 1995; Glazer 1990a; Kazamatsuri 1972; Lublin 1991). Five studies had a greater than 30% loss to follow‐up (Caroff 2011; Emsley 2004; Kane 1983; Tamminga 1994), or unbalanced loss to follow‐up between groups (Kazamatsuri 1973), and did not report outcomes for participants lost to follow‐up. These studies were rated at high risk of attrition bias. In all cases, however, we tried to ensure that every person randomised was analysed.
Selective reporting
The majority of data in this review originates from published reports. Most of the included studies reported expected outcomes (impact on TD symptoms). Only two trials fully reported outcomes outlined in protocols and were at low risk of reporting bias (Bai 2005; Chan 2010). Four trials were at unclear risk of bias, as we have had no opportunity to see protocols of these trials to compare the outcomes reported in the full publications with what was measured during the conduct of the trial (Bai 2003; Cookson 1987; Glazer 1990a; Tamminga 1994). Seven studies did not report results of all outcomes listed in the methods section fully (Caroff 2011; Chouinard 1995; Emsley 2004; Kane 1983; Kazamatsuri 1972; Kazamatsuri 1973; Lublin 1991) and were at high risk of reporting bias. Attempts to contact authors of trials for additional data were mostly unsuccessful, but we did receive some additional data from four of the trialists (Cookson 1987; Caroff 2011; Chouinard 1995; Kane 1983).
Other potential sources of bias
Four studies reported details such as baseline characteristics with sufficient detail to rate them at low risk of other bias. Four studies were at unclear risk of other bias, three (Kazamatsuri 1973; Kazamatsuri 1972; Lublin 1991) because of insufficiently detailed reporting to rule out any other bias, and one (Tamminga 1994) because the report was on a preliminary analysis with four of 49 subjects not yet having completed the study. Despite randomisation, two studies reported having unequal groups at baseline on important prognostic factors, and thus were at high risk of other bias (Cookson 1987; Glazer 1990a). Three studies randomised participants with schizophrenia, and this review included secondary reports of post hoc analyses of participants with TD. These studies were therefore at high risk of other bias (Caroff 2011; Chouinard 1995; Kane 1983).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings for the main comparison. Reduced dose of antipsychotics compared with antipsychotic maintenance for antipsychotic‐induced tardive dyskinesia.
| Reduced dose of antipsychotic compared with antipsychotic maintenance for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (schizophrenia or schizoaffective disorder) with antipsychotic‐induced tardive dyskinesia Setting: inpatients and outpatients in the UK (1 study) and the USA (1 study) Intervention: Reduced dose of antipsychotic Comparison: Antipsychotic maintenance | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with antipsychotic maintenance | Risk with reduced dose of antipsychotic | |||||
| Tardive dyskinesia: no clinically important improvement Follow‐up: 44‐48 weeks | Study population | RR 0.42 (0.17 to 1.04) | 17 (2 RCTs) | ⊕⊝⊝⊝ Very low1,2 | ||
| 875 per 1000 | 368 per 1000 (149 to 910) | |||||
| Tardive dyskinesia: deterioration of symptoms Follow‐up: 44‐48 weeks | Study population | RR 0.61 (0.11 to 3.31) | 17 (2 RCTs) | ⊕⊝⊝⊝ Very low1,2 | ||
| 250 per 1000 | 153 per 1000 (28 to 828) | |||||
| General mental state: relapse Follow‐up: 44‐48 weeks | Study population | RR 3.00 (0.16 to 57.36) | 8 (1 RCT) | ⊕⊝⊝⊝ Very low2,3 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Adverse effect: any ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Adverse effect: extrapyramidal symptoms ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Acceptability of the treatment: leaving the study early Follow‐up: 44‐48 weeks | Study population | RR 0.33 (0.06 to 1.99) | 8 (1 RCT) | ⊕⊝⊝⊝ Very low2,3,4 | ||
| 750 per 1000 | 248 per 1000 (45 to 1000) | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded one level for risk of bias: none of the studies adequately described allocation concealment, one study was a subsample from one site of an RCT, and one study's baseline characteristics were not balanced between study groups. 2Downgraded two levels for imprecision: 95% CI includes both no effect and appreciable benefit for antipsychotic reduced dose; very small sample size. 3Downgraded one level for risk of bias: allocation concealment was not adequately described, only a subsample from one site of an RCT qualified for inclusion. 4Downgraded one level for indirectness: leaving the study early can give an indication, but is not a direct measurement, of treatment acceptability.
Summary of findings 2. Antipsychotic cessation compared with antipsychotic maintenance for antipsychotic‐induced tardive dyskinesia.
| Antipsychotic cessation compared with antipsychotic maintenance for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients with antipsychotic‐induced tardive dyskinesia Setting: inpatients and outpatients in any country Intervention: Antipsychotic cessation Comparison: Antipsychotic maintenance | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Antipsychotic maintenance | Antipsychotic cessation | |||||
| There is no evidence about the effects of withdrawal of antipsychotics compared with continuation of antipsychotics; none of the included studies evaluated this comparison. | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
Summary of findings 3. Switch to another antipsychotic compared with antipsychotic cessation for antipsychotic‐induced tardive dyskinesia.
| Switch to another antipsychotic compared with antipsychotic cessation for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (schizophrenia) with antipsychotic‐induced tardive dyskinesia Setting: inpatients in Canada (1 study) and Taiwan (1 study) Intervention: Switch to another antipsychotic (risperidone, haloperidol) Comparison: Antipsychotic cessation (with placebo; from FGAs) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with antipsychotic cessation (placebo) | Risk with switch to another antipsychotic | |||||
| Tardive dyskinesia: no clinically important improvement Follow‐up: 12 weeks | Study population | RR 0.45 (0.23 to 0.89) | 42 (1 RCT) | ⊕⊕⊝⊝ Low1,2 | ||
| 700 per 1000 | 315 per 1000 (161 to 623) | |||||
| Tardive dyskinesia: deterioration of symptoms ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| General mental state: average endpoint score (BPRS, high = poor) Follow‐up: 12 weeks | The mean general mental state average endpoint score (BPRS, high = poor) was 19 | MD 4.30 lower (10.48 lower to 1.88 higher) | ‐ | 42 (1 RCT) | ⊕⊝⊝⊝ Very low1,3 | |
| Adverse effect: any ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Adverse effects: use of antiparkinsonism drugs Follow‐up: 8‐12 weeks | Study population | RR 2.08 (0.74 to 5.86) | 48 (1 RCT) 4 | ⊕⊝⊝⊝ Very low1,3 | Another study reported ESRS scale data for parkinsonism and also found little or no difference between groups (MD ‐0.4 95% CI ‐1.25 to 0.45, 42 participants). | |
| 273 per 1000 | 567 per 1000 (202 to 1000) | |||||
| Acceptability of the treatment: leaving the study early Follow‐up: 12 weeks | Study population | RR 0.60 (0.16 to 2.25) | 50 (1 RCT) | ⊕⊝⊝⊝ Very low1,3,5 | ||
| 200 per 1000 | 120 per 1000 (32 to 450) | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FGA: first‐generation antipsychotic; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded one level for risk of bias: generation of random sequence and allocation concealment not adequately described. 2 Downgraded one level for imprecision: very small sample size. 3 Downgraded two levels for imprecision: 95% CI includes appreciable benefit for both interventions as well as no effect; very small sample size. 4 Two comparisons from one study. 5 Downgraded one level for indirectness: leaving the study early can give an indication, but is not a direct measurement, of treatment acceptability.
Summary of findings 4. Switch to a specific antipsychotic compared with switch to a different specific antipsychotic for antipsychotic‐induced tardive dyskinesia.
| Switch to specific antipsychotic compared with switch to a different specific antipsychotic for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia Setting: inpatients and outpatients in Canada (1 study), Denmark and Finland (1 study), South Africa (1 study), Taiwan (2 studies) and the USA (5 studies) Interventions: switch to specific antipsychotic (amisulpride, clozapine, haloperidol, molindone, olanzapine, risperidone, thiopropazate, quetiapine, ziprasidone, zuclopenthixol) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with specific antipsychotic 1 | Risk with specific antipsychotic 2 | |||||
| Tardive dyskinesia: no clinically important improvement Follow‐up: 3‐50 weeks |
Study population | ‐ | 140 (4 RCTs) | ⊕⊝⊝⊝ Very low1,2 | No meta‐analysis, studies stratified by antipsychotic. The following comparisons found no clinically important improvement: THI vs HAL, ZUC vs HAL, OLZ vs RIS, QUE vs HAL | |
| See comment | See comment | |||||
| Tardive dyskinesia: deterioration Follow‐up: 3‐4 weeks |
Study population | ‐ | 35 (2 RCTs) | ⊕⊝⊝⊝ Very low1,2 | No meta‐analysis, studies stratified by antipsychotic. The following comparisons found no difference in deterioration: THI vs HAL, ZUC vs HAL | |
| See comment | See comment | |||||
| General mental state: deterioration Follow‐up: 3‐50 weeks |
Study population | ‐ | 120 (3 RCTs) | ⊕⊝⊝⊝ Very low 1,2 | No meta‐analysis, studies stratified by antipsychotic. The following comparisons found no difference in mental state deterioration: ZUC vs HAL, OLZ vs RIS, QUE vs HAL | |
| See comment | See comment | |||||
| Adverse events: extrapyramidal symptoms (need of antiparkinsonism drugs) Follow‐up: 8‐50 weeks |
Study population | ‐ | 53 (2 RCTs) | ⊕⊕⊝⊝ Low1,3 | No meta‐analysis, studies stratified by antipsychotic. HAL more likely to need antiparkinsonism drugs than QUE (1 RCT, 45 participants, RR 0.45, 95% CI 0.21 to 0.96). No difference: RIS vs HAL | |
| See comment | See comment | |||||
| Adverse effects: general adverse events (UKU Average change score) Follow‐up: 24 weeks |
See comment | See comment | ‐ | 80 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | No meta‐analysis, 3‐arm study comparing OLZ, ASP and unspecified FGAs found no difference in general adverse events for all pairwise comparisons. |
| Acceptability of the treatment: leaving the study early Follow‐up: 2 weeks ‐ 18 months |
Study population | ‐ | 466 (7 RCTs) | ⊕⊝⊝⊝ Very low1,2,4 | RIS more likely to leave study early than OLZ (2 RCTs, 130 participants, RR 0.73, 95% CI 0.57 to 0.95). Remaining studies no meta‐analysis, no difference (6 RCTs, 450 participants): MOL/THI/CLO/QUE vs HAL, OLZ/ASP vs unspecified FGAs, OLZ vs QUE/ZIP, QUE vs ZIP/RIS, ZIP vs RIS | |
| See comment | See comment | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | None of the included studies reported on this outcome. | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASP: amisulpride; CI: confidence interval; CLO: clozapine; FGA: first‐generation anti‐psychotic; HAL: haloperidol; MOL: molindone; OLZ: olanzapine; RCT: randomised controlled trial; RIS: risperidone; RR: risk ratio; THI: thiopropazate; QUE: quetiapine; ZIP: ziprasidone; ZUC: zuclopenthixol | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded one step for risk of bias: randomisation procedure, allocation concealment or blinding were not adequately described. 2Downgraded two steps for imprecision: small sample size, and 95% CI includes appreciable benefit for both or one of the interventions as well as no effect. 3Downgraded one step for imprecision: small sample size. 4Downgraded one step for indirectness: leaving the study early can give an indication, but is not a direct measurement, of treatment acceptability.
Summary of findings 5. Specific antipsychotic compared with other drugs for antipsychotic‐induced tardive dyskinesia.
| Specific antipsychotic compared with other drugs for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: psychiatric patients (mainly schizophrenia) with antipsychotic‐induced tardive dyskinesia Setting: inpatients in the USA (1 study) Intervention: specific antipsychotic (haloperidol) Comparison: other drugs (tetrabenazine) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with tetrabenazine | Risk with haloperidol | |||||
| Tardive dyskinesia: not improved to a clinically important extent Follow‐up: 18 weeks | Study population | RR 1.07 (0.51 to 2.23) | 13 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ||
| 667 per 1000 | 713 per 1000 (340 to 1000) | |||||
| Tardive dyskinesia: deterioration of symptoms Follow‐up: 18 weeks | Study population | RR 0.86 (0.07 to 10.96) | 13 (1 RCT) | ⊕⊝⊝⊝ Very low1,2 | ||
| 167 per 1000 | 143 per 1000 (12 to 1000) | |||||
| Mental state ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Adverse effect: any ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Adverse effect: extrapyramidal symptoms ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| Acceptability of the treatment: leaving the study early Follow‐up: 18 weeks | Study population | RR 4.38 (0.25 to 76.54) | 13 (1 RCT) | ⊕⊝⊝⊝ Very low1,2,3 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | See comment | See comment | Not estimable | (0 studies) | ‐ | None of the included studies reported on this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded one step for risk of bias: randomisation procedure, allocation concealment and blinding were not adequately described. 2Downgraded two steps for imprecision: small sample size, and 95% CI includes appreciable benefit for both interventions. 3Downgraded one step for indirectness: leaving the study early can give an indication, but is not a direct measurement, of treatment acceptability.
* indicates primary outcomes.
Comparison 1. Reduced overall dose of antipsychotic versus antipsychotic maintenance
1.1 TD symptoms
We had chosen 'No clinically important improvement in TD symptoms' for any time period as a primary outcome, with clinically important improvement defined as more than 50% improvement on any TD scale. Although the data we found in trials did not fit this exactly we feel that the outcome 'not improved to a clinically important extent' fits best with what we had hoped to find.
The authors of two RCTs of antipsychotic reduction provided additional data (Cookson 1987; Kane 1983).
1.1.1 No clinically important improvement*
No clinically important improvement in TD severity was associated with antipsychotic reduction compared with antipsychotic maintenance at 44 to 48 weeks (very low‐quality evidence, 2 RCTs, 17 people, RR 0.42 95% CI 0.17 to 1.04, Analysis 1.1).
1.1. Analysis.
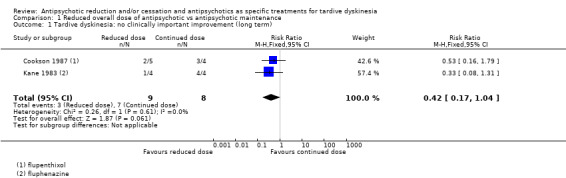
Comparison 1 Reduced overall dose of antipsychotic vs antipsychotic maintenance, Outcome 1 Tardive dyskinesia: no clinically important improvement (long term).
1.1.2 Not any improvement
When the outcome criterion was broadened from clinically significant improvement to improvement of any degree in TD severity, the result persisted at 44 to 48 weeks (very low‐quality evidence, 2 RCTs, 17 people, RR 0.42 95% CI 0.17 to 1.04, Analysis 1.2).
1.2. Analysis.
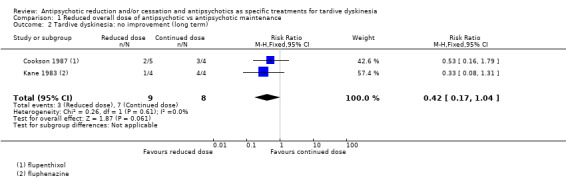
Comparison 1 Reduced overall dose of antipsychotic vs antipsychotic maintenance, Outcome 2 Tardive dyskinesia: no improvement (long term).
1.1.3 Deterioration of symptoms
Antipsychotic reduction was not associated with deterioration of TD symptoms compared with continued dose at 44 to 48 weeks (very low‐quality evidence, 2 RCTs, 17 people, RR 0.61 95% CI 0.11 to 3.31, Analysis 1.3).
1.3. Analysis.
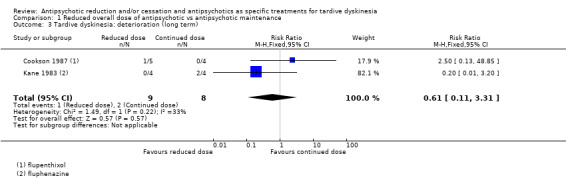
Comparison 1 Reduced overall dose of antipsychotic vs antipsychotic maintenance, Outcome 3 Tardive dyskinesia: deterioration (long term).
1.2 General mental state
1.2.1 Relapse
The number of those relapsing was equivocal over the long term (1 RCT, 8 people, RR 3.00 95% CI 0.16 to 57.36, Analysis 1.4).
1.4. Analysis.
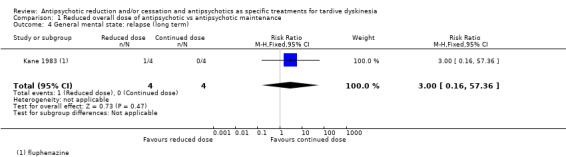
Comparison 1 Reduced overall dose of antipsychotic vs antipsychotic maintenance, Outcome 4 General mental state: relapse (long term).
1.3 Acceptability of the treatment: leaving the study early
The number of people leaving the study early was not statistically different for either the antipsychotic reduction group (1/4) or the antipsychotic maintained group (3/4) at 44 to 48 weeks (very low‐quality evidence, 1 RCT, 8 people, RR 0.33 95% CI 0.06 to 1.99, Analysis 1.5).
1.5. Analysis.
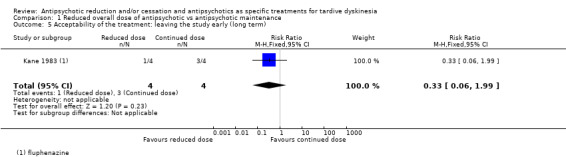
Comparison 1 Reduced overall dose of antipsychotic vs antipsychotic maintenance, Outcome 5 Acceptability of the treatment: leaving the study early (long term).
We did not identify any studies that reported on adverse events, hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
1.4 Subgroup analysis
1.4.1 Clinical stage: recent‐onset TD
It was not possible to evaluate whether those with recent‐onset TD responded differently to those with more established problems, since neither trial reported data for groups with different durations of TD that could be extracted for separate analyses.
1.4.2 Duration of follow‐up
We were unable to investigate any effects that a reduced dose of antipsychotics may have had in relation to duration of follow‐up because both studies had very similar length: 44 and 48 weeks.
1.5 Heterogeneity
Data were homogeneous. We did not detect clinical, methodological or statistical heterogeneity as described in Assessment of heterogeneity.
1.6 Sensitivity analyses
1.6.1 Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. As both studies stated that they randomised participants, we have not undertaken this sensitivity analysis.
1.6.2 Assumptions for lost binary data
At the time of updating this review, we were unable to compare the findings with completer data only, as the previous review authors held data received from study authors.
1.6.3 Risk of bias
We judged both studies in this comparison to be at high risk of bias across one or more domains. Therefore, we were unable to perform a sensitivity analysis in this instance.
1.6.4 Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from cluster‐randomised trials where we used imputed values for ICC in calculating the design effect, however, no cluster‐randomised trials were included in this comparison.
1.6.5 Fixed‐effect and random‐effects models
We also synthesised data for the primary outcome using a random‐effects model. This did not alter the significance of the results (2 RCTs, 17 people, RR 0.43, 95% CI 0.17 to 1.08).
Comparison 2. Switch to a specific antipsychotic versus placebo (cessation of antipsychotic)
2.1 TD symptoms
2.1.1 No clinically important improvement*
One study found a benefit in favour of antipsychotics against placebo for clinically important improvement in TD symptoms at 12 weeks (low‐quality evidence, 1 RCT, 42 people, RR 0.45 95% CI 0.23 to 0.89, Analysis 2.1).
2.1. Analysis.
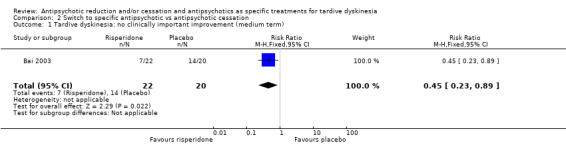
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 1 Tardive dyskinesia: no clinically important improvement (medium term).
2.1.2 Average difference in severity of TD at the end of the trial
TD symptoms were also measured using the continuous AIMS scale (see Description of studies, Outcomes). One study found a beneficial effect of antipsychotics when comparing average endpoint scores of the AIMS to placebo at 12 weeks (very low‐quality evidence, 1 RCT, 42 people, MD ‐5.50 95% CI ‐8.60 to ‐2.40, Analysis 2.2).
2.2. Analysis.
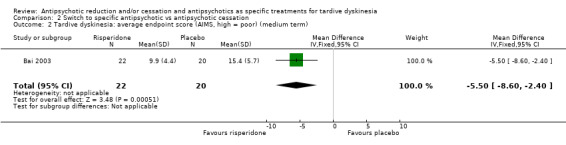
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 2 Tardive dyskinesia: average endpoint score (AIMS, high = poor) (medium term).
2.2 General mental state
2.2.1 Average difference in severity of psychiatric symptoms at the end of the trial
General mental state was measured using the continuous BPRS scale (see Description of studies, Outcomes). One study found no difference between antipsychotics and placebo on average endpoint score of the BPRS at 12 weeks (1 RCT, 42 people, MD ‐4.30 95% CI ‐10.48 to 1.88, Analysis 2.3).
2.3. Analysis.
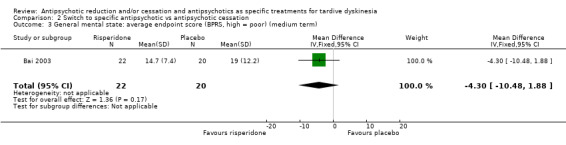
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 3 General mental state: average endpoint score (BPRS, high = poor) (medium term).
2.3 Acceptability of the treatment: leaving the study early
Using antipsychotics did not significantly increase the chances of a person leaving the study early at 12 weeks (very low‐quality evidence, 1 RCT, 50 people, RR 0.60 95% CI 0.16 to 2.25, Analysis 2.4).
2.4. Analysis.
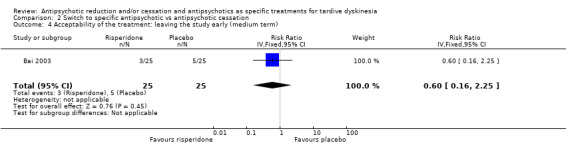
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 4 Acceptability of the treatment: leaving the study early (medium term).
2.4 Adverse effects
2.4.1 Extrapyramidal symptoms*: need of antiparkinsonism drugs
We found no difference in the use of antiparkinsonism drugs between antipsychotic and placebo at 12 weeks (1 RCT, 48 people, RR 2.08 95% CI 0.74 to 5.86, Analysis 2.5).
2.5. Analysis.
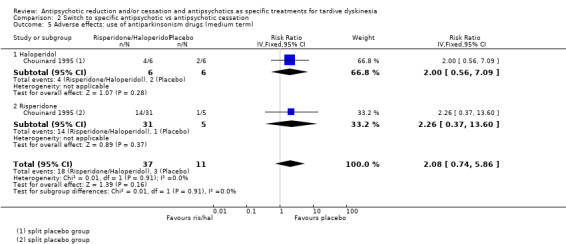
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 5 Adverse effects: use of antiparkinsonism drugs (medium term).
2.4.2 Average change in extrapyramidal adverse effects: Parkinsonism
One study measured parkinsonism on the continuous ESRS scale (see above). No difference was found between antipsychotics and placebo at 12 weeks (1 RCT, 42 people, MD ‐0.40 95% CI ‐1.25 to 0.45, Analysis 2.6).
2.6. Analysis.
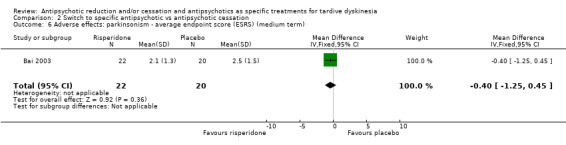
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 6 Adverse effects: parkinsonism ‐ average endpoint score (ESRS) (medium term).
2.4.3 Average change in extrapyramidal adverse effects: Dystonia
One study measured dystonia using the continuous ESRS scale. No difference was found between those allocated to antipsychotics or placebo at 12 weeks (1 RCT, 42 people, MD ‐0.70 95% CI ‐1.76 to 0.36, Analysis 2.7).
2.7. Analysis.
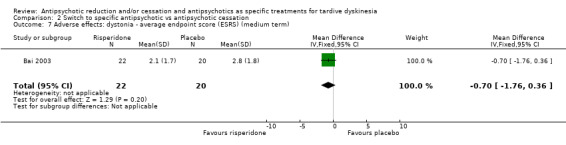
Comparison 2 Switch to specific antipsychotic vs antipsychotic cessation, Outcome 7 Adverse effects: dystonia ‐ average endpoint score (ESRS) (medium term).
We did not identify any studies that reported on hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
We also did not identify any studies that investigated the effect on TD of antipsychotic cessation compared with antipsychotic maintenance.
2.5 Subgroup and sensitivity analyses
There was only one included study in this comparison, and this study did not report on subgroups. Consequently, subgroup and sensitivity analyses were not carried out.
Comparison 3. Switch to a specific antipsychotic versus switch to another specific antipsychotic
3.1 TD symptoms
3.1.1 No clinically important improvement*
None of the four studies that reported on this outcome found a clinically important difference on improvement in TD symptoms between two specific antipsychotics (Analysis 3.1): short‐term: thiopropazate versus haloperidol (low‐quality evidence, 1 RCT, 20 people, RR 1.53, 95% CI 0.58 to 4.05), zuclopenthixol versus haloperidol (low‐quality evidence, 1 RCT, 15 people, RR 1.00, 95% CI 0.79 to 1.27); medium‐term: olanzapine versus risperidone (low‐quality evidence, 1 RCT, 60 people, RR 1.25, 95% CI 0.82 to 1.90), quetiapine versus haloperidol (low‐quality evidence, 1 RCT, 45 people, RR 0.80, 95% CI 0.52 to 1.22); long‐term: quetiapine versus haloperidol (low‐quality evidence, 1 RCT, 45 people, RR 0.88, 95% CI 0.64 to 1.21).
3.1. Analysis.
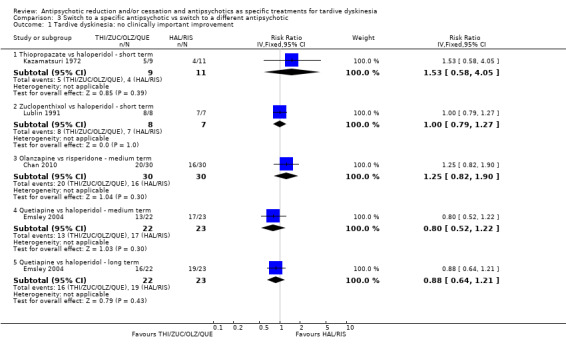
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 1 Tardive dyskinesia: no clinically important improvement.
3.1.2 Not any improvement
Neither of the two studies that reported on any improvement of TD symptoms found a difference between two specific antipsychotics (Analysis 3.2): short‐term: thiopropazate versus haloperidol (low‐quality evidence, 1 RCT, 20 people, RR 0.41, 95% CI 0.05 to 3.28), zuclopenthixol versus haloperidol (low‐quality evidence, 1 RCT, 15 people, RR 0.88, 95% CI 0.16 to 4.68).
3.2. Analysis.
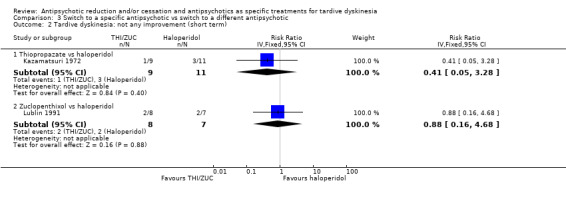
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 2 Tardive dyskinesia: not any improvement (short term).
3.1.3 Deterioration of symptoms
Neither of the two studies that reported on deterioration of TD symptoms found a difference between two specific antipsychotics (Analysis 3.3): short‐term: thiopropazate versus haloperidol (low‐quality evidence, 1 RCT, 20 people, RR 1.22, 95% CI 0.09 to 16.92), zuclopenthixol versus haloperidol (low‐quality evidence, 1 RCT, 15 people, RR 0.88, 95% CI 0.16 to 4.68).
3.3. Analysis.
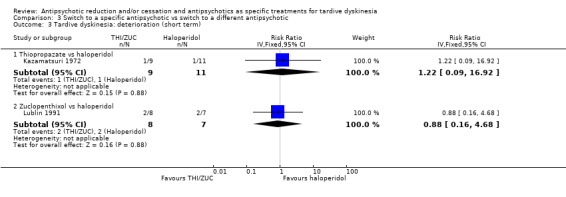
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 3 Tardive dyskinesia: deterioration (short term).
3.1.2 Average difference in severity of TD at the end of the trial
A short‐term (two weeks) study of people with established withdrawal‐exacerbated TD found no difference on endpoint AIMS scores after the first week (1 RCT, 18 people, MD 1.87, 95% CI ‐0.20 to 3.94) for the groups receiving molindone or haloperidol at the standard dosage range. Results for the second week, using higher 'masking' dosages, significantly favoured the haloperidol group (1 RCT, 18 people, MD 3.44, 95% CI 1.12 to 5.76). Another short‐term study comparing zuclopenthixol and haloperidol found no difference between groups on endpoint SHRS scores (1 RCT, 15 people, MD ‐4.81, 95% CI ‐12.15 to 2.53). Finally, a medium‐term study found no difference between olanzapine and risperidone on endpoint AIMS scores (1 RCT, 60 people, MD 2.20, 95% CI ‐0.53 to 4.93). See Analysis 3.4.
3.4. Analysis.
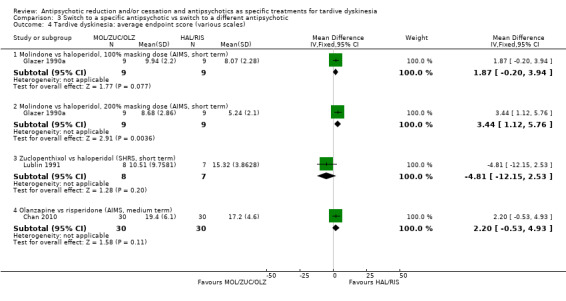
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 4 Tardive dyskinesia: average endpoint score (various scales).
3.1.4 Average change in severity of TD during the trial period
One study found amisulpride favourable to olanzapine in reducing TD symptoms measured by AIMS change from baseline scores at medium term (1 RCT, 54 people, MD 2.48, 95% CI 0.44 to 4.52). Three other comparisons found no difference between specific antipsychotics at medium term: olanzapine versus unspecified FGAs (1 RCT, 53 people, MD 1.66, 95% CI ‐0.45 to 3.77), amisulpride vs unspecified FGAs (1 RCT, 53 people, MD ‐0.82, 95% CI ‐2.85 to 1.21), and olanzapine vs risperidone (1 RCT, 60 people, MD 1.20, 95% CI ‐2.58 to 4.98, Analysis 3.5).
3.5. Analysis.
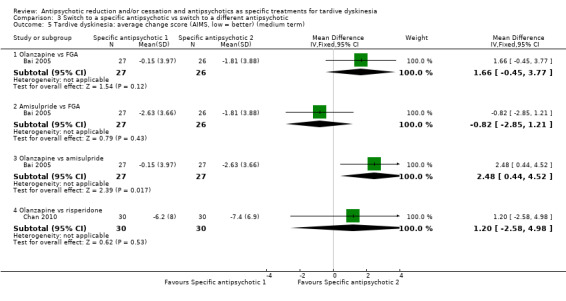
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 5 Tardive dyskinesia: average change score (AIMS, low = better) (medium term).
3.2 General mental state
3.2.1 Deterioration of mental state
No difference was found in the proportion with deterioration of mental state between specific antipsychotics: zuclopenthixol versus haloperidol at short term (1 RCT, 15 people, RR 0.30, 95% CI 0.01 to 6.29), olanzapine versus risperidone at medium term (1 RCT, 60 people, RR 1.00, 95% CI 0.15 to 6.64), quetiapine versus haloperidol at long term (1 RCT, 45 people, RR 1.83, 95% CI 0.62 to 5.39, Analysis 3.6).
3.6. Analysis.
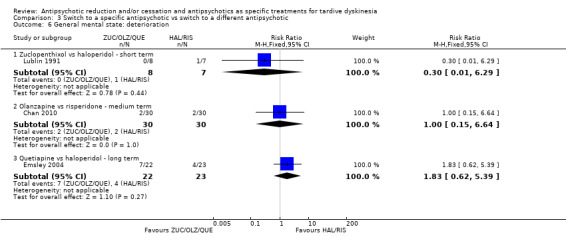
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 6 General mental state: deterioration.
3.2.2 Average difference in severity of psychiatric symptoms at the end of the trial
No difference was found in average endpoint scores of the PANSS between quetiapine and haloperidol at long term (1 RCT, 45 people, MD ‐2.20, 95% CI ‐6.02 to 1.62, Analysis 3.7).
3.7. Analysis.
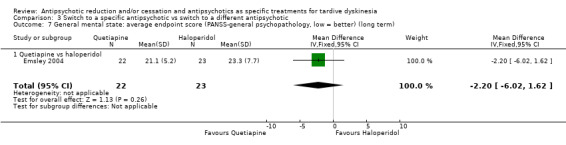
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 7 General mental state: average endpoint score (PANSS‐general psychopathology, low = better) (long term).
3.2.3 Average change in severity of psychiatric symptoms during the trial period
There was also no difference on the average endpoint scores on the BPRS between specific antipsychotics at medium term: olanzapine versus unspecified FGAs (1 RCT, 53 people, MD ‐1.14, 95% CI ‐4.79 to 2.51), amisulpride versus unspecified FGAs (1 RCT, 53 people, MD ‐2.46, 95% CI ‐6.27 to 1.35), olanzapine versus risperidone (1 RCT, 60 people, MD ‐1.70, 95% CI ‐8.37 to 4.97), and olanzapine versus amisulpride (1 RCT, 54 people, MD 1.32, 95% CI ‐1.94 to 4.58, Analysis 3.8).
3.8. Analysis.
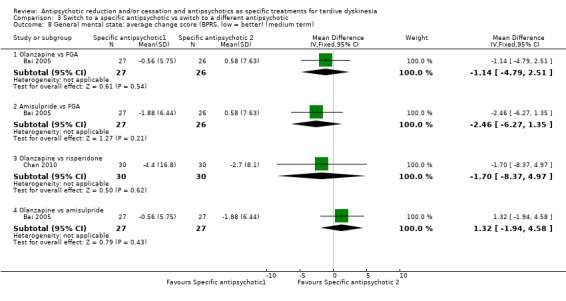
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 8 General mental state: average change score (BPRS, low = better) (medium term).
3.3 Acceptability of the treatment: leaving the study early
In the short term, a specific antipsychotic did not significantly increase the chances of a person leaving the study early compared with another specific antipsychotic: molindone versus haloperidol (1 RCT, 18 people, MD not estimable, there were no events), thiopropazate versus haloperidol (1 RCT, 20 people, MD 0.24, 95% CI 0.01 to 4.44, Analysis 3.9). In the medium term, switching to olanzapine significantly reduced the chances of a person leaving the study early compared with switching to risperidone (2 RCTs, 170 people, RR 0.73 95% CI 0.57 to 0.95, I2 = 0, Analysis 3.10) or compared with switching to quetiapine (1 RCT, 116 people, RR 0.70 95% CI 0.54 to 0.90). For all other comparisons between specific antipsychotics at medium term, there was no difference in the chances of a person leaving the study early: olanzapine versus unspecified FGAs (1 RCT, 56 people, RR 1.86, 95% CI 0.18 to 19.38), amisulpride versus unspecified FGAs (1 RCT, 55 people, RR 0.96, 95% CI 0.06 to 14.65), olanzapine versus amisulpride (1 RCT, 57 people, RR 1.93, 95% CI 0.19 to 20.12), olanzapine versus ziprasidone (1 RCT, 82 people, RR 0.77, 95% CI 0.56 to 1.05), quetiapine versus risperidone (1 RCT, 118 people, RR 1.05, 95% CI 0.88 to 1.25), quetiapine versus ziprasidone (1 RCT, 90 people, RR 1.10, 95% CI 0.86 to 1.40), ziprasidone versus risperidone (1 RCT, 84 people, RR 0.95, 95% CI 0.74 to 1.23). Finally, in the long term, there was no difference between clozapine and haloperidol (1 RCT, 39 people, RR 3.36, 95% CI 0.45 to 25.16), or between quetiapine and haloperidol on the number leaving the study early (1 RCT, 45 people, RR 1.31, 95% CI 0.63 to 2.69, see Analysis 3.11).
3.9. Analysis.
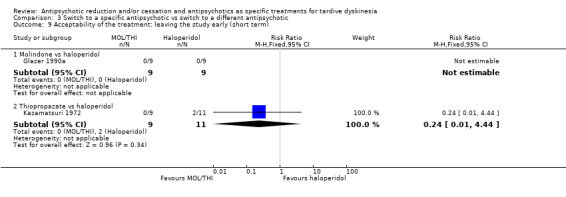
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 9 Acceptability of the treatment: leaving the study early (short term).
3.10. Analysis.
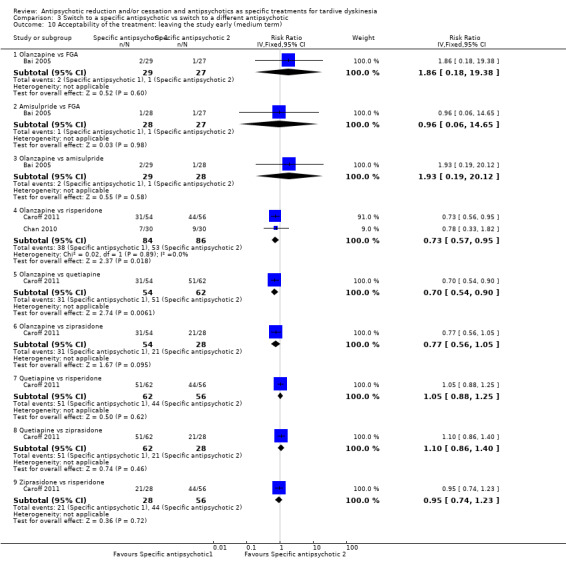
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 10 Acceptability of the treatment: leaving the study early (medium term).
3.11. Analysis.
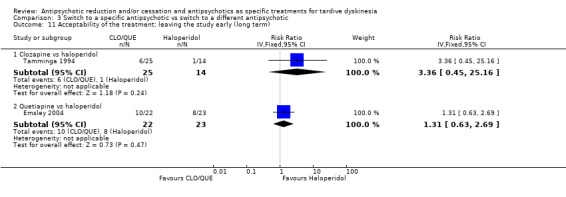
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 11 Acceptability of the treatment: leaving the study early (long term).
3.4 Adverse events
3.4.1 Extrapyramidal symptoms*: need of antiparkinsonism drugs
A study comparing risperidone and haloperidol found no difference between groups at medium term (1 RCT, 37 people, RR 0.68, 95% CI 0.34 to 1.35, Analysis 3.12). Another study found that participants allocated to haloperidol were more likely to need antiparkinsonism drugs than those allocated to quetiapine in the long term (1 RCT, 45 people, RR 0.45, 95% CI 0.21 to 2.96, Analysis 3.12).
3.12. Analysis.
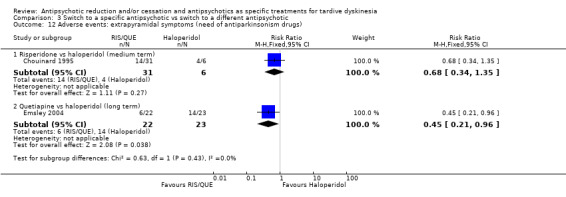
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 12 Adverse events: extrapyramidal symptoms (need of antiparkinsonism drugs).
3.4.2 Average change in extrapyramidal adverse effects: Parkinsonism
Symptoms of parkinsonism were measured using the continuous SAS, SHRS, and ESRS scales (see Description of studies, Outcomes). At short term, one study found no difference in parkinsonism symptoms measured by SHRS endpoint scores between zuclopenthixol and haloperidol (1 RCT, 15 people, MD ‐4.81, 95% CI ‐12.15 to 2.53, Analysis 3.13). One study found those allocated to olanzapine were less likely to develop symptoms of parkinsonism than those allocated to risperidone measured by ESRS change scores at medium term (1 RCT, 60 people, MD ‐0.70, 95% CI ‐1.33 to ‐0.07). For all other comparisons between specific antipsychotics at medium term, there was no difference in symptoms of parkinsonism as measured by average change scores: olanzapine versus unspecified FGAs (1 RCT, 53 people, MD ‐0.85, 95% CI ‐2.55 to 0.85), amisulpride versus unspecified FGAs (1 RCT, 53 people, MD ‐0.50, 95% CI ‐2.45 to 1.45), and olanzapine versus amisulpride (1 RCT, 54 people, MD ‐0.35, 95% CI ‐2.44 to 1.74, Analysis 3.14).
3.13. Analysis.
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 13 Adverse effects: parkinsonism (SHRS) ‐ average endpoint scores (short term).
3.14. Analysis.
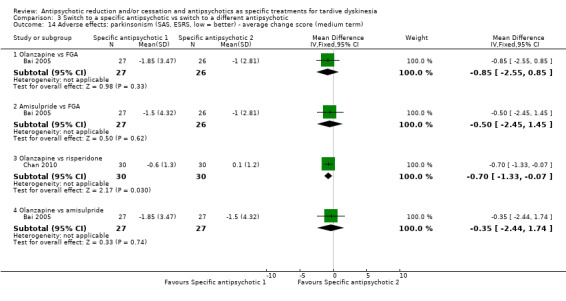
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 14 Adverse effects: parkinsonism (SAS, ESRS, low = better) ‐ average change score (medium term).
3.4.3 Average change in extrapyramidal adverse effects: Dyskinesia
Symptoms of dyskinesia were measured using the continuous ESRS scale. There was no difference in symptoms of dyskinesia between those switching to olanzapine and those switching to risperidone (1 RCT, 60 people, MD 0.30, 95% CI ‐0.91 to 1.51, Analysis 3.15).
3.15. Analysis.
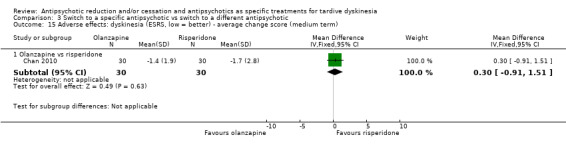
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 15 Adverse effects: dyskinesia (ESRS, low = better) ‐ average change score (medium term).
3.4.4 Average change in extrapyramidal adverse effects: Akathisia
Symptoms of akathisia were measured using the continuous BAS and ESRS scales (see Description of studies, Outcomes, Analysis 3.16). There was no difference in symptoms of akathisia for those switching to olanzapine (1 RCT, 53 people, MD 0.08 95% CI ‐0.30 to 0.46) or amisulpride (1 RCT, 53 people, MD ‐0.11 95% CI ‐0.42 to 0.20) compared with those remaining on unspecified FGAs. There was also no difference for those switching to olanzapine compared with those switching to risperidone (1 RCT, 60 people, MD ‐0.80 95% CI ‐1.76 to 0.16) or amisulpride (1 RCT, 54 people, MD 0.19 95% CI ‐0.12 to 0.50).
3.16. Analysis.
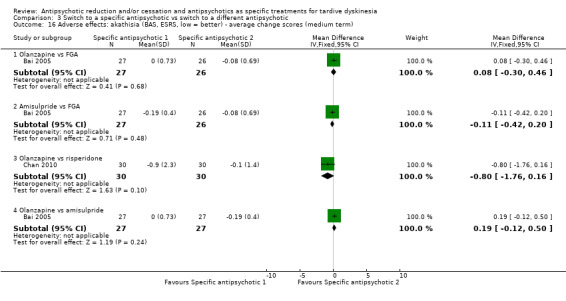
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 16 Adverse effects: akathisia (BAS, ESRS, low = better) ‐ average change scores (medium term).
3.4.5 Average change in extrapyramidal adverse effects: Dystonia
Symptoms of dystonia were measured using the continuous ESRS scale. There was no difference in symptoms of dystonia in those switching to olanzapine compared with those switching to risperidone (1 RCT, 60 people, MD ‐0.70 95% CI ‐1.41 to 0.01, Analysis 3.17).
3.17. Analysis.
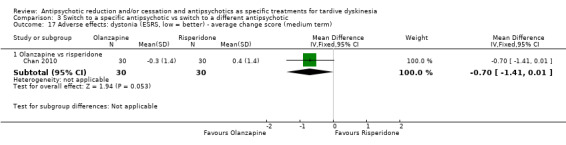
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 17 Adverse effects: dystonia (ESRS, low = better) ‐ average change score (medium term).
3.4.6 Average change in general adverse events
General adverse events were measured using the continuous UKU scale (see Description of studies, Outcomes, Analysis 3.18). There was no difference in general adverse events for those switching to olanzapine (1 RCT, 53 people, MD 0.08 95% CI ‐1.85 to 2.01) or amisulpride (1 RCT, 53 people, MD ‐0.55 95% CI ‐2.33 to 1.23) compared with those remaining on unspecified FGAs. There was also no difference for those switching to olanzapine compared with those switching to amisulpride (1 RCT, 54 people, MD 0.63 95% CI ‐0.93 to 2.19).
3.18. Analysis.
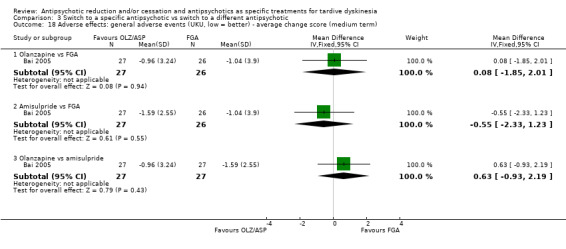
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 18 Adverse effects: general adverse events (UKU, low = better) ‐ average change score (medium term).
3.5 Global state
3.5.1 Average change in global state
Global state was measured using the continuous CGI scale (see Description of studies, Outcomes, Analysis 3.19). There was no difference in change scores of global state in those switching to olanzapine (1 RCT, 53 people, MD ‐0.07 95% CI ‐0.41 to 0.27) or amisulpride (1 RCT, 53 people, MD ‐0.19 95% CI ‐0.47 to 0.09) compared with those remaining on FGA. There was also no difference in those switching to olanzapine compared with those switching to risperidone (1 RCT, 60 people, MD 0.10 95% CI ‐0.61 to 0.81) or amisulpride (1 RCT, 54 people, MD 0.12 95% CI ‐0.19 to 0.43).
3.19. Analysis.
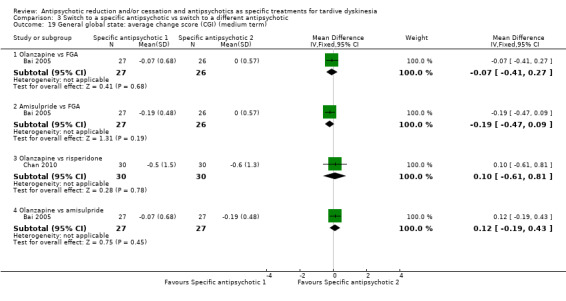
Comparison 3 Switch to a specific antipsychotic vs switch to a different antipsychotic, Outcome 19 General global state: average change score (CGI) (medium term).
We did not identify any studies that reported on hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
3.6 Subgroup and sensitivity analyses
There were no meta‐analyses for the primary outcomes of this comparison because studies were stratified by antipsychotic medication. Consequently, we did not carry out subgroup and sensitivity analyses.
Comparison 4. Specific antipsychotic versus other drug
4.1 TD symptoms
4.1.1 No clinically important improvement*
One study found no difference between haloperidol and tetrabenazine for clinically important improvement in TD symptoms at 24 weeks (very low‐quality evidence, 1 RCT, 13 people, RR 1.07 95% CI 0.51 to 2.23, Analysis 4.1).
4.1. Analysis.
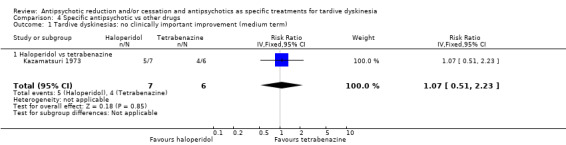
Comparison 4 Specific antipsychotic vs other drugs, Outcome 1 Tardive dyskinesias: no clinically important improvement (medium term).
4.1.2 Not any improvement
One study found no difference between haloperidol and tetrabenazine for the outcome 'no improvement in TD symptoms' at 24 weeks (1 RCT, 13 people, RR 2.57 95% CI 0.35 to 18.68, Analysis 4.2).
4.2. Analysis.
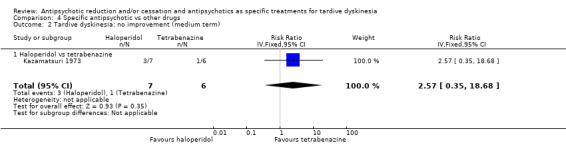
Comparison 4 Specific antipsychotic vs other drugs, Outcome 2 Tardive dyskinesia: no improvement (medium term).
4.1.3 Deterioration of symptoms
One study found no difference on deterioration of TD symptoms between haloperidol and tetrabenazine at 24 weeks (very low‐quality evidence, 1 RCT, 13 people, RR 0.86 95% CI 0.07 to 10.96, Analysis 4.3).
4.3. Analysis.
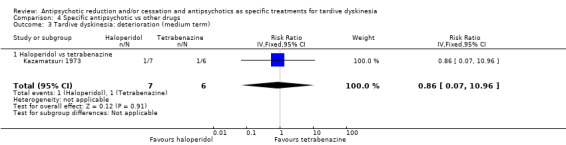
Comparison 4 Specific antipsychotic vs other drugs, Outcome 3 Tardive dyskinesia: deterioration (medium term).
4.2 Acceptability of the treatment: leaving the study early
One study found no difference in the proportion of people leaving the study early between haloperidol and tetrabenazine at 24 weeks (very low quality‐evidence, 1 RCT, 13 people, RR 4.38 95% CI 0.25 to 76.54, Analysis 4.4).
4.4. Analysis.
Comparison 4 Specific antipsychotic vs other drugs, Outcome 4 Acceptability of the treatment: leaving the study early (medium term).
We did not identify any studies that reported on mental state, adverse events, hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
4.3 Subgroup and sensitivity analyses
There was only one included study in this comparison, and this study did not report on subgroups. Consequently, we did not carry out subgroup and sensitivity analyses.
Discussion
Summary of main results
1. The search
This area of research does not seem to be active. We have identified additional data, but most trials predate the year 2000; only three were carried out after, published in 2004 to 2011. This could be due to a decreased concern with TD, or less emergence of the problem in research‐active communities because of more thoughtful use of antipsychotic drugs or loss of faith in potential treatments.
2. Few data
Only a little over 700 people were included in this review. It is possible that real and important effects have not been highlighted because of the necessarily wide CIs of the findings. Many outcomes were not measured at all by the included studies (see Overall completeness and applicability of evidence), including one of our outcome measures that was selected as important. We may have been overambitious in hoping for some of these outcomes in TD trials, but simple reporting of social impact and quality of life is of particular interest to patients and carers. Finally, we did not identify any RCTs of participants with TD that compared antipsychotic cessation with antipsychotic maintenance.
3. Outcomes
3.1 Tardive dyskinesia symptoms
We found low‐quality evidence of clinically important improvement in TD symptoms for switching antipsychotic to risperidone compared with antipsychotic cessation (with placebo) after 12 weeks (RR 0.45, 95% CI 0.23 to 0.89, 1 study, 42 participants). Because the quality of the evidence is low, we have limited confidence in the effect estimates and CIs; the true effects may be substantially different.
For the remaining comparisons: antipsychotic reduction versus maintenance, switch to a new antipsychotic versus switch to a different new antipsychotic, and specific antipsychotic versus other drug, we found low to very low‐quality evidence of little or no difference between groups for no clinically important improvement in TD symptoms and deterioration of TD symptoms, but again, our confidence in these results is limited due to the poor‐quality evidence, not least because there were few participants (13 to 60 participants per comparison).
3.2 Adverse effects
There was low‐quality evidence of fewer people that needed antiparkinsonism medication due to extrapyramidal side effects after switching to quetiapine compared with haloperidol after one year (RR 0.45, 95% CI 0.21 to 0.96, 1 study, 45 participants). Due to the low quality of this evidence, our confidence in these results is limited. For the other comparisons, switch to haloperidol or risperidone versus antipsychotic cessation, and risperidone versus haloperidol, we found very low‐quality evidence of little or no difference between groups. Therefore, we are very uncertain about these results, not least because there were few participants (37 and 48 participants per comparison).
3.3 Mental state
We found low‐ to very low‐quality evidence of little or no difference in mental state: relapse, deterioration of mental state, or average endpoint scores on scales measuring mental state, between groups of the following comparisons: antipsychotic reduction versus maintenance, switch to risperidone versus antipsychotic cessation, switch to zuclopenthixol versus haloperidol, switch to olanzapine versus risperidone, and switch to quetiapine versus haloperidol. Again, our confidence in these results is limited due to the poor quality of evidence, not least because there were few participants (8 to 60 participants per comparison).
3.4 Acceptability of treatment: leaving the study early
It is always unclear what leaving a study early means. It could be related to the participant not accepting treatment for a series of reasons, or to participants finding the trial intolerable. It also could be a function of a trial design in which willing participants are still asked to leave because of some degree of protocol violation.
There was very low‐quality evidence for most of the comparisons that the number of participants leaving the study early was no different in either group. The small number of people randomised in these comparisons (8 to 170 participants) made the likelihood of an unequivocal outcome unlikely. However, we found very low‐quality evidence that fewer participants allocated to olanzapine left the study early compared with risperidone and compared with quetiapine. Since evidence was of very low quality for both comparisons, we have very little confidence in the effect estimates and CIs; the true effects are likely to be substantially different.
3.5 Social confidence, social inclusion, social networks, or personalised quality of life
We selected this group of outcomes for the 2017 review update following a service‐user consultation, as being of importance to patients. We did not identify any studies that reported on any of these outcomes.
Overall completeness and applicability of evidence
1. Completeness
No outcomes in this review involved large numbers of people. There is a large literature of trials assessing the impact of antipsychotic reduction or cessation, or both, on outcomes such as symptoms and relapse. Much of the initial impetus for these trials was derived from concerns about antipsychotic‐induced TD. It was disappointing to note that many of these trials did not systematically assess TD, and thus were not suitable for inclusion in this review. While many trials of antipsychotic reduction and/or cessation assess TD, the baseline and endpoint means are rarely presented for those with TD at baseline. Two authors (Cookson 1987; Kane 1983) kindly provided raw data from which we could extract the impact of antipsychotic reduction on those with TD at baseline. The very small sample size limits the confidence that one can place on the equivocal results.
Post hoc analyses of the main efficacy and safety analyses for the newer atypicals have also been undertaken (for example, Caroff 2011 and Chouinard 1995). The analyses of results from these trials need to be interpreted with caution for several reasons: (a) suboptimal design for the assessment of treatment of TD (for example, pre‐entry washout or gradual conversion to study drug); (b) the analyses were post hoc; and (c) differences in results between different arms in the studies may reflect antipsychotic suppression due to differing dosing strategies.
There were no data on the patient‐designated important outcomes social confidence, social inclusion, social networks, or personalised quality of life, nor were there data on hospital and service utilisation outcomes, economic outcomes, behaviour or cognitive response.
2. Applicability
Trial participants were mostly men in their 50s with schizophrenia in hospital, but were nevertheless people who would be recognisable in everyday care.
Reducing or stopping antipsychotic medication may not be clinically prudent in some situations. The current lack of evidence showing any improvement in TD with such strategies means that clinicians may be less inclined to use them in mentally stable patients. On the other hand, high‐quality evidence in this area would be very useful and applicable in everyday clinical practice to aid in the decision making around the risk and benefits of reducing or stopping antipsychotic medication following the emergence of TD.
Quality of the evidence
Overall, the quality of the evidence is low to very low. This means that we have limited to very little confidence in the effect estimates, and the true effect may be, or is likely to be, substantially different from the estimate of the effect. The main reasons for our low confidence in the evidence were:
poor study methodology and reporting of methods resulting in downgrading evidence for risk of bias;
very small sample sizes resulting in downgrading evidence for imprecision;
wide CIs (often due to low event rates) that included appreciable benefit or harm for the intervention as well as no effect, resulting in downgrading evidence for imprecision.
Please see Table 1; Table 2; Table 3; Table 4; Table 5 for full details.
Potential biases in the review process
1. Missing studies
Every effort was made to identify relevant trials. However, these studies were all small and it is likely that we have failed to identify other studies of limited power. We do not, however, think it likely that we have failed to identify large relevant studies.
2. Missing data
We excluded 14 studies published between 1971 and 1998 that did not provide any usable data, see Excluded studies. We contacted study authors and six replied to confirm no usable data were available. Authors of three studies did not reply, and we could not find up‐to‐date contact details for authors of five studies (all over 25 years old). We find it very unlikely that we would receive a reply from study authors regarding research conducted so many years ago; therefore, we also excluded these studies.
3. Introducing bias
We have tried to be balanced in our appraisal of the evidence but could have inadvertently introduced bias. We welcome comments or criticisms. New methods and innovations now make it possible to report data where, in the past, we could not report data at all or had to report data in a different way. We believe the 'Summary of findings' tables are a valuable innovation – but problematic to those not ‘blind’ to the outcome data. It is possible to ‘cherry pick’ significant findings for presentation in this table. We have tried to decrease the chance of doing this by asking a new reviewer (HB) to select outcomes relevant for this table before becoming familiar with the data.
Agreements and disagreements with other studies or reviews
While the reduction or cessation, or both, of antipsychotics appears a rational first line of treatment for TD, the evidence from systematic reviews such as Gilbert 1995 highlights the very high risk of relapse associated with both antipsychotic cessation and antipsychotic reduction. In addition, there is evidence from the literature that intermittent therapy may be associated with more unfavourable TD outcomes (higher scores, fewer eventual remissions etc.) (Bergin 1992; Jeste 1979; NDSG 1986).
It is of interest to note that naturalistic studies suggest that TD fluctuates over time, and often improves in spite of continuing antipsychotic exposure (Casey 1986). Kane 1986 analysed data on 98 people followed up for at least seven years and reported that those on lower doses of antipsychotics were more likely to have an improvement in their TD. Gardos 1988 reported that improvement in TD was associated with lower antipsychotic doses after TD onset. Using a sophisticated epidemiological approach, Morgenstern 1993 looked at risk factors for new cases of TD. Higher average antipsychotic medication was associated with an increased risk of developing persistent TD. However, Cavallaro 1993 assessed TD prognosis (persistent, remitting etc) with respect to antipsychotic dose change (decreased, increased, unchanged) in 125 inpatients followed up for three years. They found no significant association between antipsychotic reduction and improved TD outcome. As with all uncontrolled studies, these data need to be interpreted cautiously, because those who have less responsive psychoses may require higher doses and have higher rates of TD regardless of treatment.
Also of note, in more recent years commentators are more likely to concede that no change in antipsychotic medications may be required for those with TD (Gardos 1994). Casey 1986 suggested, "therefore, antipsychotics in low to moderate doses are not contra‐indicated in patients with psychosis and will not inevitably aggravate TD" (p. 89). There is some evidence to suggest that younger people (under 60 years) who develop TD are three times more likely to recover when compared with those who develop TD after that age (Smith 1980). One interpretation of the data would suggest that, for younger people who develop TD that is not associated with significant impairment, no change to treatment is required other than regular monitoring and reassurance.
Authors' conclusions
Implications for practice.
1. For people with tardive dyskinesia
Currently, this review has no data derived from randomised controlled trials (RCTs) that can adequately support the notion that any particular antipsychotic is an effective treatment for tardive dyskinesia (TD). While products such as clozapine, and the newer 'atypical' antipsychotics such as risperidone have favourable extrapyramidal adverse effect profiles compared with classical antipsychotics, it remains to be seen if these products are associated with lower incidence rates of TD.
2. For clinicians
This review cannot provide clinicians with an effective treatment algorithm involving antipsychotic medications for the treatment of people with TD. While many practice guidelines continue to recommend antipsychotic reduction or cessation, or both (APA 1992; Barnes 1993; Jeste 1993; Shale 1996), this treatment option lacks a sound evidence‐base. There are a considerable amount of RCT‐derived data that demonstrate that this intervention is not safe because of the increased risk of relapse of psychoses (Gilbert 1995).
As outlined in the Background section of this review, there are features related to the interactions between antipsychotics and TD that need to be taken into account when assessing the evidence presented in this review. In particular, TD is thought to worsen in the short term following antipsychotic dose reduction and/or cessation, and TD is thought to be suppressed or masked in the short term following antipsychotic dose increment. These factors combine to favour antipsychotics when compared with placebo for the treatment of TD.
3. For managers and policy makers
There are no adequate data available from this review to provide evidence for managers and policy makers on best practice.
Implications for research.
1. General
The power of this review would have been greatly enhanced by better reporting of data. For example, only three studies made explicit how randomisation was undertaken, and none described allocation concealment. We realise that much of the work for these trials predates CONSORT, which was first published in 1996 (Begg 1996), and that it is only too easy to judge studies of the past by standards of today. Future studies, however, should report to a much higher standard.
2. Specific
2.1 Reviews suggested by excluded studies
As is usual with systematic reviews, we had to exclude several studies that contained comparisons that were in some way related to movement disorders and their treatment. In the case of this review, every one of these trials should have an existing Cochrane Review in which to be considered (Table 6).
2. Excluded studies relevant to schizophrenia: comparisons for existing or potential reviews.
| Study ID | Participants – people with: | Intervention | Comparison for review | Cochrane Review |
| Cai 1988 | Tardive dyskinesia | 1‐stepholidine vs placebo | 1‐stepholidine for schizophrenia | ‐ |
| Speller 1997 | Schizophrenia | Amisulpride vs haloperidol | Amisulpride versus haloperidol for schizophrenia | |
| Gerlach 1978 | Tardive dyskinesia | Biperiden vs no treatment | Anticholinergic drugs for tardive dyskinesia | |
| NDSG 1986 | Chlorprothixene versus haloperidol vs perphenazine vs haloperidol + biperiden | |||
| Greil 1984 | Biperiden vs placebo | |||
| Chouinard 1979 | Schizophrenia | Ethopropazine vs benztropine | Anticholinergics for parkinsonism | |
| Spohn 1988 | Abrupt neuroleptic cessation vs neuroleptic maintenance | Antipsychotic reduction or withdrawal for schizophrenia | ||
| Spohn 1993 | Abrupt neuroleptic cessation vs neuroleptic maintenance | |||
| Wistedt 1983 | Fluphenazine/flupenthixol decanoate continuation vs withdrawal | |||
| Goldberg 1981 | Withdrawal of fluphenazine decanoate vs continuation | |||
| Hershon 1972 | Trifluoperazine withdrawal vs trifluoperazine continuation | |||
| Johnson 1987 | Dose reduction vs maintenance (both arms used flupenthixol decanoate) | |||
| Kinon 2004 | Olanzapine with different timings of dose‐reduction periods | |||
| Levine 1980 | Fluphenazine withdrawal vs continuation | |||
| Marder 1987 | Low‐ vs conventional‐dose maintenance therapy with fluphenazine decanoate | |||
| Newcomer 1992 | Haloperidol dose reduction vs maintained dose | |||
| Singh 1990 | Abrupt neuroleptic cessation vs neuroleptic maintenance | |||
| Zeng 1994 | Tardive dyskinesia | Flunarizine vs placebo | Calcium channel blockers for neuroleptic‐induced tardive dyskinesia | |
| Jeste 1977 | Schizophrenia | Chlorpromazine schedule A vs chlorpromazine schedule B | Chlorpromazine timing of dose for schizophrenia. | |
| NDSG 1986 | Tardive dyskinesia | Chlorprothixene vs haloperidol vs perphenazine vs haloperidol + biperiden | Chlorprothixene for schizophrenia. | |
| Andia 1998 | Schizophrenia | Clozapine vs haloperidol | Clozapine versus haloperidol for schizophrenia | |
| Gerlach 1975 | Clozapine vs haloperidol | |||
| Bitter 2000 | Clozapine vs olanzapine | Clozapine versus olanzapine for schizophrenia | ||
| Jean‐Noel 1999 | Clozapine vs olanzapine | |||
| Caine 1979 | Gilles de la Tourette's, Huntington's disease and drug‐induced atypical dyskinesia | Clozapine vs placebo | Clozapine versus placebo for schizophrenia. | |
| Chouinard 1994 | Schizophrenia | Clozapine versus risperidone | Clozapine versus risperidone for schizophrenia | |
| Chouinard 1989 | Haloperidol decanoate vs fluphenazine decanoate | Depot fluphenazine for schizophrenia | ||
| Cookson 1991 | Fluphenazine decanoate vs haloperidol decanoate | |||
| Curson 1985 | Fluphenazine decanoate vs placebo | |||
| McCreadie 1980 | Fluphenazine decanoate vs intermittent pimozide | |||
| Odejide 1982 | Fluphenazine decanoate vs vitamin B complex | |||
| Chouinard 1978 | Fluphenazine ethanoate vs pipothiazine palmitate | |||
| Chouinard 1989, Cookson 1991 | Haloperidol decanoate vs fluphenazine decanoate | Depot haloperidol decanoate for schizophrenia. | ||
| Chouinard 1978 | Fluphenazine ethanoate vs pipothiazine palmitate | Depot pipothiazine for schizophrenia. | ||
| Burner 1989 | Progabide vs placebo | GABA for schizophrenia | ||
| Bateman 1979 | Tardive dyskinesia and psychiatric history | Metoclopramide (10 mg, 20 mg or 40 mg) vs haloperidol (5 mg or 10 mg) | Haloperdiol dose for schizophrenia | |
| Tran 1997, Rosenheck 2003, Tollefson 1997 | Schizophrenia | Haloperidol vs olanzapine | Haloperidol vs olanzapine for schizophrenia | |
| NDSG 1986 | Tardive dyskinesia | Chlorprothixene vs haloperidol vs perphenazine vs haloperidol + biperiden | Haloperidol vs perphenazine for schizophrenia | |
| Kopala 2004, Wirshing 1999 | Schizophrenia | Haloperidol vs risperidone | Haloperidol vs risperidone for schizophrenia | |
| Jolley 1990 | Brief intermittent antipsychotic treatment vs fluphenazine decanoate | Intermittent antipsychotic treatment for schizophrenia | ||
| McCreadie 1980 | Fluphenazine decanoate vs intermittent pimozide | |||
| Newton 1989 | Haloperidol with 'drug holiday' vs haloperidol | |||
| Goldberg 1981 | Withdrawal of fluphenazine decanoate vs continuation | |||
| MacKay 1980 | Lithium vs placebo | Lithium for schizophrenia | ||
| Borison 1987 | Molidone vs haloperidol | Molidone vs haloperidol for schizophrenia | ||
| Williamson 1995 | Olanzapine 1 mg vs olanzapine 10 mg versus placebo | Olanzapine dose for schizophrenia. | ||
| de Jesus Mari 2004 | Olanzapine vs "conventional antipsychotic drugs" | Olanzapine for schizophrenia | ||
| Peluso 2012 | First‐generation antipsychotic vs second‐generation antipsychotic | |||
| Kinon 2004 | Olanzapine with different timings of dose reduction periods | Olanzapine reduction for schizophrenia | ||
| Peluso 2012 | First‐generation antipsychotic versus second‐generation antipsychotic | Olanzapine vs other atypical antipsychotics for schizophrenia | ||
| Williamson 1995 | Olanzapine 1 mg vs olanzapine 10 mg vs placebo | Olanzapine vs placebo for schizophrenia | ||
| Peluso 2012 | First‐generation antipsychotic vs second‐generation antipsychotic | Perphenazine for schizophrenia | ||
| McCreadie 1980 | Fluphenazine decanoate vs intermittent pimozide | Pimozide for schizophrenia | ||
| Cortese 2008 | Quetiapine vs continuation of usual antipsychotic | Quetiapine vs continuation of usual antipsychotic for schizophrenia | ||
| Peluso 2012 | First generation antipsychotic vs second‐generation antipsychotic | Quetiapine vs other atypical antipsychotics for schizophrenia | ||
| Quetiapine vs typical antipsychotic medications for schizophrenia | ||||
| Risperidone vs olanzapine for schizophrenia | ||||
| Risperidone vs other atypical antipsychotics for schizophrenia | ||||
| Cortese 2008 | Quetiapine vs continuation of usual antipsychotic | Switching antipsychotic for schizophrenia. | ||
| Singer 1971 | Tardive dyskinesia | Thiopropazate vs placebo | Thiopropazate for schizophrenia | |
| Lal 1974 | Schizophrenia | Thiopropazine vs trifluoperazine vs placebo | Thiopropazine vs placebo for schizophrenia | |
| Thiopropazine vs trifluoperazine for schizophrenia | ||||
| Delwaide 1979 | Tardive dyskinesia | Thioproperazine and tiapride vs placebo | Thioproperazine for schizophrenia | |
| Tiapride for schizophrenia | ||||
| Buruma 1982 | Tiapride vs placebo | |||
| Crane 1970 | Schizophrenia | Trifluoperazine high‐dose vs trifluoperazine low‐dose vs placebo | Trifluoperazine dose for schizophrenia | |
| Trifluoperazine vs placebo for schizophrenia | ||||
| Lal 1974 | Thiopropazine vs trifluoperazine vs placebo | |||
| Odejide 1982 | Fluphenazine decanoate vs vitamin B complex | Vitamins for schizophrenia | ||
| Peluso 2012 | First‐generation antipsychotic vs second‐generation antipsychotic | Ziprasidone vs other atypical antipsychotics for schizophrenia |
2.2 Trials
There is a need to assess the utility of second‐generation antipsychotics in the management of established TD. The recent literature highlights the finding that TD can be found in people with schizophrenia who have never been treated, thus challenging basic assumptions about the causality of TD (Waddington 1988). Research is required to differentiate underlying vulnerability to movement disorders (that may be part of a latent trait underlying schizophrenia and TD) from that proportion of TD causally related to antipsychotic use. Research is required that can separate out the variable trajectory inherent in young and midlife‐onset TD from the impact of traditional and 'atypical' antipsychotics.
Well‐designed RCTs, involving a large number of participants over protracted periods of time, are needed if we are to see what role antipsychotics could have in prevention and treatment of TD. Such studies are of importance to people with the problem (Figure 1) and have long been ignored.
2.2.1 Use of cross‐over design
Trialists find it difficult to identify people with both TD and schizophrenia to participate in trials (Schmidt 1991). Randomised, cross‐over design is used in the hope of improving the power of the study to find outcomes of interest. This design initially asks participants to be randomised to one of the experimental interventions, and then, at a pre‐specified time, to be crossed over to the treatment that they did not at first receive. Conditions with a more stable time course than TD are better suited for cross‐over studies (Fleiss 1984). Further difficulties are related to the carry‐over effect. Unless cross‐over studies include a mid‐study washout period (where the person is free of treatment before starting the next arm of the study), any effect of the first intervention may continue into the second half placebo arm of the trial – the 'carry‐over effect'. Also, carry‐over may involve the re‐growth or retreat of neuroreceptors. This slow re‐balancing, if started, could continue long after all traces of intervention drugs are gone, so physiological half‐life of the experimental treatment may not be the only variable to consider when thinking though the issues of carry‐over. TD is also an unstable condition and people with TD may not remain compliant with medication. All these factors make the arguments for not using cross‐over methodology strong, despite the initial attraction (Armitage 1991; Fleiss 1984; Pocock 1983).
2.2.2 Length of study
Only five studies included in this review (Caroff 2011; Cookson 1987; Emsley 2004; Kane 1983; Tamminga 1994) used the intervention for more than six months. TD, however, is a chronic condition of insidious onset, the severity of which fluctuates spontaneously (APA 1992). Another problem in TD research is that spontaneous, age‐related, non‐neuroleptic‐induced dyskinesias occur in people with schizophrenia (Fenton 2000). The spontaneous dyskinetic movements are more prevalent in older age and appear identical to the movements of antipsychotic‐induced tardive dyskinesia (Fenton 2000). In addition, since reducing or switching antipsychotic may have a swift but reversible effect (Cavallaro 1993; Smith 1980), it is the long‐term outcomes that must be considered of most clinical value.
2.2.3 Outcomes
Scale‐derived data do have their place. Trials most commonly used the AIMS scale. This is a very widely applied tool utilised to measure the severity of symptoms of those who have TD. The use of this scale to measure change as a result of treatment is, however, problematic (Bergen 1984). It is therefore important that a scale is validated for measuring changes secondary to treatment in those with TD. In addition, many of the outcomes we initially desired when we started this review have not been investigated. Finally, a service user consultation also informed the addition of outcomes of special importance to patients. We have reconsidered all these outcomes in case they were too ambitious and tried to tailor them to a real‐world pragmatic trial design (see Table 7).
3. Suggestions for design of future study.
| Methods | Allocation: randomised, with sequence generation and concealment of allocation clearly described Blindness: double, tested Duration: 12 months beyond end of intervention at least Raters: independent |
| Participants | People with antipsychotic‐induced tardive dyskinesiaa Age: any Sex: both History: any N = 300b |
| Interventions | 1. Antipsychotic reduction/cessation (N = 150) vs antipsychotic maintenance (N = 150) OR 2. Specific antipsychotic (N = 150) vs other specific antipsychotic (N = 150) |
| Outcomes | Tardive dyskinesia: any clinically important improvement in tardive dyskinesia, any improvement, deteriorationc Adverse effects: no clinically significant extrapyramidal adverse effects ‐ any time periodc, use of any antiparkinsonism drugs, other important adverse events Leaving the study early Service outcomes: admitted, number of admissions, length of hospitalisation, contacts with psychiatric services Compliance with drugs Economic evaluations: cost‐effectiveness, cost‐benefit General state: relapse, frequency and intensity of minor and major exacerbations Social confidence, social inclusion, social networks, or personalised quality of life: binary measure Distress among relatives: binary measure Burden on family: binary measure |
| Notes |
aThis could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. bSize of study with sufficient power to highlight about a 10% difference between groups for primary outcome. cPrimary outcome. The same applies to the measure of primary outcome as for diagnosis. Not everyone may need to have operational criteria applied if clinical impression is proved to be accurate. |
Feedback
Methods and results
Summary
This comment highlights the inconsistencies in the exclusion of studies in the review. In the 'Methods of the review' section some low‐quality trials are excluded but they are included in the 'Results' section.
Reply
The review identified a marked lack of RCT‐derived data. Informative data below RCTs in the hierarchy of evidence were identified from other trials. We chose to briefly mention these data in the discussion, but clearly identify that these trials could not be included in the study. We feel that this would help the clinicians in the absence of better data.
Contributors
Comment received from Ole Olsen, Copenhagen, September 1998 Reply from John McGrath, Brisbane, October 1999
What's new
| Date | Event | Description |
|---|---|---|
| 4 October 2017 | New citation required and conclusions have changed | Results from latest searches do not change overall conclusions of this review |
| 26 April 2017 | New search has been performed | Update search run 26 April, 2017. Eight records found and assessed by editorial base Cochrane Schizophrenia, no new studies relevant to this review found. The 8 records were added to Studies awaiting classification of Miscellaneous treatments for antipsychotic‐induced tardive dyskinesia (Table 12). |
| 25 October 2016 | Amended | Title changed from 'Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for neuroleptic‐induced tardive dyskinesia'. Eight new trials added (Bai 2003; Bai 2005; Caroff 2011; Chan 2010; Chouinard 1995; Kazamatsuri 1973; Lublin 1991; Tamminga 1994), analyses and text updated, conclusions not substantially changed. |
History
Protocol first published: Issue 3, 1997 Review first published: Issue 2, 1998
| Date | Event | Description |
|---|---|---|
| 16 July 2015 | Amended | Update search run July 16, 2015. 704 records found and assessed by review authors. |
| 26 April 2008 | Amended | Converted to new review format. |
| 20 September 2005 | New citation required and conclusions have changed | Substantive amendment |
| 25 February 1998 | New search has been performed | Review first published |
| 2 June 1997 | New search has been performed | Protocol first published |
Notes
Cochrane Schizophrenia Group internal peer review complete (see Group’s Module). External peer review scheduled.
Acknowledgements
In earlier versions of this review (1998 to 2002), John McGrath was a reviewer and made a considerable contribution to the review by developing the protocol, data extracting, data assimilation, and report writing. We are grateful for his substantial input.
We are indebted to Kirsten Mason, Tracey Richardson, Geoff Davies, Carmel Meir, Leanne Roberts, Nicholas Henschke, and Loukia Spineli for assistance with this review. The following authors kindly provided additional information in order to assist this review: Dr D Bateman, Dr M Campbell, Dr S Caroff, Dr G Chouinard, Dr I Cookson, Dr J de Jesus Mari, Dr M Herz, Dr D Johnson, Dr J Kalachnik, Dr J Kane, Dr S Lal, Dr G Paulson, Dr H Spohn, Dr N Quinn, and Dr M Woerner.
We are grateful to Judy Wright (2005) and Farhad Sokraneh (2015, 2017) for the updated trial searches and to Clive Adams for his constant advice.
We thank Rosie Asher and Antonio Grande for screening literature and helping with data extraction for the 2017 update, and Ben Gray for writing the Plain language summary. We are also grateful to Dawn‐Marie Walker, Ruth Sayers, Megan Lees, and Vanessa Pinfold from McPin Foundation for organising and holding the public and patient involvement consultation with TD service users that contributed to selecting outcomes for the 'Summary of findings' tables and to guide future research. Finally, we wish to thank Sai Zhao for assessing articles in Chinese.
Appendices
Appendix 1. Previous methods
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. We included trials that were described as double‐blind, but that did not mention whether the study was randomised, in a sensitivity analysis. If there was no substantive difference within primary outcomes (see 'Types of outcome measures') when these studies were added, then we included them in the final analysis. If there was a substantive difference, we used only clearly randomised trials and described the results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people with schizophrenia or any other serious mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who required the use of neuroleptics for more than three months and who developed TD (diagnosed by any criteria) during neuroleptic treatment, and for whom the dose of neuroleptic medication had been stable for one month or more. A post hoc change was made to include studies that did not require neuroleptic medication to have been stable for one month prior to randomisation. We felt it important to include these studies as they will provide additional important information. However, these will be analysed separately, and thus will not change existing outcome data.
Types of interventions
1. Reduction or cessation of the dose of the above drugs compared with the continuation of standard dose of the same compound. For the purposes of this review, these trials were divided into those that aimed to reduce the total dosage of neuroleptic medication, for example reduced dose and intermittent dosage schedule studies, and those that cease neuroleptics (sometimes after variable periods of dose reduction).
2. Specific neuroleptic drugs proposed to have TD lessening qualities compared to placebo or no intervention. A post hoc decision was made to broaden this criteria to also include neuroleptic versus neuroleptic for the treatment of TD.
Types of outcome measures
Clinical efficacy was defined as an improvement in the symptoms of TD of more than 50% on any scale.
The outcomes of interest were:
1. Tardive dyskinesia changes: 1.1. The number of people per treatment group who did not show an improvement of more than 50% on any TD scale 1.2. The number of people per treatment group who did not show any improvement on any TD scale 1.3. The number of people per treatment group who deteriorated on any TD scale 1.4. Treatment group mean change (endpoint ‐ baseline) on any TD scale 1.5 .Treatment group mean change endpoint on any TD scale
2. Global assessment: 2.1. The number of people per treatment group who were defined as relapsed (according to any definition) 2.2. Leaving the study early
3. Adverse effects: 3.1. The number of people per treatment group who had any adverse effect (other than deterioration of symptoms of TD or relapse)
When appropriate, the outcomes were grouped into time periods ‐ short term (less than 6 weeks), medium term (between 6 weeks and 6 months) and long term (over 6 months). Clinical efficacy was defined as an improvement in the symptoms of TD of more than 50%, on any scale.
Primary outcomes
Secondary outcomes
Search methods for identification of studies
1. Electronic searching for update (July 2005).
1.1. We searched The Cochrane Schizophrenia Group's Trials Register (July 2005) using the phrase:
[((dyskine* or diskine*) in title, abstract, index terms of REFERENCE) or ((dyskine* in health care conditions of STUDY)]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module).
2. We electronically searched previous versions of the CSG library
2.1. We identified relevant randomised trials by searching several electronic databases (the Cochrane Schizophrenia Group's Register of trials), Biological Abstracts, EMBASE, LILACS, MEDLINE, PsycLIT and SCISEARCH).
2.2. We searched the The Cochrane Schizophrenia Group's Register using the following phrases:
[(dyskine*) and (*amitrex* or *deniban* or *socian* or *solian* or *sulanid* or *sulamid* or *zyprex* or *olanzapin* or *ziprasidone* or *zotepine* or *lodopin* or *nipolept* or *zopite* or *setous* or *majpin* or *cloza* or *leponex* or *71675‐85‐9* or *neuroleptic* or chlorpromazine* or chlorprothixene* or dogmatil* or droperidol* or flupenthixol* or fluperlapine* or fluphenazine* or flunarizine* or haloperidol* or lonidine* or loxapine* or molindone* or penfluridol* or perphenazine* or pimozide* or piperacetazine* or sulpiride* or thiopropazate* or thioridazine* or thiothixene* or trifluoperazine* or clozapine* or zuclopenthixol* or risperidone* or pericyazine in title) or (*reduction* or *cessation* or *withdrawal* or *decrease* or *intermittent* or *target*) and (dyskine*)] and (*amitrex* or *deniban* or *socian* or *solian* or *sulanid* or *sulamid* or *zyprex* or *olanzapin* or *ziprasidone* or *zotepine* or *lodopin* or *nipolept* or *zopite* or *setous* or *majpin* or *cloza* or *leponex* or *71675‐85‐9* or *neuroleptic* or *chlorpromazine* or *chlorprothixene* or *dogmatil* or *droperidol* or *flupenthixol* or *fluperlapine* or *fluphenazine* or *flunarizine* or *haloperidol* or *lonidine* or *loxapine* or *molindone* or *penfluridol* or *perphenazine* or *pimozide* or *piperacetazine* or *sulpiride* or *thiopropazate* or *thioridazine* or *thiothixene* or *trifluoperazine* or *clozapine* or *zuclopenthixol* or *risperidone* or *pericyazine in title, abstract, index terms of REFERENCE) or ((antipsychotics or Targeted Medication* or Treatment Frequency* or Withdrawal* or Dosage of Drug* in interventions of STUDY) and (tardive in health care conditions of STUDY)]
[(dyskine* or #30 = 28) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine or #42 = 500)]
[(reduction or cessation or withdrawal or decrease or intermittent or target*) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
2.3. We searched BIOLOGICAL ABSTRACTS (January 1982 to September 1997) using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with each of the two phrases:
[and ((tardive near (dyskine* or diskine*) or (abnormal near movement* near disorder*) or (involuntar* near movement*)) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
[and (reduction or cessation or withdrawal or decrease or intermittent or target*) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
2.4. We searched EMBASE (January 1980 to September 1997) using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with each of the two phrases:
[and ((tardive dyskinesia in thesaurus ‐subheadings, prevention, drug therapy, side effect and therapy) or (neuroleptic dyskinesia in thesaurus ‐all subheadings) or (tardive or dyskines*) or (movement* or disorder*) or (abnormal or movement* or disorder*)) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
[and (reduction or cessation or withdrawal or decrease or intermittent or target*) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
2.5. We searched LILACS (January 1982 to September 1996) using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with each of the two phrases:
[and ((tardive or (dyskinesia* or diskinesia*)) or (drug induced movement disorders in thesaurus)) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
[and (reduction or cessation or withdrawal or decrease or intermittent or target*) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
2.6. We searched MEDLINE (January 1966 to September 1997) using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with each of the two phrases:
[and ((movement‐disorders in MeSH / explode all subheadings) or (anti‐dyskinesia‐agents in MeSH / explode all subheadings) or (dyskinesia‐drug‐induced in MeSH / explode all subheadings) and (psychosis in MeSH / explode all subheadings) or (schizophrenic disorders in MeSH / explode all subheadings) or (tardive near (dyskine* or diskine*)) or (abnormal* near movement* near disorder*) or (involuntar* near movement*)) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
[and (reduction or cessation or withdrawal or decrease or intermittent or target*) and (antipsychotic‐agents / all subheadings or neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
2.7. We searched PsycLIT (January 1974 to September 1997) using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with each of the two phrases:
[and ((explode movement‐disorders in DE) or (explode tardive‐dyskinesia in DE) or (tardive near (dyskine* or diskine*) or (abnormal* near movement* near disorder*) or (involuntar* near movement*)) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)] [and (reduction or cessation or withdrawal or decrease or intermittent or target*) and (neuroleptic* or chlorpromazine or chlorprothixene or dogmatil or droperidol or flupenthixol or fluperlapine or fluphenazine or flunarizine or haloperidol or lonidine or loxapine or molindone or penfluridol or perphenazine or pimozide or piperacetazine or sulpiride or thiopropazate or thioridazine or thiothixene or trifluoperazine or clozapine or zuclopenthixol or risperidone or pericyazine)]
3. Reference searching 3.1. SCISEARCH ‐ Science Citation Index We sought each of the included studies as a citation on the SCISEARCH database. We inspected reports of articles that had been cited from these studies in order to identify further trials.
3.2. We inspected the references of all identified studies for more studies.
4. Personal contact We contacted the first author of each included study for information regarding unpublished trials.
Electronic searches
Searching other resources
Data collection and analysis
[For definitions of terms used in this, and other sections, please refer to the Glossary].
1. Selection of trials We (KSW and JR) independently inspected citations from the searches and identified relevant abstracts. Where doubts arose, we acquired the full report for more detailed scrutiny. We obtained the full report of the abstracts that met the review criteria. Again, when resolving disputes by discussion was not possible, the article was added to those awaiting assessments and we contacted the authors of the study for clarification.
2. Assessment of methodological quality KSW allocated trials to three quality criteria as described in the Cochrane Collaboration Handbook (Higgins 2005) and only trials meeting category A or B were included. In addition, we rated trials using the Jadad Scale (Jadad 1996) with a cut‐off of two points used to check the assessment made by the handbook criteria. However, the latter were not used to exclude trials in this review.
3. Data management 3.1. Data extraction We (KSW and JR) independently extracted data from included studies. Again, any disagreements were discussed, the decisions documented and, where necessary, we contacted the authors of the studies for clarification. Justifications for excluding references from the review were documented. We anticipated that many trials would have inadequate reporting and therefore, for those who dropped out of studies, we assumed they had no change in their TD symptoms. When insufficient data were provided to identify the original group size (prior to drop outs), we contacted the authors and the trials were allocated to the list of those awaiting assessment.
3.2. Intention to treat analysis We excluded data from studies where more than 50% of participants in any group were lost to follow up (this does not include the outcome of 'leaving the study early'). In studies with less than 50% dropout rate, people leaving early were considered to have had the negative outcome, except for the event of death.
3.3. Dichotomous yes/no ‐ data We analysed dichotomous outcomes by calculating the relative risk (RR) (Fixed effects) with a 95% confidence interval (CI) to express the uncertainty of each result. The relative risk ratios from the individual trials were combined using appropriate methods of meta‐analysis. When overall results were significant, the number needed to treat (NNT) to produce (or prevent) one outcome was calculated by combining the overall (RR) with an estimate of the prevalence of the event in the control groups of the trials. If heterogeneity was found (see section 5) we used a random effects model.
3.4 Continuous data 3.4.1. Skewed data: continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, the following standards were applied to all data before inclusion: (a) standard deviations and means were reported in the paper or were obtainable from the authors; (b) when a scale started from the finite number zero, the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); (c) if a scale started from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied to them. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale), it is difficult to tell whether data are non‐normally distributed (skewed) or not.
For change data (endpoint minus baseline), the situation is even more problematic. In the absence of individual patient data it is impossible to know if data are skewed, though this is likely. After consulting the ALLSTAT electronic statistics mailing list, we presented change data in MetaView in order to summarise available information. In doing this, it was assumed either that data were not skewed or that the analyses could cope with the unknown degree of skew. Without individual patient data it is impossible to test this assumption. Where both change and endpoint data were available for the same outcome category, we only presented endpoint data. We acknowledge that by doing this much of the published change data were excluded, but argue that endpoint data is more clinically relevant and that if change data were to be presented along with endpoint data, it would be given undeserved equal prominence. We are contacting authors of studies reporting only change data for endpoint figures. Non‐normally distributed data were reported in the 'other data types' tables.
3.4.2. Rating Scales A wide range of instruments are available to measure outcomes in mental health studies. These instruments vary in quality and many are not validated, or are even ad hoc. As a minimum standard, we did not include data from an instrument in this review unless the instrument and its properties had been published in a peer‐reviewed journal. In addition, the following minimum standards for instruments were set: The instrument should either be (a) a self‐report or (b) completed by an independent rater or relative (not the therapist) and (c) the instrument should be a global assessment of an area of functioning.
3.4.3. Intention to treat analysis Where possible we analysed data on an intention‐to‐treat basis and assumed that those who had not been accounted for had the negative outcome e.g. global assessment 'not improved' and relapse. This rule did not include the outcomes death or adverse effects. We tested this assumption with a sensitivity analysis. For continuous data it is impossible to manage the data in this way therefore we presented 'completer' data. Where feasible, we converted continuous scores to dichotomous data.
If, for a given outcome, more than 50% of the total numbers randomised were not accounted for, we did not present the results (except for leaving the study early) as such data are impossible to interpret with authority. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked data with '*' to indicate that the result may well be prone to bias.
4. Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
5. Investigation for heterogeneity
Firstly, we considered all the included studies within any comparison to judge clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. This was supplemented, primarily, by employing the I‐squared statistic. This provides an estimate of the percentage of inconsistency thought to be due to chance. Where the I‐squared estimate was greater than or equal to 75%, this was interpreted as evidence of high levels of heterogeneity (Higgins 2003). Data were then re‐analysed using a random effects model to see if this made a substantial difference. If it did, and results became more consistent, i.e. falling below 75% in the estimate, the studies were added to the main body of trials. If using the random effects model did not make a difference and inconsistency remained high, data were not summated, but were presented separately and reasons for heterogeneity investigated.
6. Addressing publication bias We used funnel plots (trial effect versus trial size) for outcomes where more than five trials reported usable data, and visually inspected for asymmetry in an attempt to investigate the likelihood of overt publication bias (Egger 1997).
Data and analyses
Comparison 1. Reduced overall dose of antipsychotic vs antipsychotic maintenance.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: no clinically important improvement (long term) | 2 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.17, 1.04] |
| 2 Tardive dyskinesia: no improvement (long term) | 2 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.17, 1.04] |
| 3 Tardive dyskinesia: deterioration (long term) | 2 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.11, 3.31] |
| 4 General mental state: relapse (long term) | 1 | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.16, 57.36] |
| 5 Acceptability of the treatment: leaving the study early (long term) | 1 | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.06, 1.99] |
Comparison 2. Switch to specific antipsychotic vs antipsychotic cessation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: no clinically important improvement (medium term) | 1 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.23, 0.89] |
| 2 Tardive dyskinesia: average endpoint score (AIMS, high = poor) (medium term) | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐5.5 [‐8.60, ‐2.40] |
| 3 General mental state: average endpoint score (BPRS, high = poor) (medium term) | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐4.30 [‐10.48, 1.88] |
| 4 Acceptability of the treatment: leaving the study early (medium term) | 1 | 50 | Risk Ratio (IV, Fixed, 95% CI) | 0.6 [0.16, 2.25] |
| 5 Adverse effects: use of antiparkinsonism drugs (medium term) | 1 | 48 | Risk Ratio (IV, Fixed, 95% CI) | 2.08 [0.74, 5.86] |
| 5.1 Haloperidol | 1 | 12 | Risk Ratio (IV, Fixed, 95% CI) | 2.0 [0.56, 7.09] |
| 5.2 Risperidone | 1 | 36 | Risk Ratio (IV, Fixed, 95% CI) | 2.26 [0.37, 13.60] |
| 6 Adverse effects: parkinsonism ‐ average endpoint score (ESRS) (medium term) | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐1.25, 0.45] |
| 7 Adverse effects: dystonia ‐ average endpoint score (ESRS) (medium term) | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐1.76, 0.36] |
Comparison 3. Switch to a specific antipsychotic vs switch to a different antipsychotic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: no clinically important improvement | 4 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Thiopropazate vs haloperidol ‐ short term | 1 | 20 | Risk Ratio (IV, Fixed, 95% CI) | 1.53 [0.58, 4.05] |
| 1.2 Zuclopenthixol vs haloperidol ‐ short term | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 1.0 [0.79, 1.27] |
| 1.3 Olanzapine vs risperidone ‐ medium term | 1 | 60 | Risk Ratio (IV, Fixed, 95% CI) | 1.25 [0.82, 1.90] |
| 1.4 Quetiapine vs haloperidol ‐ medium term | 1 | 45 | Risk Ratio (IV, Fixed, 95% CI) | 0.80 [0.52, 1.22] |
| 1.5 Quetiapine vs haloperidol ‐ long term | 1 | 45 | Risk Ratio (IV, Fixed, 95% CI) | 0.88 [0.64, 1.21] |
| 2 Tardive dyskinesia: not any improvement (short term) | 2 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Thiopropazate vs haloperidol | 1 | 20 | Risk Ratio (IV, Fixed, 95% CI) | 0.41 [0.05, 3.28] |
| 2.2 Zuclopenthixol vs haloperidol | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 0.88 [0.16, 4.68] |
| 3 Tardive dyskinesia: deterioration (short term) | 2 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Thiopropazate vs haloperidol | 1 | 20 | Risk Ratio (IV, Fixed, 95% CI) | 1.22 [0.09, 16.92] |
| 3.2 Zuclopenthixol vs haloperidol | 1 | 15 | Risk Ratio (IV, Fixed, 95% CI) | 0.88 [0.16, 4.68] |
| 4 Tardive dyskinesia: average endpoint score (various scales) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Molindone vs haloperidol, 100% masking dose (AIMS, short term) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 1.87 [‐0.20, 3.94] |
| 4.2 Molindone vs haloperidol, 200% masking dose (AIMS, short term) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 3.44 [1.12, 5.76] |
| 4.3 Zuclopenthixol vs haloperidol (SHRS, short term) | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐4.81 [‐12.15, 2.53] |
| 4.4 Olanzapine vs risperidone (AIMS, medium term) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 2.20 [‐0.53, 4.93] |
| 5 Tardive dyskinesia: average change score (AIMS, low = better) (medium term) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 1.66 [‐0.45, 3.77] |
| 5.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.82 [‐2.85, 1.21] |
| 5.3 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 2.48 [0.44, 4.52] |
| 5.4 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.20 [‐2.58, 4.98] |
| 6 General mental state: deterioration | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Zuclopenthixol vs haloperidol ‐ short term | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.01, 6.29] |
| 6.2 Olanzapine vs risperidone ‐ medium term | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.15, 6.64] |
| 6.3 Quetiapine vs haloperidol ‐ long term | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.62, 5.39] |
| 7 General mental state: average endpoint score (PANSS‐general psychopathology, low = better) (long term) | 1 | 45 | Mean Difference (IV, Fixed, 95% CI) | ‐2.20 [‐6.02, 1.62] |
| 7.1 Quetiapine vs haloperidol | 1 | 45 | Mean Difference (IV, Fixed, 95% CI) | ‐2.20 [‐6.02, 1.62] |
| 8 General mental state: average change score (BPRS, low = better) (medium term) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐1.14 [‐4.79, 2.51] |
| 8.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐2.46 [‐6.27, 1.35] |
| 8.3 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐8.37, 4.97] |
| 8.4 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 1.32 [‐1.94, 4.58] |
| 9 Acceptability of the treatment: leaving the study early (short term) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Molindone vs haloperidol | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 Thiopropazate vs haloperidol | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.01, 4.44] |
| 10 Acceptability of the treatment: leaving the study early (medium term) | 3 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 Olanzapine vs FGA | 1 | 56 | Risk Ratio (IV, Fixed, 95% CI) | 1.86 [0.18, 19.38] |
| 10.2 Amisulpride vs FGA | 1 | 55 | Risk Ratio (IV, Fixed, 95% CI) | 0.96 [0.06, 14.65] |
| 10.3 Olanzapine vs amisulpride | 1 | 57 | Risk Ratio (IV, Fixed, 95% CI) | 1.93 [0.19, 20.12] |
| 10.4 Olanzapine vs risperidone | 2 | 170 | Risk Ratio (IV, Fixed, 95% CI) | 0.73 [0.57, 0.95] |
| 10.5 Olanzapine vs quetiapine | 1 | 116 | Risk Ratio (IV, Fixed, 95% CI) | 0.70 [0.54, 0.90] |
| 10.6 Olanzapine vs ziprasidone | 1 | 82 | Risk Ratio (IV, Fixed, 95% CI) | 0.77 [0.56, 1.05] |
| 10.7 Quetiapine vs risperidone | 1 | 118 | Risk Ratio (IV, Fixed, 95% CI) | 1.05 [0.88, 1.25] |
| 10.8 Quetiapine vs ziprasidone | 1 | 90 | Risk Ratio (IV, Fixed, 95% CI) | 1.10 [0.86, 1.40] |
| 10.9 Ziprasidone vs risperidone | 1 | 84 | Risk Ratio (IV, Fixed, 95% CI) | 0.95 [0.74, 1.23] |
| 11 Acceptability of the treatment: leaving the study early (long term) | 2 | Risk Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1 Clozapine vs haloperidol | 1 | 39 | Risk Ratio (IV, Fixed, 95% CI) | 3.36 [0.45, 25.16] |
| 11.2 Quetiapine vs haloperidol | 1 | 45 | Risk Ratio (IV, Fixed, 95% CI) | 1.31 [0.63, 2.69] |
| 12 Adverse events: extrapyramidal symptoms (need of antiparkinsonism drugs) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 Risperidone vs haloperidol (medium term) | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.34, 1.35] |
| 12.2 Quetiapine vs haloperidol (long term) | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.21, 0.96] |
| 13 Adverse effects: parkinsonism (SHRS) ‐ average endpoint scores (short term) | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐4.81 [‐12.15, 2.53] |
| 13.1 Zuclopenthixol vs haloperidol | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐4.81 [‐12.15, 2.53] |
| 14 Adverse effects: parkinsonism (SAS, ESRS, low = better) ‐ average change score (medium term) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.85 [‐2.55, 0.85] |
| 14.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐2.45, 1.45] |
| 14.3 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.7 [‐1.33, ‐0.07] |
| 14.4 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐2.44, 1.74] |
| 15 Adverse effects: dyskinesia (ESRS, low = better) ‐ average change score (medium term) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.1 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.91, 1.51] |
| 16 Adverse effects: akathisia (BAS, ESRS, low = better) ‐ average change scores (medium term) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 16.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 0.08 [‐0.30, 0.46] |
| 16.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.42, 0.20] |
| 16.3 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.8 [‐1.76, 0.16] |
| 16.4 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 0.19 [‐0.12, 0.50] |
| 17 Adverse effects: dystonia (ESRS, low = better) ‐ average change score (medium term) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.1 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.7 [‐1.41, 0.01] |
| 18 Adverse effects: general adverse events (UKU, low = better) ‐ average change score (medium term) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 0.08 [‐1.85, 2.01] |
| 18.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.55 [‐2.33, 1.23] |
| 18.3 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 0.63 [‐0.93, 2.19] |
| 19 General global state: average change score (CGI) (medium term) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 19.1 Olanzapine vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.41, 0.27] |
| 19.2 Amisulpride vs FGA | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.19 [‐0.47, 0.09] |
| 19.3 Olanzapine vs risperidone | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.61, 0.81] |
| 19.4 Olanzapine vs amisulpride | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.19, 0.43] |
Comparison 4. Specific antipsychotic vs other drugs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesias: no clinically important improvement (medium term) | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 1.07 [0.51, 2.23] |
| 1.1 Haloperidol vs tetrabenazine | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 1.07 [0.51, 2.23] |
| 2 Tardive dyskinesia: no improvement (medium term) | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 2.57 [0.35, 18.68] |
| 2.1 Haloperidol vs tetrabenazine | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 2.57 [0.35, 18.68] |
| 3 Tardive dyskinesia: deterioration (medium term) | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 0.86 [0.07, 10.96] |
| 3.1 Haloperidol vs tetrabenazine | 1 | 13 | Risk Ratio (IV, Fixed, 95% CI) | 0.86 [0.07, 10.96] |
| 4 Acceptability of the treatment: leaving the study early (medium term) | 1 | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.38 [0.25, 76.54] |
| 4.1 Haloperidol vs tetrabenazine | 1 | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.38 [0.25, 76.54] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bai 2003.
| Methods | Allocation: "randomly assigned", not described
Blindness: "double blind", partially described
Design: parallel groups
Setting: inpatients, Taiwan Duration: 12 weeks |
|
| Participants | Diagnosis: schizophrenia with persistent severe TD (DSM IV, Kane criteria)
N = 49 randomised, 42 completed
Age: 50.2 (SD 9.7) years
Sex: 28 men and 14 female History: maintenance on conventional antipsychotics for > 1 year with an equivalent dosage of < 200 mg/d of chlorpromazine; duration of TD not reported |
|
| Interventions | After a 4‐week washout period with all original conventional antipsychotics discontinued: 1. Risperidone: started at 2 mg/d and increased, with a 2‐mg increase every 2 weeks, to 6 mg/d over 6 weeks; then maintenance dose 6 mg/d for 12 weeks. N = 22 2. Placebo: placebo for 12 weeks. N = 20 Concomitant medication included benzodiazepines (86%‐90%) and antiparkinsonism drugs (50%‐86%) |
|
| Outcomes | TD symptoms: AIMS Adverse effects: extrapyramidal symptoms (parkinsonism) (ESRS) Adverse effects: dystonia (ESRS) TD symptoms: clinical efficacy (decrease in AIMS of 3 or 4 = responder) BPRS |
|
| Notes | Sponsorship source: supported by Janssen‐Cilag Taiwan, Johnson & Johnson Taiwan Ltd | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "subjects were randomly assigned to the risperidone or placebo groups", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double‐blind" "A placebo with an identical appearance to the risperidone dose was prescribed for the placebo group using the same dose schedule." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The TD condition was evaluated blindly by a psychiatrist with the Abnormal Involuntary Movement Scale (AIMS) every 2 weeks" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Forty‐two patients completed the 12‐week study and 7 subjects withdrew. Four subjects dropped out due to psychotic symptom exacerbation (2 subjects during the washout period: 1 subject in the placebo group and 1 subject in the risperidone group). Another 3 subjects withdrew due to a medical condition (infectious disease, heart condition, and lung carcinoma)." |
| Selective reporting (reporting bias) | Unclear risk | Unclear if all pre‐defined outcomes were reported. A protocol is not available for verification. |
| Other bias | Low risk | The study seems to be free of other sources of bias. |
Bai 2005.
| Methods | Allocation: "randomized", not described
Blindness: "single blind", partially described
Design: parallel groups
Setting: Inpatients, Taiwan Duration: 24 weeks |
|
| Participants | Diagnosis: schizophrenia (DSM IV), Schooler and Kane's criteria for persistent TD
N = 80
Age: 50.2 (SD 7.1) years
Sex: 39 men and 41 women History: duration of TD not reported; treatment with conventional antipsychotics for > one year |
|
| Interventions | No washout period on the discontinuation of all conventional antipsychotics was reported 1. Olanzapine: dose not reported, 24 weeks. N = 27 2. Amisulpride: dose not reported, 24 weeks. N = 27 3. First generation antipsychotic (FGA): dose not reported, 24 weeks. N = 26 |
|
| Outcomes | TD symptoms: AIMS Adverse effects: extrapyramidal side effects (SAS) Adverse effects: akathisia (BAS) Adverse effects: general (UKU) General mental state (BPRS) Leaving the study early |
|
| Notes | Sponsorship source: the study was supported by grants from National Science Council, Taiwan. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The subjects were randomized to three groups", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "single‐blind and controlled study" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "single‐blind and controlled study" Blinding details of outcome assessors not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Finally 76 cases (95%) completed the 24‐week study, 2 cases in the olanzapine groups withdrew due to impaired liver function, 1 case in the amisulpride group due to infectious disease, and 1 case in the FGA controlled groups withdrew due to unstable psychiatric condition" “All data were analyzed on an intent‐to‐treat basis, and endpoint data were generated with the last observation carried forward (LOCF).” |
| Selective reporting (reporting bias) | Low risk | All outcomes appear to have been reported |
| Other bias | Low risk | The study seems to have been free of other sources of bias. |
Caroff 2011.
| Methods | Allocation: "randomly assigned", not described
Blindness: "double blind", partially described.
Design: post hoc analysis of parallel‐group RCT
Setting: inpatients, USA Duration: 18 months |
|
| Participants | Diagnosis: schizophrenia and TD (DSM IV, Schooler‐Kane criteria)
N = 200
Age: 47.2 (SD 9.4) years (18‐65 years)
Sex: 158 men and 42 women History: duration of TD not reported |
|
| Interventions | Overlap in administration of the antipsychotic drugs that participants received before study entry was permitted for the first 4 weeks after randomisation to allow a gradual transition to study medication: 1. Oolanzapine: flexible dose of 7.5 mg each day/twice a day/3 times a day/4 times a day for 18 months. N = 54 2. quetiapine: flexible dose of 200 mg each day/twice a day/3 times a day/4 times a day for 18 months. N = 62 3. Rrisperidone: flexible dose of 1.5 mg each day/twice a day/3 times a day/4 times a day for 18 months. N = 56 4. ziprasidone: flexible dose of 40 mg each day/twice a day/3 times a day/4 times a day for 18 months. N = 28 Medications were flexibly dosed with 1‐4 capsules daily, as judged by the study doctor. Concomitant medications were permitted, except for additional antipsychotic agents. |
|
| Outcomes | Leaving the study early Unable to use ‐ AIMS, PANSS, SAS, BAS, cogitive composite score (not reported in means and SDs for the separate intervention groups)* |
|
| Notes | Sponsorship source: supported by the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project, National Institute of Mental Health. This article was based on results from the CATIE project, supported by the National Institute of Mental Health. Astra Zeneca Pharmaceuticals LP, Bristol‐Myers Squibb Company, Forest Pharmaceuticals, Inc., Janssen Pharmaceutica Products, L.P., Eli Lilly and Company, Otsuka Pharmaceutical Co., Ltd., Pfizer Inc., and Zenith Goldline Pharmaceuticals, Inc., provided medications for the studies. This material is based upon work also supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research Development, with resources and the use of facilities at the Philadelphia Veterans Affairs Medical Center. *Study author kindly replied to our request for data. At the time of preparing this review no more outcome data were available. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were initially randomly assigned", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "...double‐blind conditions..." "Identical‐appearing capsules contained olanzapine (7.5 mg), quetiapine (200 mg), risperidone (1.5 mg), perphenazine (8 mg), or ziprasidone (40 mg)." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of outcome assessors not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary clinical outcome measure was time to all‐cause treatment discontinuation. Total population (N = 200): 74% discontinuation olanzapine: 31/54 (57%); quetiapine: 51/62 (82%); risperidone: 44/56 (79%); zipraidone: 21/28 (75%). Reasons for withdrawal reported |
| Selective reporting (reporting bias) | High risk | Original CATIE study: "The primary clinical outcome measure was time to all‐cause treatment discontinuation. Secondary outcomes included discontinuations for intolerability, inefficacy, and patient decision; rates of discontinuations; mean modal dose; and change from baseline in the PANSS and neurocognitive composite scores/".TD: "The primary outcome measure used to evaluate the course of TD was change from baseline in total AIMS score. Secondary outcome measures included change in global, distress, and impairment of function items on the AIMS; percentage of patients meeting Schooler‐Kane criteria for at least 2 consecutive visits post baseline; percentage of visits at which patients met modified Schooler‐Kane criteria; and percentage of patients with at least a 50% change in AIMS score (excluding month 1). In addition, treatment differences with respect to all cause discontinuation are described for patients with TD at baseline." |
| Other bias | High risk | Post hoc analysis; modified diagnostic criteria for TD were applied at baseline, and a 3‐month history of antipsychotics exposure was not required. |
Chan 2010.
| Methods | Allocation: "randomly assigned by coin method"
Blindness: single‐blind (outcome assessor)
Design: parallel groups
Setting: inpatients, Taiwan Duration: 24 weeks |
|
| Participants | Diagnosis: schizophrenia (58) and schizoaffective disorder (2) (DSM‐IV criteria); antipsychotic‐induced TD
N = 60
Age: 45.3 (SD 11.6) years (range 18‐70 years)
Sex: 21 men and 39 women History: duration of TD not reported. Antipsychotic exposure ˜10 years. All of the participants received FGAs prior to participation in this study. |
|
| Interventions | Following a washout period of 3‐7 days: 1. risperidone: flexible dose of 1.9 ± 0.7 (baseline) to 4.1 ± 1.4 (end point) mg/d for 24 weeks. N = 30 2. olanzapine: flexible dose of 8.1 ± 2.0 (baseline) to 12.6 ± 5.4 (end point) mg/d for 24 weeks. N = 30 |
|
| Outcomes | TD symptoms: no clinical improvement > 50% (AIMS) TD symptoms: AIMS Adverse effect: dyskinesia Adverse effect: parkinsonism Adverse effect: dystonia Adverse effect: akathisia Adverse effects: general adverse events (39/30 vs 31/30) General mental state: BPRS Leaving the study early |
|
| Notes | Sponsorship source: supported by research grant from the Taoyuan Mental Hospital and from the Department of Health, Executive Yuan, Taiwan | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomly assigned to receive either olanzapine or risperidone with a 1‐to‐1 ratio by coin method with a 6‐block design". |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "...primary care physicians and patients were not blinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | “Two investigators (C.‐H.C. and J.‐J.C.) served as blinded raters.” "The BPRS, CGI‐S, AIMS and global impression of ESRS were performed at baseline and at weeks 1, 2, 3, 4, 8, 12, 16, 20, and 24 or at end point visit by blinded‐rater" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 9/30 in the risperidone and 7/30 in the olanzapine groups dropped out from the study; reasons reported. "All patients who were randomly assigned and had at least 1 post‐baseline assessment were included in the intent‐to‐treat (ITT) analysis. If the ITT subjects withdrew from the study earlier than scheduled, then the last observation carried forward method was employed to extend the end point scores." |
| Selective reporting (reporting bias) | Low risk | Data for all outcomes in the trial registry, NCT00621998, have been reported. |
| Other bias | Low risk | The study seems to be free of other sources of bias. |
Chouinard 1995.
| Methods | Allocation: "randomly assigned", not described
Blindness: "double blind", partially described
Design: post hoc analysis of parallel, 6‐group RCT
Setting: inpatients, Canada Duration: 8 weeks |
|
| Participants | Diagnosis: chronic schizophrenia (DSM‐III R criteria)
N = 135
Age: mean 39 years, range 19‐60 years
Sex: 34 men and 14 women History: duration TD not reported; the most common pre‐study medications were haloperidol, procyclidine, lorazepam, benztropine and chlorpromazine; the most commonly used depot antipsychotic agents were haloperidol decanoate, fluphenazine decanoate, flupenthixol decanoate and pipothiazine palmitate. |
|
| Interventions | Mean duration of washout phase 6 days. 1. Risperidone: dose 2 mg/d for 8 weeks. N = 8 2. Risperidone: dose 6 mg/d for 8 weeks. N = 6 3. Risperidone: dose 10 mg/d for 8 weeks. N = 6 4. Risperidone: dose 16 mg/d for 8 weeks. N = 11 5. Haloperidol: dose 20 mg/d. N = 6 6. Placebo: N = 11 "At the time of selection, all psychotropic and antiparkinsonism medications were discontinued"; "no other psychotropic medication was administered except for chloral hydrate or a benzodiazepine if a sedative/hypnotic was required.", "An antiparkinsonian medication (biperiden or procyclidine) was given in case of the emergence of clinically significant drug‐induced parkinsonism and dystonia" |
|
| Outcomes | Adverse events: use of antiparkinsonism medication Unable to use (data does not have variability measures, and only reports differences from baseline to worst scores) ‐ ESRS: dyskinesia symptoms total score, CGI severity dyskinesia, buccolinguomasticatory factor, choreoathetoid factor |
|
| Notes | Sponsorship source: not reported. Study author kindly replied to our request for data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical tablets" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of raters not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 33% of participants terminated the study early due to insufficient therapeutic response. All early terminations were included in the ITT analysis |
| Selective reporting (reporting bias) | High risk | Outcomes not fully reported |
| Other bias | High risk | Subgroup with TD |
Cookson 1987.
| Methods | Allocation: "allocated randomly", not described
Blindness: "double blind", not described
Design: parallel groups
Setting: inpatients, UK Duration: 44 weeks |
|
| Participants | Diagnosis: hebephrenic or paranoid schizophrenia (ICD‐9 and Feighner criteria) N = 18 (only 9 people had TD at baseline) Age: mean 44.5 years Sex: 12 men and 6 women History: duration of TD not reported; patients resistant to low doses of antipsychotics but improved with higher dosages and maintained this improvement for at least 3 months | |
| Interventions | No washout period before study entry 1. Antipsychotic reduction: dose 50% previous dose of cis(z)‐flupenthixol decanoate, bi‐weekly. N = 5 2. Antipsychotic maintenance: dose standard dosage of cis(z)‐flupenthixol decanoate. N = 4 Procyclidine allowed during study. Supplementary antipsychotics allowed were haloperidol (oral) or zuclopenthixol decanoate (depot). Amitriptyline used for depression |
|
| Outcomes | TD (AIMS derived) Unable to use ‐ Adverse effects: GSES (no usable data) General mental state: BPRS (no usable data) |
|
| Notes | Dr Cookson kindly provided additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomized in blocks of 4 and stratified by antipsychotic dose and gender", implies adequate random sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind", no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind", no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants seem to have completed the study |
| Selective reporting (reporting bias) | Unclear risk | All outcomes proposed in the methods were reported, but some were not presented adequately. No protocol available to check |
| Other bias | High risk | "The randomised allocation of the small number of patients in the pilot study results in inequalities between the 2 groups at entry and confounded comparisons of group mean values during the study" |
Emsley 2004.
| Methods | Allocation: "randomly assigned", not described
Blindness: investigators blinded
Design: parallel group Setting: inpatients and outpatients, South Africa Duration: 50 weeks |
|
| Participants | Diagnosis: schizophrenia (DSM IV), TD (Schooler and Kane criteria) N = 45 Age: 49.2 (SD 14.5) years, range 18‐65 years Sex: 16 men and 29 women History: duration of TD not reported; at least 3 months' antipsychotic exposure; patients with established psychiatric disorder who did not receive clozapine | |
| Interventions | After an initial screening visit, subjects were tapered from all psychotropic medication over a 2‐week period. 1. Quetiapine: dose 100 mg/d increased to 400 mg/d. N = 22 2. Haloperidol: dose 5 mg/d increased to 10 mg/d. N = 23 Concomitant medications allowed were benzodiazepines for agitation or insomnia and anticholinergic agents in the event of treatment‐emergent or worsening EPS. Medications not allowed were other antipsychotics or other medication known to improve or exacerbate movement disorders. |
|
| Outcomes | TD symptoms: no clinical improvement
Leaving the study early General mental health PANSS Unable to use ‐ Adverse effects: ESRS, EPS (no usable data) Global assessment: CGI. (data in graphs, no variability) |
|
| Notes | Sponsorship source: supported in part by the Medical Research Council of South Africa, Cape Town, and the University of Stellenbosch. Trial medication and monitoring of the study were provided by AstraZeneca, Wilmington, Del. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Subjects were then randomly assigned", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "investigator‐blinded", further blinding details not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "investigator‐blinded", further blinding details not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 43% dropouts (including the 2 participants excluded in the early stages). 10/22 (45%) quetiapine and 8/23 (35%) haloperidol participants dropped out. |
| Selective reporting (reporting bias) | High risk | Adverse effects: extrapyramidal symptoms (other than dyskinesia) not fully reported |
| Other bias | Low risk | The study seems to be free of other sources of bias. Baseline characteristics were balanced in the compared groups. |
Glazer 1990a.
| Methods | Allocation: "randomly assigned", not described
Blindness: double, "identical capsules"
Design: parallel groups Setting: outpatients, USA Duration: 2 weeks |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder with TD (operational criteria) and criterion for withdrawal‐exacerbated TD N = 18 Age: mean 47 years Sex: 55% women History: duration of TD at least 3 months; "Patients had been receiving at least a year of continuous treatment with antipsychotics other than molindone or haloperidol"; "After a week on one of these two medications at pre‐established doses equivalent to that of the pre‐study neuroleptic" | |
| Interventions | Antipsychotic medications were tapered over a 7‐10 d period and then withdrawn, with single‐blind substitution of placebo for 7‐14 d. Study medication was administered when there was a demonstrable increase in involuntary movements. 1. Molindone: dose 75 mg (mean) during the first week (100% of pre‐trial dose equivalent) and 145 mg (mean) (200% of pre‐trial dose equivalent) during the second week. N = 9 2. Haloperidol: dose 19.3 mg (mean) (100% of pre‐trial dose equivalent) during the first week and 34.3 mg (mean) (200% of pre‐trial dose equivalent) during the second week. N = 9 Concomitant medications: psychoactive medications including antiparkinsonism agents, within 6 months before study entry were not allowed |
|
| Outcomes | Clinical improvement: AIMS
Leaving the study early Unable to use ‐ Mental state: BPRS Websters Parkinson Rating Scale: no usable data |
|
| Notes | Sponsorship source: supported in part by a grant from E.I. DuPont Pharmaceutical Company and National Institute of Mental Health Grant | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned to receive either molindone or haloperidol in a double‐blind fashion", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind" "Medication was supplied in identical‐appearing red capsules containing 25 mg and 5 mg, respectively, of molindone and haloperidol" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Details of blinding of outcome assessment not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants completed the trial |
| Selective reporting (reporting bias) | Unclear risk | Unclear if psychiatric symptoms, dyskinetic movements and parkinsonism measured by BPRS and Webster Parkinsonism Rating Scale were defined as outcomes. Only AIMS results were reported. |
| Other bias | High risk | The two groups were comparable except for a greater past hospitalisation duration in the molindone as compared with haloperidol‐treated group |
Kane 1983.
| Methods | Allocation: randomised using random numbers table
Blindness: double
Design: parallel groups Setting: outpatients, USA Duration: 48 weeks |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (RDC) N = 8 Age: range 17‐60 years Sex: not reported History: in a state of remission or at a stable clinical plateau | |
| Interventions | 1. Fluphenazine decanoate: low dose 1.25 mg‐5 mg/2 weeks. N = 4 2. Fluphenazine decanoate: antipsychotic maintenance: standard dose 12.5 mg‐50 mg/2 weeks. N = 4 Procyclidine, 5 mg‐20 mg/d, was allowed if needed to treat extrapyramidal side effects. No other psychotropic medication except flurazepam or diazepam was allowed (these benzodiazepines were used sparingly for insomnia). |
|
| Outcomes | TD ('no clinical improvement'; 'no improvement'; 'deterioration'), reported as adverse effects Incidence of TD (modified versions of SDS)
Leaving the study early General mental state: relapse Unable to use ‐ GAS, BPRS, CGI, SAS |
|
| Notes | Sponsorship source: this investigation was supported in part by grants from the National Institute of Mental Health. Dr Woerner kindly provided unpublished data for one site of the main study and only these are used in this review; the sex ratios are not available. If people in this study developed TD, participation was stopped and they were classified as leaving the study early. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised using random numbers table |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind". Details not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Details not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4/8 participants left the study early |
| Selective reporting (reporting bias) | High risk | Not all data were reported |
| Other bias | High risk | Only subsample with TD from one site included in this review |
Kazamatsuri 1972.
| Methods | Allocation: randomised
Blindness: raters blind
Design: parallel groups Setting: inpatients, USA Duration: 4 weeks |
|
| Participants | Diagnosis: chronic schizophrenia (15), chronic brain syndrome (3), and mental retardation (2) all manifesting typical buccolingual‐masticatory oral dyskinesia due to prolonged antipsychotic medication. N = 20 Age: average 56.9 years; range 44‐70 years Sex: 11 men and 9 women History: duration of TD not reported; "Before beginning this study, all patients had received tetrabenazine (56 to 156 mg daily) for six weeks " |
|
| Interventions | 4‐week washout using placebo medication, then: 1. haloperidol: dose 2 mg/d, increased to maximum of 16 mg/d. N = 11 2. thiopropazate: dose 10 mg/d, increased to maximum of 80 mg/d. N = 9 Concomitant medication not reported |
|
| Outcomes | TD symptoms Leaving the study early Unable to use ‐ Ward behaviour NOSIE (no SD) |
|
| Notes | Sponsorship source: supported in part by Public Health Service Research grant from the National Institute of Mental Health | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "20 patients were randomly divided", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants not reported. Ward nurses were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Quantitative evaluation of oral dyskinesia... was carried out every two weeks, by a psychiatrist... using a blind basis". Ward nurses were also blind to the treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two participants dropped out from haloperidol with reasons reported. |
| Selective reporting (reporting bias) | High risk | Adverse effects (except for 2 participants who were discontinued from the study) were not reported. Also, oral dyskinesia and reversible EPS scores reported only as mean for each arm. |
| Other bias | Unclear risk | Insufficient information to make a judgement |
Kazamatsuri 1973.
| Methods | Allocation: "randomly"
Blindness: rater blind
Duration: 24 weeks (4‐week antipsychotic and antiparkinsonism drug cessation and placebo administration, 18‐week intervention and then 2 weeks placebo)
Design: parallel Setting: inpatients, USA |
|
| Participants | Diagnosis: chronic psychotic patients: chronic schizophrenia (10), mentally deficient (2), chronic brain syndrome (1); all manifesting typical buccolinguomasticatory oral dyskinesia associated with long‐term antipsychotic medication
N = 13
Sex: 5 women and 8 men
Age: mean 55.8 years, range 41‐63 years History: duration of TD not reported |
|
| Interventions | 4 week washout from antiparkinsonism and antipsychotic medication (all replaced by placebo), then:. 1. haloperidol: dose 4 mg twice/d. From week 15 dose was doubled to 16 mg/d. N = 7 2. tetrabenazine: dose 50 mg twice/d. From week 15 onwards, dose was doubled to 200 mg/d. N = 6 Concomitant medications: "Other medications, such as antidiabetic or anticonvulsant drugs, were continued unchanged." |
|
| Outcomes | TD symptoms: not clinically improved TD symptoms: no improvement TD symptoms: deterioration Leaving the study early Unable to use ‐ TD scale scores and adverse effects: EPS Ward behaviour (NOSIE) (means, SDs not reported) |
|
| Notes | Sponsorship source: supported in part by Public Health Service grant from the National institute of Mental Health. Tetrabenazine and placebo tablets were provided by Hoffman‐La Roche. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The 13 patients were divided randomly into two groups." further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and personnel not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A frequency count of mouth movements (18), done by a psychiatrist blind to the study design was used to assess oral dyskinesia." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2/7 (29%) participants dropped out from the haloperidol group; no further details are provided for addressing the outcomes of these participants. No participants dropped out from the tetrabenazine group. |
| Selective reporting (reporting bias) | High risk | TD scale scores and extrapyramidal symptoms scale scores not fully reported |
| Other bias | Unclear risk | Insufficient information to make a judgement |
Lublin 1991.
| Methods | Allocation: "randomised", no details
Blindness: rater blind
Design: cross‐over Setting: inpatients, Denmark and Finland Duration: 18 weeks (3 weeks treatment, followed by 6 weeks washout, then crossed to another 3 weeks treatment followed by 6 weeks washout) |
|
| Participants | Diagnosis: psychotic patients with TD
N = 20
Sex: 12 women and 3 men
Age: mean 64.5, range 47‐79 years History: duration of TD on average 5.1 years (range 0.5‐11 years); duration of antipsychotic treatment on average 18.8 years (range 5‐34 years) |
|
| Interventions | Participants were down‐titrated to the lowest possible dose of haloperidol and kept stable for 4 weeks in order to keep them in an optimal mental condition, then: 1. haloperidol: dose 5.4 mg/d‐6.2 mg/d for 3 weeks (followed by 6 weeks washout and 3 weeks zuclopenthixol). N = 15 (7 during first period and 8 during second period) 2. zuclopenthixol: dose of 16.5 mg/d‐zuclopenthixol 26.6 mg/d for 3 weeks (followed by 6 weeks washout and 3 weeks haloperidol). N = 15 (8 during first period and 7 during second period) Concomitant medication: no antiparkinsonism medication was given. 4 participants (2 from each group) received benzodiazepines without changes during the study. "Other neuroleptic drugs, antidepressants and antiparkinsonian medication were not allowed" |
|
| Outcomes | TD symptoms: improvement 50% TD symptoms: not any improvement TD symptoms: deterioration Sct. Hans Rating Scale for Extrapyramidal Side Effects: parkinsonism and TD symptoms (data extracted from figure using digitisation software) Unable to use ‐ Leaving the study early (not reported for the phase before crossing over) Adverse events: UKU (not reported) Mental state: BPRS (not reported) |
|
| Notes | Sponsorship source: sponsorship source not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were then randomized to receive either HAL or ZPT", further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and personnel not reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "blind evaluation of TD and parkinsonism by means of video recordings."; "All the videotapes were later randomly sequenced and blindly scored by two of the same three raters" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 20 participants entered the study, 4 dropped out during randomised phases with reasons provided |
| Selective reporting (reporting bias) | High risk | Side effects and mental state were measured (UKU, BPRS) but not fully reported |
| Other bias | Unclear risk | Insufficient information to judge |
Tamminga 1994.
| Methods | Allocation: randomised
Blindness: double
Design: parallel groups Setting: not reported, USA Duration: 12 months |
|
| Participants | Diagnosis: schizophrenia; diagnosis of TD of a moderate or severe degree
N = 32*
Age: mean 35.57 (SD 7.60) years
Sex: 20 men and 12 women History: duration of TD not reported; "Before beginning the protocol, each participant was treated with a clinically optimal dose of haloperidol for an initial 1‐ to 6‐month stabilization period" |
|
| Interventions | After the stabilisation period, each participant was withdrawn from antipsychotic treatment for 4 weeks to allow an antipsychotic‐free assessment of their dyskinetic symptoms, then: 1. clozapine plus placebo: mean dose at 293.8 ± 171.9 mg/d for 12 months. N = 25 2. haloperidol plus benztropine: mean dose at 28.5 ± 23.8 mg/d for 12 months. N = 14 |
|
| Outcomes | Leaving the study early Unable to use ‐ TD symptoms (reported means only in graph) |
|
| Notes | Sponsorship source: sponsorship source not reported We contacted study authors for updated data but at the time of preparing this review no more information had been received *49 were recruited for this study but only 32 completed the blind protocol. The study authors reported only on these 32 participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Subjects were then blindly randomised to two different drug groups," further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Staff, patients, and all raters were blind to the drug group; one non rating physician and one nurse were non blind to dispense medication and monitor safety"; no further details were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Staff, patients, and all raters were blind to the drug group; one non rating physician and one nurse were non blind to dispense medication and monitor safety"; no further details were provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Forty‐three patients have entered the 15‐month blinded study, and 4 have not yet finished. Seven participants have been withdrawn from the protocol (6 taking clozapine; one taking haloperidol). One subject from each treatment group was dropped for leukopenia.The other 5 clozapine subjects were dropped for noncompliance (1 patient), decompensation (1 patient), seizure (1 patient), hypotension (1 patient), and ECG changes (1 patient)." Data were reported for completers only. |
| Selective reporting (reporting bias) | Unclear risk | Unclear if all predefined outcomes were reported. Efficacy data reported in graphs as means only. A study protocol is needed for firm conclusions. |
| Other bias | Unclear risk | Preliminary results as 4 subjects had not completed the study. |
DSM: Diagnostic and Statistical Manual of Mental Disorders EPS: Extrapyramidal symptoms FGA: first‐generation antipsychotic ICD‐9 ‐ International Classification of Diseases 9th edition ITT: intention‐to‐treat RDC ‐ Research Diagnostic Criteria TD: tardive dyskinesia
Rating Scales:
Global impression CGI ‐ Clinical Global Impression
Mental state BPRS ‐ Brief Psychiatric Rating Scale
Adverse events AIMS ‐ Abnormal Involuntary Movement Scale EPS ‐ Extrapyramidal Symptoms Scale ESRS ‐ Extrapyramidal side effect rating scale GSES ‐ General Side Effects Scale NOSIE ‐ Nurses Observational Scale of Inpatients Evaluation SAS ‐ Simpson Angus Scale SDS ‐ Simpson Dyskinesia Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Albus 1985 | Allocation: not randomised |
| Ananth 1977 | Allocation: not randomised |
| Andersson 1988 | Allocation: not randomised |
| Andia 1998 | Allocation: randomised Participants: schizophrenia (DSM‐III‐R), N = 26, 14 with TD Intervention: haloperidol vs clozapine Outcomes: no data available for AIMS in clozapine group, the study also reported on plasma homovanillic acid levels, an outcome not relevant for this review We contacted the study authors but no information was received. This study is over 15years old and was excluded. |
| Asnis 1979 | Allocation: not randomised |
| Auberger 1985 | Allocation: not randomised |
| Barnes 2002 | Allocation: random not mentioned in this short trial registration Participants: elderly patients, many started treatment with antipsychotics at start of study (no baseline TD) |
| Bateman 1979 | Allocation: random
Participants: psychiatric patients with TD
Interventions: metoclopramide (10 mg, 20 mg or 40 mg) vs haloperidol (5 mg or 10 mg) Outcomes: no outcome data was provided for the first period before cross‐over. We were unable to find contact details for the study authors; study is over 35 years old and was excluded |
| Bitter 2000 | Allocation: random Participants: people with schizophrenia, no TD symptoms at baseline Interventions: olanzapine vs clozapine |
| Blaha 1980 | Allocation: not randomised |
| Borison 1987 | Allocation: randomised
Participants: schizophrenia (DSM‐III criteria); TD (Schooler and Kane criteria)
Interventions: molindone versus haloperidol Outcomes: no usable efficacy data; only P values were reported. We were unable to find contact details for the study authors; study is over 25 years old and was excluded |
| Branchey 1981 | Allocation: not randomised |
| Brecher 1999 | Allocation: randomised Participants: people with dementia, not schizophrenia, not TD at baseline |
| Buchanan 1992 | Allocation: not randomised |
| Burner 1989 | Allocation: 'randomly assigned' Participants: people with schizophrenia, no TD symptoms at baseline Interventions: progabide vs placebo |
| Buruma 1982 | Allocation: randomised, cross‐over
Participants: "patients with tardive dyskinesia" ‐ no further details Interventions: tiapride vs placebo Outcomes: doppler ratings, none before cross‐over |
| Cai 1988 | Allocation: randomisation not mentioned
Participants: patients with antipsychotic‐induced TD Intervention: 1‐stepholidine (herbal product that has shown antipsychotic properties in animals) versus placebo Assessed and data extracted by Sai Zhao |
| Caine 1979 | Allocation: "allocated by toss of a coin"
Participants: Gilles de la Tourette's, Huntington's disease and drug‐induced atypical dyskinesia, no TD symptoms at baseline Interventions: clozapine vs placebo |
| Calne 1974 | Allocation: not randomised |
| Campbell 1988 | Allocation: not randomised |
| Carpenter 1980 | Allocation: not randomised |
| Casey 1977 | Allocation: not randomised |
| Casey 1979 | Allocation: not randomised |
| Casey 1981 | Allocation: not randomised |
| Casey 1983 | Allocation: not randomised |
| Cassady 1992 | Allocation: not randomised |
| Chouinard 1978 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: fluphenazine ethanoate vs pipothiazine palmitate |
| Chouinard 1979 | Allocation: random
Participants: people with schizophrenia, and parkinsonism, no TD symptoms at baseline Interventions: ethopropazine vs benztropine |
| Chouinard 1989 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: haloperidol decanoate vs fluphenazine decanoate |
| Chouinard 1994 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: clozapine vs risperidone |
| Claveria 1975 | Allocation: not randomised |
| Cookson 1991 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: haloperidol decanoate vs fluphenazine decanoate |
| Cortese 2008 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: quetiapine vs continuation of usual antipsychotic |
| Cowen 1997 | Allocation: not randomised |
| Crane 1968 | Allocation: not randomised, review article |
| Crane 1969 | Allocation: not randomised |
| Crane 1970 | Allocation: random
Participants: people with schizophrenia, only 2%‐3% with TD at baseline Interventions: trifluoperazine high dose vs trifluoperazine low dose vs placebo |
| Curran 1973 | Allocation: not randomised |
| Curson 1985 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: fluphenazine decanoate vs placebo |
| Davidson 2000 | Allocation: not randomised |
| de Jesus Mari 2004 | Allocation: randomised
Participants: diagnosis of schizophrenia or related disorders (DSM‐IV criteria). < 50% of participants had TD at baseline Interventions: olanzapine vs "conventional antipsychotic drugs" Outcomes: author contacted for data regarding people with TD ‐ data no longer available |
| Delwaide 1979 | Allocation: randomised
Participants: hospitalised patients with TD on a psychogeriatric ward
Intervention: thioproperazine vs tiapride vs placebo
Outcome: all data unusable, unable to extract data from first arm of cross‐over The study is over 35 years old and we were unable to identify contact details for the author |
| Diamond 1986 | Allocation: not randomised |
| Dixon 1993 | Allocation: not randomised |
| Fahn 1983 | Allocation: not randomised |
| Fahn 1985 | Allocation: not randomised |
| Freeman 1980 | Allocation: not randomised |
| Gardos 1984 | Allocation: not randomised |
| Gerlach 1975 | Allocation: random Participants: schizophrenia, no established, stable TD diagnosis at baseline Interventions: clozapine vs haloperidol |
| Gerlach 1978 | Allocation: the randomisation was just in one arm of the study “Haloperidol + biperiden for 4 weeks (phase 2 and phase 3 in randomized sequence)”. All other arms thioridazine for 3 months, haloperidol for 4 weeks; thioridazine for 4 weeks, clozapine for 4 weeks were not Participants: elderly people with psychiatric history and neuroleptic‐induced TD Interventions: biperiden vs no treatment as an adjunct to haloperidol |
| Gerlach 1984a | Allocation: not randomised, cohort study |
| Gerlach 1984b | Allocation: not randomised |
| Gibson 1980 | Allocation: not randomised |
| Glazer 1984 | Allocation: not randomised |
| Glazer 1989 | Allocation: not randomised |
| Goldberg 1981 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: withdrawal of fluphenazine decanoate vs continuation |
| Greil 1984 | Allocation: "randomly assigned" Participants: people with schizophrenia Interventions: biperiden vs placebo |
| Haggstrom 1980 | Allocation: not randomised |
| Heresco‐Levy 1993 | Allocation: not randomised |
| Hershon 1972 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: trifluoperazine withdrawal vs trifluoperazine continuation |
| Herz 1991 | Allocation: randomised
Participants: people with schizophrenia
Interventions: neuroleptic reduction (intermittent treatment) vs maintenance neuroleptic
Outcomes: no usable data Dr Herz kindly replied to our request for more information. Unfortunately, individual baseline and endpoint AIMS score are no longer available |
| Hogarty 1976 | Allocation: not randomised |
| Hogarty 1988 | Allocation: quasi‐randomised |
| Inada 2003 | Allocation: not randomised |
| Inderbitzin 1994 | Allocation: Not randomised ("by alternate allocation") |
| Jean‐Noel 1999 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: clozapine vs olanzapine |
| Jeste 1977 | Allocation: not randomised Participants: chronic schizophrenia with TD, N = 2 Interventions: chlorpromazine schedule A vs chlorpromazine schedule B. Treatments in the 2 groups were the same except for timing of the doses (frequency and withdrawal) |
| Jeste 1979 | Allocation: not randomised |
| Johnson 1983 | Allocation: not randomised |
| Johnson 1987 | Allocation: randomised
Participants: people with schizophrenia
Interventions: neuroleptic dose reduction vs maintenance dose (both arms used flupenthixol decanoate)
Outcomes: no usable data Dr Johnson kindly replied to our letter. No further data available from the first author |
| Jolley 1990 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: brief intermittent antipsychotic treatment vs fluphenazine decanoate |
| Jus 1979 | Allocation: not randomised |
| Kalachnik 1984 | Allocation: not randomised, case‐control study Dr Kalachnick kindly provided additional information. After randomisation clinicians reviewed group allocations and re‐assigned selected individuals on clinical grounds |
| Kane 1993 | Allocation: not randomised, 2 case series |
| Kinon 2004 | Allocation: randomised Participants: schizophrenia and TD (Schooler and Kane criteria) Interventions: olanzapine (5 mg‐20 mg/d) with 1 set of intermittent dose‐reduction periods versus olanzapine (5 mg‐20 mg/d) with a different set of intermittent dose reduction periods |
| Kirch 1983 | Allocation: not randomised |
| Kopala 2004 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: haloperidol vs risperidone |
| Lal 1974 | Allocation: randomised, cross‐over. Participants: people with schizophrenia. Interventions: thiopropazine vs trifluoperazine vs placebo. Outcomes: no usable data. Dr Lal kindly replied to inquiry. Unable to extract data from the first segment. Jadad score = 4/5 |
| Leblanc 1994 | Allocation: not randomised, cohort study |
| Leblhuber 1987 | Allocation: not randomised |
| Levine 1980 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: fluphenazine withdrawal vs continuation |
| Lieberman 1988 | Allocation: randomised
Participants: TD according to the criteria of Schooler and Kane, schizophrenia, schizoaffective disorder, major affective disorder and attention deficit disorder Intervention: physostigmine vs bromocriptine vs benztropine vs haloperidol for 1 day, then crossed over. Outcomes: no outcome data provided for the first period before cross‐over. We contacted the study author but no information received. Study is over 25 years old years old and was excluded |
| Lieberman 1989 | Allocation: not randomised, cohort study |
| Lin 2006 | Allocation: not randomised: naturalistic observational study |
| Littrell 1993 | Allocation: not randomised |
| MacKay 1980 | Allocation: "patients were divided into pairs" Participants: people with schizophrenia Intervention: lithium vs placebo |
| Marder 1987 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: low vs conventional‐dose maintenance therapy with fluphenazine decanoate |
| McCreadie 1980 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: intermittent pimozide vs fluphenazine decanoate |
| Meco 1989 | Allocation: not randomised |
| Miller 1994 | Allocation: not randomised |
| NDSG 1986 | Allocation: randomised cross‐over
Participants: psychiatric inpatients with TD Intervention: chlorprothixene vs haloperidol vs perphenazine vs haloperidol + biperiden Outcomes: no outcome data provided for the first period before cross‐over We contacted the study author but no reply. Study is 30 years old and was excluded. |
| Newcomer 1992 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: haloperidol dose reduction vs maintained dose |
| Newton 1989 | Allocation: randomised Participants: people with schizophrenia (no history of TD) Interventions: haloperidol with 'drug holiday' versus haloperidol |
| Odejide 1982 | Allocation: randomisation not mentioned Participants: people with schizophrenia (no history of TD) Interventions: fluphenazine decanoate vs vitamin B complex |
| Pai 2001 | Allocation: not randomised Participants: people with schizophrenia and TD Interventions: risperidone vs placebo |
| Paulson 1975 | Allocation: not randomised Dr Paulsen kindly provided additional information about this double‐blind study |
| Peacock 1996 | Allocation: not randomised |
| Peluso 2012 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: FGA vs second generation antipsychotic |
| Perry 1985 | Allocation: randomised Participants: children with autism without history of TD Dr Campbell kindly provided all published and in‐press data. The authors found no difference in TD between the intermittent and continuous treatment groups but further details required for this review were not available |
| Pyke 1981 | Allocation: not randomised |
| Quinn 1984 | Allocation: randomised, double‐blind, cross‐over study Participants: people with schizophrenia Intervention: sulpiride (Dogmatil) 300 mg‐ 1200 mg/d Outcomes: no usable data. Drs Marsden and Quinn kindly replied to our letter, but no data suitable for this review could be provided |
| Quitkin 1977 | Allocation: not randomised |
| Rapoport 1997 | Allocation: not described |
| Ringwald 1978 | Allocation: not random |
| Rosenheck 2003 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: haloperidol vs olanzapine |
| Roxburgh 1970 | Allocation: not randomised |
| Schultz 1995 | Allocation: not randomised |
| Schwartz 1990 | Allocation: randomised
Participants: psychiatric inpatients with TD Interventions: sulpiride vs placebo Outcomes: no outcome data provided for the first period before cross‐over. We were unable to find contact details for the study authors; this study is over 25 years old and was excluded |
| Seeman 1981 | Allocation: not randomised |
| Simpson 1978 | Allocation: not randomised, cohort study |
| Singer 1971 | Allocation: randomised
Participants: psychiatric inpatients with persistent TD Interventions: thiopropazate vs placebo Outcomes: no outcome data provided for the first period before cross‐over. We were unable to find contact details for the study authors; this study is 45 years old and was excluded |
| Singh 1990 | Allocation: randomised
Participants: people with schizophrenia (majority did not have TD) Intervention: abrupt antipsychotic withdrawal versus continuation of antipsychotic medication |
| Small 1987 | Allocation: not randomised, cohort study |
| Smith 1979 | Allocation: not randomised, cohort study |
| Soni 1984 | Allocation: not randomised |
| Speller 1997 | Allocation: randomised Participants: schizophrenia (DSM‐III‐R), majority with TD Intervention: amisulpride versus haloperidol Outcomes: schizophrenia symptom changes, especially negative symptoms, adverse events, and TD as adverse event. We excluded this reference because, although the majority had TD at baseline and the intervention drugs qualified, the drugs were not examined as a treatment for TD (as our inclusion criteria demand), but for negative symptoms of schizophrenia |
| Spivak 1997 | Allocation: not randomised, cohort study |
| Spohn 1988 | Allocation: randomised
Participants: people with schizophrenia
Interventions: abrupt neuroleptic cessation versus neuroleptic maintenance
Outcomes: no usable data Dr Spohn kindly replied to our request for further information. Data on baseline and endpoint TD not available |
| Spohn 1993 | Allocation: randomised
Participants: people with schizophrenia
Interventions: abrupt neuroleptic withdrawal versus maintenance
Outcomes: no usable data Dr Spohn kindly replied to our letter, but no further data were available |
| Suh 2004 | Allocation: randomised Participants: dementia and not TD |
| Thapa 1994 | Allocation: randomised Participants: nursing home staff Interventions: education about neuroleptic prescribing vs no specific additional education |
| Tollefson 1997 | Allocation: random Participants: no TD at baseline, investigates incidence of TD with long‐term treatment with olanzapine vs haloperidol |
| Tran 1997 | Allocation: random
Participants: people with schizophrenia, no TD symptoms at baseline Interventions: olanzapine versus haloperidol |
| Turek 1972 | Allocation: not randomised ‐ allocated to treatment group in a "nonsystematic" fashion, but then participants were re‐allocated to alternate groups based on clinical judgement |
| Williamson 1995 | Allocation: random
Participants: schizophrenia, not TD Interventions: olanzapine 1 mg vs olanzapine 10 mg versus placebo |
| Wirshing 1999 | Allocation: random
Participants: people with treatment‐resistant schizophrenia, no TD symptoms at baseline Interventions: haloperidol vs risperidone |
| Wistedt 1983 | Allocation: randomised
Participants: people with schizophrenia (no history of TD) Interventions: fluphenazine/flupenthixol decanoate continuation vs withdrawal |
| Wolf 1991 | Allocation: not randomised, cohort study |
| Wright 1998 | Allocation: not randomised |
| Zander 1981 | Allocation: not randomised |
| Zarebinski 1990 | Allocation: not randomised, cohort study |
| Zeng 1994 | Allocation: randomised
Participants: patients with antipsychotic‐induced TD Intervention: flunarizine (calcium channel antagonist) vs placebo Assessed and data extracted by Sai Zhao |
FGA: first‐generation antipsychotic IV = intravenous TD: tardive dyskinesia
Characteristics of ongoing studies [ordered by study ID]
N0546099389.
| Trial name or title | A six month, rater blind comparison of quetiapine and risperidone in the treatment of tardive dyskinesia in patients with schizophrenia |
| Methods | Allocation: randomised Blindness: rater blind Design: not reported Setting: not reported Duration: 6 months |
| Participants | People with schizophrenia with TD |
| Interventions | 1. Quetiapine 2. Risperidone |
| Outcomes | Prevalence and severity of abnormal involuntary movements |
| Starting date | |
| Contact information | |
| Notes | Very limited information from two trials registries. We were unable to locate author contact details. |
Differences between protocol and review
The protocol as published with this review has evolved over time. The revisions of protocol are in line with the development of Review Manager and in keeping with Cochrane guidance. We think the revisions have greatly improved and enhanced this review. We do not think, however, that it has materially affected our conduct of the review or interpretation of the results.
There was a substantial update to the protocol in the 2017 review update. The biggest changes to affect the review were to:
broaden the inclusion criteria, and add the comparison 'Specific antipsychotic versus other drug';
change the title from 'Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia' to 'Antipsychotic reduction and/or cessation and antipsychotics as specific treatments for tardive dyskinesia';
update list of outcomes following consultation with consumers; and
add 'Summary of findings' tables.
Previous methods are reproduced in Appendix 1.
Contributions of authors
HB ‐ study selection, data extraction and assimilation, GRADE and Summary of Findings tables, report writing (2017 update)
JR ‐ study selection, data extraction and assimilation, report writing (original version).
VA ‐ report writing (2017 update).
KSW ‐ protocol development, searching, study selection, data extraction and assimilation, report writing (original version and 2017 update).
Sources of support
Internal sources
Queensland Health, Australia.
CAPES ‐ Ministry of Education, Brazil.
Universidade Federal de São Paulo, Brazil.
-
Enhance Reviews Ltd., UK.
Logistics support for Hanna Bergman
External sources
-
NIHR HTA Project Grant, reference number: 14/27/02, UK.
Salary support for Hanna Bergman. Support for patient involvement consultation. Support for accessible, traceable data.
Declarations of interest
HB worked for Enhance Reviews Ltd. during preparation of this review and was paid for her contribution to this review. Enhance Reviews Ltd. is a private company that performs systematic reviews of literature. HB works for Cochrane Response, an evidence consultancy that takes commissions from healthcare guideline developers and policy makers.
JR is not aware of any conflicts of interest for this review.
VA has declared no known conflicts of interest for this review.
KSW is the Deputy Editor‐in‐Chief for Cochrane and Cochrane Innovations. When the NHIR HTA programme grant relevant to this review update was awarded, KSW was the Managing Director of Enhance Reviews Ltd.
One of the earlier reviewers (JJM) is a member of the following advisory boards: Janssen‐Cilag Australia, Eli Lilly Australia, Lundbeck Australia. In addition, JJM has been a co‐investigator on studies of neuroleptic medications produced by the following companies: Astra Zeneca (ICI), Janssen‐Cilag, Eli Lilly, Sandoz, and Pfizer. The same companies have provided travel and accommodation expenses for JJM to attend relevant investigator meetings and scientific symposia. No funds have been paid directly to JJM. Payments related to participation in drug trials and board attendance has been paid to a Government‐audited trust account to support schizophrenia research.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Bai 2003 {published data only}
- Bai YM, Lin CC, Yu SC. Risperidone for severe tardive dyskinesia: one year follow up study. International Journal of Neuropsychopharmacology 2002;5:S165. [Google Scholar]
- Bai YM, Yu SC, Chen JY, Lin CY, Chou P, Lin CC. Risperidone for pre‐existing severe tardive dyskinesia: a 48‐week prospective follow‐up study. International Clinical Psychopharmacology 2005;20(2):79‐85. [DOI] [PubMed] [Google Scholar]
- Bai YM, Yu SC, Lin CC. Risperidone for severe tardive dyskinesia: a 12‐week randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2003;64:1342‐8. [PubMed] [Google Scholar]
- Pai YM, Yu SC, Lin CC. Risperidone in reducing tardive dyskinesia: a double‐blind, placebo‐controlled study. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, Pennsylvania, USA. 2002.
- Pai YM, Yu SC, Lin CC. Risperidone in reducing tardive dyskinesia: a double‐blind, placebo‐controlled study. Proceedings of the 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans, Louisiana, USA. 2001.
Bai 2005 {published data only}
- Bai YM. Tardive dyskinesia and cognitive function. clinicaltrials.gov/ct2/show/record/NCT00926965 accessed on 20 June 2016.
- Bai YM, Ping LY, Lin CC, Wang YC, Liou YJ, Wu BJ, et al. Comparative effects of atypical antipsychotic on tardive dyskinesia and neurocognition: a 24‐week randomized, single‐blind, controlled study. 8th World Congress of Psychiatry; 2005 Sep 10‐15; Cairo, Egypt. 2005.
- Bai YM, Ping LY, Lin CC, Wang YC, Liou YJ, Wu BJ, et al. Comparative effects of atypical antipsychotic on tardive dyskinesia and neurocognition: a 24‐week randomized, single‐blind, controlled study. European Neuropsychopharmacology 2005;15(Suppl 3):S473. [Google Scholar]
Caroff 2011 {published data only}
- Caroff SN, Davis VG, Miller DD, Davis SM, Rosenheck RA, McEvoy JP, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. Journal of Clinical Psychiatry 2011;72(3):295‐303. [DOI: 10.4088/JCP.09m05793yel] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, McEvoy JP, Saltz BL, et al. Extrapyramidal side‐effects of antipsychotics in a randomised trial. British Journal of Psychiatry 2008; Vol. 193, issue 4:279‐88. [DOI] [PMC free article] [PubMed]
- Miller DD, McEvoy JP, Davis SM, Caroff SN, Saltz BL, Chakos MH, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophrenia Research 2005; Vol. 80, issue 1:33‐43. [DOI] [PubMed]
Chan 2010 {published data only}
- Chan HY, Chiang SC, Chang CJ, Gau SS, Chen JJ, Chen CH, et al. A randomized controlled trial of risperidone and olanzapine for schizophrenic patients with neuroleptic‐induced tardive dyskinesia. Journal of Clinical Psychiatry 2010;71(9):1226‐33. [DOI] [PubMed] [Google Scholar]
- NCT00621998. Risperidone and olanzapine for the schizophrenic patients with neuroleptic‐induced tardive dyskinesia. www.clinicaltrials.gov 2008. [DOI] [PubMed]
Chouinard 1995 {published data only}
- Chouinard G, Arnott W. An antidyskinetic effect of risperidone. 9th World Congress of Psychiatry. 1993; Vol. Jun 6‐12:22.
- Chouinard G, Jones B, Remington G, Bloom D, Addington D, MacEwan GW, et al. A Canadian multicenter placebo‐controlled study of fixed doses of risperidone and haloperidol in the treatment of chronic schizophrenic patients. Journal of Clinical Psychopharmacology 1993;13(1):25‐40. [PubMed] [Google Scholar]
- Chouinard, G. Effects of risperidone in tardive dyskinesia: an analysis of the Canadian multicenter risperidone study. Journal of Clinical Psychopharmacology 1995;15(Suppl 1):S36‐S44. [DOI] [PubMed] [Google Scholar]
- Chouinard, G. Arnott W. Antidyskinetic effect of risperidone in chronic schizophrenic patients. Clinical Neuropharmacology 1992;15 (Suppl 1 Pt B):266. [Google Scholar]
- Chouinard, G. Arnott W. The effect of risperidone on extrapyramidal symptoms in chronic schizophrenic patients. Biol Psychiatry 1992;31:158. [Google Scholar]
Cookson 1987 {published and unpublished data}
- Cookson IB. The effects of a 50% reduction of cis(z)‐flupenthixol decanoate in chronic schizophrenic patients maintained on a high dose regime. International Clinical Psychopharmacology 1987;2:141‐9. [DOI] [PubMed] [Google Scholar]
Emsley 2004 {published data only}
- Emsley R, Turner HJ, Schronen J, Botha K, Smit R, Oosthuizen PP. A single‐blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. Journal of Clinical Psychiatry 2004;65(5):696‐701. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Emsley RA, Turner J, Schronen J, Botha K, Smit R, Oosthuizen PP. Quetiapine: greater improvements in tardive dyskinesia versus haloperidol. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, USA. 2004. [APA Conference Abstracts NR574]
Glazer 1990a {published data only}
- Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophrenia Research 1990;3(5‐6):315‐20. [MEDLINE: ; PMID 1980827] [DOI] [PubMed] [Google Scholar]
- Glazer WM, Hafez HM, Benarroche CL. Molindone and haloperidol in tardive dyskinesia. Journal of Clinical Psychiatry 1985;46(8):4‐7. [MEDLINE: ; PMID 1980827] [PubMed] [Google Scholar]
Kane 1983 {published and unpublished data}
- Kane JM, Rifkin A, Woerner M, Reardon G, Sarantokos S, Schiebel D, et al. Low‐dose neuroleptic treatment of outpatient schizophrenics. I. Preliminary results for relapse rates. Archives of General Psychiatry 1983;40(8):893‐6. [DOI] [PubMed] [Google Scholar]
Kazamatsuri 1972 {published data only}
- Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. II. Short‐term efficacy of dopamine‐blocking agents haloperidol and thiopropazate. Archives of General Psychiatry 1972;27(1):100‐3. [DOI] [PubMed] [Google Scholar]
Kazamatsuri 1973 {published data only}
- Kazamatsuri H, Chien CP, Cole JO. Long‐term treatment of tardive dyskinesia with haloperidol and tetrabenazine. American Journal of Psychiatry 1973;130(4):479‐83. [DOI] [PubMed] [Google Scholar]
Lublin 1991 {published data only}
- Lublin H, Gerlach J, Hagert U, Meidahl B, Molbjerg C, Pedersen V, et al. Zuclopenthixol, a combined dopamine D1/D2 antagonist, versus haloperidol, a dopamine D2 antagonist, in tardive dyskinesia. European Neuropsychopharmacology 1991;1(4):541‐8. [DOI] [PubMed] [Google Scholar]
Tamminga 1994 {published data only}
- Tamminga CA, Thaker GK, Moran M, Kakigi T, Gao XM. Clozapine in tardive dyskinsia: observations from human and animal model studies. Journal of Clinical Psychiatry 1994;55(9, Suppl B):102‐6. [PubMed] [Google Scholar]
References to studies excluded from this review
Albus 1985 {published data only}
- Albus M, Naber D, Muller‐Spahn F, Douillet P, Reinertshofer T, Ackenheil M. Tardive dyskinesia: relation to computer‐tomographic, endocrine, and psychopatholgical variables. Biological Psychiatry 1985;20:1082‐9. [DOI] [PubMed] [Google Scholar]
Ananth 1977 {published data only}
- Ananth JV, Ban TA, Lehmann HE. An uncontrolled study with thiopropazate in the treatment of persistent dyskinesia. Psychopharmacology Bulletin 1977;13(3):9. [PubMed] [Google Scholar]
Andersson 1988 {published data only}
- Andersson U, Haggstrom JE, Nilsson MJ, Widerlov E. Remoxipride, a selective dopamine D2 receptor antagonist, in tardive dyskinesia. Psychopharmacology 1988;94(2):167‐71. [DOI] [PubMed] [Google Scholar]
Andia 1998 {published data only}
- Andia I, Zumarraga M, Zabalo MJ, Bulbena A, Davila R. Differential effect of haloperidol and clozapine on plasma homovanillic acid in elderly schizophrenic patients with or without tardive dyskinesia. Biological Psychiatry 1998;43:20‐3. [DOI] [PubMed] [Google Scholar]
Asnis 1979 {published data only}
- Asnis GM, Sachar EJ, Langer G, Halpern FS, Fink M. Normal prolactin responses in tardive dyskinesia. Psychopharmacology 1979;66(3):247‐50. [DOI] [PubMed] [Google Scholar]
Auberger 1985 {published data only}
- Auberger S, Greil W, Ruther E. Tiapride in the treatment of tardive dyskinesia. A double‐blind study. Pharmacopsychiatry 1985;18(1):61‐2. [EMBASE 1998088014] [Google Scholar]
Barnes 2002 {published data only}
- Barnes T. A prospective study of tardive dyskinesia in the elderly; single‐blind comparison of amisulpride and risperidone. National Research Register 2002. [N0195113456]
Bateman 1979 {published data only}
- Bateman DN, Dutta DK, McClelland HA, Rawlins MD. The effect of metoclopramide and haloperidol on tardive dyskinesia. British Journal of Pharmacology 1979;66(3):475‐6. [PMC free article] [PubMed] [Google Scholar]
Bitter 2000 {published data only}
- Bitter I, Slabber M, Pretorius J, Bartko GY, Danics Z, Dossenbach M, et al. Olanzapine versus clozapine in patients non responsive or intolerant to standard acceptable treatment of schizophrenia. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):S141. [Google Scholar]
Blaha 1980 {published data only}
- Blaha L. A strategy to reduce the frequency of early dyskinesias under high‐dosed haloperidol treatment [Therapiekonzept zur reduktion der fruehdyskinesie‐frequenz bei hochdosierter haloperidolbehandlung]. Arzneimittel Forschung 1980;30(8):1208. [N0195113456] [Google Scholar]
Borison 1987 {published data only}
- Borison RL, Shah C, White TH, Diamond BI. Atypical and typical neuroleptics and tardive dyskinesia. Psychopharmacology Bulletin 1987;23(1):218‐20. [PubMed] [Google Scholar]
Branchey 1981 {published data only}
- Branchey MH, Branchey LB, Richardson MA. Effects of gradual decrease and discontinuation of neuroleptics on clinical condition and tardive dyskinesia. Psychopharmacology Bulletin 1981;17(1):118‐20. [PubMed] [Google Scholar]
- Branchey MH, Branchey LB, Richardson MA. Effects of neuroleptic adjustment on clinical condition and tardive dyskinesia in schizophrenic patients. American Journal of Psychiatry 1981;138(5):608‐12. [DOI] [PubMed] [Google Scholar]
Brecher 1999 {published data only}
- Brecher M. Follow‐up study of risperidone in the treatment of patients with dementia: interim results on tardive dyskinesia and dyskinesia severity. 11th European College of Neuropsychopharmacology Congress (ECNP). Paris: European College of Neuropsychopharmacology, 1998 Oct 31‐Nov 4, Paris:P4005.
- Brecher M, Okamoto A, Napolitano J, Kane JM, The Risperidone Study Group. Low frequency of tardive dyskinesia in elderly patients with dementia exposed to risperidone for up to one year. Schizophrenia Research 1999;1‐3:362. [Google Scholar]
- Brecher M, Okamoto A, Napolitano J, Kane JM, Risperidone Study Group. Low frequency of tardive dyskinesia in elderly patients with dementia exposed to risperidone for up to one year. American Journal of Geriatric Psychiatry 1999;7:53‐4. [Google Scholar]
Buchanan 1992 {published data only}
- Buchanan RW, Kirkpatrick B, Summerfelt A, Hanlon TE, Levine J, Carpenter TW Jr. Clinical predictors of relapse following neuroleptic withdrawal. Biological Psychiatry 1992;32:72‐8. [DOI] [PubMed] [Google Scholar]
Burner 1989 {published data only}
- Burner M, Giroux C, L'Heritier C, Garreau M, Morselli PL. Preliminary observations on the therapeutic action of progabide in tardive dyskinesia. Brain Dysfunction 1989;2(6):289‐96. [MEDLINE: ; PMID 2886223]2886223 [Google Scholar]
Buruma 1982 {published data only}
- Buruma OJ, Roos RA, Bruyn GW, Kemp B, Velde EA. Tiapride in the treatment of tardive dyskinesia. Acta Neurologica Scandinavica 1982;65(1):38‐44. [MEDLINE: ; PMID 6121443] [DOI] [PubMed] [Google Scholar]
Cai 1988 {published data only}
- Cai N. A controlled study on the treatment of tardive dyskinesia using 1‐stepholidine. Chinese Journal of Neurology and Psychiatry 1988;21(5):281‐3. [PubMed] [Google Scholar]
Caine 1979 {published data only}
- Caine ED, Polinsky RJ, Kartzinel R, Ebert MH. The trial use of clozapine for abnormal involuntary movement disorders. American Journal of Psychiatry 1979;136:317‐20. [DOI] [PubMed] [Google Scholar]
Calne 1974 {published data only}
- Calne DB, Claveria LE, Teychenne PF, Haskayne L, Lodge‐Patch IC. Pimozide in tardive dyskinesia. Transcripts of the American Neurology Association 1974;99:166‐70. [PubMed] [Google Scholar]
Campbell 1988 {published data only}
- Armenteros JL, Adams PB, Campbell M, Eisenberg ZW. Haloperidol‐related dyskinesias and pre‐ and perinatal complications in autistic children. Psychopharmacology Bulletin 1995;31(2):363‐9. [PubMed] [Google Scholar]
- Campbell M, Adams P, Perry R, Spencer EK, Overall JE. Tardive and withdrawal dyskinesia in autistic children: a prospective study. Psychopharmacology Bulletin 1988;24(2):251‐5. [PubMed] [Google Scholar]
- Campbell M, Armenteros JL, Malone RP, Adams PB, Eisenberg ZW, Overall JE. Neuroleptic‐related dyskinesias in autistic children: a prospective longitudinal study. Journal of the American Acadamy of Child and Adolescent Psychiatry 1997;36(6):835‐43. [DOI] [PubMed] [Google Scholar]
Carpenter 1980 {published data only}
- Carpenter WT, Rey AC, Stephens JH. Covert dyskinesia in ambulatory schizophrenia. The Lancet 1980;July:212‐3. [DOI] [PubMed] [Google Scholar]
Casey 1977 {published data only}
- Casey DE, Denney D. Pharmacological characterization of tardive dyskinesia. Psychopharmacology Berlin 1977;54(1):1‐8. [DOI] [PubMed] [Google Scholar]
Casey 1979 {published data only}
- Casey DE, Gerlach J, Simmelsgaard H. Sulpiride in tardive dyskinesia. Psychopharmacology 1979;66:73‐7. [N0195113456] [DOI] [PubMed] [Google Scholar]
Casey 1981 {published data only}
- Casey DE, Korsgaard S, Gerlach J, Jorgensen A, Simmelsgaard H. Effect of des‐tyrosine‐gamma‐endorphin in tardive dyskinesia. Archives of General Psychiatry 1981;38(2):158‐60. [DOI] [PubMed] [Google Scholar]
Casey 1983 {published data only}
- Casey DE. Tardive dyskinesia: what is the natural history?. International Drug Therapy Newsletter 1983;18:13‐6. [Google Scholar]
Cassady 1992 {published data only}
- Cassady SL, Thaker GK, Moran M, Birt A, Tamminga CA. GABA agonist‐induced changes in motor, oculomotor, and attention measures correlate in schizophrenics with tardive dyskinesia. Biological Psychiatry 1992;32(4):302‐11. [DOI] [PubMed] [Google Scholar]
Chouinard 1978 {published data only}
- Chouinard G, Annable L, Kropsky M. A double‐blind controlled study of pipothiazine palmitate in the maintenance treatment of schizophrenic outpatients. Journal of Clinical Pharmacology 1978;18(2‐3):148‐54. [DOI] [PubMed] [Google Scholar]
Chouinard 1979 {published data only}
- Chouinard G, Annable L, Ross‐Chouinard A, Kropsky ML. Ethopropazine and benztropine in neuroleptic‐induced parkinsonism. Journal of Clinical Psychiatry 1979;40(3):147‐52. [PubMed] [Google Scholar]
Chouinard 1989 {published data only}
- Chouinard G, Annable L, Campbell W. A randomized clinical trial of haloperidol decanoate and fluphenazine decanoate in the outpatient treatment of schizophrenia. Journal of Clinical Psychopharmacology 1989;9(4):247‐53. [PubMed] [Google Scholar]
Chouinard 1994 {published data only}
- Chouinard G, Vainer JL, Belanger MC, Turnier L, Beaudry P, Roy JY, et al. Risperidone and clozapine in the treatment of drug‐resistant schizophrenia and neuroleptic‐induced supersensitivity psychosis. Progress in Neuro‐psychopharmacology & Biological Psychiatry 1994;18(7):1129‐41. [DOI] [PubMed] [Google Scholar]
Claveria 1975 {published data only}
- Claveria LE, Teychenne PF, Calne DB, Haskayne L, Petrie A, Pallis CA, et al. Tardive dyskinesia treated with pimozide. Journal of the Neurological Sciences 1975;24(4):393‐401. [DOI] [PubMed] [Google Scholar]
Cookson 1991 {published data only}
- Cookson JC. Side effects during long‐term treatment with depot antipsychotic medication. Clinical Neuropharmacology 1991;14(Suppl 2):S24‐32. [PubMed] [Google Scholar]
Cortese 2008 {published data only}
- Cortese L, Caligiuri MP, Williams R, Schieldrop P, Manchanda R, Malla A, et al. Reduction in neuroleptic‐induced movement disorders after a switch to quetiapine in patients with schizophrenia. Journal of Clinical Psychopharmacology 2008;28(1):69‐73. [DOI] [PubMed] [Google Scholar]
Cowen 1997 {published data only}
- Cowen MA, Green M, Bertollo DN, Abbott K. A treatment for tardive dyskinesia and some other extrapyramidal symptoms. Journal of Clinical Psychopharmacology 1997;17(3):190‐3. [DOI] [PubMed] [Google Scholar]
Crane 1968 {published data only}
- Crane GE. Tardive dyskinesia in schizophrenic patients treated with psychotropic drugs. Agressologie 1968;9(2):209‐16. [PubMed] [Google Scholar]
Crane 1969 {published data only}
- Crane GE, Ruiz P, Kernohan J, Wilson W, Royalty N. Effects of drug withdrawal on tardive dyskinesia. Activitas Nervosa Superior 1969;11:30‐5. [PubMed] [Google Scholar]
Crane 1970 {published data only}
- Crane GH. High doses of trifluperazine and tardive dyskinesia. Archives of Neurology 1970;22(2):176‐80. [DOI] [PubMed] [Google Scholar]
Curran 1973 {published data only}
- Curran JP. Management of tardive dyskinesia with thiopropazate. American Journal of Psychiatry 1973;130:925‐7. [DOI] [PubMed] [Google Scholar]
Curson 1985 {published data only}
- Curson DA, Barnes TR, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long term depot maintenance of chronic schizophrenic out patients: the seven year follow up of the Medical Research Council fluphenazine/placebo trial. II. The incidence of compliance problems, side effects, neurotic symptoms and depression. British Journal of Psychiatry 1985;146:469‐74. [DOI] [PubMed] [Google Scholar]
Davidson 2000 {published data only}
- Davidson M, Harvey PD, Vervarcke J, Gagiano CA, Hooge JD, Bray G, et al. A long‐term, multicenter, open‐label study of risperidone in elderly patients with psychosis. On behalf of the Risperidone Working Group. International Journal of Geriatric Psychiatry 2000;15(6):506‐14. [DOI] [PubMed] [Google Scholar]
de Jesus Mari 2004 {published data only}
- Jesus Mari J, Lima MS, Costa AN, Alexandrino N, Rodrigues‐Filho S, Oliveira IR, et al. The prevalence of tardive dyskinesia after a nine month naturalistic randomized trial comparing olanzapine with conventional treatment for schizophrenia and related disorders. European Archives of Psychiatry and Clinical Neuroscience 2004;254(6):356‐61. [DOI: 10.1007/s00406-004-0514-1] [DOI] [PubMed] [Google Scholar]
Delwaide 1979 {published data only}
- Delwaide PJ, Desseilles M. Controlled therapeutic study of spontaneous bucco‐linguo‐facial dyskinesias [Etude therapeutique controlee des dyskinesies bucco‐linguo‐faciales spontanees]. Semaine des Hopitaux 1979;55(35‐6):1585‐9. [MEDLINE: ; PMID 9248874] [PubMed] [Google Scholar]
Diamond 1986 {published data only}
- Diamond BI, Borison RL. Basic and clinical studies of neuroleptic‐induced supersensitivity psychosis and dyskinesia. Psychopharmacology Bulletin 1986;22(3):900‐5. [PubMed] [Google Scholar]
Dixon 1993 {published data only}
- Dixon L, Thaker G, Conley R, Ross D, Cascella N, Tamminga C. Changes in psychopathology and dyskinesia after neuroleptic withdrawal in a double‐blind design. Schizophrenia Research 1993;10(3):267‐71. [DOI] [PubMed] [Google Scholar]
Fahn 1983 {published data only}
- Fahn S. Long term treatment of tardive dyskinesia with presynaptically acting dopamine‐depleting agents. Advances in Neurology 1983;37:267‐76. [PubMed] [Google Scholar]
Fahn 1985 {published data only}
- Fahn S. A therapeutic approach to tardive dyskinesia. Journal of Clinical Psychiatry 1985;46:19‐24. [PubMed] [Google Scholar]
Freeman 1980 {published data only}
- Freeman H, Soni SD. Oxypertine for tardive dyskinesia. British Journal of Psychiatry 1980;136:522‐3. [MEDLINE: ; PMID 6104524] [DOI] [PubMed] [Google Scholar]
- Freeman HL, Soni SD. Treatment of tardive dyskinesia with oxypertine. Supplement to Progress in Neuropsychopharmacology. Proceedings of the 12th Congress of Collegium Internationale Neuro‐psychopharmacologicum; 1980. 1980. [MEDLINE: ; PMID 1020682]1020682
- Freeman HL, Soni SD, Carpenter L. A controlled trial of oxypertine in tardive dyskinesia. International Pharmacopsychiatry 1980;15(5):281‐91. [MEDLINE: ; PMID 6114934] [DOI] [PubMed] [Google Scholar]
Gardos 1984 {published data only}
- Gardos G, Cole JO, Rapkin RM, LaBrie A, Baquelod E, Moore P, et al. Anticholinergic challenge and neuroleptic withdrawal. Changes in dyskinesia and symptom measures. Archives of General Psychiatry 1984;41(11):1030‐5. [DOI] [PubMed] [Google Scholar]
Gerlach 1975 {published data only}
- Gerlach J, Thorsen K, Fog R. Extrapyramidal reactions and amine metabolites in cerebrospinal fluid during haloperidol and clozapine treatment of schizophrenic patients. Psychopharmacologia 1975;40(4):341‐50. [DOI] [PubMed] [Google Scholar]
Gerlach 1978 {published data only}
- Gerlach J, Simmelsgaard H. Tardive dyskinesia during and following treatment with haloperidol, haloperidol and biperiden, thioridazine and clozapine. Psychopharmacology 1978;59:105‐12. [DOI] [PubMed] [Google Scholar]
Gerlach 1984a {published data only}
- Bjorndal N, Casey D, Gerlach J. Dopamine antagonist and agonist treatment of tardive dyskinesia. Advances in Biochemical Psychopharmacology 1980;24:541‐3. [PubMed] [Google Scholar]
- Gerlach J, Casey DE. Dogmatil in delayed dyskinesia [Le Dogmatil dans les dyskinesies tardives]. Semaine des Hopitaux 1985;61(19):1369‐75. [Google Scholar]
- Gerlach J, Casey DE. Sulpiride in tardive dyskinesia. Acta Psychiatrica Scandinavica 1984;69(S311):93‐102. [DOI] [PubMed] [Google Scholar]
Gerlach 1984b {published data only}
- Gerlach J, Korsgaard S, Noring U. Primary (initial) and secondary (tardive) dyskinesia: effect of fluperlapine, a new atypical neuroleptic drug. Catecholamines: Neuropharmacology and Central Nervous System ‐ Therapeutic Aspects. New York: Liss, 1984. [Google Scholar]
- Korsgaard S, Noring U, Gerlach J. Fluperlapine in tardive dyskinesia and parkinsonism. Psychopharmacology 1984;84(1):76‐9. [DOI] [PubMed] [Google Scholar]
Gibson 1980 {published data only}
- Gibson AC. Depot fluphenazine and tardive dyskinesia in an outpatient population. In: Fann WE, Smith RC, Davis JM, Domino EF editor(s). Tardive Dyskinesia: Research and Treatment. New York: Spectrum Publications, 1980. [Google Scholar]
- Gibson AC. Effect of drug holidays on tardive dyskinesia. In: Usdine E, Forrest IS editor(s). Phenothiazines and structurally related drugs: basic and clinical studies. New York: Elsevier‐North Holland, 1980. [Google Scholar]
Glazer 1984 {published data only}
- Glazer WM, Morre DC, Schooler NR, Brenner LM, Morgenstern H. Tardive dyskinesia: a discontinuation study. Archives of General Psychiatry 1984;41:623‐7. [DOI] [PubMed] [Google Scholar]
Glazer 1989 {published data only}
- Glazer WM, Bowers MB, Charney DS, Heninger GR. The effect of neuroleptic discontinuation on psychopathology, involuntary movements, and biochemical measures in patients with persistent tardive dyskinesia. Biological Psychiatry 1989;26:224‐33. [DOI] [PubMed] [Google Scholar]
Goldberg 1981 {published data only}
- Goldberg SC, Shenoy RS, Sadler A, Hamer R, Ross B. The effects of a drug holiday on relapse and tardive dyskinesia in chronic schizophrenics. Psychopharmacology Bulletin 1981;17(1):116‐7. [MEDLINE: ; PMID 7232638] [PubMed] [Google Scholar]
- Shenoy RS, Sadler AG, Goldberg SC, Hamer RM, Ross B. Effects of a six‐week drug holiday on symptom status, relapse, and tardive dyskinesia in chronic schizophrenics. Journal of Clinical Psychopharmacology 1981;1(3):141‐5. [DOI] [PubMed] [Google Scholar]
Greil 1984 {published data only}
- Greil W, Haag H, Rossnagl G, Ruther E. Effect of anticholinergics on tardive dyskinesia. A controlled discontinuation study. British Journal of Psychiatry 1984;145:304‐10. [MEDLINE: ; PMID 6148119] [DOI] [PubMed] [Google Scholar]
Haggstrom 1980 {published data only}
- Haggstrom J. Sulpride in tardive dyskinesia. Current Therapeutic Research 1980;27(2):164‐9. [Google Scholar]
Heresco‐Levy 1993 {published data only}
- Heresco‐Levy U, Greenberg D, Lerer B, Dasberg H, Brown WA. Trial of maintenance neuroleptic dose reduction in schizophrenic outpatients: two‐year outcome. Journal of Clinical Psychiatry 1993;54(2):59‐62. [PubMed] [Google Scholar]
- Heresco‐Levy U, Greenberg D, Lerer B, Dasberg H, Brown WA. Two‐year trial of maintenance neuroleptic dose reduction in schizophrenic out‐patients: predictors of relapse. Israel Journal of Psychiatry and Related Science 1995;32(4):268‐75. [PubMed] [Google Scholar]
Hershon 1972 {published data only}
- Hershon HI, Kennedy PF, McGuire RJ. Persistence of extra‐pyramidal disorders and psychiatric relapse after withdrawal of long‐term phenothiazine therapy. British Journal of Psychiatry 1972;120(554):41‐50. [DOI] [PubMed] [Google Scholar]
Herz 1991 {published and unpublished data}
- Herz MI, Glazer WM, Mostert MA, Sheard MA, Szymanski HV, Hafez H, et al. Intermittent versus maintenance medication in schizophrenia: two‐year results. Archives of General Psychiatiry 1991;48:333‐9. [DOI] [PubMed] [Google Scholar]
Hogarty 1976 {published data only}
- Hogarty GE, Ulrich RF, Mussare F, Aristigueta N. Drug discontinuation among long term, successfully maintained schizophrenic outpatients. Diseases of the Nervous System 1976;37:494‐500. [PubMed] [Google Scholar]
Hogarty 1988 {published data only}
- Hogarty GE, McEvoy JP, Munetz M, DiBarry AL, Bartone P, Cather R, et al. Dose of fluphenazine, familial expressed emotion, and outcome in schizophrenia ‐ results of a two‐year controlled study. Archives of General Psychiatry 1988;45(September):797‐805. [DOI] [PubMed] [Google Scholar]
Inada 2003 {published data only}
- Inada T, Beasley CM Jr, Tanaka Y, Walker DJ. Extrapyramidal symptom profiles assessed with the Drug‐Induced Extrapyramidal Symptom Scale: comparison with Western scales in the clinical double‐blind studies of schizophrenic patients treated with either olanzapine or haloperidol. International Clinical Psychopharmacology. United States of America: Lippincott Williams & Wilkins, 2003; Vol. 18, issue 1:39‐48. [DOI] [PubMed]
Inderbitzin 1994 {published data only}
- Inderbitzin LB, Lewine RRJ, SchellerGilkey G, Swofford CD, Egan GJ, Gloersen BA, et al. A double‐blind dose‐reduction trial of fluphenazine decanoate for chronic, unstable schizophrenic patients. American Journal of Psychiatry 1994;151(12):1753‐9. [DOI] [PubMed] [Google Scholar]
Jean‐Noel 1999 {published data only}
- Jean‐Noel B, Wood AJ, Kiesler GM, Birkett M, Tollefson GD. Olanzapine vs. clozapine: an international double blind study in the treatment of resistant schizophrenia. 11th World Congress of Psychiatry; 1999 Aug 6‐11; Hamburg, Germany. 1999; Vol. 2:143.
Jeste 1977 {published data only}
- Jeste DV, Olgiati SG, Ghali AY. Masking of tardive dyskinesia with four times‐a‐day administration of chlorpromazine. Diseases of the Nervous System 1977;38(9):755‐8. [PubMed] [Google Scholar]
Jeste 1979 {published data only}
- Jeste DV, Potkin SG, Sinha S, Feder S, Wyatt RJ. Tardive dyskinesia ‐ reversible and persistent. Archives of General Psychiatry 1979;36(May):585‐90. [DOI] [PubMed] [Google Scholar]
Johnson 1983 {published data only}
- Johnson DAW, Pasterski G, Ludlow JM, Street K, Taylor RDW. The discontinuance of maintenance neuroleptic therapy in chronic schizophrenic patients: drug and social consequences. Acta Psychiatrica Scandinavica 1983;67:339‐52. [DOI] [PubMed] [Google Scholar]
Johnson 1987 {published and unpublished data}
- Johnson DA, Ludlow JM, Street K, Taylor RD. Double‐blind comparison of half‐dose and standard‐dose flupenthixol decanoate in the maintenance treatment of stabilised out‐patients with schizophrenia. British Journal of Psychiatry 1987;151:634‐8. [DOI] [PubMed] [Google Scholar]
Jolley 1990 {published data only}
- Jolley AG, Hirsch SR, McRink A, Manchanda R. Trial of brief intermittent neuroleptic prophylaxis for selected schizophrenic outpatients: clinical outcome at one year. BMJ 1989;298(6679):985‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley AG, Hirsch SR, Morrison E, McRink A, Wilson L. Trial of brief intermittent neuroleptic prophylaxis for selected schizophrenic outpatients: clinical and social outcome at two years. BMJ 1990;301(6756):837‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jus 1979 {published data only}
- Jus A, Jus K, Fontaine P. Long term treatment of tardive dyskinesia. Journal of Clinical Psychiatry 1979;40(2):72‐7. [PubMed] [Google Scholar]
Kalachnik 1984 {published and unpublished data}
- Kalachnik JE, Harder SR, Kidd‐Nielsen P, Errickson E, Doebler M, Sprague RL. Persistent tardive dyskinesia in randomly assigned neuroleptic reduction, neuroleptic nonreduction, and no‐neuroleptic history groups: preliminary results. Psychopharmacology Bulletin 1984;20(1):27‐32. [PubMed] [Google Scholar]
Kane 1993 {published data only}
- Kane JM, Woerner MG, Pollack S, Safferman AZ, Lieberman JA. Does clozapine cause tardive dyskinesia?. Journal of Clinical Psychiatry 1993;54(9):327‐30. [PubMed] [Google Scholar]
Kinon 2004 {published data only}
- Kinon B, Stauffer VL, Wang L, Thi KT, Kollack‐Walker S. Olanzapine improves tardive dyskinesia in patients with schizophrenia in a controlled prospective study. International Journal of Neuropsychopharmacology. Abstracts of the 23rd Congress of the Collegium Internationale Neuro‐Psychopharmacologicum 2002;5(Suppl 1):S165. [Google Scholar]
- Kinon BJ, Jeste DV, Kollack‐Walker S, Stauffer V, Liu‐Seifert H. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Progress in neuro‐psychopharmacology & biological psychiatry 2004;28(6):985‐96. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Stauffer VL, Wang L, Thi K, Niewoehner J, Kollack‐Walker S. Olanzapine improves dyskinesia in patients with schizophrenia. International Congress on Schizophrenia Research (ICSR) March 29‐April 2, 2003, Colorado Springs, Colorado. 2003.
- Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine improves tardive dyskinesia in patients with schizophrenia. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, PA, USA. 2002.
- Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine improves tardive dyskinesia in patients with schizophrenia. Schizophrenia Research 2002;53(3 Suppl.1):191. [MEDLINE: ; PMID 11580001]11580001 [Google Scholar]
- Kollack‐Walker S, Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine improves tardive dyskinesia in patients with schizophrenia. Schizophrenia Research 2003;60:359. [PsycINFO 2003‐06156‐011; PMID 12765745]12765745 [Google Scholar]
Kirch 1983 {published data only}
- Kirch D, Hattox S, Bell J, Murphy R, Freedman R. Plasma homovanillic acid and tardive dyskinesia during neuroleptic maintenance and withdrawal. Psychiatry Research 1983;9:217‐23. [DOI] [PubMed] [Google Scholar]
Kopala 2004 {published data only}
- Kopala L, Rabinowitz J, Emsley R, McGorry P. Extra‐pyramidal signs and symptoms (EPS) in recent onset schizophrenia: a comparison of risperidone and haloperidol. Schizophrenia Research 2004;67(1):187. [Google Scholar]
Lal 1974 {published data only}
- Lal S, Ettigi P. Comparison of thiopropazate and trifluoperazine on oral dyskinesia ‐ a double blind study. Current Therapeutic Research, Clinical and Experimental 1974;16(9):990‐7. [PubMed] [Google Scholar]
Leblanc 1994 {published data only}
- Leblanc G, Cormier H, Gagne MA, Vaillancourt S. Effects of neuroleptic reduction in schizophrenic outpatients receiving high doses. Canadian Journal of Psychiatry 1994;39(4):223‐9. [DOI] [PubMed] [Google Scholar]
Leblhuber 1987 {published data only}
- Leblhuber F. Treatment of permanent tardive dyskinesia with tiapride, a selective D2‐receptor blocking agent. Clinical Neuropharmacology 1987;10(5):458‐61. [MEDLINE: ; EMBASE 1989028406; PMID 2902921] [DOI] [PubMed] [Google Scholar]
Levine 1980 {published data only}
- Levine J, Schooler NR, Severe J, Escobar J, Gelenberg A, Mandel M, et al. Discontinuation of oral and depot fluphenazine in schizophrenic patients after one year of continuous medication: a controlled study. Advances in Biochemical Psychopharmacology 1980;24:483‐93. [PubMed] [Google Scholar]
Lieberman 1988 {published data only}
- Lieberman J, Pollack S, Lesser M, Kane J. Pharmacologic characterization of tardive dyskinesia. Journal of Clinical Psychopharmacology 1988;8(4):254‐60. [PubMed] [Google Scholar]
Lieberman 1989 {published data only}
- Lieberman JA, Saltz BL, Johns CA, Pollack S, Borenstein M, Kane J. The effects of clozapine on tardive dyskinesia. British Journal of Psychiatry 1991;158:503‐10. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Saltz BL, Johns CA, Pollack S, Kane, JM. Clozapine effects on tardive dyskinesia. Psychopharmacology Bulletin 1989;25(1):57‐62. [PubMed] [Google Scholar]
Lin 2006 {published data only}
- Lin CC, Chen JY, Bai YM, Chen TT, Wang YC, Liou YJ, et al. Remission of tardive dyskinesia following the switch from clozapine to zotepine: 2‐year follow‐up. European Neuropsychopharmacology 2006;16(Suppl 4):S410. [Google Scholar]
Littrell 1993 {published data only}
- Littrell K, Magill AM. The effect of clozapine ‐ on preexisting tardive dyskinesia. Journal of Psychosocial Nursing and Mental Health Services 1993;31(9):14‐8. [DOI] [PubMed] [Google Scholar]
MacKay 1980 {published data only}
- MacKay AV, Sheppard GP, Saha BK, Motley B, Johnson A, Marsden CD. Failure of lithium treatment in established tardive dyskinesia. Psychological Medicine 1980;10(3):583‐7. [DOI] [PubMed] [Google Scholar]
Marder 1987 {published data only}
- Marder SR, Putten T, Mintz J, Lebell M, McKenzie J, May PRA. Low‐ and conventional‐dose maintenance therapy with fluphenazine decanoate: two‐year outcome. Archives of General Psychiatry 1987;44:518‐21. [DOI] [PubMed] [Google Scholar]
- Marder SR, Putten T, Mintz J, McKenzie J, Lebell M, Faltico G, et al. Costs and benefits of two doses of fluphenazine. Archives of General Psychiatry 1984;41:1025‐9. [DOI] [PubMed] [Google Scholar]
McCreadie 1980 {published data only}
- McCreadie RG, Dingwall JM, Wiles DH, Heykants JJ. Intermittent pimozide versus fluphenazine decanoate as maintenance therapy in chronic schizophrenia. British Journal of Psychiatry 1980;137(6):510‐7. [DOI] [PubMed] [Google Scholar]
Meco 1989 {published data only}
- Meco G, Bedini L, Bonifati V, Sonsini U. Risperidone in the treatment of chronic schizophrenia with tardive dyskinesia. A single‐blind crossover study versus placebo. Current Therapeutic Research ‐ Clinical and Experimental 1989;46(5):876‐83. [Google Scholar]
Miller 1994 {published data only}
- Miller DD, Flaum M, Arndt S, Fleming F, Andreasen NC. Effect of antipsychotic withdrawal on negative symptoms in schizophrenia. Neuropsychopharmacology 1994;11(1):11‐20. [DOI] [PubMed] [Google Scholar]
NDSG 1986 {published data only}
- Gerlach J. Tardive dyskinesia: pathophysiological mechanisms and clinical trials. L'Encephale 1988;XIV:227‐32. [PubMed] [Google Scholar]
- Nordic Dyskinesia Study Group. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video‐controlled multicenter study with chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden. Psychopharmacology 1986;90(4):423‐9. [PubMed] [Google Scholar]
- Povlsen UJ, Noring U, Meidahl B, Korsgaard S, Waehrens J, Gerlach J. [Neuroleptikas virkning pa tardive dyskinesier]. Ugeskr Laeger 1987;149(25):1682‐5. [PubMed] [Google Scholar]
Newcomer 1992 {published data only}
- Newcomer JW, Riney SJ, Vinogradov S, Csernansky JG. Plasma prolactin and homovanillic acid as markers for psychopathology and abnormal movements after neuroleptic dose decrease. Psychopharmacology Bulletin 1992;28(1):101‐7. [PubMed] [Google Scholar]
Newton 1989 {published data only}
- Newton JE, Cannon DJ, Couch L, Fody EP, McMillan DE, Metzer WS, et al. Effects of repeated drug holidays on serum haloperidol concentrations, psychiatric symptoms, and movement disorders in schizophrenic patients. Journal of Clinical Psychiatry 1989;50:132‐5. [PubMed] [Google Scholar]
Odejide 1982 {published data only}
- Odejide OA, Aderounmu AF. Double‐blind placebo substitution: withdrawal of fluphenazine decanoate in schizophrenic patients. Journal of Clinical Psychiatry 1982;43:195‐6. [PubMed] [Google Scholar]
Pai 2001 {published data only}
- Pai Y‐M, Yu S‐C, Lin C‐C. Risperidone in reducing tardive dyskinesia: a double‐blind, placebo‐controlled study. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, PA, USA. 2002. [NR710 Wednesday, May 9, 3:00 p.m.‐5:00 p.m]
- Pai YM, Yu SC, Lin CC. Risperidone in reducing tardive dyskinesia: a double‐blind, placebo‐controlled study. Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; LA, USA. Marathon Multimedia, 2001. [MEDLINE: ; PMID 11580001]11580001
Paulson 1975 {published data only}
- Paulson GW, Rizvi CA, Crane GE. Tardive dyskinesia as a possible sequel of long‐term therapy with phenothiazines. Clinical Pediatrics 1975;14(10):953‐5. [DOI] [PubMed] [Google Scholar]
Peacock 1996 {published data only}
- Peacock L, Solgaard T, Lublin H, Gerlach J. Clozapine versus typical antipsychotics. A retro‐ and prospective study of extrapyramidal side effects. Psychopharmacologia 1996;124(1‐2):188‐96. [DOI] [PubMed] [Google Scholar]
Peluso 2012 {published data only}
- Peluso MJ, Lewis SW, Barnes TRE, Jones PB. Extrapyramidal motor side‐effects of first‐ and second‐generation antipsychotic drugs. British Journal of Psychiatry 2012;200(5):387‐92. [DOI] [PubMed] [Google Scholar]
Perry 1985 {published data only}
- Perry R, Campbell M, Adams P, Lynch N, Spencer EK, Curren EL, et al. Long‐term efficacy of haloperidol in autistic children: continuous versus discontinuous drug administration. Journal of the American Academy of Child and Adolescent Psychiatry 1989;28(1):87‐92. [DOI] [PubMed] [Google Scholar]
- Perry R, Campbell M, Green WH, Small AM, Die Trill ML, Meiselas K, et al. Neuroleptic‐related dyskinesias in autistic children: a prospective study. Psychopharmacology Bulletin 1985;21(1):140‐3. [PubMed] [Google Scholar]
Pyke 1981 {published data only}
- Pyke J, Seeman M. Neuroleptic‐free intervals in the treatment of schizophrenia. American Journal of Psychiatry 1981;138(12):1620‐1. [DOI] [PubMed] [Google Scholar]
Quinn 1984 {published data only}
- Quinn N, Marsden CD. A double blind trial of sulpiride in Huntington's disease and tardive dyskinesia. Journal of Neurology Neurosurgery and Psychiatry 1984;47(8):844‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn N, Marsden, CD. Double blind trial of Dogmatil in Huntington chorea and tardive dyskinesia [Essai en double insu du Dogmatil dans la choree de Huntington et la dyskinesie tardive]. Semaine des Hopitaux 1985;61(19):1376‐80. [Google Scholar]
Quitkin 1977 {published data only}
- Quitkin F, Rifkin A, Gochfeld L, Klein DF. Tardive dyskinesia: are first signs reversible?. American Journal of Psychiatry 1977;134:84‐7. [DOI] [PubMed] [Google Scholar]
Rapoport 1997 {published data only}
- Rapoport J, Kumra S, Jacobsen LK. The spectrum of extrapyramidal symptoms in children and young adults. 150th Annual Meeting of the American Psychiatric Association; 1997 May 17‐22; San Diego, USA. 1997. [PsycINFO 78‐16286]
Ringwald 1978 {published data only}
- Ringwald E. Dopamine‐receptor stimulators and neuroleptic‐induced dyskinesia. Pharmakopsychiatrie und Neuropsychopharmakologie 1978;11:294‐8. [DOI] [PubMed] [Google Scholar]
Rosenheck 2003 {published data only}
- Rosenheck R, Perlick D, Bingham S, Liu‐Mares W, Collins J, Warren S, et al. Effectiveness and cost of olanzapine and haloperidol in the treatment of schizophrenia: a randomized controlled trial. JAMA 2003;290(20):2693‐702. [DOI] [PubMed] [Google Scholar]
Roxburgh 1970 {published data only}
- Roxburgh PA. Treatment of persistent phenothiazine‐induced oral dyskinesia. British Journal of Psychiatry 1970;116:277‐80. [DOI] [PubMed] [Google Scholar]
Schultz 1995 {published data only}
- Schultz SK, Miller DD, Arndt S, Ziebell S, Gupta S, Andreasen NC. Withdrawal‐emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biological Psychiatry 1995;38:713‐9. [DOI] [PubMed] [Google Scholar]
Schwartz 1990 {published data only}
- Schwartz M, Moguillansky L, Lanyi G, Sharf B. Sulpiride in tardive dyskinesia. Journal of Neurology, Neurosurgery & Psychiatry 1990;53(9):800‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Seeman 1981 {published data only}
- Seeman MV. Tardive dyskinesia: two‐year recovery. Comprehensive Psychiatry 1981;22:189‐92. [DOI] [PubMed] [Google Scholar]
Simpson 1978 {published data only}
- Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology 1978;56:75‐80. [DOI] [PubMed] [Google Scholar]
Singer 1971 {published data only}
- Singer K, Cheng MN. Thiopropazate hydrochloride (Dartalan) in persistent dyskinesia with phenothiazine therapy. 5th World Congress of Psychiatry; 1971 Nov 28 ‐ Dec 4; Ciudad de Mexico, Mexico. 1971:528. [MEDLINE: ; PMID 215095]215095
- Singer K, Cheng MN. Thiopropazate hydrochloride in persistent dyskinesia. British Medical Journal 1971;4:22‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 1990 {published data only}
- Singh H, Hunt JI, Vitiello B, Simpson GM. Neuroleptic withdrawal in patients meeting criteria for supersensitivity psychosis. Journal of Clinical Psychiatry 1990;51:319‐21. [PubMed] [Google Scholar]
Small 1987 {published data only}
- Small JG, Milstein V, Marhenke JD, Hall DD, Kellams JJ. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment‐resistant psychosis. Journal of Clinical Psychiatry 1987;48:263‐7. [PubMed] [Google Scholar]
Smith 1979 {published data only}
- Smith JS, Kiloh LG. Six month evaluation of thiopropazate hydrochloride in tardive dyskinesia. Journal of Neurology, Neurosurgery and Psychiatry 1979;42(6):576‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Soni 1984 {published data only}
- Soni SD, Freeman HL, Hussein EM. Oxypertine in tardive dyskinesia: an 8‐week controlled study. British Journal of Psychiatry 1984;144:48‐52. [MEDLINE: ; PMID 6140973] [DOI] [PubMed] [Google Scholar]
Speller 1997 {published data only}
- Barnes TRE, Speller JC, Curson DA, Pantelis C, Alberts JL. A one‐year dose reduction study in chronic schizophrenic inpatients: amisulpride vs haloperidol. Schizophrenia Research 1992;6(2):107. [EMBASE 1997383387] [Google Scholar]
- Speller JC, Barnes TRE, Curson DA, Pantelis C, Alberts JL. One‐year, low‐dose neuroleptic study of in‐patients with chronic schizophrenia characterised by persistent negative symptoms. Amisulpride v. haloperidol. British Journal of Psychiatry 1997;171(December):564‐8. [EMBASE 1997383387] [DOI] [PubMed] [Google Scholar]
Spivak 1997 {published data only}
- Spivak B, Mester R, Abesgaus J, Wittenberg N, Adlersberg S, Gonen N, et al. Clozapine treatment for neuroleptic‐induced tardive dyskinesia, parkinsonism, and chronic akathisia in schizophrenic patients. Journal of Clinical Psychiatry 1997;58:318‐22. [DOI] [PubMed] [Google Scholar]
Spohn 1988 {published data only}
- Spohn HE, Coyne L, Spray J. The effect of neuroleptics and tardive dyskinesia on smooth‐pursuit eye movement in chronic schizophrenia. Archives of General Psychiatry 1988;45:833‐40. [DOI] [PubMed] [Google Scholar]
Spohn 1993 {published data only}
- Spohn HE, Coyne L. The effect of attention/information processing impairment of tardive dyskinesia and neuroleptics in chronic schizophrenics. Special Issue: tardive dyskinesia and cognitive dysfunction. Brain and Cognition 1993;23(1):28‐39. [DOI] [PubMed] [Google Scholar]
Suh 2004 {published data only}
- Suh GH, Son HG, Ju YS, Jcho KH, Yeon BK, Shin Y, et al. A randomized, double‐blind, crossover comparison of risperidone and haloperidol in Korean dementia patients with behavioral disturbances. American Journal of Geriatric Psychiatry 2004;12:509‐16. [DOI] [PubMed] [Google Scholar]
Thapa 1994 {published data only}
- Thapa PB, Meador KG, Gideon P, Fought RL, Ray WA. Effects of antipsychotic withdrawal in elderly nursing home residents. Journal of the American Geriatrics Society 1994;42:280‐6. [DOI] [PubMed] [Google Scholar]
Tollefson 1997 {published data only}
- Beasley CM, Dellva MA, Tamura RN, Morgenstern H, Glazer WM, Ferguson K, et al. A randomised double‐blind comparison of the incidence of tardive dyskinesia in patients with schizophrenia during long‐term treatment with olanzapine or haloperidol. British Journal of Psychiatry 1999;174:23‐30. [DOI] [PubMed] [Google Scholar]
- Tamura RN, Tollefson GD, Dellva MA, Beasley CM, Glazer WM, Morgenstern H. What is the differential risk of tardive dyskinesia with the novel antipsychotic olanzapine?. Schizophrenia Research 1998;29:176. [Google Scholar]
- Tollefson GD, Beasley CM, Tamura RN, Tran PV, Potvin JH. Blind, controlled, long term study of the comparative incidence of treatment emergent tardive dyskinesia with olanzapine or haloperidol. American Journal of Psychiatry 1997;154:1248‐54. [DOI] [PubMed] [Google Scholar]
Tran 1997 {published data only}
Turek 1972 {published data only}
- Turek I, Kurland AA, Hanlon TE, Bohm M. Tardive dyskinesia: its relation to neuroleptic and antiparkinson drugs. British Journal of Psychiatry 1972;121:605‐12. [DOI] [PubMed] [Google Scholar]
Williamson 1995 {published data only}
- Williamson DJ, Tran PV, Beasley CM, Tollefson GD, Sanger T, Satterlee WG. Clinical efficacy and safety of olanzapine, a new atypical antipsychotic agent. Journal of Psychopharmacology 1995;9:A47. [Google Scholar]
Wirshing 1999 {published data only}
- Wirshing DA, Marshall BD Jr, Green MF, Mintz J, Marder SR, Wirshing WC. Risperidone in treatment‐refractory schizophrenia. American Journal of Psychiatry 1999;156(9):1374‐9. [DOI] [PubMed] [Google Scholar]
Wistedt 1983 {published data only}
- Wistedt B, Wiles D, Jorgensen A. A depot neuroleptic withdrawal study neurological effects. Psychopharmacology 1983;80:101‐5. [DOI] [PubMed] [Google Scholar]
Wolf 1991 {published data only}
- Wolf MA, Bailly L, Diener JM, Martinet JP, Peretti S, Garneau Y. [Sevrage neuroleptique complet chez des patients schizophrenes presentant une symptomatologie intense et resistant au traitement]. Encephale 1991;17(4):255‐61. [PubMed] [Google Scholar]
Wright 1998 {published data only}
- Wright P, Tollefson GD, Beasley CM, Tamura RN, Tran PV, Potvin JII. A blinded, controlled, long‐term study of the comparative incidence of treatement‐emergent tardive dyskinesia with olanzapine or haloperidol. Schizophrenia Research 1998;29(1‐2):206. [No. 84E] [DOI] [PubMed] [Google Scholar]
Zander 1981 {published data only}
- Zander KJ, Fischer B, Zimmer R, Ackenheil M. Long‐term neuroleptic treatment of chronic schizophrenic patients: clinical and biochemical effects of withdrawal. Psychopharmacology 1981;73:43‐7. [DOI] [PubMed] [Google Scholar]
Zarebinski 1990 {published data only}
- Zarebinski JM, Royds JNA. Sulpiride in tardive dyskinesia. South African Medical Journal 1990;78(6):374‐5. [PubMed] [Google Scholar]
Zeng 1994 {published data only}
- Zeng ZX, Li ZC, Yu XW, Zhang YD, Cao ZC. A double‐blind trial of flunarizine therapy for tardive dyskinesia. Chinese Journal of Pharmaco Epidemiology 1994;3(4):183‐4. [Google Scholar]
References to ongoing studies
N0546099389 {published data only}
- N0546099389. A six month, rater blind comparison of quetiapine and risperidone in the treatment of tardive dyskinesia in patients with schizophrenia. National Research Register (first received 16 July 2015). [N0546099389]
Additional references
Alabed 2011
- Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma‐aminobutyric acid agonists for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 4. [DOI: 10.1002/14651858.CD000203.pub3] [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1992
- American Psychiatry Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC, 1992.
Armitage 1991
- Armitage P. Should we cross off the crossover?. Journal of Clinical Pharmacology 1991;32:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ascher‐Svanum 2008
- Ascher‐Svanum H, Zhu B, Faries D, Peng X, Kinon BJ, Tohen M. Tardive dyskinesia and the 3‐year course of schizophrenia: results from a large, prospective, naturalistic study. Journal of Clinical Psychiatry 2008;69(10):1580‐8. [DOI] [PubMed] [Google Scholar]
Ballesteros 2000
- Ballesteros J, Gonzalez‐Pinto A, Bulbena A. Tardive dyskinesia associated with higher mortality in psychiatric patients: results of a meta‐analysis of seven independent studies. Journal of Clinical Psychopharmacology 2000;20(2):188‐94. [DOI] [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR, Charing C. Westminster Medical School HHES. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Barnes 1993
- Barnes TRE, Edwards JG. The side‐effects of antipsychotic drugs. I. CNS and neuromuscular effects. In: Barnes TRE editor(s). Antipsychotic drugs and their side‐effects. London: Academic Press/Harcourt Brace & Company, 1993. [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276(8):637‐9. [PUBMED: 8773637] [DOI] [PubMed] [Google Scholar]
Bergen 1984
- Bergen JA, Griffiths DA, Rey JM, Beumont PJV. Tardive dyskinesia: fluctuating patient or fluctuating rater. British Journal of Psychiatry 1984;144:498‐502. [DOI] [PubMed] [Google Scholar]
Bergen 1989
- Bergen JA, Eyland EA, Campbell JA. The course of tardive dyskinesia in patients on long‐term neuroleptics. British Journal of Psychiatry 1989;154:523‐8. [DOI] [PubMed] [Google Scholar]
Bergin 1992
- Bergin J, Kitchin R, Berry G. Predictors of the course of tardive dyskinesia in patients receiving neuroleptics. Biological Psychiatry 1992;32:580‐94. [DOI] [PubMed] [Google Scholar]
Bergman 2017
- Bergman H, Walker DM, Nikolakopoulou A, Soares‐Weiser K, Adams CE. Systematic review of interventions for treating or preventing antipsychotic‐induced tardive dyskinesia. Health Technol Assess 2017 Aug;21(43):1‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhoopathi 2006
- Bhoopathi PS, Soares‐Weiser K. Benzodiazepines for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD000205.pub2] [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Cadet 1989
- Cadet JL, Lohr JB. Possible involvement of free radical in neuroleptic‐induced movement disorders. Annals of the New York Academy of Sciences 1989;570:176‐85. [DOI] [PubMed] [Google Scholar]
Casey 1986
- Casey D, Gerlach J. Tardive dyskinesia: what is the long‐term outcome?. In: Casey D, Gardos G editor(s). Tardive dyskinesia and neuroleptics: from dogma to reason. Washington DC: American Psychiatric Press, 1986. [Google Scholar]
Casey 1994
- Casey DE. Tardive dyskinesia: pathophysiology. In: Bloom FE, Kupfer DJ editor(s). Psychopharmacology. The Fourth Generation of Progress. New York: Raven Press, 1994. [Google Scholar]
Casey 1999
- Casey DE. Tardive dyskinesia and atypical antipsychotic drugs. Schizophrenia Research 1999;35:S31‐S36. [DOI] [PubMed] [Google Scholar]
Cavallaro 1993
- Cavallaro R, Regazzetti MG, Mundo E, Brancato V, Smeraldi E. Tardive dyskinesia outcomes: clinical and pharmacologic correlates of remission and persistence. Neuropsychopharmacology 1993;8(3):233‐9. [DOI] [PubMed] [Google Scholar]
Chong 2009
- Chong SA, Tay JA, Subramaniam M, Pek E, Machin D. Mortality rates among patients with schizophrenia and tardive dyskinesia. Journal of Clinoca; Psychopharmacology 2009;29:5‐8. [DOI] [PubMed] [Google Scholar]
Chouinard 2005
- Chouinard G, Margolese HC. Manual for the Extrapyramidal Symptom Rating Scale (ESRS). Schizophrenia Research 2005;76(2‐3):247‐65. [DOI] [PubMed] [Google Scholar]
Chouinard 2008
- Chouinard G, Chouinard VA. Atypical antipsychotics: CATIE study, drug‐induced movement disorder and resulting iatrogenic psychiatric‐like symptoms, supersensitivity rebound psychosis and withdrawal discontinuation syndromes. Psychotherapy and Psychosomatics 2008;77(2):69‐77. [DOI] [PubMed] [Google Scholar]
Cloud 2014
- Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics 2014;11(1):166‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Correll 2004
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second‐generation antipsychotics: a systematic review of 1‐year studies. American Journal of Psychiatry 2004;161(3):414‐25. [DOI] [PubMed] [Google Scholar]
Covidence [Computer program]
- Veritas Health Innovation. Covidence systematic review software. Melbourne, Australia: Veritas Health Innovation.
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Sayeh 2006
- El‐Sayeh HG, Lyra da Silva JP, Rathbone J, Soares‐Weiser K. Non‐neuroleptic catecholaminergic drugs for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000458.pub2] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthington HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2011
- Essali A, Deirawan H, Soares‐Weiser K, Adams CE. Calcium channel blockers for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD000206.pub3] [DOI] [PubMed] [Google Scholar]
Fenton 2000
- Fenton WS. Prevalence of spontaneous dyskinesia in schizophrenia. Journal of Clinical Psychiatry 2000;61(suppl 4):10‐14. [PubMed] [Google Scholar]
Fernandez 2001
- Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14‐year follow‐up. Neurology 2001;56:805‐7. [DOI] [PubMed] [Google Scholar]
Fleiss 1984
- Fleiss JL. The crossover study. The Design and Analysis of Clinical Experiments. Chichester: John Wiley & Sons, 1984. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gardos 1988
- Gardos G, Cole JO, Haskell D, Marby D, Schniebolk Paine S, Moore P. The natural history of tardive dyskinesia. Journal of Clinical Psychopharmacology 1988;8:31S‐7S. [PubMed] [Google Scholar]
Gardos 1994
- Gardos G, Casey DE, Cole JO, Perenyi A, Kocsis E, Arato M, et al. Ten‐year outcome of tardive dyskinesia. American Journal of Psychiatry 1994;151:836‐41. [DOI] [PubMed] [Google Scholar]
Gerlach 1988
- Gerlach J, Casey DE. Tardive dyskinesia. Acta Psychiatrica Scandinavica 1988;77:369‐78. [DOI] [PubMed] [Google Scholar]
Gerlach 1993
- Gerlach J, Korsgaard S, Clemmesen P, Lauersen AML, Magelund G, Noring U, et al. The St. Hans Rating Scale for extrapyramidal syndromes: reliability and validity. Acta Psychiatrica Scandinavica 1993;87(4):244‐52. [DOI] [PubMed] [Google Scholar]
Gilbert 1995
- Gilbert PL, Harris J, McAdam LA, Jeste DV. Neuroleptic withdrawal in schizophrenic patients ‐ a review of the literature. Archives of General Psychiatry 1995;52:173‐88. [DOI] [PubMed] [Google Scholar]
Glazer 1990
- Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. British Journal of Psychiatry 1990;157:585‐92. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1970
- Guy W, Bonato RR. Manual for the ECDEU Assessment Battery. National Institute of Mental Health. In: Guy W, Bonato RR, editor(s). Washington DC: NIMH, 1970. [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Revised Edition. Washington, DC: Department of Health, Education and Welfare, 1976. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. In: Higgins JPT, Green S editor(s). The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd., 2005. [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 (updated September 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011a
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Jadad 1996
- Jadad A, Moore A, Carroll D, Jenkinson C, Reynolds DJM, Gavanagh DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Jeste 1993
- Jeste DV, Caligiuri MP. Tardive dyskinesia. Schizophrenia Bulletin 1993;19(2):303‐15. [DOI] [PubMed] [Google Scholar]
Kane 1986
- Kane JM, Woerner M, Satantakos S, Kinon B, Lieberman J. Do low‐dose neuroleptics prevent or ameliorate tardive dyskinesia?. In: Casey D, Gardos G editor(s). Tardive Dyskinesia and Neuroleptics: From Dogma to Reason. Washington DC: American Psychiatric Press, 1986. [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Kissling W, Davis JM. Second‐generation antipsychotics for schizophrenia: can we resolve the conflict?. Psychological Medicine 2009;39(10):1591‐602. [DOI] [PubMed] [Google Scholar]
Lieberman 1996
- Lieberman JA, Fleishhacker W. Introduction. British Journal of Psychiatry 1996;168(Supplement 29):7‐8. [Google Scholar]
Lingjaerde 1987
- Lingjærde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychiatrica Scandinavica 1987;76:Suppl. 334. [DOI] [PubMed] [Google Scholar]
Maher 2012
- Maher AR, Theodore G. Summary of the comparative effectiveness review on off‐label use of atypical antipsychotics. Journal of Managed Care Pharmacy 2012;18(5 Suppl B):S1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Margolese 2005
- Margolese HC, Chouinard G, Kolivakis TT, Beauclair L, Miller R. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Canadian Journal of Psychiatry. Revue Canadienne de Psychiatrie 2005;50(9):541‐7. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Martins 2011
- Martins ES, Rosso A, Coutinho E, Adams C, Huf G. Prevalence of tardive dyskinesia and all‐cause mortality amongst patients in a large psychiatric institute in Rio de Janeiro. Revista de Psiquiatria Clínica 2011;38:44. [Google Scholar]
Miller 2007
- Miller DD, Eudicone JM, Pikalov A, Kim E. Comparative assessment of the incidence and severity of tardive dyskinesia in patients receiving aripiprazole or haloperidol for the treatment of schizophrenia: a post hoc analysis. The Journal of Clinical Psychiatry 2007;68(12):1901‐6. [DOI] [PubMed] [Google Scholar]
Miller 2008
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, McEvoy JP, Saltz BL, et al. Extrapyramidal side‐effects of antipsychotics in a randomised trial. British Journal of Psychiatry 2008;193(4):279‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. The PRISMA Group (2009). Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. PLoS Medicine 6;7:e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Morgenstern 1993
- Morgenstern H, Glazer WM. Identifying risk factors for tardive dyskinesia among long‐term outpatients maintained with neuroleptic medication. Results of the Yale Tardive Dyskinesia Study. Archives of General Psychiatry 1993;50:723‐33. [DOI] [PubMed] [Google Scholar]
NICE 2014
- NICE. Psychosis and schizophrenia in adults: treatment and management. NICE clinical guideline 178 (guidance.nice.org.uk/cg178) 2014.
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pocock 1983
- Pocock SJ. Crossover trials. Clinical trials. A practical approach. Chichester: John Wiley & Sons, 1983. [Google Scholar]
Rosenheck 2007
- Rosenheck RA. Evaluating the cost‐effectiveness of reduced tardive dyskinesia with second‐generation antipsychotics. British Journal of Psychiatry 2007;191:238‐45. [DOI] [PubMed] [Google Scholar]
Schmidt 1991
- Schmidt M, Meister P, Baumann P. Treatment of tardive dyskinesias with vitamin E. European Psychiatry 1991;6:201‐7. [Google Scholar]
Schooler 1993
- Schooler NR, Keith SJ. Clinical research for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Shale 1996
- Shale H, Tanner C. Pharmacological options for the management of dyskinesias. Drugs 1996;52(6):849‐60. [DOI] [PubMed] [Google Scholar]
Shokraneh 2017
- Shokraneh F, Adams CE. Study‐based registers of randomized controlled trials: starting a systematic review with data extraction or meta‐analysis. BioImpacts 2017;InPress:InPress. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212:11‐19. [DOI] [PubMed] [Google Scholar]
Smith 1980
- Smith JM, Balessarini RJ. Changes in prevalence, severity and recovery in tardive dyskinesia with age. Archives of General Psychiatry 1980;37:1368‐73. [DOI] [PubMed] [Google Scholar]
Soares 2000
- Soares K, McGrath J. Anticholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000204; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 1997
- Soares‐Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1997, Issue 2. [DOI: 10.1002/14651858.CD000204] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2003
- Soares‐Weiser K, Joy C. Miscellaneous treatments for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000208] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2011
- Soares‐Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD000209.pub2] [DOI] [PubMed] [Google Scholar]
Tammenmaa 2002
- Tammenmaa I, McGrath J, Sailas E, Soares‐Weiser K. Cholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD000207; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tarsy 2011
- Tarsy D, Lungu C, Baldessarini RJ. Epidemiology of tardive dyskinesia before and during the era of modern antipsychotic drugs. Handbook of Clinical Neurology 2011;100:601‐16. [DOI] [PubMed] [Google Scholar]
Taylor 2009
- Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines (10th Edition). London: Informa Healthcare, 2009. [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Waddington 1988
- Waddington JL, Crow TJ. Abnormal involuntary movements and psychosis in the preneuroleptic era and in unmedicated patients: implications for the concept of tardive dyskinesia. In: Wolf ME, Mosnaim AD editor(s). Tardive Dyskinesia: Biological Mechanisms and Clinical Aspects. Washington DC: American Psychiatric Press, 1988. [Google Scholar]
Woods 2010
- Woods SW, Morgenstern H, Saksa JR, Walsh BC, Sullivan MC, Money R, et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications: a prospective cohort study. Journal of Clinical Psychiatry 2010;71(4):463‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
McGrath 1998
- McGrath JJ, Soares‐Weiser KVS. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 1998, Issue 2. [DOI: 10.1002/14651858.CD000459] [DOI] [PubMed] [Google Scholar]
McGrath 2000
- McGrath J, Soares K. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000459.pub2] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2006
- Soares‐Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000459.pub2] [DOI] [PubMed] [Google Scholar]