

Abstract
Background
Up to 75% of people with serious mental illness (SMI) such as schizophrenia and bipolar disorder have co‐occurring substance use disorders (dual diagnosis). Dual diagnosis can have an adverse effect on treatment and prognosis of SMI.
Objectives
To evaluate the effects of risperidone compared to treatment with other antipsychotics (first‐generation and other second‐generation antipsychotics) used in people with serious mental illness and co‐occurring substance misuse.
Search methods
On 6 January 2016 and 9 October 2017, we searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials (including trial registers).
Selection criteria
We selected randomised trials of risperidone versus any other antipsychotic in people with SMI and substance abuse (dual diagnosis). We included trials meeting our inclusion criteria and reporting useable data. We excluded trials that either did not meet our inclusion criteria or met our inclusion criteria but did not report any useable data.
Data collection and analysis
We independently inspected citations and selected studies. For included studies, we independently extracted data and appraised study quality. For binary outcomes we calculated the risk ratios (RRs) and their 95% confidence intervals. For continuous outcomes we calculated the mean differences (MDs) and their 95% confidence intervals. We pooled data using random‐effects meta‐analyses and assessed the quality of evidence, creating a 'Summary of findings' table using the GRADE approach.
Main results
We identified eight randomised trials containing a total of 1073 participants with SMI and co‐occurring substance misuse. Seven of these contributed useable data to the review. There was heterogeneity in trial design and measurement. Risperidone was compared to clozapine, olanzapine, perphenazine, quetiapine and ziprasidone. Few trials compared risperidone with first‐generation agents. Few trials examined participants with a dual diagnosis from the outset and most trials only contained separate analyses of subgroups with a dual diagnosis or were secondary data analyses of subgroups of people with a dual diagnosis from existing larger trials.
For risperidone versus clozapine we found no clear differences between these two antipsychotics in the reduction of positive psychotic symptoms (1 randomised controlled trial (RCT), n = 36, mean difference (MD) 0.90, 95% CI −2.21 to 4.01, very low quality evidence), or reduction in cannabis use (1 RCT, n = 14, risk ratio (RR) 1.00, 95% CI 0.30 to 3.35, very low quality evidence), improvement in subjective well‐being (1 RCT, n = 36, MD −6.00, 95% CI −14.82 to 2.82, very low quality evidence), numbers discontinuing medication (1 RCT, n = 36, RR 4.05, 95% CI 0.21 to 78.76, very low quality evidence), extrapyramidal side‐effects (2 RCTs, n = 50, RR 2.71, 95% CI 0.30 to 24.08; I² = 0%, very low quality evidence), or leaving the study early (2 RCTs, n = 45, RR 0.49, 95% CI 0.10 to 2.51; I² = 34%, very low quality evidence). Clozapine was associated with lower levels of craving for cannabis (1 RCT, n = 28, MD 7.00, 95% CI 2.37 to 11.63, very low quality evidence).
For risperidone versus olanzapine we found no clear differences in the reduction of positive psychotic symptoms (1 RCT, n = 37, MD −1.50, 95% CI −3.82 to 0.82, very low quality evidence), reduction in cannabis use (1 RCT, n = 41, MD 0.40, 95% CI −4.72 to 5.52, very low quality evidence), craving for cannabis (1 RCT, n = 41, MD 5.00, 95% CI −4.86 to 14.86, very low quality evidence), parkinsonism (1 RCT, n = 16, MD −0.08, 95% CI −1.21 to 1.05, very low quality evidence), or leaving the study early (2 RCT, n = 77, RR 0.68, 95% CI 0.34 to 1.35; I² = 0%, very low quality evidence).
For risperidone versus perphenazine, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 281, RR 1.05, 95% CI 0.92 to 1.20, low‐quality evidence).
For risperidone versus quetiapine, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 294, RR 0.96, 95% CI 0.86 to 1.07, low‐quality evidence).
For risperidone versus ziprasidone, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 240, RR 0.96, 95% CI 0.85 to 1.10, low‐quality evidence).
For many comparisons, important outcomes were missing; and no data were reported in any study for metabolic disturbances, global impression of illness severity, quality of life or mortality.
Authors' conclusions
There is not sufficient good‐quality evidence available to determine the effects of risperidone compared with other antipsychotics in people with a dual diagnosis. Few trials compared risperidone with first‐generation agents, leading to limited applicability to settings where access to second‐generation agents is limited, such as in low‐ and middle‐income countries. Moreover, heterogeneity in trial design and measurement of outcomes precluded the use of many trials in our analyses. Future trials in this area need to be sufficiently powered but also need to conform to consistent methods in study population selection, use of measurement scales, definition of outcomes, and measures to counter risk of bias. Investigators should adhere to CONSORT guidelines in the reporting of results.
Plain language summary
Risperidone versus other antipsychotics for people with dual diagnosis of a psychiatric disorder and an alcohol or drug use disorder
What is dual diagnosis?
Dual diagnosis is a term used to describe people who have both a psychiatric disorder and an alcohol or drug use disorder. Up to 75% of people with a serious mental illness (SMI) are dual diagnosis. It has been suggested that one of the reasons behind the high levels of substance use in people with SMI is due to 'self‐medication', with patients taking additional drugs in order to counter their distressing symptoms. People with a dual diagnosis have been shown to have more complications in their treatment, including higher rates of relapse and re‐hospitalisation, more contact with legal and forensic services, higher levels of psychotic symptoms, more risk‐taking behaviour, greater levels of side‐effects to antipsychotics and lower medication adherence. Antipsychotics are the main treatment for SMI. It has been suggested that second‐generation antipsychotics (SGAs) such as risperidone may be superior to older, first‐generation antipsychotics (FGAs) in improving negative affective states, reducing drug craving, improving subjective well‐being, and may lead to fewer side‐effects and hence greater medication adherence. Such improvements in symptoms may lead to less self‐medication with alcohol and drugs, and improved overall mental states. However it remains unclear to what extent risperidone, one of the first atypical antipsychotics to be manufactured, is superior to other antipsychotics for dual diagnosis.
Who may be interested in this review?
Mental health care practitioners who treat people with SMI and dual diagnosis, and who prescribe antipsychotics for these conditions. People who use mental health services and their families who may be involved in their treatment and care.
What does this review aim to answer?
How effective and safe is risperidone compared to other antipsychotics for treating people with a dual diagnosis?
Which studies were included in the review?
We conducted searches for relevant randomised studies in January 2016 and October 2017. We found eight randomised controlled trials with 1073 participants who had a dual diagnosis. The majority of participants were adults over 18 years (4 participants were 17 years). Risperidone was compared to clozapine, olanzapine, perphenazine, quetiapine and ziprasidone.
What does the evidence from the review tell us?
We found no great effect favouring risperidone over any of the other comparison medications. Very limited data were available for side‐effects; and again, we found no real differences between risperidone and other antipsychotics. Overall the quality of the evidence available was graded as low to very low, and currently there is not sufficient evidence to indicate risperidone is superior or inferior to other antipsychotics in the treatment of people with severe mental illness and co‐occurring substance misuse.
What should happen next?
More high‐quality research is needed. Future research should include samples sufficiently large to detect meaningful clinical differences in outcomes.
Summary of findings
Summary of findings for the main comparison. RISPERIDONE versus CLOZAPINE ‐ all data short term for people with severe mental illness and co‐occurring substance misuse.
| RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: for people with serious mental illness and co‐occurring substance misuse Setting: Intervention: Risperidone Comparison: Clozapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with Clozapine | Risk with Risperidone | |||||
| Mental state: positive symptoms ‒average endpoint score (PANSS positive subscale, lower = better) | The mean positive symptoms (PANSS positive subscale, lower = better) in the intervention group was 0.9 higher (2.21 lower to 4.01 higher) | ‐ | 36 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | No trial reported "improvement in symptoms of severe mental illness" ‒ this continuous measure is the nearest proxy for this. | |
| Substance use: improvement ‒ (at least 20% reduction in use, TLFB scale) | Study population | RR 1.00 (0.30 to 3.35) | 14 (1 RCT) | ⊕⊝⊝⊝ very low3 4 | ||
| 429 per 1000 | 429 per 1000 (129 to 1000) | |||||
| Moderate | ||||||
| 429 per 1000 | 429 per 1000 (129 to 1000) | |||||
| Subjective well‐being: Subjective well‐being under neuroleptics scale ‒ average endpoint scores (SWN scale, higher = better) | The mean subjective well‐being under neuroleptics scale score (SWN scale, higher = better) in the intervention group was 6 lower (14.82 lower to 2.82 higher) | ‐ | 36 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Craving for substances: Marijuana Craving Questionnaire ‒ average endpoint scores (MCQ, lower = better) | The mean craving for substances score on the Marijuana Craving Questionairre (MCQ, lower = better) in the intervention group was 7 higher (2.37 higher to 11.63 higher) | ‐ | 28 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Adherence to antipsychotic medication: discontinued medication | Study population | RR 4.05 (0.21 to 78.76) | 36 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 0 per 1000 | 0 per 1,000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1,000 | 0 per 1,000 (0 to 0) | |||||
| Adverse effects. 1. Movement disorders ‐ any extrapyramidal | Study population | RR 2.71 (0.30 to 24.08) | 50 (2 RCTs) | ⊕⊝⊝⊝ very low5 6 | Many adverse effects reported ‒ none designated 'clinically important' (extrapyramidal used as proxy). | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Leaving the study early ‒ any reason | Study population | RR 0.49 (0.10 to 2.51) | 45 (2 RCTs) | ⊕⊝⊝⊝ very low5 6 | ||
| 318 per 1000 | 156 per 1000 (32 to 799) | |||||
| Moderate | ||||||
| 386 per 1000 | 189 per 1000 (39 to 968) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 High risk of performance bias and detection bias
2 Sample size is very small, optimal information size (OIS) not met to detect 25% difference
3 Performance bias, attrition bias, selective outcome reporting
4 Sample size is very small (n = 14)
5 High risk of performance bias, detection bias, attrition bias and selective outcomes reporting
6 Total sample size is very small (n<300), total event rate is very low and optimum information size (OIS) is not met
Summary of findings 2. RISPERIDONE versus OLANZAPINE ‒ short‐ and long‐term data for people with severe mental illness and co‐occurring substance misuse.
| RISPERIDONE versus OLANZAPINE‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with serious mental illness and co‐occurring substance misuse Setting: In and outpatients, United States Intervention: Risperidone Comparison: Olanzapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with Olanzapine | Risk with Risperidone | |||||
| Mental state: 2. Specific‐ Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower = better) | The mean positive symptoms total score at endpoint (SADS‐C‐PD scale, lower = better) in the intervention group was 1.5 lower (3.82 lower to 0.82 higher) | ‐ | 37 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week) | The reduction of cannabis joints smoked (number of joints smoked/week‐short term data, up to 6 months) in the intervention group was 0.4 higher (4.72 lower to 5.52 higher) | ‐ | 41 (1 RCT) | ⊕⊝⊝⊝ very low3 4 | ||
| Subjective well‐being | ‐ | ‐ | ‐ | No trial reported on this important outcome for participants with a co‐occurring substance use disorder | ||
| Craving for substances: 2. Drug Desires Questionnaire‐ average endpoint scores (DDQ, lower = better) | The mean endpoint. Drug Desires Questionnaire‐ endpoint scores (DDQ, lower = better), short term, up to 6 months‐in the intervention group was5 higher (4.86 lower to 14.86 higher) | ‐ | 41 (1 RCT) | ⊕⊝⊝⊝ very low2 3 | ||
| Adherence to antipsychotic medication: number of missed doses, average endpoint data, short term (up to 6 months) | ‐ | ‐ | ‐ | ‐ | no useable data available for this outcome | |
| Adverse effects: Parkinsonism ‐ average endpoint score (SAS, high = worse) | The mean adverse effects: ‐ Parkinsonism‐ average endpoint score (SAS, high = worse)‐ short‐term‐ up to 6 months in the intervention group was 0.08 lower (1.21 lower to 1.05 higher) | ‐ | 16 (1 RCT) | ⊕⊝⊝⊝ very low2 5 | ||
| Leaving study early: any reason | Study population | RR 0.68 (0.34 to 1.35) | 77 (2 RCTs) | ⊕⊝⊝⊝ very low4 6 | ||
| 357 per 1000 | 243 per 1000 (121 to 482) | |||||
| Moderate | ||||||
| 411 per 1000 | 279 per 1000 (140 to 554) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 High risk for performance bias, allocation concealment, unknown risk for attrition and slective reporting
2 Very low sample size, optimal information size (OIS) not met
3 High risk of attrition bias, study sponsored by pharmaceutical industry
4 Very low sample size, optimal information criterion not met, CI crosses both appreciable harm and benefit
5 High attrition risk, high other risk of funding by pharmaceutical industry, all other risk items unclear risk of bias
6 High risk of performance, attrition and funding bias. Several domains with unclear risk of bias
Summary of findings 3. RISPERIDONE versus PERHENAZINE ‒ long‐term data for people with severe mental illness and co‐occurring substance misuse.
| RISPERIDONE versus PERPHENAZINE‐long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse Setting: Outpatients, United States Intervention: RISPERIDONE Comparison: PERPHENAZINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with PERPHENAZINE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 1.05 (0.92 to 1.20) | 281 (1 RCT) | ⊕⊕⊝⊝ low1 2 | ||
| 750 per 1000 | 788 per 1000 (690 to 900) | |||||
| Moderate | ||||||
| 750 per 1000 | 788 per 1000 (690 to 900) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 High risk of attrition bias, but this does not affect this particular outcomes
2 Optimal information size criterion is met but the estimate includes no effect with both appreciable harm and benefit
Summary of findings 4. RISPERIDONE versus QUETIAPINE ‒ short‐ and long‐term data for people with severe mental illness and co‐occurring substance misuse.
| RISPERIDONE versus QUETIAPINE‐ short and long term data (up to 6months and > 12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse Setting: Outpatients, United States Intervention: RISPERIDONE Comparison: QUETIAPINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with QUETIAPINE | Risk with RISPERIDONE | |||||
| Leaving the study early: 1. any reason, long term (>12 months) | Study population | RR 0.96 (0.86 to 1.07) | 294 (1 RCT) | ⊕⊕⊝⊝ low3 4 | ||
| 825 per 1000 | 792 per 1000 (709 to 883) | |||||
| Moderate | ||||||
| 825 per 1000 | 792 per 1000 (709 to 883) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias unclear across all groups and with high risk of funding bias
2 Sample size meets optimal information threshold/ required sample size to detect 25% difference from control group in in PANSS score; at alpha of 0.05 and power of 80%.
3 Outcome not affected by risk of attrition bias
4 Optimal information criterion not met, estimate includes both appreciable harm and benefit
Summary of findings 5. RISPERIDONE versus ZIPRASIDONE ‒ long‐term data (> 12 months) for people with severe mental illness and co‐occurring substance misuse.
| RISPERIDONE versus ZIPRASIDONE‐ all data long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse Setting: Outpatients, United States Intervention: RISPERIDONE Comparison: ZIPRASIDONE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with ZIPRASIDONE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 0.96 (0.85 to 1.10) | 240 (1 RCT) | ⊕⊕⊝⊝ low1 2 | ||
| 819 per 1000 | 787 per 1000 (696 to 901) | |||||
| Moderate | ||||||
| 819 per 1000 | 787 per 1000 (696 to 901) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of attrition bias high but this does not affect this outcome
2 Optimal information size criterion met but estimate includes both appreciable harm and benefit. Total sample size small
Background
Description of the condition
Serious mental illness is characterised by severe and persisting psychiatric disorder associated with significant functional impairment, and includes disorders such as schizophrenia and bipolar disorder, but is not limited to these conditions (Ruggeri 2000). The prevalence of serious mental illness varies according to survey methods and has been reported to range from 0.4% to 7.7% between different countries (Demyttenaere 2004).
Comorbid substance use frequently occurs in people with severe mental illness such as schizophrenia and major affective disorders. The terms 'dual diagnosis' and more recently 'co‐occurring disorders' have been used to describe persons who have a diagnosis of both a severe mental illness and a drug or alcohol use disorder (Buckley 2006). Depending on the sample, population and assessment method, prevalence rates of drug and alcohol use disorders in persons with serious mental illness have been reported to vary from 25% to 74% in developed countries (Barnett 2007; Fioritti 1997; Fowler 1998; Jablensky 2000; Kessler 2004; Lambert 2005; Menezes 1996; Modestin 1997; Regier 1990; Soyka 1993; van Mastrigt 2004). The prevalence of co‐occurring substance use disorders in persons with severe mental illness from low‐ and middle‐income countries has been found to be in a similar range, with some studies reporting prevalence rates of 51% and up to 68% (Hauli 2011; Weich 2009). Patients participating in studies investigating the use of atypical antipsychotics such as risperidone in the treatment of dual diagnosis have a variety of substance use disorder diagnoses including cocaine, cannabis, opioid and alcohol use disorders; and most have a mental illness diagnosis of schizophrenia or schizoaffective disorder (Kelly 2012; Stuyt 2006). Comorbidity adversely affects the treatment and prognosis of both drug use disorders and mental illness. In comparison to people without substance use disorders, persons with a dual diagnosis have been reported to have higher levels of positive psychotic symptoms (Katz 2010), shorter time to relapse, higher rates of readmission to hospital, lower remission rates (Lambert 2005; Swofford 1996; Wade 2006), higher levels of medication non‐compliance (Ascher‐Svanum 2006; Lacro 2002; Owen 1996), and worse clinical outcomes (Lambert 2005). In addition, dual diagnosis patients with psychotic or manic symptoms treated with antipsychotics are more susceptible to extrapyramidal side‐effects such as akathisia (Salyers 2001).
Description of the intervention
Risperidone is an antipsychotic medication described as a serotonergic and dopaminergic antagonist (SDA) (Horacek 2006, Figure 1). It was the first medication in the second‐generation class of antipsychotics (SGAs), also described as 'atypical' antipsychotics, to be synthesised (Moller 2005). Risperidone is an antagonist on dopamine type 2 (D₂) receptors, and has an even higher affinity for serotonin type 2A receptors, where it also acts as an antagonist (Janssen 1988). These properties result in a relatively low propensity to induce extrapyramidal side‐effects when compared with first‐generation antipsychotics (FGAs) or 'typical' antipsychotics such as haloperidol (Hunter 2003). Important non‐neurological side‐effects include a higher risk of weight gain compared with certain typical and other atypical antipsychotics (Newcomer 2007).
1.
Risperidone
Risperidone is available in an oral as well as a long‐acting injectable formulation (Fleischhacker 2003; Moller 2007). There are no specific recommendations for any particular type of antipsychotic medication to be used preferentially in the dual diagnosis population, and it has been recommended that the same guidance for persons with only a single severe mental illness diagnosis be used in persons with a dual diagnosis (NICE 2011). Accordingly, some treatment guidelines recommend that second‐generation — or 'atypical' — antipsychotics such as risperidone be considered as potential first‐line treatment for persons with a first episode of psychosis and schizophrenia, following discussion with patients and their caregivers (Hasan 2012; Hasan 2013; Lehman 2004; NICE 2009).
How the intervention might work
A number of findings from pre‐clinical and clinical studies have suggested various mechanisms and effects through which second‐generation antipsychotics may result in improved outcomes in the treatment of dual diagnosis.
Preclinical studies have shown that risperidone blocks cocaine‐induced dopamine and serotonin release in the nucleus accumbens in the rat brain, and reduces locomotion and stereotypical behaviour in rats (Broderick 2003; Tsibulsky 1998). In turn, treatment of humans with atypical antipsychotics such as risperidone, which act antagonistically on both serotonergic and dopaminergic receptors, may translate to a reduction of conditioned responses such as cue‐elicited craving mediated via dopamine release. This may consequently lead to lower levels of drug use (Drevets 2001; Smelson 1997; Volkow 2006). In addition, research in rats has demonstrated increased sensitivity to cocaine‐induced hyper‐locomotion and increased conditioned place preference following withdrawal from haloperidol, but not from the atypical agent ziprasidone. This serves as a model of dopamine supersensitivity and induced craving which is potentially analogous to situations of non‐compliance with antipsychotic medication in humans (Fukushiro 2007; Fukushiro 2008). Treatment non‐compliance is a phenomenon that can occur in up to three‐quarters of people treated with antipsychotics (Lieberman 2005).
Despite the acute reinforcing effects of drug use via increased dopamine release during the early phases of addiction, neuro‐imaging studies in persons with drug dependence have demonstrated lower concentrations of dopaminergic receptors following chronic use (Volkow 1997; Volkow 2007). Lower dopamine receptor concentration may be due to persistently increased dopamine release in the nucleus accumbens induced by drugs of abuse (Kuhar 1996). In turn, increased craving has been associated with lower dopamine receptor levels during protracted withdrawal (Volkow 2011). Consequently, in comparison to drugs with a lower affinity for D₂ receptors such as risperidone, drugs with a high affinity for dopamine receptors, such as the first‐generation antipsychotics, may exacerbate or prolong hypodopaminergic states leading to increased drug craving (Siris 1990).
Craving has been shown to be an important predictor of relapse into substance use (Sinha 2006; Sinha 2011). Uncontrolled trials have demonstrated a decrease in craving and drug use in persons treated with risperidone (Smelson 2002). Consequently, the use of risperidone may result in lower scores on craving measures (Rosenberg 2009), and consequently decreased drug use.
Results from uncontrolled clinical trials have shown lower levels of psychotic symptoms and depression in persons treated with risperidone when compared to first‐generation antipsychotics (Smelson 1997; Smelson 2002). Risperidone also acts on the serotonergic system by means of 5HT2A receptor antagonism, which may in turn mediate its antidepressant effect (McIntyre 2007; Sajatovic 2002; Yatham 2005). This may affect the 5HT1A system resulting in fewer depressive symptoms and improved cognitive symptoms in persons with serious mental illness such as schizophrenia (Kuroki 2008; Sajatovic 2002). Altogether, these effects may translate into improved ratings on measures of psychosis, mood and quality of life in persons treated with risperidone (Smelson 2002).
Clinical studies have demonstrated comparatively lower rates of extrapyramidal side‐effects in participants treated with risperidone (Hunter 2003). As people with dual diagnosis have been shown to be more sensitive to the neurological side‐effects of neuroleptic drugs (Potvin 2009; Salyers 2001), the low propensity of risperidone to induce extrapyramidal side‐effects offers a potentially favourable side‐effect profile. This may result in improved adherence (Perkins 2002; Perkins 2006); and consequently lower rates of symptom re‐emergence and of relapse into illness (Sun 2007).
It has been postulated that one of the properties that differentiate atypical antipsychotics from typical antipsychotics may be their lower affinity for D₂ receptors and higher affinity for 5HT2A receptors (Seeman 2002). However, it remains uncertain to what extent potent 5HT2A receptor‐antagonism is responsible for the 'atypical features' of SGAs, given alternative theories of D₂ receptor binding profiles and dissociation rates conferring "atypicality" (Kapur 1995; Kapur 1996; Kapur 2001; Seeman 2002; Tort 2006). In addition, higher levels of D₂ receptor occupancy in the context of neuroleptic treatment has also been associated with lower levels of subjective well‐being (de Haan 2000). Second‐generation agents such as risperidone that have lower D₂ receptor occupancy rates (and higher dissociation rates), therefore have a more favourable profile in that they have been demonstrated to produce higher levels of subjective well‐being, and are associated with less negative affective states and anhedonia compared to first‐generation, typical medications (Smelson 2002; Vothknecht 2011). Therefore, the degree of D₂ binding and D₂ receptor dissociation of antipsychotics may have a differential impact on measures used to assess subjective well‐being. However, within the class of serotonin dopamine antagonists various agents differ in their degree of D₂ receptor affinity and dissociation, with clozapine and olanzapine having the lowest affinities (and highest dissociation rates) and risperidone having higher D₂ affinity. Therefore uncertainty remains as to how medications such as risperidone, that differ only marginally from FGAs with regards to D₂ receptor‐binding profiles (Seeman 2002), offer additional benefit over typical agents.
Figure 2 contains a diagram of a model within a Baxter 2010 logic framework, outlining the relationship between antipsychotic treatment with risperidone, potential effect modifiers and the final target clinical outcomes in a complex causal pathway.
2.
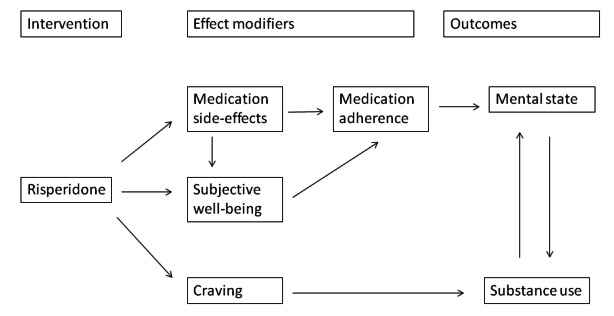
Logic framework model with potential causal pathways: risperidone treatment in persons with dual diagnosis.
Why it is important to do this review
Despite preclinical animal studies and observational research in humans, there remains a paucity of randomised trials on the efficacy of risperidone compared with other antipsychotics in the treatment of people with a dual diagnosis. In fact, most randomised controlled trials that investigate the efficacy of risperidone in the treatment of serious mental disorders such as schizophrenia have excluded people with comorbid drug or alcohol use disorders (Hunter 2003). As comorbid drug and alcohol use disorders occur commonly in people with severe mental illness, it is important to evaluate the impact of risperidone on drug use and symptom control, as well as on other functional outcomes in people with dual diagnosis. Moreover other atypical medications such as olanzapine and clozapine, that have higher D₂ receptor dissociation rates and greater activity on D₁ receptors (Seeman 2002; Tort 2006), have been postulated to reduce drug use to a greater extent compared to risperidone (Akerele 2007). This review aims to assess whether risperidone offers any additional advantage over FGAs or other SGAs in the treatment of people with serious mental illness and co‐occurring substance misuse.
Objectives
To evaluate the effects of risperidone compared to treatment with other antipsychotics (first‐generation and other second‐generation antipsychotics) used in people with serious mental illness and co‐occurring substance misuse.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs), in which risperidone was the antipsychotic treatment that was randomised. If a trial was described as 'double blind' but randomisation was implied, we included such trials after confirming randomisation with the study investigators. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week.
Types of participants
Adults, aged 17 to 65 years, with a severe mental illness and a co‐occurring substance use disorder, where severe mental illness is defined to include the following disorders as diagnosed according to the method specified in each individual trial.
Schizophrenia, schizoaffective disorder, schizophreniform disorder.
Major affective disorders such as bipolar disorder and major depressive disorder with psychotic features.
Severe mental illness is defined to exclude the following disorders.
Organic mental disorders.
Moderate to severe intellectual disability.
Drug‐induced mental disorders.
Personality disorders.
Non‐severe mental disorders such as anxiety disorders, mild depression, and somatoform disorders.
Factitious disorders or malingering.
Co‐occurring substance use disorder is diagnosed by any means or scale and includes:
alcohol or drug abuse or dependence.
Co‐occurring substance use disorder is defined to exclude:
nicotine dependence, volatile solvent abuse or dependence, and caffeine use disorders, where the trial has been designed to primarily measure the impact of risperidone on these substance use disorders and related outcomes;
studies in which drug use was not classified as either abuse or dependence.
We included RCTs that compared treatment with risperidone with another antipsychotic in people with a dual diagnosis of severe mental disorder and a co‐occurring substance use disorder.
We placed no restrictions on co‐morbid conditions such as anxiety and depressive disorders.
As indicated in the description of the condition, the phenomenon of dual diagnosis is prevalent across various settings as reflected in data from a variety of studies. Therefore we included all settings, embracing those from both the developed and the developing world, as well as where dual diagnosis treatments were delivered in various models of care (such as integrated or parallel service models).
We included studies with participants in any clinical state or stage of illness.
Types of interventions
1. Experimental interventions
Treatment with risperidone in any formulation and any dose.
2. Comparator interventions
Any other antipsychotic, divided into first‐generation ('typical') and second‐generation ('atypical').
In addition to antipsychotic treatment, we included studies in which co‐administration with a mood stabiliser (lithium, valproate, lamotrigine, carbamazepine, topiramate) or antidepressant (serotonin re‐uptake inhibitors, tricyclic, tetracyclic or new generation anti‐depressants) occurred in both treatment arms. Furthermore, studies comparing risperidone with another antipsychotic, where both treatment arms receive the same additional psychosocial intervention, were also included. Studies where only one treatment arm received an additional psychopharmacological or psychosocial intervention were excluded. Where imbalances exist between the treatment arms in terms of the dose, timing or duration of the additional treatments, we intended to note and discuss the impact of such imbalances in the section on assessment of risk of bias (Table 6).
1. Assessment of risk of bias.
| Sequence generation |
|
| Allocation concealment |
|
| Blinding of participants and personnel |
|
| Blinding of outcome assessment |
|
| Incomplete outcome data |
|
| Selective reporting |
|
| Other forms of bias |
|
Types of outcome measures
We categorised outcomes into short‐term, medium‐term and long‐term outcomes. Short‐term outcomes included outcomes measured in the first six months, medium‐term outcomes include outcomes from six months to one year, and long‐term outcomes include outcomes beyond one year.
Primary outcomes
1. Mental state
1.1 General
1.1.1 Clinically important change in general mental state ‒ as defined by trial authors.
1.2 Specific symptoms
1.2.1 Clinically important change in positive symptoms ‒ as defined by trial authors. 1.2.2 Clinically important change in negative symptoms ‒ as defined by trial authors. 1.2.3 Clinically important change in anxiety symptoms ‒ as defined by trial authors.
2. Substance use (determined according to how it was assessed, i.e. whether by means of self‐report, or determined biochemically, either by positive urine or blood tests, or gas chromatography mass spectroscopy on hair follicles)
2.1 A reduction in substance use (drug or alcohol). 2.2 The presence or absence of drug or alcohol use at the end of the study follow‐up period.
3. Adverse effects *
3.1 Clinically important adverse effect ‒ as defined by trial authors.
Secondary outcomes
1. Mental state
1.1 General
1.1.1 Any change in general mental state ‒ as defined by trial authors. 1.1.2 Average change/endpoint scores mental state scale. 1.1.3 Change in co‐morbid psychopathology.
1.2 Specific symptoms
1.2.1 Any change in positive symptoms ‒ as defined by trial authors. 1.2.2 Average change/endpoint scores positive mental state scale. 1.2.3 Any change in negative symptoms ‒ as defined by trial authors. 1.2.4 Average change/endpoint scores negative mental state scale. 1.2.5 Average change/endpoint scores anxiety scale.
2. Substance use (determined according to how it was assessed, i.e. whether by means of self‐report, or determined biochemically, either by positive urine or blood tests, or gas chromatography mass spectroscopy on hair follicles)
2.1 Time to relapse into drug or alcohol use. 2.2 Frequency of use over the study period.
3. Subjective well‐being as measured by validated rating scales
3.1 Average endpoint/change scores on subjective well‐being scales.
4. Craving for substances
4.1 Average endpoint/change scores on craving scales.
5. Adherence to antipsychotic medication
5.1 Average endpoint/change scores on medication adherence rating scales. 5.2 An improvement in adherence to antipsychotics ‒ as defined by trial authors.
6. Adverse effects
6.1 Any general adverse effects. 6.2 Specific adverse effects. 6.2.1 Allergic reactions. 6.2.2 Blood dyscrasia such as agranulocytosis. 6.2.3 Central nervous system (ataxia, nystagmus, drowsiness, fits, diplopia, tremor). 6.2.4 Metabolic adverse events (weight changes, serum glucose measures, serum triglyceride measures, serum high density lipoprotein (HDL) measures). 6.2.5 Endocrinological dysfunction (hyperprolactinaemia, disturbance of reproductive organ and sexual performance functioning). 6.2.6 Gastrointestinal (nausea, vomiting, diarrhoea). 6.2.7 Kidney dysfunction. 6.2.8 Movement disorders (extrapyramidal side‐effects, including neuroleptic malignant syndrome and tardive dyskinesia).
7. Leaving the study early
7.1 A reduction in numbers leaving study early: any reason. 7.2 A reduction in the proportion of participants lost to follow‐up at the study end‐point. 7.3 An increase in the proportion of participants attending the first follow‐up visit. 7.4 An increase in time to attrition. 7.5 An increase in the mean number of clinic visits.
8. Mortality
8.1 Due to natural causes. 8.2 Due to drug overdose. 8.3 Due to suicide. 8.4 Due to any other unnatural cause.
9. Quality of life
9.1 General quality of life
9.1.1 Clinically important change in general quality of life ‒ as defined by trial authors. 9.1.2 Any change in quality of life ‒ as defined by trial authors. 9.1.3 Average change/endpoint scores quality of life scale.
9.2 Physical health
9.2.1 Clinically important change in physical health ‒ as defined by trial authors. 9.2.2 Any change in physical health ‒ as defined by trial authors. 9.2.3 Average change/endpoint scores physical health. scale
10. 'Summary of findings' table
We used the GRADE approach to interpret findings (Guyatt 2011; Schünemann 2011); and used GRADEpro to export data from our review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Mental state: General — clinically important change ‒ at study endpoint.
Substance use: a reduction in substance use ‒ at study endpoint.
Subjective well‐being ‒ improvement in measures of subjective well‐being.
Craving for substances ‒ improvement in measures of substance craving.
Adherence to antipsychotic medication ‒ improvement in measures of medication adherence.
Adverse effects ‒ clinically important adverse effect.
Leaving the study early ‒ a reduction in the proportion of participants leaving the study early: any reason.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On 6 January 2016 and 9 October 2017, the Information Specialist searched the register using the following search strategy:
*Risperidone* in Intervention AND *Substance Abuse* in Healthcare Condition Fields of STUDY
In such study‐based registers, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017).
This register is compiled by systematic searches of major resources (AMED, BIOSIS, CINAHL, ClinicalTrials.Gov, Embase, MEDLINE, PsycINFO, PubMed, WHO ICTRP) and their monthly updates; ProQuest Dissertations and Theses A&I and its quarterly update; Chinese databases (CBM, CNKI, and Wanfang) and their annual updates; handsearches; grey literature; and conference proceedings (see Group’s Module). There are no language, date, document type, or publication status limitations for inclusion of records into the register.
2. Cochrane Common Mental Disorders Group's Trials Register
On 8 January 2016 and 10 October 2017, the Information Specialist of Cochrane's Common Mental Disorders Group searched the trials register of their group using the following search strategy:
Search 1:
#1. (*Risp* AND (*Substance* OR *Cannabis* OR *Amphetamine* OR *Alcohol* OR *Cocaine* OR *Opioid* OR *Drug Dependence* OR *Addict*)) [in Register]
Search 2:
#2. (*Risp*)
#3. (“substance use disorder*” or SUD or SUDs)
#4. “drug abuse”
#5. (abuser* or abusing or addict* or depend* or habit* or misuse or user*)
#6. (abuse not (child* or sex*))
#7. (adinazolam or aerosol* or alcohol* or alprazolam or amphetamin* or anthramycin or anxiolytic* or ativan or barbituat* or bentazepam or benzodiazepin* or bromazepan or brotizolam or buprenorphin* or camazepam or cannabi* or chlordiazepoxid* or cinolazepam or clobazam or clonazepam or clorazepam or clotiazepam or cloxazolam or cocaine* or codeine or crack or crystal or cyprazepam or depressant* or diacetylmorphin* or diazepam* or doxefazepam or ecstasy or estazolam or etizolam or fentanyl or flunitrazepam or flurazepam or flutazoram or flutoprazepam or fosazepam or gases or GHB or girisopam or halazepam or hallucinogen* or haloxazepam or heroin* or hydromorphone or hydroquinone or hypnotic* or inhalant* or ketamin* or ketazolam or librium or loflazepate or loprazolam or lorazepam or lormetazepam or LSD or marihuana* or marijuana* or MDMA or meclonazepam or medazepam or meperidine or mephedrone or mescalin* or metaclazepam or methadone or methamphetamin* or methaqualone or mexazolam or midazepam or midazolam or morphine* or narcotic* or nerisopam or nimetazepam or nitrazepam or nitrites or "nitrous oxide" or "n‐methyl‐3,4‐methylenedioxyamphetamine" or nordazepam or opiate* or opiod* or opium or oxazepam or oxazolam or oxazypam or oxycodone or oxzepam or painkiller* or "pain killer*" or PCP or pethidin* or phencyclidin* or pinasepam or prazepam or propazepam or propoxyphene or psilocybin or psychedelic* or psychoactive* or psychostimulant* or quinazolinone or ripazepam or ritalin or sedative* or serazepin* or solvent* or steroid* or stimulant* or substance* or temazepam or tetrazepam or tofisopam or tramadol or triazolam or triflubazam or valium or vicodin)
#8. (drug* and (recreational or street))
#9. #2 and (#3 or #4 or #5 or #6 or #7 or #8)
#10. #9 not #1 [in Register]
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected reference lists of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials. In addition we contacted pharmaceutical companies regarding unpublished trials.
Data collection and analysis
Selection of studies
HT and TW independently inspected citations from the searches and identified relevant abstracts. NS independently re‐inspected included abstracts to ensure reliability. HT and TW obtained and inspected full reports of the abstracts meeting the review criteria. Where disputes arose, NS re‐inspected reports in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Review authors HT and TW extracted data from all included studies. Again, any disagreements were discussed; decisions documented; and, if necessary, authors of studies were contacted for clarification. NS helped to clarify issues with remaining problems and we documented these final decisions. If data had been presented only in graphs we would have used data in analyses only when both reviewers derived similar results from extraction. One study reported data in graphs only, but we were unable to extract as data were reported for subgroups and the total number of participants at study endpoint for each subgroup was not known, precluding a calculation of standard deviations (SDs) (Akerele 2007). In case of multi‐centre studies we attempted to get information from authors so as to extract data from each component centre separately. In one multi‐centre study, authors were unable to provide this level of data and the data across all participating centres were reported.
2. Management
2.1 Forms
We extracted data onto a standardised form and piloted it on one study prior to use.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and
the measuring instrument had not been written or modified by one of the trial investigators for that particular trial.
Ideally the measuring instrument should either be i) a self‐report or ii) completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly and recorded if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand calculation of change needs two assessments (baseline and endpoint) which can be difficult to measure in unstable conditions such as schizophrenia. We decided to primarily use endpoint data, and only used change data if the former were not available. We decided that in cases where change data had been used, endpoint and change data would be combined in the analysis. If this had occurred we would have used mean differences (MD) rather than standardised mean differences (Higgins 2011). As pooling of continuous data in a meta‐analysis was not possible for this review we did not use this method.
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion.
Standard deviations and means are reported in the paper or obtainable from the authors.
When a scale starts from the finite number zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996).
If a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) which can have values from 30 to 210) we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S − Smin), where S is the mean score and Smin is the minimum score.
Endpoint scores on scales often have a finite start and end point, and the above rules can be applied. Had we found skewed data from studies of fewer than 200 participants we would have entered such data in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at means if the sample size is large, and had we found such studies we would have entered them into syntheses.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We included change data in statistical analyses regardless of the size of the study.
2.5 Common measure
To facilitate comparisons between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary data
Where possible, we converted outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS, Kay 1986, this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors. As the definition of a clinically significant treatment response may differ across various patient populations, we accepted a 50% symptom reduction as an adequate response in acute, non‐refractory schizophrenia and a 25% reduction in chronic, refractory patients (Leucht 2009), and used these different definitions depending on the population studied. Where possible we also attempted to analyse continuous data.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for risperidone. Where keeping to this makes it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not unimproved') we reported data where the left of the line indicates an unfavourable outcome. This was noted in the relevant graphs.
Assessment of risk of bias in included studies
Review authors HT and TW worked independently to assess risk of bias by using criteria described in theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, and includes assessment of sequence generation, allocation concealment, blinding, incomplete outcome data, selective reporting and other forms of bias. Measures to prevent risk of bias were assessed as either 'high', 'low' or 'unclear', according to the definitions of these ratings in the Cochrane Handbook for Systematic Reviews of Interventions (Table 6).
If the raters disagreed, they came to a consensus over the final rating by involving another review author (NS). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information.
We noted the level of risk of bias for each included study in both the text of the review and within 'Risk of bias' tables and incorporated them in the judgement of overall quality across studies in the 'Summary of findings' tables.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive than odds ratios (Boissel 1999); and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes we estimated the mean difference (MD) and the standard deviation (SD) between groups. However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated the standardised mean difference (SMD) and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster randomised control trials
Analysis of multi‐level data may pose problems and failure to account for clustering in data may result in unit of analysis errors (Divine 1992), whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC (Design effect = 1 + (m − 1) * ICC) (Donner 2002). If the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
If we had found cluster randomised trials in this review and had been able to analyse them appropriately, taking into account ICCs and relevant data documented in the report, we could then have included them in a synthesis with other studies using the generic inverse variance technique. However, we found no cluster randomised trials.
2. Repeated measurements
Multi‐level data may also arise where multiple measurements are conducted on one participant, i.e. counts of the number of urine tests over a period of time per participant that screen positive for drug use. In such cases, unit of analysis issues may also occur. Outcomes could also be reported as the percentage of participants per treatment group with positive urine drug tests, which represents another form of count data that is not compatible with Cochrane Review Manager 5 (RevMan 5) software, that analyses data derived at an individual participant level. In such cases, we contacted the authors to provide actual count data on the number of individual urine tests that screened positive or negative per participant over the study period. In accordance with methods previously described by Mattick 2014, we asked authors to derive a mean and SD of positive urine tests per treatment group over the study period. We would have then analysed differences between treatment groups as continuous data. In cases where the denominator differed for individual participants due to missed study visits and missing data for urine testing, we would have omitted studies where missing data occurred in more than 50% visits per individual participant and for more than 50% of overall participants in either treatment arm. In studies where no data on number of urine tests were obtained, we would have dealt with missing outcome data by the methods described under Dealing with missing data for continuous outcomes. We found one such study where urine tests were measured over time, generating longitudinal data (Akerele 2007). No information was provided by study authors despite attempts to contact them. In a second study authors did report means and standard deviations (van Nimwegen 2008).
3. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, if we obtained data from cross‐over trials we would have only used data from the first phase of such studies. However, we included no cross‐over studies in this review.
4. Studies with multiple treatment groups
When a study involved more than two treatment arms, if relevant we presented the additional treatment arms in comparisons. If data were binary these would have simply been added and combined within a two‐by‐two table, in order to avoid double counting a common comparison group in the same meta‐analysis. If data had been continuous we would have combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011). If the additional treatment arms were not relevant, these data would not have been reproduced.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss to follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, if more than 50% of data were unaccounted for, we did not reproduce these data or use them within analyses (except for the outcome 'leaving the study early'). If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
In cases where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once randomised always analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were all assumed to have had the same rates of negative outcome as those who completed, with the exception of the outcomes of death and adverse effects. For these outcomes, the rate of those who stayed in the study — in that particular arm of the trial — was used for those who did not. However, due to the small study sample sizes we were not able to conduct such a sensitivity analysis in order to test how prone the primary outcomes are to change when 'completer' data only are compared to the intention‐to‐treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
Where attrition for a continuous outcome is between 0% and 50%, and completer‐only data were reported, we reproduced these.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the study authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and CIs available for group means, and either P value or t value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). When only the standard error (SE) is reported, SDs are calculated by the formula SD = SE * √(n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formula for estimating SDs from P values, t or F values, CIs, ranges or other statistics. If these formulae did not apply, we would have calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome, and thus to lose information. We nevertheless would have examined the validity of the imputations in a sensitivity analysis excluding imputed values. However, as our search only yielded two studies with two different comparisons, we were unable to use this method of imputation.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would have been be employed in the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, when LOCF data had been used in the trial and if less than 50% of the data had been assumed, we would have reproduced these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We inspected all studies to determine whether the studies were similar enough in terms of participant profile and intervention comparisons to combine them, and discussed such differences if any were found.
2. Methodological heterogeneity
We simply inspected all studies for clearly outlying methods and discussed any outlying methods in full.
3. Statistical heterogeneity
3.1 Visual inspection
Where possible we visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I² statistic
We investigated heterogeneity between studies by considering the I² statistic alongside the Chi² test P value. The I² statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I² depends on i) magnitude and direction of effects and ii) strength of evidence for heterogeneity (e.g. P value from Chi² test, or a CI for I²). I² values between 0% to 40% were interpreted as possibly unimportant, 30% to 60% as possibly significant, 50% to 90% as possibly substantial, and 75% to 100% as possibly considerable (Deeks 2011). Had substantial levels of heterogeneity been found in the primary outcome, we would have explored reasons for heterogeneity (see Subgroup analysis and investigation of heterogeneity). As no study data was combined in a meta‐analysis for any primary outcomes, we did not conduct any exploration of heterogeneity.
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included RCTs by searching clinical trials registries and contacting authors. If the trial protocol was available, outcomes in the protocol and in the published report were compared. If the protocol was not available, outcomes listed in the Methods section of the trial report were compared with actual reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are again described in section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. However, we were not able to generate funnel plots as there were no outcomes including more than two studies. Should more studies become available in future, and funnel plots become possible, we will seek statistical advice in their interpretation.
Data synthesis
As we anticipated clinical and methodological heterogeneity we used random‐effects models for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We planned to conduct subgroup analyses based on a number of factors (substance abuse versus dependence, the type and formulation of risperidone, and the presence of additional pharmacological or psychosocial treatment in some studies), but due to the small number of studies included were unable to carry out such subgroup analyses.
Sensitivity analysis
We would have applied the following sensitivity analyses in our review to primary outcome data. However, due to the small number of studies, the low sample size within included studies and single studies within each comparison, none of these sensitivity analyses were possible.
1. Risk of bias
We would have analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available), allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. In case the exclusion of trials at high risk of bias would have not substantially altered the direction of effect or the precision of the effect estimates, then we would have included data from these trials in the analysis.
2. Assumptions for lost binary data
Where we would have had to make assumptions regarding people lost to follow‐up (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption with completer data only. If there had been substantial difference, we would have reported results and discussed them but would have continued to employ our assumption.
3. Assumptions for lost continuous data
If we had to make assumptions, such those used in imputation methods, regarding missing SD data (see Dealing with missing data), we intended to compare the findings on primary outcomes when we applied these assumptions with completer data only. We intended to conduct a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using different assumptions (LOCF, imputation from other studies). If there had been a substantial difference, we would have reported results and discussed them but would have continued to employ our assumption.
4. Imputed values
We would have also undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If substantial differences had been noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but would have presented them separately.
5. Fixed effect and random effects
We expected substantial clinical and methodological heterogeneity in the studies included in the review. We therefore used a random‐effects model to combine data in a meta‐analysis. We intended to carefully inspect the results of our meta‐analysis and if it turned out that smaller studies received a higher weighting, we did intend to conduct a sensitivity analysis using a fixed‐effect model. If a fixed‐effect meta‐analysis did not show a similar beneficial effect compared to the random‐effects analysis, we would have carefully considered whether the conclusions of the random‐effects model were justified in light of the methodological rigour and risk of bias assessments of the larger compared to the smaller studies. If it had turned out that larger studies were indeed more rigorous, we would have restricted our report to the results of the meta‐analysis of the larger studies.
Results
Description of studies
For description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
We identified 197 records from the searches of the Cochrane Schizophrenia Group’s Study‐Based Register of Trials (including trial registers) and the Cochrane Common Mental Disorders' database (Figure 3). In addition we obtained four records from other sources (two through searches of a clinical trials register, one from communication with an author and one from search of reference lists). After removal of 145 abstracts that did not meet eligibility criteria, a total of 56 full text records containing 37 studies that appeared to meet eligibility criteria were inspected in detail. After excluding 23 studies which did not meet our inclusion criteria (see Characteristics of excluded studies) and a further six studies either awaiting classification or ongoing (Characteristics of studies awaiting classification; Characteristics of ongoing studies), we were left with eight studies meeting our inclusion criteria (see Characteristics of included studies).
3.
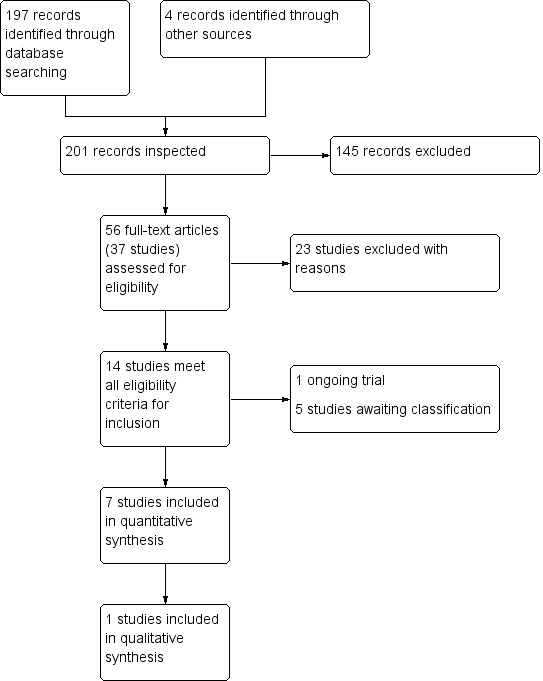
PRISMA flow diagram of study selection from 2016 and 2017 searches
Included studies
A total of eight studies met criteria for inclusion (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008); however, Brunette 2011 did not provide any useable data for analyses.
1. Design and duration
Four of the included studies were parallel‐group superiority trials that randomised participants to risperidone and a comparator drug. These studies all had pre‐specified hypotheses about the comparator drugs. In the study by Akerele 2007 the authors hypothesised that olanzapine would be superior to risperidone in reducing cocaine and cannabis use in people with schizophrenia in a 14‐week trial. In Noordsy 2010 the authors hypothesised that clozapine would be superior to risperidone in reducing substance use and psychiatric symptoms over 24 weeks of study. In a 4‐week trial, Machielsen 2014 hypothesised that clozapine would be superior to risperidone in reducing brain activation in regions associated with attentional bias on fMRI, reduce craving and increase subjective well‐being in people with first episode schizophrenia with comorbid cannabis use disorders. In a 12‐week trial Brunette 2011 randomised participants to either continue their treatment with their treatment as usual (TAU) or to switch participants to clozapine, with the hypothesis that clozapine would reduce cannabis use. In this study five participants in the TAU group were taking risperidone.
Three trials were post‐hoc, secondary data analyses of existing larger parent randomised trials (Sevy 2011; Smelson 2006; Swartz 2008). In Sevy 2011 the authors studied a group of 49 participants from a larger randomised trial of 120 participants (Robinson 2006) over 16 weeks who were randomised to risperidone and olanzapine. Smelson 2006 analysed data on 236 out of 632 substance users from a parent study (Tunis 2006) randomised to risperidone, olanzapine and conventional agents (including perphenazine, loxapine, haloperidol, fluphenazine, thiothixene) over a 12‐month study period. Swartz 2008 studied 643 out of 1432 participants from the CATIE study (Stroup 2003) randomised to risperidone, olanzapine, quetiapine, perphenazine, and ziprasidone, continued over 18 months of study.
One study was a randomised parallel‐group superiority trial comparing risperidone to a comparator drug and reported outcomes on a subgroup of participants with co‐occurring substance use disorders (van Nimwegen 2008).
2. Participants
Participants in all of the included studies had a diagnosis of either schizophreniform disorder, schizophrenia or schizoaffective disorder and co‐occurring substance misuse. In Noordsy 2010 and Sevy 2011, participants were experiencing their first episode of psychosis (schizophrenia or schizoaffective disorder). In Swartz 2008, participants were multi‐episode.
Five studies exclusively randomised participants with a co‐occurring cannabis use disorder (Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008). The other three studies included participants with other substance use disorders such as alcohol, amphetamine, cocaine and opioid use disorders (Akerele 2007; Smelson 2006; Swartz 2008).
In Akerele 2007, Smelson 2006 and Swartz 2008, participants were adults of mixed ethnicity and in Noordsy 2010 all participants were Caucasian (understood to be white participants). Brunette 2011 had 83.9% Caucasian participants; and in Machielsen 2014, van Nimwegen 2008 and Sevy 2011 ethnicity was not stated. Seven of the studies included adults over the age of 18 years. In the study by Noordsy 2010 four participants (28% of the total sample) were reported to be 17 years; the age range for this study was 17 to 45 years. Both males and females were included in studies but male participants predominated and all participants were male in the Machielsen 2014 study.
3. Settings
Studies were conducted either in the USA or the Netherlands. Most studies included outpatients from single or a small number of sites (1 to 4), and a few recruited from inpatient sites. One study was a large, multi‐centre trial conducted over 57 sites (Swartz 2008).
4. Study size
A total of 2466 people were randomised after giving informed consent to participate in the trials. Of these 1073 had a dual diagnosis. The number of participants with SMI and co‐occurring substance misuse varied from study to study: six studies randomised fewer than 50 dual diagnosis participants (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008); one study randomised between 100 and 300 (Smelson 2006); and Swartz 2008 randomised 643 dual diagnosis participants.
5. Interventions
Risperidone (dose range: 1 mg to 9 mg) was compared to olanzapine (dose range: 2.5 mg to 30 mg) in five different studies (Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008). Three studies compared risperidone (dose range: 3.5 mg to 5 mg daily) to clozapine (dose range: 12.5 mg to 400 mg daily) (Brunette 2011; Machielsen 2014; Noordsy 2010). One study compared risperidone (dose range: 1.5 mg to 6 mg) to quetiapine (dose range: 200 mg to 800 mg) (Swartz 2008). Two studies compared risperidone (dose range: 1 mg to 6 mg daily) with first‐generation antipsychotics (perphenazine, loxapine, haloperidol, fluphenazine, thiothixene, various dosages) (Smelson 2006; Swartz 2008). One study compared risperidone (dose range: 1.5 mg to 6 mg) to ziprasidone (dose range: 40 mg to 160 mg daily) (Swartz 2008).
Concomitant psychosocial interventions were delivered in a number of studies. In the trial by Akerele 2007 all participants received weekly psychotherapy over the study period and were asked to nominate a "significant other" to assist with attendance and follow‐up. In Brunette 2011 all participants received weekly individual substance abuse and mental health counselling and attended weekly Alcoholics Anonymous meetings. In Machielsen 2014 it is reported that participants had "supportive treatment as usual". In the trial by Noordsy 2010 participants also received a "Lifestyle Intervention" to help prevent metabolic side‐effects and assist with recovery. In the trial by Sevy 2011 all participants received psychoeducation about schizophrenia, were seen on a regular basis by allocated social workers and also had access to the ancillary treatment service available from two large departments of psychiatry. In Smelson 2006 it is unclear what psychosocial interventions participants received. In Swartz 2008 the investigators did not account for substance abuse treatments received, but they noted that very few were actively engaged in such treatments.
6. Sources of funding
The study by Akerele 2007 received funding from the National Institute on Drug Abuse (NIDA), the National Alliance for Research on Schizophrenia and Depression (NARSAD) and Eli Lilly, the pharmaceutical company. The study by Brunette 2011 was sponsored by Novartis and Janssen; and Machielsen 2014 by the Dutch health research council. Sponsors and collaborators for the study by Noordsy 2010 included the National Institute of Mental Health and the Dartmouth‐Hitchcock Medical Centre; no funding was received from the pharmaceutical industry and the investigators were not employed by the sponsors. In Sevy 2011 several authors declared ties with the pharmaceutical industry but it is stated that the study was sponsored by the NIMH. Funding for Smelson 2006 and van Nimwegen 2008 was received from Eli Lilly; and in Swartz 2008, study medications were provided by several pharmaceutical companies.
7. Outcomes
Scales reported to have been used in the included studies are summarised in Table 7. A description of the scales for which results have been reported is included below.
2. Scales, diagnostic instruments and other outcome measures used in included studies.
| Diagnostic tools | Abbreviation | Source of scale/ instrument | Study using instrument | Results reported or usable data for re‐analysis/ quantitative synthesis or qualitative results/data only |
| Structured Clinical Interview for DSM Disorders | SCID‐I | First 1994 | Akerele 2007; Sevy 2011; Swartz 2008; van Nimwegen 2008 | Not an outcome measure |
| Mental state scales | ||||
| Brief Psychiatric Rating Scale | BPRS | Lukoff 1986 | Noordsy 2010 | No results or usable data reported or obtained |
| Clinical Global Impression scale | CGI | Guy 1976 | Akerele 2007; Noordsy 2010 | No results or usable data reported or obtained |
| Hamilton Depression Rating Scale | HAM‐D | Hamilton 1960 | Akerele 2007 | Results reported; usable data for quantitative synthesis |
| Positive and Negative Syndrome Scale | PANSS | Kay 1986 | Akerele 2007;Greenspan 2005; Machielsen 2014; Swartz 2008 | Results reported; usable data for quantitative synthesis |
| Schedule for Affective Disorders and Schizophrenia ‒ Change Version with Psychosis and Disorganization items | SADS‐C‐PD | Endicott 1978 | Sevy 2011 | Results reported; usable data for quantitative synthesis |
| Schedule for the Assessment of Negative Symptoms | SANS | Andreasen 1982 | Noordsy 2010 | No results or usable data reported or obtained |
| Substance use scales | ||||
| Addiction Severity Index | ASI | McLellan 1980; McLellan 1992 | Akerele 2007 | No results or usable data reported or obtained |
| Composite International Diagnostic Interview | CIDI | Robins 1988 | Machielsen 2014 | Not an outcome measure |
| Substance Use Questionnaire | SUQ | Sevy 2011, Locally derived instrument/ non‐validated | Sevy 2011 | Results reported, used together with urine testing; usable data for quantitative synthesis |
| Time‐Line Follow‐Back | TLFB | Sobell 1992 | Noordsy 2010 | Results reported in dichotomised format for quantitative synthesis |
| Quantitative Substance Use Inventory | Locally derived instrument/ non‐validated | Akerele 2007 | Non‐validated scale | |
| Subjective‐Wellbeing Scales | ||||
| Subjective Well‐being Under Neuroleptics Scale | SWN | de Haan 2002; Naber 1995 | Machielsen 2014; van Nimwegen 2008 | Results reported; usable data for quantitative synthesis |
| Craving for substances measures | ||||
| Cocaine Craving Report | Weddington 1990 | Akerele 2007 | No usable data for quantitative synthesis | |
| Desires for Drug Questionnaire | DDQ | Franken 2002 | van Nimwegen 2008 | Results reported; usable data for quantitative synthesis |
| Marijuana Craving Report | Weddington 1990 | Akerele 2007 | No usable data for quantitative synthesis | |
| Marijuana Craving Questionnaire | MCQ | Heishman 2009 | Machielsen 2014 | Results reported; usable data for quantitative synthesis |
| Obsessive Compulsive Drug Use Scale | OCDUS | Dekker 2012 | Machielsen 2014;van Nimwegen 2008 | Results reported; usable data for quantitative synthesis |
| Adverse effect scales | ||||
| Abnormal Involuntary Movement Scale | AIMS | National Institute of Mental Health 1988 | Akerele 2007 | No results or usable data reported or obtained |
| Barnes Akathisia Rating Scale | BARS | Barnes 1989 | Brunette 2011 | No results or usable data reported or obtained |
| Simpson Angus Scale | SAS | Simpson 1970 | Akerele 2007 | Results reported; usable data for quantitative synthesis |
| Other measures (categorical or time to event) | ||||
| Urine assay for cannabis and cocaine use (proportion of treatment group positive per week) | Akerele 2007 | No usable data for quantitative synthesis | ||
| Number of participants with improvement in substance use (categorised as improved or not‐improved versus unchanged) | Noordsy 2010 | Results reported; usable data for quantitative synthesis (dichotomised) | ||
| Days of self‐reported drug use in past week | Akerele 2007 | No usable data for quantitative synthesis | ||
| Weeks in treatment | Akerele 2007; Smelson 2006 | Results reported; usable data for quantitative synthesis | ||
| Number of participants not completing the study | Akerele 2007; Machielsen 2014; Noordsy 2010; Sevy 2011; Swartz 2008 | Results reported, usable data for quantitative synthesis | ||
| Compliance with medication (missed doses) | Akerele 2007 | No usable data for quantitative synthesis | ||
7.1 Mental state scales
a. Hamilton Depression Rating Scale (HAM‐D)
This scale has a 17‐item and 21‐item version. The severity of depression is rated for the past week on separate items with 2‐ to 5‐point severity scales. A total of eight items are scored on a 5‐point scale ranging from 0 "absent" to 4 "severe" and 9‐items on a 2‐point scale. Scoring ranges from 0 to 50, with scores on the first 17 items contributing to the final score. A further four items (items 18 to 21) provide additional information on the characteristics of the depressive symptoms (Hamilton 1960). This scale was used by Akerele 2007.
b. Positive and Negative Syndrome Scale (PANSS)
This scale was used in Akerele 2007, Machielsen 2014 and Swartz 2008 and measures positive psychotic symptoms (delusions, thought disorganization, hallucinations, excitement, grandiosity, hostility, persecutory ideation) and negative symptoms (affective blunting, poor rapport, social and emotional withdrawal, stereotypical thinking, poverty of speech, difficulty in abstract thinking) and a range of general psychopathology symptoms. The scale includes seven items measuring positive psychotic symptoms, seven items measuring negative symptoms and 14 items measuring general psychopathology on a 1 to 7 point scale ranging from "absent" to "extreme". It has a range of 30 to 210 (Kay 1986; Kay 1987).
c. Schedule for Affective Disorders and Schizophrenia—Change Version with psychosis and disorganization items (SADS‐C+PD) (Endicott 1978)
This instrument was used to measure positive psychotic symptoms (delusions, hallucinations, thought disorder, and bizarre behaviour) and negative symptoms (affective flattening/blunting, alogia, avolition‐apathy, asociality‐anhedonia) in the trial by Sevy 2011.
7.2 Substance use scales
a. Timeline Follow‐back (TLFB)
This scale is a calendar‐based method that assesses the frequency of drug use over a period of time (usually the past week) (Sobell 1992). This scale can either be self‐administered by participants or clinician‐completed. In the Noordsy 2010 study this scale was used together with other sources of information such as urine tests and reports from collateral sources of information to determine cannabis use. At the end of the study graphs were plotted showing days of cannabis use per week as rated by the TLFB method and were then rated as "Improved", "Unchanged", or "Worse" by a pair of expert judges (Noordsy 2010). Raters were instructed to rate the graph "Improved" or "Worsened" if it appeared to be more than 20% better or worse and to rate it "Unchanged" if there was little or no change (less than ˜20%) (See under "Other measures: categorical and time to event data").
b. Substance Use Questionnaire:
This instrument was used in conjunction with urine testing to derive best estimate of substance discontinuation in Sevy 2011.
7.3 Subjective well‐being scales
a. Subjective Well‐being Under Neuroleptics Scale (SWN): This self‐rated scale (used in the study by Machielsen 2014) measures various aspects of self‐perceived symptoms, treatment experience and quality of life and functioning over the past 7 days. The SWN (short‐version) consists of 20 statements (10 positive and 10 negative) assessing five domains (mental functioning, self‐control, emotional regulation, physical functioning, and social integration), with each domain containing four questions. Rating is on a 6‐point Likert‐type scale yielding total scores varying from 20 to 120 (de Haan 2002; Naber 1995).
7.4 Craving for substances scales
a. Cocaine and Marijuana Craving Report (Weddington 1990)
This scale measures craving of cocaine or cannabis on a 100‐point, 10 cm visual analogue scale with instructions to participants to rate their desire for cocaine or cannabis in the past 24 hours from "not at all" to "more than ever". This scale was used in Akerele 2007.
b. Desires for Drug Questionnaire (DDQ) (Franken 2002)
This instrument measures instantaneous, immediate craving on 14 different items phrased in a positive manner and assessing desires to use, control over using and use to ameliorate negative emotions (negative reinforcement).
c. Marijuana Craving Questionnaire (MCQ) (Heishman 2009)
This instrument measures current cannabis craving on four domains (compulsivity, emotionality, expectancy and purposefulness) containing three questions each using a 7‐point Likert type scale.
d. Obsessive‐Compulsive Drug Use Scale (OCDUS) (Dekker 2012; Franken 2002)
This instrument measures drug craving over the past 7 days on 12 items containing a 5‐point Likert type rating.
7.5 Adverse effects scales
a. Simpson‐Angus Scale (SAS)
In one study — Akerele 2007 — the Simpson‐Angus Scale was used to measure the presence of parkinsonism (Simpson 1970).This scale has 10 items and measures bradykinesia, rigidity and tremor in different body regions on a scale of 0 to 4. It has a range of 0 to 28.
7.6 Leaving the study early: Time to leaving
In the study by Akerele 2007 the time to study attrition was measured in weeks.
7.7 Categorical outcomes and time‐to‐event data
a. Substance use: urine assay for cannabis and cocaine use
In the study by Akerele 2007 urine samples were collected at each of the three meetings per week. Each week over the 10 weeks of follow‐up was classified as either positive or negative for drug use for each participant if any one of the three samples tested positive. Urine tests for cannabis were classified as positive if the cannabis concentration was above a cut‐off of 100 ng/ml; and for cocaine, if the sample concentration was above the cut‐off of 300 ng/ml.
b. Substance use: number of participants with improvement in substance use
In the study by Noordsy 2010 days of cannabis use per week as gathered by the Time‐Line Follow‐Back method were plotted on graphs at the end of the study. A pair of expert judges were then instructed to rate cannabis use as "improved" or "worsened" if it appeared to be more than 20% better or worse and to rate it as "unchanged" if there was little or no change (less than 20% change). Results were reported as the number of participants per intervention group that had "improved" cannabis use versus "unchanged" or "worsened". Machielsen 2014 reports on the number of participants who continued or stopped using cannabis during the study.
c. Adherence with medication
In the study by Akerele 2007 the proportion of missed doses of all administered doses was reported.
d. Leaving the study early
For six studies the number of participants who did not complete the study was reported (Akerele 2007; Machielsen 2014; Noordsy 2010; Sevy 2011; Swartz 2008; van Nimwegen 2008).
Excluded studies
We excluded 23 studies from the 37 studies examined for inclusion. Table 8 contains a summary of the various excluded studies. A total of 12 studies were excluded as there was no measure or reporting of substance use comorbidity (Blin 1996; Gaebel 2010; Harvey 2007; Ikuta 2014; Liemburg 2011; Perlis 2006; Rezayat 2014; Sachs 2002; Sajatovic 2002; Smulevich 2005; van Nimwegen 2008a; Yatham 2007). Three studies were excluded as they were records of study protocols for which no unpublished data were provided after authors were contacted (Green 2001; NCT00169026; NCT00498550). Three studies were excluded due to the use of quasi‐randomisation (Rubio 2006a; Rubio 2006b; Zhangyue 2005). One study was excluded as only 8.3% of participants had a severe mental illness (Nejtek 2008). One study was excluded as it compared risperidone in oral versus depot formulation (NCT00130923). One study was excluded as it included only mental disorders which were due to alcohol use (Liu 2008). One study was excluded as it did not contain any data comparing risperidone with other medications but examined the impact of substance use on prognosis (Kerfoot 2011). Another study was excluded as it was an observational study (NCT00063349).
3. Excluded randomised and quasi randomised studies and their relevant comparisons.
| Study tag | Participants | Comparison | Relevant review | ||
| Primary problem | Co‐morbidity | Note (reasons for exclusion) | |||
| Blin 1996 | schizophrenia | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, haloperidol, methotrimeprazine | ‐ |
| Gaebel 2010 | schizophrenia, schizoaffective disorder | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, olanzapine, conventional antipsychotics vs. risperidone long‐acting injectable (RLAI),or quetiapine | ‐ |
| Green 2001 | schizophrenia | cannabis use disorder | Excluded as this was a study protocol, authors contacted for unpublished data, no response | risperidone vs. clozapine | ‐ |
| Harvey 2007 | bipolar type I disorder | No measure of substance use | Excludes comorbidity | risperidone, quetiapine | ‐ |
| Ikuta 2014 | schizophrenia, schizophreniform, psychosis not otherwise specified | No measure of substance use | Excludes comorbidity | risperidone, aripirazole | ‐ |
| Kerfoot 2011 | schizophrenia | Co‐occurring substance use in subgroup of 44.9% | No comparison of study medication, mainly a prognostic study of the impact of substance use | risperidone, quetiapine, perphenazine, olanzapine, ziprasidone | ‐ |
| Liemburg 2011 | schizophrenia | No measure of substance use | Excludes comorbidity | risperidone, aripiprazole | ‐ |
| Liu 2008 | mental disorders due to alcohol use | substance induced mental disorders | Exclusion criterion | risperidone, olanzapine | Suggested review: risperidone versus other antipsychotics for substance induced psychosis |
| NCT00063349 | schizophrenia | Co‐occurring cannabis use disorder | Study protocol, authors contacted for unpublished data no response | risperidone, clozapine | |
| NCT00130923 | schizophrenia, schizoaffective disorder | Co‐occurring alcohol use disorder (abuse or dependence) | Comparison of same medication (risperidone) in different preparations (oral vs depot) | oral risperidone, depot risperidone | ‐ |
| NCT00169026 | schizophrenia, schizoaffective disorder | Co‐occurring alcohol and substance use disorder | Study protocol, study terminated, authors contacted for unpublished data no response | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ |
| NCT00498550 | schizophrenia, schizoaffective disorder | Co‐occurring cannabis use disorder | Study protocol, authors contacted no data provided | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ |
| Nejtek 2008 | bipolar I and II disorder, recent manic or mixed episode with or without psychosis | co‐occurring cocaine or methamphetamine use disorder | Only 8.3% of total sample had psychotic features and 15.9% had bipolar type II disorder. | risperidone, quetiapine | Suggested review: Risperidone versus other antipsychotics in bipolar disorder with co‐occurring substance use disorders |
| Perlis 2006 | bipolar I disorder with mania or mixed states | Excludes participants with recent substance use | Excludes comorbidity. Excludes bipolar disorder with psychotic features. | risperidone or olanzapine | ‐ |
| Rezayat 2014 | bipolar disorder, acute mania | Excludes participants with drug or alcohol use in past 3 months | Excludes comorbidity | aripiprazole, risperidone | ‐ |
| Rubio 2006a | schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone long‐acting depot, zuclopenthixol long‐acting depot | ‐ |
| Rubio 2006b | schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone (oral), zuclopenthixol (oral), zuclopenthixol long acting depot | ‐ |
| Sachs 2002 | bipolar with current manic or mixed episode | Study excludes participants with drug or alcohol in past 1 months | Excludes comorbidity | mood stabiliser augmentation with risperidone, haloperidol or placebo | ‐ |
| Sajatovic 2002 | psychotic disorders: schizoaffective disorder, bipolar I disorder, major depressive disorder, delusional disorder, Alzheimer's dementia, schizophreniform disorder, vascular dementia, and substance abuse dementia | No subgroups with substance use reported | Excludes comorbidity. Authors contacted for unpublished data, no response. | risperidone, quetiapine | ‐ |
| Smulevich 2005 | bipolar I disorder | Excludes participants with recent substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ |
| van Nimwegen 2008a | schizophrenia, schizophreniform, schizoaffective disorder | No measure of substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ |
| Yatham 2007 | bipolar I and II | Excludes participants with drug or alcohol use in past 3 months. | Excludes comorbidity | continuation of oral risperidone, olanzapine, quetiapine or switch to long‐acting injectable LAI risperidone | ‐ |
| Zhangyue 2005 | schizophrenia or schizophrenia‐like illnesses | Excludes participants with alcohol or substance dependence | Quasi‐randomisation.Excludes comorbidity. | aripiprazole versus risperidone | ‐ |
Studies awaiting classification
One study appeared to meet inclusion criteria but no results were reported hence it could not be assessed (NCT00208143). This study is described as an open (no masking), randomised, parallel, superiority trial, comparing the efficacy of quetiapine with risperidone in adults with a diagnosis of schizophrenia or schizoaffective disorder and co‐occurring cocaine or methamphetamine abuse or dependence. We contacted the investigators for information and results but received no response. Another study appeared to meet eligibility criteria, but was described only as "double blind" (Greenspan 2005). Authors were contacted to confirm randomisation but did not respond. A third study appeared to meet eligibility criteria but the authors responded after being contacted that no data for the subgroup with a dual diagnosis are available (Yatham 2003). It was also not clear how many participants in this study had psychotic bipolar disorders. Two studies contained subgroups of participants with substance use disorders, but no data were provided for the subgroup after authors were contacted (Johnsen 2010; San 2012).
Ongoing studies
We found one study currently recruiting participants (NCT01639872). This study is described as a double‐blind, randomised, parallel assignment, superiority trial, comparing the efficacy of clozapine with risperidone in people with schizophrenia and a cannabis use disorder. The estimated sample size is 132.
Risk of bias in included studies
We assessed risk of bias for the eight included studies. Overall the risk of bias was unclear across most domains assessed, as reporting of study results lacked particulars of how potential bias was handled and how potential bias could have influenced results. Figure 4 and Figure 5 contains a graphical overview of the risk of bias. The details of the studies are included under Characteristics of included studies.
4.
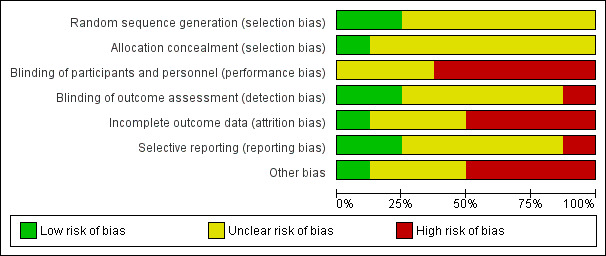
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
5.
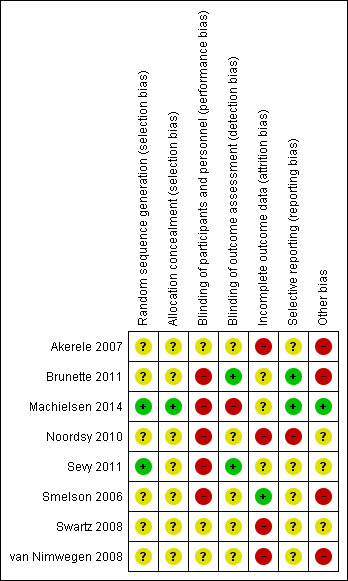
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
1. Random generation
The method of random sequence generation was unclear in all but two studies for which a description of random sequence generation was provided and therefore assessed as low risk of bias (Machielsen 2014; Sevy 2011).
2. Allocation concealment
The method of allocation concealment was unclear in all the included studies, except for one study for which allocation concealment was adequately described, with low risk of bias (Machielsen 2014).
Blinding
1. Performance bias
There was high risk of performance bias in five of the eight included studies, as many studies were described as open label and both participants and personnel were aware of the medications they received (Brunette 2011; Noordsy 2010; Machielsen 2014; Sevy 2011; Smelson 2006). For Noordsy 2010, Brunette 2011 and Machielsen 2014 we assessed risk of unmasking of participants and personnel as high, as the monitoring requirements for clozapine (weekly blood tests) differs from that of risperidone and no description was given how blinding was maintained. No description of blinding of personnel or participants was provided for the remainder of the included studies and risk of bias was assessed as unclear (Akerele 2007; Swartz 2008; van Nimwegen 2008).
2. Detection bias
We judged detection bias to be low for two studies where outcome assessors were reported to be blinded and independent of study physicians (Brunette 2011; Sevy 2011). One study was judged as high risk of bias as outcome assessors were not blinded and were aware of allocation (Machielsen 2014). We judged the risk of detection bias as unclear for five of the included studies as no description or an incomplete description of blinding was reported (Akerele 2007; Noordsy 2010; Smelson 2006; Swartz 2008; van Nimwegen 2008).
Incomplete outcome data
For four studies we judged the risk of attrition bias as being high, due to high levels of attrition (Swartz 2008), unbalanced numbers in leaving between treatments (Akerele 2007), differing reasons for attrition between medications (Noordsy 2010), the use of only single imputation methods (Swartz 2008; van Nimwegen 2008), and the absence of data on baseline differences in groups that dropped out (van Nimwegen 2008). For one study (Smelson 2006), for which the primary — and only outcome — was attrition, we judged the risk to be low. For three studies (Brunette 2011; Sevy 2011; Machielsen 2014), risk of attrition bias was judged as unclear as the number of participants that failed to complete the study was either not reported across the randomised groups, or only simple imputation methods were used to account for missing data in analyses, or the impact of attrition on the study specific outcomes was not clear.
Selective reporting
Risk of selective reporting was judged to be low for two studies for which a study protocol was available and where there were no changes between the protocol‐defined outcomes and outcomes in the study report (Brunette 2011; Machielsen 2014). For five studies risk of selective reporting was judged as unclear as there was either no study protocol available (Akerele 2007), or outcomes stated in the protocol were not mentioned in the study report (van Nimwegen 2008), or the study was a secondary analysis of an existing parent randomised trial, making it unclear whether the outcome selection could have been influenced by preliminary results and analysis of the primary study (Sevy 2011; Smelson 2006; Swartz 2008). In the original protocol for the study by Noordsy 2010, the primary outcome was specified as days of substance use as measured by the Time Line Follow Back method. However, changes were made to the protocol prior to the end of the study and substance use outcomes were judged by experts based on graphs derived from data collected by TLFB method and dichotomised into the proportion of participants with an improvement (20% or more reduction in substance use) versus no improvement or unchanged and worsened groups. This process appeared to have occurred following the collection of data and a change to the protocol was added. Risk of selective reporting was judged to be high for Noordsy 2010.
Other potential sources of bias
Four studies were judged as having high risk of other bias due to sponsorship of the study by the pharmaceutical industry (Akerele 2007; Brunette 2011; Smelson 2006; van Nimwegen 2008). In three studies the risk of other bias was judged as unclear as sponsorship was by academic centres and national funding agencies such as the NIMH but other potential conflicts of interests are not reported and therefore remain unclear, or many authors were supported by the pharmaceutical industry even though the study sponsorship was stated as being independent of industry (Noordsy 2010; Sevy 2011; Swartz 2008). One study was judged to be at low risk of bias as sponsorship was through national funding bodies and university departments with no other potential conflicts of interest declared (Machielsen 2014).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
See: Table 1; Table 2; Table 3; Table 4; Table 5.
Comparison 1: risperidone versus clozapine
Only two studies containing data on 50 participants contributed data to this comparison (Machielsen 2014; Noordsy 2010). See Table 1. All data are short term ‒ up to 6 months.
1.1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower = better)
One trial reported data on these outcomes (Machielsen 2014). There were no clear differences between risperidone and clozapine at study endpoint in general psychopathology symptoms (1 RCT, n = 36, MD 2.70, 95% CI −2.14 to 7.54), Analysis 1.1.
1.1. Analysis.
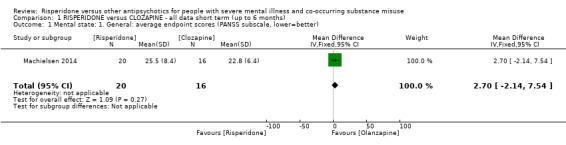
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower=better).
1.2 Mental state: 2. General: any change in general symptoms
Only one study reported results for 'worsening of psychotic symptoms' (non‐systematic assessment) as an adverse effect (Noordsy 2010). There were no clear differences between the risperidone and clozapine groups for this outcome (1 RCT, n = 14, RR 0.14, 95% CI 0.01 to 2.34), Analysis 1.2.
1.2. Analysis.
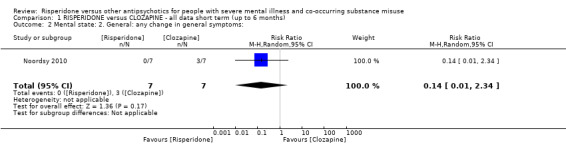
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 2 Mental state: 2. General: any change in general symptoms:.
1.3 Mental state: 3. Specific: positive, negative symptoms ‒ average endpoint scores (PANSS subscales, lower = better)
One trial reported data on these outcomes (Machielsen 2014). There were no differences between risperidone and clozapine at study endpoint in positive symptoms (1 RCT, n = 36, MD 0.90, 95% CI −2.21 to 4.01). For negative symptoms participants with clozapine had significantly lower scores (1 RCT, n = 36, MD 4.00, 95% CI 0.79 to 7.21), Analysis 1.3.
1.3. Analysis.
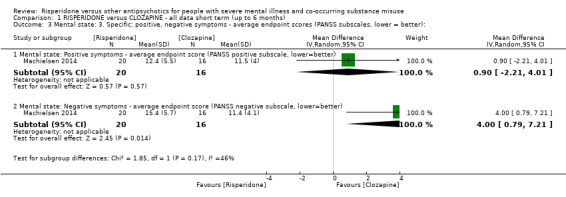
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 3 Mental state: 3. Specific: positive, negative symptoms ‐ average endpoint scores (PANSS subscales, lower = better):.
1.4 Mental state: 4. Specific: anxiety symptoms
The Noordsy 2010 study reported emergence of anxiety symptoms (non‐systematic assessment). No clear differences were found between the risperidone and clozapine groups (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15), Analysis 1.4.
1.4. Analysis.
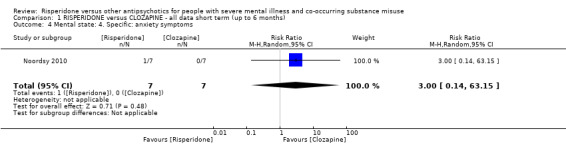
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 4 Mental state: 4. Specific: anxiety symptoms.
1.5.1 Substance use: 1. Improvement (at least 20% reduction in use, TLFB scale)
One study provided data with this outcome (Noordsy 2010). No significant differences in this measure were demonstrated between the risperidone and clozapine groups (1 RCT, n = 14, RR 1.00, 95% CI 0.30 to 3.35), Analysis 1.5.
1.5. Analysis.
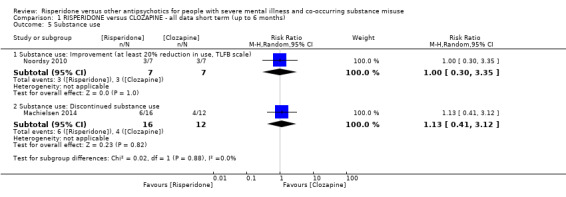
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 5 Substance use.
1.5.2 Substance use: 2. Discontinued substance use
One study provided data for this outcome (Machielsen 2014). There were no significant differences between risperidone and clozapine in the number of participants who stopped using cannabis (1 RCT, n = 28, RR 1.13, 95% CI 0.41 to 3.12), Analysis 1.5.
1.6 Subjective well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher = better)
One study provided data for this outcome (Machielsen 2014). There were no significant differences in the endpoint scores on the Subjective Well‐being under Neuroleptics scale for risperidone compared to clozapine (1 RCT, n = 36, MD −6.00, 95% CI −14.82 to 2.82), Analysis 1.6.
1.6. Analysis.
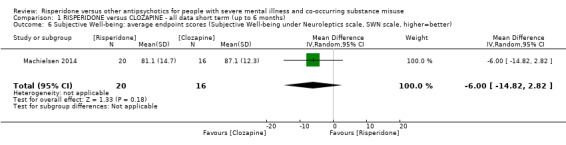
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 6 Subjective Well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher=better).
1.7 Craving for substances
1.7.1 Specific: current craving ‒ average endpoint scores (Marijuana Craving Questionnaire, MCQ, lower = better)
One study included data on this outcome (Machielsen 2014). Participants treated with clozapine had significantly lower scores on the Marijuana Craving Questionnaire (MCQ) compared to participants treated with risperidone (1 RCT, n = 28, MD 7.00, 95% CI 2.37 to 11.63, P = 0.003), Analysis 1.7.
1.7. Analysis.
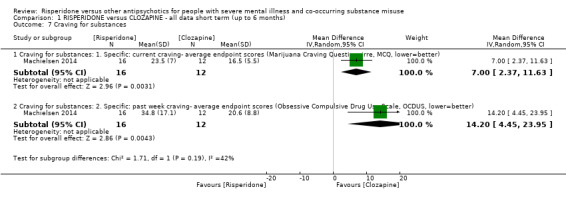
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 7 Craving for substances.
1.7.2 Specific: past week craving ‒ average endpoint scores (Obsessive Compulsive Drug Use Scale, OCDUS, lower = better)
One study included data on this outcome (Machielsen 2014). Participants treated with clozapine had significantly lower craving scores on the Obsessive Compulsive Craving Scale (OCDUS), compared to participants treated with risperidone (1 RCT, n = 28, MD 14.20, 95% CI 4.45 to 23.95, P = 0.004), Analysis 1.7.
1.8 Adherence to antipsychotic medication: discontinued medication
One study included data on this outcome (Machielsen 2014). There were no significant differences between risperidone‐ and clozapine‐treated participants in the number of participants who discontinued antipsychotic treatment (1 RCT, n = 36, RR 4.05, 95% CI 0.21 to 78.76), Analysis 1.8.
1.8. Analysis.
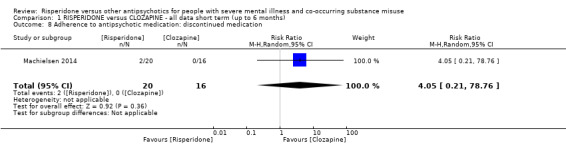
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 8 Adherence to antipsychotic medication: discontinued medication.
1.9 Adverse effects: 1. Movement disorders
1.9.1 Any extrapyramidal side‐effects
Two studies reported data on extrapyramidal side‐effects (Machielsen 2014; Noordsy 2010). There were no significant differences between risperidone‐ and clozapine‐treated participants in terms of the number of participants who experienced any extrapyramidal side‐effects (2 RCTs, n = 50, RR 2.71, 95% CI 0.30 to 24.08; I² = 0%), Analysis 1.9.
1.9. Analysis.
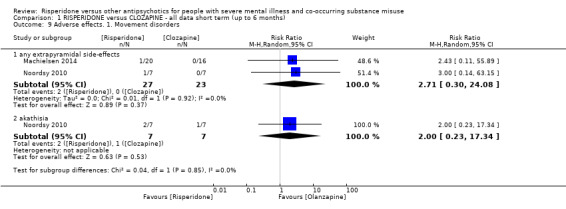
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 9 Adverse effects. 1. Movement disorders.
1.9.2 Akathisia
One study provided data for this outcome (Noordsy 2010). There were no statistically significant differences in the risperidone compared to the clozapine groups for akathisia (1 RCT, n = 14, RR 2.00, 95% CI 0.23 to 17.34), Analysis 1.9.
1.10 Adverse effects: 2. Non‐movement disorder related side‐effects
One study — (Noordsy 2010) — provided data for non‐movement disorder‐related adverse effects. In all cases no statistically significant differences were found in the proportion of participants with side‐effects in the risperidone compared to the clozapine groups. Cardiovascular side‐effects: palpitations (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); hypotension (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Central nervous system side‐effects: headache (1 RCT, n = 14, RR 0.20, 95% CI 0.01 to 3.54); somnolence (1 RCT, n = 14, RR 0.20, 95% CI 0.03 to 1.30). Dermatological side‐effects: acne (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Endocrinological side‐effects: decreased libido (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Ear and labyrinthine: ear canal blockage (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Gastrointestinal: abdominal pain (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); elevated liver function tests (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); hypersalivation (1 RCT, n = 14, RR 0.11, 95% CI 0.01 to 1.74). General adverse effects: fatigue (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Injuries: sprain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Metabolic side‐effects: increased appetite (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); weight gain (1 RCT, n = 14, RR 1.00, 95% CI 0.19 to 5.24). Musculoskeletal: ankle pain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); knee and foot pain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); muscle twitch (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Renal side‐effects: retention (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); urgency (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02) (Analysis 1.10).
1.10. Analysis.
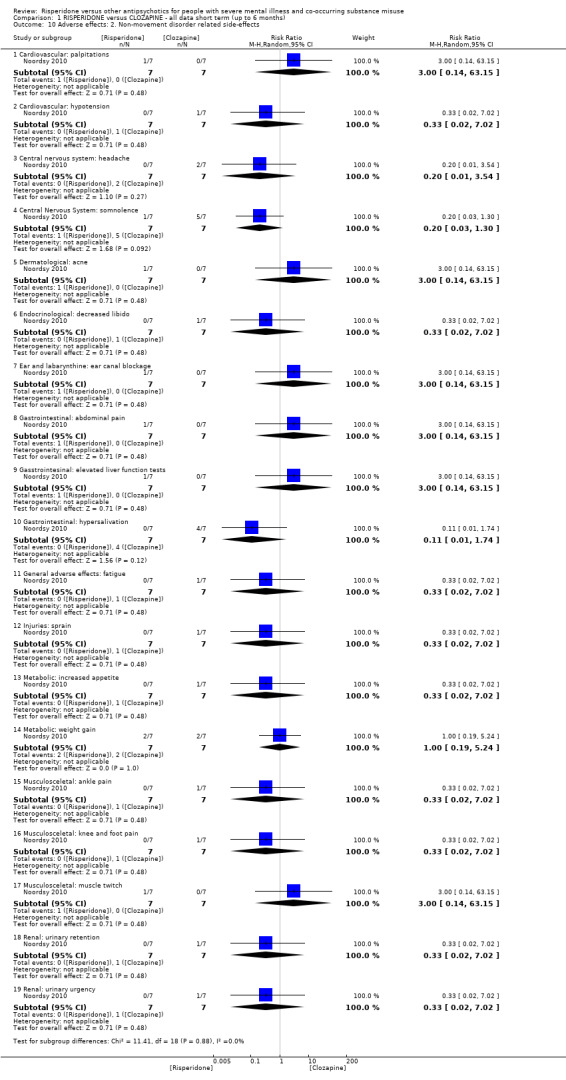
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 10 Adverse effects: 2. Non‐movement disorder related side‐effects.
1.11 Leaving the study early
1.11.1 Any reason
Two studies reported data on this outcome (Machielsen 2014; Noordsy 2010). There were no statistically significant differences between risperidone and clozapine groups (2 RCT, n = 45, RR 0.49, 95% CI 0.10 to 2.51; I² = 34%), Analysis 1.11.
1.11. Analysis.
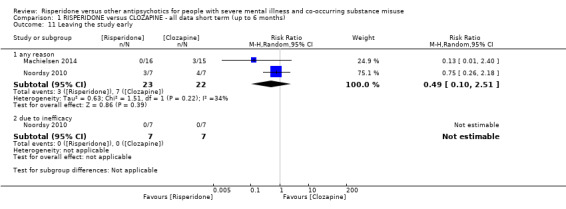
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 11 Leaving the study early.
1.11.2 Due to inefficacy
One study provided data for this outcome (Noordsy 2010). There were no participants who left the study due to inefficacy.
Missing outcomes (risperidone versus clozapine)
There were no studies reporting data on mortality or quality of life.
Comparison 2: risperidone versus olanzapine
A total of five studies contributed data to this comparison (Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008). See Table 2.
2.1 Mental state: 1. Specific: Depression ‒ change scores (HAM‐D, higher = better), short term (up to 6 months)
Only Akerele 2007 included data on this outcome. There were no significant differences between the risperidone and olanzapine groups in the reduction of depressive symptoms from baseline to the end of the study (1 RCT, n = 22, MD −0.11, 95% CI −0.78 to 0.56), Analysis 2.1.
2.1. Analysis.
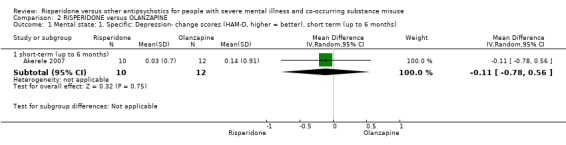
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 1 Mental state: 1. Specific: Depression‐ change scores (HAM‐D, higher = better), short term (up to 6 months).
2.2 Mental state: 2. Specific: Positive symptoms, total score ‒ average endpoint scores (SADS‐C‐PD scale, lower = better), short term (up to 6 months)
Only one study provided analysable data for this outcome (Sevy 2011). There were no significant differences between the risperidone and olanzapine groups in positive symptoms scores (1 RCT, n = 37, MD −1.50, 95% CI −3.82 to 0.82), Analysis 2.2.
2.2. Analysis.
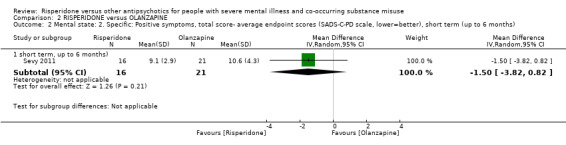
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 2 Mental state: 2. Specific: Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower=better), short term (up to 6 months).
2.3 Mental state: 3. Specific: Positive symptom subscales ‒ average endpoint scores (SADS‐C‐PD scale, lower = better), short term (up to 6 months) ‒ skewed data
In addition to total scores for positive symptoms, one study reported on endpoint data for specific types of positive symptoms, namely delusions, hallucinations and thought disorder (Sevy 2011). Data were skewed and best viewed in an additional table (Analysis 2.3). No significant differences between risperidone and olanzapine were reported.
2.3. Analysis.
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 3 Mental state: 3. Specific: Positive symptom subscales‐ average endpoint scores (SADS‐C‐PD subscores, lower=better), short term (up to 6 months)‐ skewed data.
| Mental state: 3. Specific: Positive symptom subscales‐ average endpoint scores (SADS‐C‐PD subscores, lower=better), short term (up to 6 months)‐ skewed data | |||||
|---|---|---|---|---|---|
| Study | Intervention | Outcome (symptom subscore) | Mean | SD | N |
| Sevy 2011 | Risperidone | Delusions | 2.6 | 1.7 | 16 |
| Sevy 2011 | Olanzapine | 2.7 | 1.6 | 21 | |
| Sevy 2011 | Risperidone | Hallucinations | 1.8 | 1.2 | 16 |
| Sevy 2011 | Olanzapine | 2 | 1.6 | 21 | |
| Sevy 2011 | Risperidone | Thought disorder | 3.6 | 0.8 | 16 |
| Sevy 2011 | Olanzapine | 4.5 | 2.6 | 21 | |
2.4 Mental state: 4. Specific: Negative symptoms, subscales ‒ average endpoint scores (SANS subscales, lower = better), short term (up to 6 months)
Only one study provided analysable data (Sevy 2011). There were no significant differences between the risperidone and olanzapine groups in terms of different subscales for negative symptoms, i.e. affective flattening (1 RCT, n = 39, MD 0.50, 95% CI −0.17 to 1.17); alogia (1 RCT, n = 39, MD 0.40, 95% CI −0.22 to 1.02); avolition apathy (1 RCT, n = 39, MD −0.10, 95% CI −0.73 to 0.53), asociality‐anhedonia (1 RCT, n = 39, MD −0.10, 95% CI −0.80 to 0.60), Analysis 2.4.
2.4. Analysis.
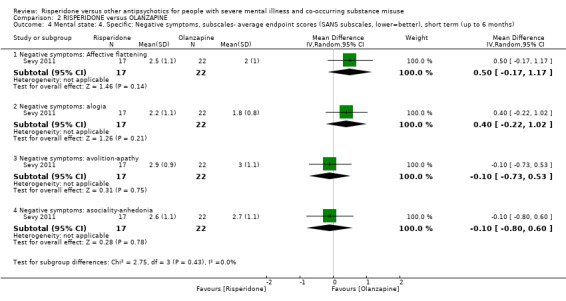
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 4 Mental state: 4. Specific: Negative symptoms, subscales‐ average endpoint scores (SANS subscales, lower=better), short term (up to 6 months).
2.5. Substance use: 1. Substance use: Reduction of cannabis use‐ change data (number of joints smoked/week, LOCF data, higher = better), short‐term data (up to 6 months)
One study provided data (van Nimwegen 2008). There were no significant differences between risperidone and olanzapine in the number of joints smoked (1 RCT, n = 41, MD 0.40, 95% CI −4.72 to 5.52), Analysis 2.5.
2.5. Analysis.
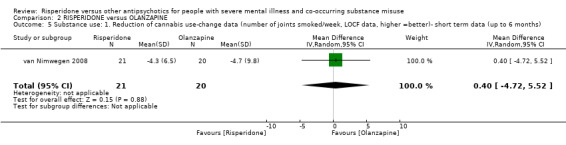
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 5 Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week, LOCF data, higher =better)‐ short term data (up to 6 months).
2.6 Substance use: 2. Discontinued substance use, short term (up to 6 months)
2.6.1 Substance use: stopped using cannabis (Urine testing and Substance Use Questionnaire)
Only one study provided analysable data (Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants who discontinued cannabis (1 RCT, n = 37, RR 1.19, 95% CI 0.68 to 2.08), Analysis 2.6.
2.6. Analysis.
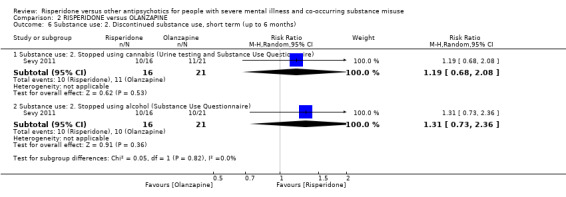
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 6 Substance use: 2. Discontinued substance use, short term (up to 6 months).
2.6.2 Substance use: stopped using alcohol (Substance Use Questionnaire)
One study provided analysable data (Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants who discontinued alcohol (1 RCT, n = 28, RR 1.31, 95% CI 0.73 to 2.36), Analysis 2.6.
2.7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale ‒ average endpoint score (OCDUS, lower = better), short term (up to 6 months)
One study provided data for this outcome (van Nimwegen 2008). There were no significant differences between the risperidone and olanzapine groups in craving as measured by this scale (1 RCT, n = 41, MD 1.30, 95% CI −3.51 to 6.11), Analysis 2.7.
2.7. Analysis.
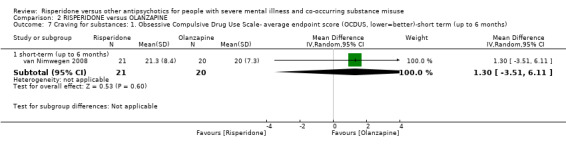
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale‐ average endpoint score (OCDUS, lower=better)‐short term (up to 6 months).
2.8 Craving for substances: 2. Desires for Drug Questionnaire ‒ average endpoint scores (DDQ, LOCF data, lower = better), short term (up to 6 months)
One study provided data for this outcome (van Nimwegen 2008). There were no significant differences between the risperidone and olanzapine groups in craving as measured by this scale (1 RCT, n = 41, MD 5.00, 95% CI −4.86 to 14.86), Analysis 2.8.
2.8. Analysis.
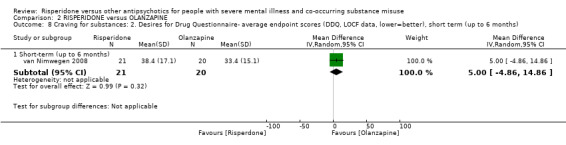
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 8 Craving for substances: 2. Desires for Drug Questionnaire‐ average endpoint scores (DDQ, LOCF data, lower=better), short term (up to 6 months).
2.9 Adverse effects
2.9.1 Movement disorders: Parkinsonism ‒ average endpoint score (SAS, high = worse), short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). There were no significant differences in the scores on this scale between the risperidone and olanzapine groups (1 RCT, n = 16, MD −0.08, 95% CI −1.21 to 1.05), Analysis 2.9.
2.9. Analysis.
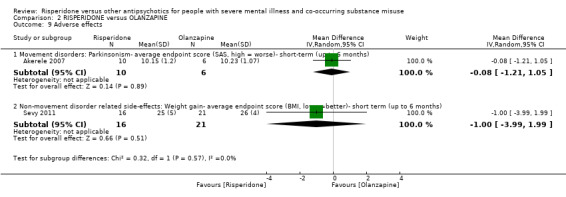
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 9 Adverse effects.
2.9.2 Non‐movement disorder related side‐effects: weight gain‐ average endpoint score (BMI, lower = better), short term (up to 6 months)
Only one study provided data on this outcome (Sevy 2011). There were no significant differences in the scores on this scale between the risperidone and olanzapine groups (1 RCT, n = 37, MD −1.00, 95% CI −3.99 to 1.99), Analysis 2.9.
2.10 Leaving the study early: 1. Various reasons
2.10.1 Any reason, short term (up to 6 months)
Two studies provided data for this outcome (Akerele 2007; Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants leaving the studies early (2 RCT, n = 77, RR 0.68, 95% CI 0.34 to 1.35; I² = 0%), Analysis 2.10.
2.10. Analysis.
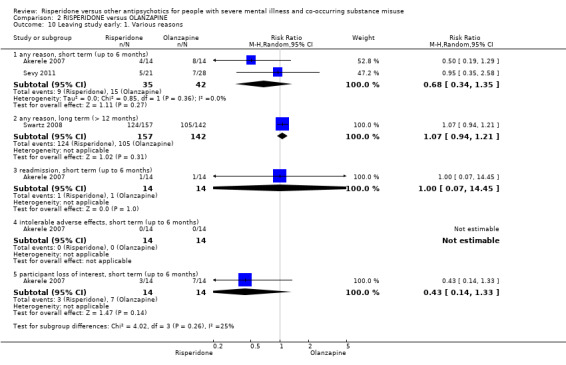
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 10 Leaving study early: 1. Various reasons.
2.10.2 Any reason, long‐term data ( > 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and olanzapine in the number of participants leaving the study early (1 RCT, n = 299, RR 1.07, 95% CI 0.94 to 1.21), Analysis 2.10.
2.10.3 Readmission, short term (up to 6 months)
Only one study provided data on this outcome (Akerele 2007). In Akerele 2007 participants leaving the study early as a result of readmission to an inpatient unit were similar across the risperidone and olanzapine groups, with no significant differences (1 RCT, n = 28, RR 1.00, 95% CI 0.07 to 14.45), Analysis 2.10.
2.10.4 Intolerable adverse effect, short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). In Akerele 2007 no participants left the study early in either treatment group due to adverse medication effects (Analysis 2.10).
2.10.5 Participant loss of interest, short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). In Akerele 2007 there were no significant differences between risperidone and olanzapine in leaving the study early due to lack of interest, although the risperidone group were less likely to drop out due to lack of interest (1 RCT, n = 28, RR 0.43, 95% CI 0.14 to 1.33), Analysis 2.10.
2.11 Leaving the study early: 2. Weeks in the study ‒ average endpoint data (high = good), short term (up to 6 months)
Only one study provided data on this outcome (Akerele 2007). In Akerele 2007 there were no differences in time remaining in the study treatment between the risperidone and olanzapine groups (1 RCT, n = 28, MD 0.00, 95% CI −3.35 to 3.35), Analysis 2.11.
2.11. Analysis.
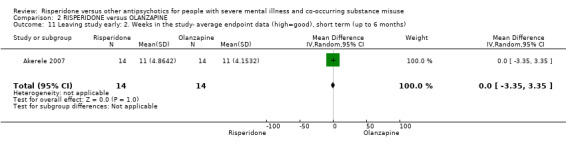
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 11 Leaving study early: 2. Weeks in the study‐ average endpoint data (high=good), short term (up to 6 months).
2.12 Leaving the study early: 3. Weeks in study ‒ average endpoint data (high = good), short term (up to 6 months) ‒ skewed data
2.12.1 Weeks remained in study ‒ average endpoint data (high = good), short term (up to 6 months) ‒ skewed data
One study provided data for this outcome (Smelson 2006). Data was skewed so is best inspected in an additional table (Analysis 2.12).
2.12. Analysis.
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 12 Leaving study early: 3. Weeks in study‐ average endpoint data (high=good), short term (up to 6 months)‐ skewed data.
| Leaving study early: 3. Weeks in study‐ average endpoint data (high=good), short term (up to 6 months)‐ skewed data | ||||
|---|---|---|---|---|
| Study | Intervention | Mean (number of weeks) | SD | N |
| Smelson 2006 | Risperidone | 207 | 142.9 | 76 |
| Smelson 2006 | Olanzapine | 267.9 | 127.4 | 85 |
Comparison 3: risperidone versus perphenazine
3.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and perphenazine for this outcome (1 RCT, n = 281, RR 1.05, 95% CI 0.92 to 1.20), Analysis 3.1.
3.1. Analysis.
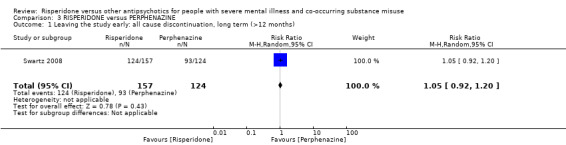
Comparison 3 RISPERIDONE versus PERPHENAZINE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).
Comparison 4: risperidone versus quetiapine
4.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There was no statistically significant difference between risperidone and quetiapine (1 RCT, n = 294, RR 0.96, 95% CI 0.86 to 1.07), Analysis 4.1.
4.1. Analysis.
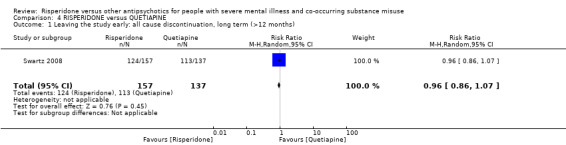
Comparison 4 RISPERIDONE versus QUETIAPINE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).
Comparison 5: risperidone versus ziprasidone
5.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and ziprasidone (1 RCT, n = 240, RR 0.96, 95% CI 0.85 to 1.10), Analysis 5.1.
5.1. Analysis.
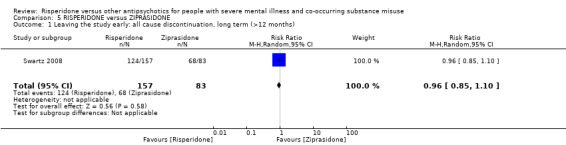
Comparison 5 RISPERIDONE versus ZIPRASIDONE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).
6. Missing outcomes (risperidone versus olanzapine)
There were no studies reporting data for general mental state (CGI), anxiety symptoms, subjective well‐being, adverse effects such as metabolic syndrome (other than parkinsonism and weight gain), mortality and quality of life.
Discussion
Summary of main results
Comparison 1: risperidone versus clozapine
Only two studies (Noordsy 2010; Machielsen 2014), containing 50 participants, provided usable data for this comparison (Table 1). A third study, Brunette 2011, met criteria for inclusion but we were unable to extract any usable data from this study. Studies with useable data reported no statistically significant differences between risperidone and clozapine in terms of worsening of general psychotic symptoms, general anxiety symptoms, reduction in substance use, subjective well‐being, medication adherence, movement disorder‐related side‐effects, other side‐effects or leaving the study early (Machielsen 2014; Noordsy 2010). However, in the study by Machielsen 2014, lower negative symptoms were found in the clozapine group as compared to the risperidone‐treated group. In addition, lower immediate and 'past 1 week' craving scores were found for participants treated with clozapine as opposed to risperidone (Machielsen 2014). No results were reported on mortality or quality of life, and side‐effects such as weight gain and metabolic syndrome were inconsistently reported. The sample size in this study was very low (N = 36), limiting the conclusions that can be drawn from the analysis of data (Machielsen 2014). Moreover in the study by Noordsy 2010 participants in the clozapine group reported the emergence or worsening of psychotic symptoms and more participants in the risperidone group reported the emergence of anxiety symptoms, although these differences did not reach statistical significance.
Comparison 2: risperidone versus olanzapine
Five studies — Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008 — comprising 997 participants provided data for this comparison (Table 2). Overall the quality of evidence was low to very low. Studies differed in terms of design and outcomes measured, precluding pooling of outcome data. In addition sample size was low for most of the individual studies, with Akerele 2007, Sevy 2011 and van Nimwegen 2008 having sample sizes below N = 50, limiting the conclusions that can be drawn from them. There were no statistically significant differences between risperidone and olanzapine for a reduction in any mental symptoms (depression, positive or negative symptoms). In one study there was some evidence of significantly fewer days of any substance use for the olanzapine group compared to the risperidone group, whereas the risperidone group showed significantly greater reductions in craving for cannabis use (Akerele 2007). In turn, olanzapine‐treated participants had a significantly longer time to all‐cause antipsychotic treatment discontinuation in one study (Smelson 2006). However, there were no significant differences between the risperidone and olanzapine groups in the proportion of participants with urine‐positive weeks for the cannabis and cocaine use subgroups, cocaine craving, parkinsonism, weight gain or leaving the study early. Results for many outcomes were incompletely reported or not suitable for re‐analysis and we were only able to report in a qualitative, narrative format. Of note, in many studies authors did not report on any measures relating to metabolic side‐effects such as weight gain, abnormalities in glucose or lipid metabolism. For subjective well‐being, authors reported only total sample findings and did not report on subgroups who used substances. Furthermore, no results were reported on mortality or quality of life.
Comparison 3: risperidone versus perphenazine
One study (Swartz 2008), with data on 281 participants, reported on leaving the study early, with no differences between risperidone‐ or perphenazine‐treated participants.
Comparison 4: risperidone versus quetiapine
One study (Swartz 2008), with data on 294 participants, provided data for this comparison. There was no significant difference between the risperidone and quetiapine groups in leaving the study early.
Comparison 5: risperidone versus ziprasidone
One study (Swartz 2008), with data on 240 participants, reported on leaving the study early, with no differences between risperidone‐ or ziprasidone‐treated participants.
Overall completeness and applicability of evidence
1. Completeness
The reporting of outcome data was incomplete and generally poor. In addition, some trials often failed to report on outcomes that were mentioned to have been measured in the protocol and Methods sections (Akerele 2007; Noordsy 2010). Primary outcomes of improvement in mental state and substance use were poorly reported, with many studies not reporting on changes in mental state or reporting incompletely (Noordsy 2010; Smelson 2006; van Nimwegen 2008). Substance use was also not reported on in Smelson 2006 and Swartz 2008.
Many studies also failed to report on important outcomes such as weight gain, metabolic changes and endocrinological adverse effects. No outcomes on craving were reported in Noordsy 2010, Sevy 2011, Smelson 2006, and Swartz 2008; and adherence was not reported by Noordsy 2010, Sevy 2011, Swartz 2008 and van Nimwegen 2008. Many studies failed to report on subjective well‐being, mortality or quality of life (Akerele 2007, Noordsy 2010; Sevy 2011; Smelson 2006; Swartz 2008).
2. Applicability of evidence
Six of the included trials — Akerele 2007, Brunette 2011, Noordsy 2010, Sevy 2011, Smelson 2006 and Swartz 2008 — were conducted in the USA and two in the Netherlands (Machielsen 2014; van Nimwegen 2008). We found no trials conducted in developing countries. All trials involved comparisons of risperidone with SGAs or clozapine; and only two trials included comparison with FGAs (Smelson 2006; Swartz 2008), only one with useable data (Swartz 2008). Some SGAs may have lower availability due to cost in middle‐ and low‐income countries, potentially leading to lower applicability of findings to these settings. In most studies males and females were eligible for inclusion, but the study by Akerele 2007 included mostly men and only males were included in Machielsen 2014, potentially limiting applicability to female populations. Moreover, mixed ethnic groups were included in most studies. Noordsy 2010 and Brunette 2011 included only Caucasians (understood to be white participants).
The sample size of many included trials was very low and below N = 50 (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008). One trial had sample sizes between 100 and 300 (Smelson 2006); and one study had a sample size of more than 400 (Swartz 2008).
Quality of the evidence
1. General
Overall, of the eight studies included in this review, across most studies reporting of results was poor and did not adhere to CONSORT standards (1996). As a result many outcomes yielded unusable data.
2. Specific
From the eight included studies there were five comparisons of risperidone with other antipsychotics. For most studies sample sizes were small. Risk of bias was unclear in most domains across the included studies, however in some instances there was high risk of bias particularly with regards to performance bias, attrition bias, selective outcome reporting and other forms of bias. Moreover, due to heterogeneity in design and poor outcome reporting, pooling of data in meta‐analyses was not possible for most outcomes leading to imprecise effect estimates and precluding any meaningful analysis of results and yielding low to very low quality evidence for the main outcomes.
Potential biases in the review process
We conducted a wide search, including a grey literature search, and searches of clinical trial registers and screening of recent relevant conference abstracts. Study selection and data extraction was done in duplicate to minimise the potential for selection and information bias. Our search was conducted within the Cochrane Schizophrenia Group’s Trials Register and the Cochrane Depression, Anxiety and Neurosis Controlled Trials Register. We did not include searches of the Drugs and Alcohol group register. This may have lead us to miss some studies examining treatments for substance use disorders that contain some participants with serious mental illness, but this is unlikely.
We also contacted authors to clarify reporting of studies and to provide missing data. Following our extensive search we are not aware of any additional studies in this field, but we are open to review the evidence and would call upon authors to contact us should there be any trial that warrant consideration for inclusion.
Agreements and disagreements with other studies or reviews
There have been a number of systematic reviews evaluating the efficacy of antipsychotics in people with a dual diagnosis. In Baker 2010 and Baker 2012 the authors reached similar conclusions regarding the evidence for risperidone versus other antipsychotics. In addition Wobrock 2009 reached conclusions similar to ours, although the methods of grading evidence differed from our review. In Lazary 2012, authors suggest certain agents such as olanzapine and clozapine may be superior to others (including risperidone) but express reservations on the quality of evidence as they based their findings on a mixture of observational and randomised trial evidence. Furthermore, Lazary 2012 used different methods from our review to assess study quality and strength of evidence. In Machielsen 2009 the authors included two studies that we excluded in our review (containing mixed single and dual diagnosis groups and a mixed comparator medication group) and reached a conclusion that clozapine may be superior to risperidone, although they caution against a firm conclusion because of the paucity of well‐designed trials. McLoughlin 2014 compared the use of different antipsychotics (classified as antipsychotic A versus B) in people with schizophrenia and co‐occurring cannabis disorders and reached similar conclusion to our review, despite different methods and inclusion of different studies.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Although there are theories postulating risperidone to be more efficacious in the treatment of people with a dual diagnosis, all evidence currently available is low in quality and does not favour risperidone over any other antipsychotic medication. We found risperidone did not lead to significantly greater reductions of substance use or improvements in mental state compared to clozapine. In addition, risperidone did not lead to significantly greater reductions in substance use or improvements in mental state compared to olanzapine. Risperidone did not significantly improve study retention compared to perphenazine, quetiapine or ziprasidone. Of note: patient‐relevant outcomes such as quality of life were not measured in any of the included studies. Due to the small sample size of the included studies and the overall low quality of evidence, limited conclusions can be drawn regarding these findings.
2. For clinicians
Overall the quality of the existing evidence is very low and includes a very small number of studies with very small sample sizes. We did not find evidence suggesting superiority of risperidone over clozapine, olanzapine, perphenazine, quetiapine or ziprasidone for any of the primary or secondary outcomes in our review. Of note: studies were confined to short‐term outcomes and studies with comparisons of risperidone with first‐generation antipsychotics were limited. All studies were conducted in a developed world setting.
3. For managers or policy makers
Reviews and pharmacological treatment guidelines for people with a dual diagnosis that suggest that second‐generation antipsychotics (SGAs) such as risperidone may be the preferred treatment in this population need to be interpreted with caution. In particular, we found no evidence that risperidone was superior to clozapine, olanzapine, perphenazine, quetiapine or ziprasidone. Due to the paucity of comparisons with first‐generation antipsychotics (e.g. haloperidol and chlorpromazine), little can be said about the treatment effects of risperidone compared to first‐generation antipsychotics. Therefore, it may be beneficial to include less expensive medications such as first‐generation antipsychotics (such as haloperidol and chlorpromazine) in future in clinical trials in people with a dual diagnosis.
Implications for research.
1. General
Future studies should include larger samples and comparisons of risperidone to both first‐ and second‐generation antipsychotics. Investigators should adhere to CONSORT guidelines in reporting results. Adverse effects including those related to weight gain and metabolic disturbances should be reported.
2. Specific
2.1 Reviews
Many of the excluded studies could, perhaps, be included in other reviews; and titles or existing reviews are suggested in Table 8.
2.2 Trials
People with a dual diagnosis are likely to face substantial adversity across clinical, social, legal and financial areas of their lives. Whereas participation in clinical trials investigating the impact of pharmacological treatments is encouraged, it is likely that the recruitment and retention of people with a dual diagnosis in such trials may be difficult at best. The conduct of well‐designed, multi‐centre randomised adequately powered trials is therefore important. Furthermore it is critical that consistency be maintained in trial design in order to minimize heterogeneity in design, clinical populations studied and methods of outcome assessment. Consistency across studies and adherence to CONSORT guidelines in order to minimise risk of bias should allow for more robust pooling of results in meta‐analyses. Engaging with the AllTrials initiative would allow liberation of all relevant data.
In addition, although the mechanisms underlying the differential treatment efficacy of antipsychotics is likely to be complex, and although the role of causal mechanisms is likely to be controversial in evidence‐based assessments such as systematic reviews, we would encourage trialists to include a study component that investigates potential underlying neural mechanisms that may underlie the treatment effects of antipsychotics in people with a dual diagnosis. We realise that much care and thought goes into trial design but we also have given this some consideration and, therefore, suggest a design in Table 9.
4. Suggested design for future trial.
| Methods | Allocation: centralised sequence generation with table of random numbers or computer‐generated code, stratified by severity of illness, sequence concealed till interventions assigned. Blinding: could be optional, depending on choice of outcome. Duration: 12 months. |
| Participants | Diagnosis: schizophrenia and co‐occurring ongoing substance misuse (clinical criteria). N = 300*. Age: adults. Sex: men and women. Setting: any. |
| Interventions | 1. Risperidone: clinically indicated dose. N = 150. 2. Olanzapine: clinically indicated dose. N = 150. |
| Outcomes | Global state: CGI‐I and CGI‐S. Substance use: pragmatic binary/continuous measure. Well‐being: pragmatic binary/continuous measure. Craving: pragmatic binary/continuous measure. Service outcomes: re‐hospitalisation, days in hospital, time attending psychiatric outpatient clinic. Quality of life: important change. Adverse effects: including mortality, weight change and extrapyramidal symptoms. Satisfaction with care: patients/carers. Leaving the study early. Economic data. Other routine data, such as incidents with the police, |
| Notes | * size of study to detect a 10% difference in improvement with 80% certainty. For all outcomes there should be binary cut‐off points of clinically important improvement, defined before the study starts. |
What's new
| Date | Event | Description |
|---|---|---|
| 6 February 2018 | Amended | Addition of affiliations for review author Nandi Siegfried. |
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required. We would like to thank Professor Clive Adams, Claire Irving, Jun Xia and Farhad Shokraneh for their patience, editorial assistance and efficient responses to questions.
We would also like to thank Trentham Furness for peer reviewing the protocol and Eleni‐Maria Kokkoli, Joseph Wakeman and Zein Hussain for peer reviewing this version of the review.
We also express our gratitude to Juanjuan Ren, who extracted the data from Chinese studies; and to Sarah Dawson, Information Specialist of Cochrane Common Mental Disorders Group, for developing the search strategies and searching their group's register for this review.
Appendices
Appendix 1. Previous Searches
1.1 Search in 2014
1.1.1 Electronic searches
On April 25, 2014, we searched the Cochrane Schizophrenia Review Group’s Register with the phrase:¨[(*rispe* or *9‐OH‐risperid* or *r 64766* in abstract, index terms of REFERENCE] or [*rispe* in interventions of STUDY] AND [(*polydrug* or *substanc* or *alcoh* or *tranquiliz* or *narcot*or * abus* or *opiat* or *street drug* or *intoxi*) in REFERENCE) and (substance abus* or drug abus*or *alcohol*or dual* and diagnos*) in STUDY)]
The Schizophrenia Review Group's trials register is based on regular searches of: BIOSIS Inside; CENTRAL; CINAHL; EMBASE; MEDLINE and PsycINFO; the hand searching of relevant journals and conference proceedings; and searches of several key grey literature sources. A full description is given in the Group's module.
1.1.2 Searching other resources
1.1.2.1 Reference searching
We inspected reference lists of all included studies for further relevant studies.
1.1.2.2 Personal contact
We contacted the first author of each included study for information regarding unpublished trials. In addition we contacted pharmaceutical companies regarding unpublished trials.
Data and analyses
Comparison 1. RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower=better) | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | 2.70 [‐2.14, 7.54] |
| 2 Mental state: 2. General: any change in general symptoms: | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.34] |
| 3 Mental state: 3. Specific: positive, negative symptoms ‐ average endpoint scores (PANSS subscales, lower = better): | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Mental state: Positive symptoms ‐ average endpoint score (PANSS positive subscale, lower=better) | 1 | 36 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐2.21, 4.01] |
| 3.2 Mental state: Negative symptoms ‐ average endpoint score (PANSS negative subscale, lower=better) | 1 | 36 | Mean Difference (IV, Random, 95% CI) | 4.0 [0.79, 7.21] |
| 4 Mental state: 4. Specific: anxiety symptoms | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 5 Substance use | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Substance use: Improvement (at least 20% reduction in use, TLFB scale) | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.30, 3.35] |
| 5.2 Substance use: Discontinued substance use | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.41, 3.12] |
| 6 Subjective Well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher=better) | 1 | 36 | Mean Difference (IV, Random, 95% CI) | ‐6.0 [‐14.82, 2.82] |
| 7 Craving for substances | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Craving for substances: 1. Specific: current craving‐ average endpoint scores (Marijuana Craving Questionairre, MCQ, lower=better) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 7.00 [2.37, 11.63] |
| 7.2 Craving for substances: 2. Specific: past week craving‐ average endpoint scores (Obsessive Compulsive Drug Use Scale, OCDUS, lower=better) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 14.20 [4.45, 23.95] |
| 8 Adherence to antipsychotic medication: discontinued medication | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 4.05 [0.21, 78.76] |
| 9 Adverse effects. 1. Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 any extrapyramidal side‐effects | 2 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [0.30, 24.08] |
| 9.2 akathisia | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.23, 17.34] |
| 10 Adverse effects: 2. Non‐movement disorder related side‐effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Cardiovascular: palpitations | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.2 Cardiovascular: hypotension | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.3 Central nervous system: headache | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 3.54] |
| 10.4 Central Nervous System: somnolence | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.03, 1.30] |
| 10.5 Dermatological: acne | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.6 Endocrinological: decreased libido | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.7 Ear and labarynthine: ear canal blockage | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.8 Gastrointestinal: abdominal pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.9 Gasstrointesinal: elevated liver function tests | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.10 Gastrointestinal: hypersalivation | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 1.74] |
| 10.11 General adverse effects: fatigue | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.12 Injuries: sprain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.13 Metabolic: increased appetite | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.14 Metabolic: weight gain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.19, 5.24] |
| 10.15 Musculosceletal: ankle pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.16 Musculosceletal: knee and foot pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.17 Musculosceletal: muscle twitch | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.18 Renal: urinary retention | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.19 Renal: urinary urgency | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 11 Leaving the study early | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 any reason | 2 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.10, 2.51] |
| 11.2 due to inefficacy | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. RISPERIDONE versus OLANZAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. Specific: Depression‐ change scores (HAM‐D, higher = better), short term (up to 6 months) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 short‐term (up to 6 months) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐0.78, 0.56] |
| 2 Mental state: 2. Specific: Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower=better), short term (up to 6 months) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 short term, up to 6 months) | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐3.82, 0.82] |
| 3 Mental state: 3. Specific: Positive symptom subscales‐ average endpoint scores (SADS‐C‐PD subscores, lower=better), short term (up to 6 months)‐ skewed data | Other data | No numeric data | ||
| 4 Mental state: 4. Specific: Negative symptoms, subscales‐ average endpoint scores (SANS subscales, lower=better), short term (up to 6 months) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Negative symptoms: Affective flattening | 1 | 39 | Mean Difference (IV, Random, 95% CI) | 0.5 [‐0.17, 1.17] |
| 4.2 Negative symptoms: alogia | 1 | 39 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐0.22, 1.02] |
| 4.3 Negative symptoms: avolition‐apathy | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.73, 0.53] |
| 4.4 Negative symptoms: asociality‐anhedonia | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.80, 0.60] |
| 5 Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week, LOCF data, higher =better)‐ short term data (up to 6 months) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐4.72, 5.52] |
| 6 Substance use: 2. Discontinued substance use, short term (up to 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Substance use: 2. Stopped using cannabis (Urine testing and Substance Use Questionnaire) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.68, 2.08] |
| 6.2 Substance use: 2. Stopped using alcohol (Substance Use Questionnaire) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.73, 2.36] |
| 7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale‐ average endpoint score (OCDUS, lower=better)‐short term (up to 6 months) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 short‐term (up to 6 months) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 1.30 [‐3.51, 6.11] |
| 8 Craving for substances: 2. Desires for Drug Questionnaire‐ average endpoint scores (DDQ, LOCF data, lower=better), short term (up to 6 months) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Short‐term (up to 6 months) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 5.0 [‐4.86, 14.86] |
| 9 Adverse effects | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Movement disorders: Parkinsonism‐ average endpoint score (SAS, high = worse)‐ short‐term (up to 6 months) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐1.21, 1.05] |
| 9.2 Non‐movement disorder related side‐effects: Weight gain‐ average endpoint score (BMI, lower=better)‐ short term (up to 6 months) | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐3.99, 1.99] |
| 10 Leaving study early: 1. Various reasons | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 any reason, short term (up to 6 months) | 2 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.34, 1.35] |
| 10.2 any reason, long term (> 12 months) | 1 | 299 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.94, 1.21] |
| 10.3 readmission, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.45] |
| 10.4 intolerable adverse effects, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.5 participant loss of interest, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.14, 1.33] |
| 11 Leaving study early: 2. Weeks in the study‐ average endpoint data (high=good), short term (up to 6 months) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐3.35, 3.35] |
| 12 Leaving study early: 3. Weeks in study‐ average endpoint data (high=good), short term (up to 6 months)‐ skewed data | Other data | No numeric data |
Comparison 3. RISPERIDONE versus PERPHENAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) | 1 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.92, 1.20] |
Comparison 4. RISPERIDONE versus QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.07] |
Comparison 5. RISPERIDONE versus ZIPRASIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) | 1 | 240 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.85, 1.10] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Akerele 2007.
| Methods | Allocation: randomised. Blindness: not described. Duration: 14 weeks, 4‐week medication taper and 10‐week follow‐up. Design: superiority, parallel group, randomised trial, single site. Setting: outpatients attending day treatment, USA. | |
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) schizophrenia or schizoaffective disorder and either current cannabis or cocaine abuse or dependence.
N = 28. All with co‐occurring substance use disorders. Age: mean ˜36 years. Sex: 25 M, 3 F. History: cannabis use at least twice/week or cocaine once/week in 3 months prior to enrolment. Ethnicity: 54% African American, 32% Hispanic, 14% Caucasian Exclusion: pregnant, unstable psychiatric or medical condition, current physiological dependence on alcohol or another substance for which had experienced significant withdrawal symptoms in past (caffeine, nicotine dependence was acceptable), history of seizures or neuroleptic malignant syndrome, enzyme levels 3 times upper normal limit, violent crime committed in past 2 years, non‐response to either olanzapine or risperidone in the past, score of more than 30 on PANSS positive and negative subscales. |
|
| Interventions | 1. Risperidone: fixed dose escalation of 3 mg/day for 3 days followed by 6 mg for 4 days and then 9 mg for remainder of study. N = 14 2. Olanzapine: fixed dose escalation of 5 mg/day for 3 days then 10 mg/day for 4 days and then 15 mg/day for 5 days followed by 20 mg/day for remainder of study. N = 14 All participants received weekly psychotherapy over the study period and were asked to nominate a "significant other" to assist with attendance and follow‐up. |
|
| Outcomes | Mental state: change scores HAM‐D scale Adverse effects: parkinsonism endpoint score SAS. Leaving the study early: any reason Unable to use: Mental state: PANSS positive and PANSS negative subscales (no means or SD, longitudinal data), CGI (no data reported). Substance use: proportion of positive urine tests for cannabis and cocaine weekly over 10‐week study period (no means or SD, longitudinal data), days of self‐reported substance use (no SD); Quantitative Substance Use Inventory (psychometric properties of instrument not validated). Craving for substances: Marijuana Craving Report, Cocaine Craving Report (no means or SD, longitudinal data). Adherence to antipsychotic medication: number of medication doses missed (no means or SD). Adverse effects: tardive dyskinesia (AIMS)(not reported by group), sedation (no data provided). |
|
| Notes | Funding: support for this study was provided in part by grants from the National Institute on Drug Abuse and the National Alliance for Research on Schizophrenia and Depression (NARSAD, currently known as The Brain and Behavior Research Foundation) and Eli Lilly and Co. Declarations of interest made by researchers conducting this study include support from a number of pharmaceutical companies, i.e. Ortho‐McNeil Pharmaceuticals, Eli Lilly & Company, UCB Pharma and consultancy to Shire, Pharmaceuticals Inc, AstraZeneca Pharmaceutical, Eli Lilly and Company. Contact of authors: we contacted the study primary and co‐authors by e‐mail to clarify items of study design and to obtain study data. The authors did not respond to these attempts. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised but there was insufficient information on the method used to randomise the participants. Quote: "Randomization was not stratified, but was a 50=50 uniform distribution of groups of 4". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No description of blinding is provided in the study report. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No mention is made of whether the outcome assessors were indeed blinded and independent. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Twice as many people withdrew from the olanzapine group (8/14; 57%) compared to the risperidone group (4/14; 29%). The most common reasons for withdrawal were that the participants were not interested (N = 10) or that they were admitted to inpatient units (N = 3). There were no other significant differences between the groups with respect to demographic and baseline clinical characteristics. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. Subgroups are reported as primary outcomes. |
| Other bias | High risk | Funding for study provided in part by industry (Eli Lilly and Co). No other sources of bias were identified. |
Brunette 2011.
| Methods | Allocation: randomised
Blindness: single‐blind (outcome assessor)
Duration: 12 weeks
Design: superiority, parallel group, randomised trial
Setting: outpatients, two treatment sites: New Hampshire and South Carolina, USA Funding: National Institute on Drug Abuse |
|
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) diagnosis of schizophrenia or schizoaffective disorder and a current cannabis use disorder (abuse or dependence).
N = 31. All with co‐occurring substance use disorders. Age: range 18 to 65 years; mean ˜36 years. Sex: 24 M, 7 F History: cannabis use on at least 5 days in the 3 weeks prior to screening. Ethnicity: 26 (83.9%) were Caucasian. Inclusion: outpatient status prior to randomisation and on current antipsychotic treatment other than clozapine. Exclusion: patients with serious, active medical illness, suicidality, severe psychiatric instability, on treatment with medications that could affect alcohol use such as naltrexone, topiramate, disulfiram, low white cell counts (< 3.500/mm³), seizure disorder. |
|
| Interventions | Clozapine: titrated to 400 mg daily in 4 weeks. n = 15 Treatment as usual (TAU): i.e. continue on existing antipsychotic treatment. n = 16; (n = 5 on risperidone) All participants received weekly individual substance abuse and mental health counselling and attended weekly Alcoholics Anonymous meetings. |
|
| Outcomes |
Unable to use: Mental state: (BPRS, CGI, SANS) (no data reported) Substance use: cannabis use (TLFB) number of "average joints" used per week, assessed weekly for 12 weeks; (no data on subgroup with risperidone) Substance Abuse Treatment Scale (SATS) (1 to 8) measuring treatment involvement, Single‐Item Contemplation ladder (0 to 10) motivation to stop using cannabis (no data on subgroup with risperidone) Adverse effects: SAS scale, BARS scale, AIMS scale (no data reported) |
|
| Notes | Further data requested. Author responded and indicated that 5/16 patients in TAU group were on risperidone. Authors decided not to provide requested data as they were advised by their methodological consultant that using subgroup data will in effect interfere with the randomisation given the specific design of this study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was blocked by site. No description of how sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was blocked by site. No description of how sequence was generated or how allocation concealment was maintained. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Unblinded clinicians prescribed and adjusted study medications weekly. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinded raters assessed patients weekly, independent of study physicians. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Longitudinal random‐effects modelling was used that could have accounted for missing data; however no indication as to the extent of missing data; |
| Selective reporting (reporting bias) | Low risk | Study outcomes are identical to protocol‐defined outcomes. |
| Other bias | High risk | Protocol indicates study was sponsored by Janssen, Novartis. |
Machielsen 2014.
| Methods | Allocation: randomised.
Blindness: no description.
Duration: 4 weeks.
Design: superiority, parallel group, randomised trial.
Setting: Inpatients and outpatients recruited from the Early Psychosis Department of The Academic Medical Centre of the University of Amsterdam between April 2009 and June 2012. Funding: Dutch Health Research Council. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophreniform, schizophrenia or schizoaffective disorder. CIDI diagnosis of cannabis use disorder (abuse or dependence, N = 35).
N = 39 randomised, N = 31 with co‐occurring substance use disorders. Age: range 18 to 50 years; mean ˜22.4 years (risperidone), mean ˜22.3 years (clozapine). Sex: all participants were male. Ethnicity: not stated. Inclusion: males, aged 18 to 30 with DSM‐IV diagnosis of schizophreniform, schizophrenia or schizoaffective disorder. Exclusion: previous contraindication or unsuccessful treatment with risperidone or clozapine, depot antipsychotic use in the 3 months prior to recruitment, treatment with medication or than biperiden or benzodiazepines. |
|
| Interventions | Risperidone: titrated to initial dose of 3.5 mg/day, then according to treatment response. N = 16 Clozapine: titrated to initial dose of 350 mg/day, then according to treatment response. N = 15 Participants had "supportive treatment as usual". |
|
| Outcomes | Mental state: positive psychotic symptoms (average endpoint score, PANSS positive sub‐scale), negative symptoms (average endpoint score, PANSS negative sub‐scale), general psychopathology (average endpoint score, PANSS general sub‐scale). Substance use: number discontinuing cannabis use. Subjective well‐being: Subjective well‐being under neuroleptics scale (SWN scale) Craving for substances: Marijuana Craving Questionnaire (MCQ), Obsessive Compulsive Drug Use Scale (OCDUS). Adherence to medication: discontinuing medication. Adverse effects: any extrapyramidal side‐effects (no data for subgroups with specific extrapyramidal side‐effects). Leaving the study early. |
|
| Notes | Authors e‐mailed for additional information: response was given to questions about randomisation, and a flow diagram of study attrition was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number generator software was used through the ALEA program: randomisation has been performed on‐line via a secure internet facility by the TENALEA Clinical Trial Data Management System. Randomisation has been performed in a 1:1 ratio, using randomly permuted blocks with maximum blocksize of 4, within strata formed by use of drugs (Cannabis use, no drugs use). |
| Allocation concealment (selection bias) | Low risk | The physician states the patient’s date of birth and the stratification factor and receives treatment allocation when submitting this information to the website from central trial office. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Due to feasibility and ethical considerations this was an open label study over a relatively short period of time in which dosage of medication could be adjusted in case of side‐effects or lack of efficacy. Clozapine required blood monitoring which differs from risperidone requirements. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcome assessment was not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Authors provided flow diagram on request reporting differential drop‐out (with reasons stated) in two treatment arms (20%, 3/15 cannabis users in clozapine arm, 0/16 in risperidone arm). The impact of not including these participants in the endpoint analysis is unclear. |
| Selective reporting (reporting bias) | Low risk | Outcomes in pre‐published protocol are identical to study reported outcomes. |
| Other bias | Low risk | No clear evidence for bias. |
Noordsy 2010.
| Methods | Allocation: randomised
Blindness: single‐blind (outcome assessor)
Duration: 24 weeks
Design: superiority, parallel group, randomised trial
Setting: New Hampshire, United States including: New Hampshire Hospital (Concord), Dartmouth–Hitchcock Medical Center (Lebanon), West Central Behavioral Health (Lebanon), Mental Health Center of Greater Manchester (Manchester), Center for Psychiatric Advancement & Community Council of Nashua (Nashua). Funding: Sponsors and collaborators stated as Dartmouth–Hitchcock Medical Center and National Institute of Mental Health. |
|
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) schizophrenia or schizoaffective disorder and either current cannabis abuse or dependence.
N = 14. All with co‐occurring substance use disorders.
Age: 17 to 45 years. Mean ˜ 22.4 years. 4 participants were 17 years old.;(see amendments to protocol). Sex: 8 M, 6 F History: first episode of schizophrenia, cannabis use within the five weeks prior to recruitment Ethnicity: Caucasian Exclusion: Medical contraindications to treatment with clozapine or risperidone, including previous paralytic ileus. Cumulative treatment with antipsychotic medication in excess of 16 weeks prior to hospital admission (or case identification if an outpatient), unless waived by the medication adjustment group (MAG). History of allergic reaction to clozapine or risperidone. History of seizure disorder or blood dyscrasia. Note: if participants had a history of seizures, but not a diagnosed seizure disorder, they could be admitted to the study if approved by the medication adjustment group. Current treatment with clozapine. Currently pregnant, planning to become pregnant, or unwilling to use an acceptable form of birth control. Currently residing in a residential programme designed to treat substance use disorders. Participants who required treatment at baseline with a psychotropic agent proposed to curtail substance use (e.g. disulfiram, naltrexone, valproic acid, topiramate, acamprosate or benzodiazepines) were reviewed by the medication adjustment group before entering into the study. Participants who, in the opinion of the investigator, are judged unsuitable to participate in the study (for example, are actively homicidal or have a pending incarceration that would prevent them from participating in the study). History of, or current breast cancer. People who are doing well on current therapy. Lack of an identifiable primary family/support person, and unable to come to a study site for weekly visits. Treatment with serotonin re‐uptake inhibitors did not mean exclusion but required a review by the MAG prior to randomisation. Participants with current cocaine dependence required review by the MAG to determine stability for the study.Treatment with multiple antipsychotics or long‐acting injectable antipsychotic at baseline not excluded, but reviewed by the MAG to assess appropriateness for the study. |
|
| Interventions | 1. Clozapine: tablets ‒ 12.5 mg to maximum 100 mg daily for 24 weeks. N = 7 2. Risperidone: tablets ‒ 0.5 mg to maximum 5 mg daily for 24 weeks. N = 7 All participants received a Lifestyle Intervention to manage metabolic side‐effects and to assist with recovery. |
|
| Outcomes | Mental state: worsening of psychotic symptoms, emergence of anxiety symptoms reported as trial adverse events. Substance use: cannabis use (TLFB), urine tests, collateral reports, and monthly clinician ratings, final expert clinician rating ‒ dichotomised Adverse effects: movement disorder, various adverse effects. Leaving the study early. Unable to use: Mental state: (BPRS, CGI, SANS ‒ no data reported). |
|
| Notes | Contact of authors: no response from authors to e‐mails sent requesting clarification on study design and to obtain missing data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given as to how sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No details given as to whether allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study described as single‐blind with outcome assessors blind. Knowledge of treatment allocation and monitoring procedures of clozapine could have influenced participants or personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Outcome assessors described as masked, but no mention is made of whether the outcome assessors were indeed blinded and independent. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | One participant in both groups did not receive treatment but were randomised and were not included in the outcome report. "About half of participants in the clozapine group have discontinued treatment early, a rate similar to previous first episode schizophrenia studies in the US. There were no discontinuations due to lack of efficacy in either group, but several discontinuations due to inability to tolerate medication side‐effects in clozapine group". It is also mentioned that 2 participants in risperidone group terminated early and 2 study completers in the risperidone group elected to discontinue medication and 1 to switch to a different antipsychotic at the end of the study. |
| Selective reporting (reporting bias) | High risk | The manner in which the primary outcome was determined changed in later versions of the protocol (from 2007 to following completion of data collection in 2011, with a change in 2013). The initial outcome was marijuana use measured weekly by means of TLFB method, but this changed to improvement as judged by experts at a particular cut‐point of 20% improvement and then dichotomised, assessed at the end of the study. |
| Other bias | Unclear risk | Study did not receive funding from pharmaceutical industry and principal Investigators are not employed by the organization sponsoring the study. Declarations of interest made by researchers conducting this study: "Principal Investigators are not employed by the organization sponsoring the study". |
Sevy 2011.
| Methods | Allocation: randomised.
Blindness: single‐blind (outcome assessor).
Duration: 4 months' acute treatment phase of 32 month study.
Design: secondary data analysis of existing superiority, parallel group, randomised trial.
Setting: all new patients referred to acute care at the Zucker Hillside, Bronx Lebanon Hospital were screened. Funding: National Institutes for Health, Feinstein Institute for Medical Research |
|
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) diagnosis of current schizophrenia, schizophreniform disorder, or schizoaffective disorder and a lifetime or current 3 months' history of cannabis abuse or dependence.
N = 49 (post‐hoc analysis of a subgroup of 49 patients with co‐occurring substance use disorders from larger study of 120 participants). Age: range 16 to 40 years; mean ˜21.7 years (risperidone), mean ˜21.7 years (olanzapine). Sex: 40 (81.6%) M, 9 (18.4%) F. Ethnicity: not stated. Inclusion criteria: Less than 12 weeks of lifetime antipsychotic medication treatment. Current positive symptoms evidenced by a rating of 4 or more on the severity of delusions, hallucinations, or thought disorder items of the Schedule for Affective Disorders and Schizophrenia Change Version with psychosis and disorganization items (SADS‐C+PD) or current negative symptoms demonstrated by a rating of 4 or more on the affective flattening, alogia, avolition, or anhedonia global items of the Hillside Clinical Trials version of the Scale for Assessment of Negative Symptoms (SANS). For women, a negative pregnancy test and agreement to use a medically accepted method of birth control. Competent and willing to sign informed consent. Exclusion criteria: 1) meeting DSM‐IV criteria for a current substance‐induced psychotic disorder, psychotic disorder due to a general medical condition, or mental retardation; 2) medical condition/treatment known to affect the brain; 3) any medical condition requiring treatment with a medication with psychotropic effects; 4) medical contraindications to treatment with olanzapine or risperidone; or 5) significant risk of suicidal or homicidal behaviour. |
|
| Interventions | Risperidone: mean modal daily dose 4 mg. N = 21 Olanzapine: mean modal daily dose 15 mg. N = 28 All participants received psychoeducation about schizophrenia, were seen on a regular basis by allocated social workers and also had access to the ancillary treatment service available from 2 large departments of psychiatry. |
|
| Outcomes | Mental state: positive psychotic symptoms ‒ average endpoint scores (SADS‐C‐PD scale, lower = better), negative symptoms (SADS‐C‐PD scale, lower = better). Substance use: stopped using cannabis (Urine testing and Substance Use Questionnaire). Substance use: stopped using alcohol (Substance Use Questionnaire). Leaving the study early. |
|
| Notes | Authors e‐mailed for additional data and information: no response. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer pre‐generated block randomization list provided by the department of biostatistics and only accessible to the biostatisticians and dedicated research coordinators. |
| Allocation concealment (selection bias) | Unclear risk | Unclear whether "research coordinators" were involved in patient recruitment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label, both patients and staff were aware of treatments received. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Diagnosis and psychopathology assessments were performed by masked ("blind") assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Although attrition was equal across groups (approximately 25%) no method of accounting for missing outcomes was present, i.e. ITT analysis with imputation. |
| Selective reporting (reporting bias) | Unclear risk | Secondary data analysis directed by analysis of primary study. Not clear if this could have influenced selection of outcomes. |
| Other bias | Unclear risk | Several authors have ties to drug companies; however unclear whether this could have an impact on the results as the parent study was supported by NIH grants K23 DA015541 (SS), MH60004 (DR), MH41960, and RR018535. It is stated that the NIH had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. |
Smelson 2006.
| Methods | Allocation: randomised. Blindness: open‐label trial, not blinded Duration: 12 months Design: secondary data analysis of subgroup with substance use from existing superiority, parallel group, randomised trial. Setting (parent study): academic and community treatment settings mainly outpatient clinics, USA. Multicenter trial. | |
| Participants | Diagnosis: DSM‐IV diagnosis of schizophreniform, schizophrenia or schizoaffective disorder, illicit drug or alcohol use 30 days prior to study entry as measured by the quantity/frequency sub‐scale of the Addiction Severity Index. N = 664 (236 with analysable data were substance users). Age: > 18 years; mean age ˜43 years. Sex (parent study): 420 (63%) M, 244 (37%) F Ethnicity (parent study): Caucasian 361 (54%), African American 224 (34%), Other 79 (12%) Inclusion: psychotic symptom threshold of 18 or more on the Brief Psychiatric Rating Scale (BPRS). Individuals recently experiencing an adverse event attributable to current antipsychotic treatment (unless olanzapine or risperidone) were also eligible, although the vast majority met symptom criteria. Exclusion criteria: patients with very serious, unstable physical illnesses and other medical conditions or histories contraindicating use of any study medication. |
|
| Interventions | Risperidone: suggested initiating dose 1 mg twice daily with flexible dosing and titration by study clinicians. N = 76 Olanzapine: suggested initiating dose 10 mg daily with flexible dosing and titration by study clinicians. N = 85 Conventional antipsychotics: 2 conventional agents (from: perphenazine, loxapine, haloperidol, fluphenazine, thiothixene) for minimum of 8 weeks consecutively as decided by study physicians based on prior history. N = 75 It is unclear what psychosocial interventions participants received. |
|
| Outcomes | Unable to use: Time to discontinuation (skewed data) Numbers discontinuing treatment (no data) |
|
| Notes | Authors e‐mailed for additional data and information: no response received | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description of how sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No description of how allocation sequence was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Treatment was described as open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No description of blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary outcome and only outcome reported is time to all‐cause medication discontinuation, including leaving study for any reason. |
| Selective reporting (reporting bias) | Unclear risk | Secondary data analysis of existing trial. Analysis for primary trial not fully reported so unclear if this informed the aims and hypothesis of the secondary data analysis. |
| Other bias | High risk | Several authors have relationships with the pharmaceutical industry. Parent study funded by Eli Lilly. |
Swartz 2008.
| Methods | Allocation: randomised Blindness: double‐blind Duration: 18 months Design: secondary data‐analysis of subgroup with and without substance use from an existing superiority, parallel group, randomised trial. Setting (parent study): multicenter trial, 57 sites in USA. | |
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) diagnosis of schizophrenia, with past history of more than one episode. Alcohol and illicit drug use was determined by a combination of self‐reported use, SCID‐I interviews, urine and hair samples, ratings on Clinician Alcohol and Drug Use Scale. N = 1432 cases available from the parent study for analysis; 643 were substance users. Age: 18 to 65 years; substance user group mean age ˜38.1 years, non‐substance user group mean age ˜42.6 years. Sex: 1062 (74.1%) M, 370 (25.8%) F. Ethnicity: White 722 (50.4%), Non‐white 710 (49.5%). Inclusion: multi‐episode schizophrenia with or without illicit substance use disorder. Exclusion criteria: People with schizoaffective disorder, mental retardation or other cognitive disorders. A history of serious adverse reactions to the proposed treatments. Patients with only 1 schizophrenic episode or a history of treatment resistance, including non‐response to one of the proposed treatments or prior treatment with clozapine. Pregnant, breast‐feeding or presence of an unstable medical condition. |
|
| Interventions | Risperidone: flexible dosing, allowable daily dose 1.5 mg to 6 mg, mean dose 3.8 mg/day. N = 157 Olanzapine: flexible dosing, allowable daily dose 7.5 mg to 30 mg, mean dose 20.0 mg/day. N = 142 Perphenazine: flexible dosing, allowable daily dose 8 mg to 32 mg, mean dose 20.4 mg/day. N = 124 Quetiapine: flexible dosing, allowable daily dose 200 mg to 800 mg, mean dose 515.1 mg/day. N = 137 Ziprasidone: flexible dosing, allowable daily dose 40 mg to 160 mg, mean dose 113.3 mg/day. N = 83 The investigators did not account for substance abuse treatments received, but they noted that very few were actively engaged in such treatments. |
|
| Outcomes | Leaving the study early (any reason) Unable to use: Mental state: psychotic symptoms, positive psychotic symptoms, negative symptoms and general psychopathology (PANSS total and sub‐scales) (N and SD not available) Clinical Global Impression of Severtiy of illness (CGI‐severity) (N and SD not available) Readmission rate (no data) Adherence to antipsychotic medication (no SD) Adverse events: weight gain (no data), neurological side‐effects (no data) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised but no details given as to how the sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No description of how sequence was kept concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study described as double blind with identically appearing capsules. Different medications had different side‐effect profiles and some overlap in side‐effect profiles for some medications (i.e. weight gain and sedation). Unclear if this could have favoured one or more medications over others. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No clear description of how blinding was maintained. Different side‐effect profiles may have unblinded medication and symptom severity ratings could have been influenced by this. Nevertheless, outcomes such as discontinuation would unlikely have been affected by blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High attrition rates with up to 69% of risperidone patients discontinuing treatment and high numbers (56% to 81%) in other treatment groups. Simple imputation with LOCF was used which could have biased results given such a large attrition rate. This would however not have affected the outcome of time to medication all‐cause discontinuation (as all patients were counted for medication discontinuation outcome), but could have impacted on measurement of mental state (i.e. LOCF imputations). |
| Selective reporting (reporting bias) | Unclear risk | The results from earlier analysis directed the current hypothesis in this study. Nevertheless the outcomes (time to all‐cause discontinuation), were similar to the original study protocol. It is unclear how earlier analyses could have directed results. |
| Other bias | Unclear risk | Several pharmaceutical companies provided medication for the study. A number of authors had ties to the pharmaceutical industry. The NIMH was responsible for the study was design, data collection, analysis, writing up and decision to publish the study. |
van Nimwegen 2008.
| Methods | Allocation: randomised Blindness: double‐blind Duration: 6 weeks Design: subgroup with substance (cannabis) reported, superiority, parallel group, randomised trial. Setting: outpatients. Multisite across 4 sites in the Netherlands. (Academic Medical Centre University of Amsterdam, Erasmus Medical Centre Rotterdam, Panassia Psychomedical Centre in the Hague, Mediant in Enschede). | |
| Participants | Diagnosis: Structured Clinical Interview for DSM (SCID‐I) diagnosis of schizophreniform disorder, schizophrenia or schizoaffective disorder, cannabis self‐report and urine testing for cannabis. N = 138 (subgroup of 41 (29.7%) used cannabis). Age: 18 to 30 years, mean age ˜25 years Sex: 80% male. Ethnicity: not reported Exclusion criteria: pregnant or lactating, no adequate contraception, known hypersensitivity to any ingredient of olanzapine or risperidone. Concomitant use of any other antipsychotic drug than olanzapine or risperidone. Use of depot anti‐psychotics for a period of at least three months prior to the study or the use of other psychotropic medication other than oxazepam or biperiden. Narrow‐angle glaucoma, neurological or endocrine disease. |
|
| Interventions | Risperidone: flexible dosing, 1.25 mg, 2.5 mg, 3.75 mg, 5 mg, titrated to a fixed dose within the first week. N = 21 Olanzapine: flexible dosing, 5 mg, 10 mg, 15 mg, 20 mg, titrated to fixed dose within first week. N = 20 |
|
| Outcomes | Substance use: cannabis use self‐report scores ‒ change data (joints per week) Craving for substances: Obsessive Compulsive Drug Use Scale (OCDUS), Desires for Drug Questionairre (DDQ) ‒ endpoint data. Leaving the study early Unable to use: Subjective Well‐being: Subjective Well‐being Under Neuroleptics (SWN) score (no subgroup mean, SD or N) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description of how sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No description of where sequence was kept and who allocated participants. Nevertheless tablets were described as identical‐looking. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study described as "double blind" with identically appearing capsules, although no description is given as to how blinding was achieved. Different side‐effect profiles of the two medications could have lead to unblinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as "double blind" with identically appearing capsules, although no description is given as to how outcome assessors were kept masked from treatment. Different side‐effect profiles of the two medications could have lead to unblinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis with single imputation method (LOCF). Although attrition was comparable across groups it is unclear if groups differed with regards to other factors such as symptoms severity and other baseline measures. |
| Selective reporting (reporting bias) | Unclear risk | Only some outcomes stated in protocol are reported. Unclear how some factors, such as symptom severity that was not reported, could have impacted on reported outcomes of SWN and craving. |
| Other bias | High risk | Study funded by Eli‐Lilly |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Blin 1996 | Allocation: randomised Participants: people with schizophrenia who do not have co‐occurring substance misuse. |
| Gaebel 2010 | Allocation: randomised Participants: people with schizophrenia, schizoaffective disorder; people with schizophrenia who do not have co‐occurring substance misuse. |
| Green 2001 | Allocation: randomised. Participants: patients with both schizophrenia and a cannabis use disorder. Intervention: risperidone vs. clozapine. Outcomes: cannabis use, negative symptoms, psychotic symptoms, neuropsychological function and quality of life. No data available: only published as study protocol, authors contacted for unpublished data: no response. |
| Harvey 2007 | Allocation: randomised. Participants: people with bipolar I disorder who do not have co‐occurring substance misuse. |
| Ikuta 2014 | Allocation: randomised Participants: people with schizophrenia, schizophreniform, or psychosis not otherwise specified who do not have co‐occurring substance misuse. |
| Kerfoot 2011 | Allocation: randomised.
Participants: schizophrenia with and without co‐occurring substance use.
Intervention: risperidone versus quetiapine versus perphenazine versus olanzapine versus ziprasidone.
Outcomes: psychotic symptoms, depression, quality of life, neurocognition No data available comparing risperidone with other medications. Study examined the impact of substance use on prognosis. |
| Liemburg 2011 | Allocation: randomised. Participants: people with schizophrenia who do not have co‐occurring substance misuse. |
| Liu 2008 | Allocation: randomised. Participants: not people with dual diagnosis ‒ people with 'mental disorders due to alcohol use'. |
| NCT00063349 | Allocation: trial suspended, reported as non‐randomised, retrospective observational study. Participants: schizophrenia, schizoaffective disorder, cannabis and/or alcohol use disorder Intervention: risperidone, clozapine. Outcomes: cessation of substance use. |
| NCT00130923 | Allocation: randomised Participants: schizophrenia, schizoaffective disorder and alcohol use disorder (abuse or dependence) Intervention: risperidone oral formulation compared to risperidone long‐acting injectable formulation (RLAI). Not risperidone versus another antipsychotic. |
| NCT00169026 | Allocation: randomised
Participants: schizophrenia, schizoaffective disorder and alcohol or substance use disorder.
Intervention: clozapine, conventional antipsychotics, atypical antipsychotics.
Outcomes: substance and alcohol use (breathalyzer, urine tests, TLFB, Alcohol and Drug Use Scale), mental state (BPRS), SANS, CGI, neurological side‐effects, cognitive function, quality‐of‐life measure No data available. study protocol of terminated study. |
| NCT00498550 | Allocation: randomised
Participants: schizophrenia, schizoaffective disorder and cannabis use disorder
Intervention: clozapine, conventional antipsychotics, atypical antipsychotics.
Outcomes: substance use measures (urine testing, breathalyzer), medication side‐effects, physical and psychological symptoms, substance use, treatment services received, and living situation, quality of life. Study protocol only, authors contacted but no data provided. |
| Nejtek 2008 | Allocation: randomised Participants: bipolar I and II disorder, recent manic or mixed episode with or without psychosis and with co‐occurring cocaine‐ or methamphetamine‐use disorder. Only 8.3% of total sample had psychotic features and 15.9% had bipolar type II disorder. |
| Perlis 2006 | Allocation: randomised Participants: bipolar I disorder with mania or mixed states. Patients with psychosis excluded. Patients with recent substance use excluded. |
| Rezayat 2014 | Allocation: randomised Participants: acute mania (bipolar disorder). Study excludes participants with drug or alcohol use in past 3 months |
| Rubio 2006a | Allocation: quasi‐randomisation (participants allocated "alternately"). |
| Rubio 2006b | Allocation: quasi‐randomisation (participants allocated "alternately"). |
| Sachs 2002 | Allocation: randomised Participants: bipolar with current manic or mixed episode. Study excludes participants with drug or alcohol in past 1 months. |
| Sajatovic 2002 | Allocation: randomised
Participants: psychotic disorders: schizoaffective disorder, bipolar I disorder, major depressive disorder, delusional disorder, Alzheimer's dementia, schizophreniform disorder, vascular dementia, and substance abuse dementia.
Intervention: risperidone, quetiapine
Outcomes: psychotic symptoms (PANSS), depression (HAM‐D), extrapyramidal symptoms (ESRS) No subgroups with substance use reported, authors contacted for unpublished data, no response. |
| Smulevich 2005 | Allocation: randomised Participants: bipolar I disorder who do not have recent drug or alcohol use. |
| van Nimwegen 2008a | Allocation: randomised Participants: schizophrenia, schizophreniform, schizoaffective disorder. No co‐occurring substance use disorders. Intervention: haloperidol, risperidone, placebo Outcomes: Obsessions and compulsions (Y‐BOCS), PANSS scores, CDSS scores. Authors contacted to determine if there were participants with co‐occurring substance use disorders. Authors clarified that there were no participants with co‐occurring substance use disorders. |
| Yatham 2007 | Allocation: randomised Participants: bipolar I and II. Excludes participants with drug or alcohol use in past 3 months. |
| Zhangyue 2005 | Allocation: quasi‐randomisation (allocation based on admission order) |
Characteristics of studies awaiting assessment [ordered by study ID]
Greenspan 2005.
| Methods | Allocation: described as "double‐blind" * Blindness: described as "double‐blind" * Duration: 6 weeks, 2‐week monotherapy phase Design: "double‐blind" efficacy study Setting: unclear |
| Participants | Patients with schizophrenia and co‐occurring alcohol, cocaine, amphetamine, marijuana, opiate use disorder. N = 111 with substance use disorders |
| Interventions | Risperidone; (dose and delivery method unclear) N = 51. Quetiapine; (dose and delivery method unclear ) N = 40. Placebo. N = 20** |
| Outcomes | Mental state: psychotic symptoms, PANSS scale |
| Notes | * Randomisation could not be confirmed from authors, no response to e‐mails sent. ** Data from placebo group not used for this review. |
Johnsen 2010.
| Methods | Allocation: randomised, rater blinded, prospective head‐to‐head trial |
| Participants | Participants: schizophrenia, schizoaffective disorder, delusional disorder, affective psychosis (supplementary data with sample characteristics indicate that 3.8% of risperidone group had alcohol use disorder at baseline and 21.2% of risperidone group had drug misuse at baseline). |
| Interventions | risperidone, clinician determined dose, N = 53 (2 alcohol misuse in past 6 months, 11 drug misuse in past 6 months) olanzapine, clinician determined dose, N = 52 (5 alcohol misuse in past 6 months, 9 drug misuse in past 6 months) quetiapine, clinician determined dose, N = 50 (10 alcohol misuse in past 6 months, 7 drug misuse in past 6 months) ziprasidone, clinician determined dose, N = 58 (5 alcohol misuse in past 6 months, 11 drug misuse in past 6 months) |
| Outcomes | Outcomes: time to antipsychotic discontinuation, discharge and readmission. Improvement in PANSS, Calgary Depression Scale for Schizophrenia, CGI‐S, GAF,adverse effects, UKU Side Effect Rating Scale (UKU‐SERS). Baseline, 6 weeks, 3‐, 6‐, 12‐ and 24‐month measures. |
| Notes | Authors contacted for any subgroup data or analysis, no response to e‐mails sent. |
NCT00208143.
| Methods | Open (no masking), randomised, parallel assignment, superiority trial |
| Participants | Adults age 19 to 65 years with a diagnosis of schizophrenia or schizoaffective disorder and co‐occurring cocaine or methamphetamine abuse or dependence as diagnosed by Structured Clinical Interview for DSM‐IV. |
| Interventions | quetiapine or risperidone oral formulation |
| Outcomes | Primary: 50% or greater decrease in the drug use determined by the Time Line Follow Back (TLFB) method versus baseline Secondary: psychiatric symptoms assessed with the CGI, PANSS, BPRS, HAM‐D, and HAM‐A. Safety and tolerability assessed by patient‐ and physician‐reported adverse events and AIMS. Quality of life assessed with QoLI. |
| Notes | Authors were contacted via e‐mail but no response received. |
San 2012.
| Methods | Allocation: randomised |
| Participants | Participants: schizophrenia, schizophreniform, schizoaffective, bipolar, psychotic disorder NOS. Substantial subgroup used substances (cannabis: N = 64, 56.1%; alcohol: N = 87, 76.3%; cocaine: N = 24, 21.1%). |
| Interventions | Open‐label flexible‐doses of antipsychotic treatment with the following dose ranges:
haloperidol 1.5 mg to 8.5 mg, N = 21 (cannabis = 14, alcohol = 17, cocaine = 7) olanzapine 7.5 mg to 40 mg, N = 25 (cannabis = 15, alcohol = 19, cocaine = 4) risperidone 1.5 mg to 7.0 mg, N = 25 (cannabis = 14, alcohol = 17, cocaine = 5) quetiapine 100 mg to 1500 mg, N = 23 (cannabis = 11, alcohol = 17, cocaine = 2) ziprasidone 40 mg to 240 mg, N = 20 (cannabis = 10, alcohol = 17, cocaine = 6) |
| Outcomes | Time to medication discontinuation, PANSS scores, CDSS scores, Adverse effects |
| Notes | No data provided for substance misuse subgroup ‒ authors contacted and responded, no data provided. |
Yatham 2003.
| Methods | Allocation: multicentre, randomised, placebo‐controlled trial Blindness: described as "double blind" Duration: 52 weeks Setting: Canadain and Brazilian academic centres |
| Participants | Bipolar I disorder in remission from recent manic or mixed episode on treatment with mood stabiliser (valproate or lithium) and either risperidone or olanzapine (N = 159, not clear how many had psychotic features). Total of 39% (62/159) of total sample had co‐occurring alcohol or substance use disorder. |
| Interventions | Discontinuation of risperidone or olanzapine at either 0 weeks, 24 weeks or 52 weeks and substitution with placebo. |
| Outcomes | Time to any mood episode, YMRS, HAMD‐21, MADRS, CGI‐BP, CGI‐S, Side‐effects UKU scale, ESRS, weight, metabolic measures (glucose, lipid profile). |
| Notes | Authors contacted. Responded that no data or analyses available at present for subgroups. No information provided on how many participants had bipolar type I with psychotic features. |
Characteristics of ongoing studies [ordered by study ID]
NCT01639872.
| Trial name or title | Clozapine for Cannabis Use in Schizophrenia (CLOCS) |
| Methods | Double blind (subject, caregiver, investigator, outcomes assessor), randomised, parallel assignment, superiority trial, comparing the efficacy of clozapine with risperidone, Estimated recruitment target N = 132 |
| Participants | Adults 18 to 55 years, males and females, clinical diagnosis of schizophrenia and a co‐occurring cannabis use disorders (abuse or dependence) |
| Interventions | clozapine with target dose of 400 mg/day and maximum of 550 mg/day; risperidone with target dose of 4 mg/day and maximum of 6 mg/day |
| Outcomes | Primary: intensity (amount of cannabis used); frequency (number of days in past week) Secondary: symptoms of schizophrenia as measured by the BPRS, SANS, CGI; neuropsychological function by means of MATRICS Consensus Cognitive Battery; and reward responsiveness by means of a computerised Probablistic Reward Task. |
| Starting date | April 2013 |
| Contact information | alan.i.green@dartmouth.edu; christopher.okeefe@dartmouth.edu |
| Notes | Estimated completion in Oct 2016 (recruitment); Oct 2017 (results) |
Differences between protocol and review
The search revealed very few studies that met inclusion criteria as per the original protocol. This was due to the original protocol requiring that all participants randomised had to have both a severe mental illness and a substance use disorder and that the primary pre‐specified study aims should be to examine the efficacy of risperidone against a comparator drug. Studies that included subgroup and post hoc, secondary data analyses were also excluded.
Our original protocol followed this approach due to a concern that amongst studies that randomised mixed populations (single and dual diagnosis patients), the analysis of a subgroup with particular characteristics may lead to the selection of participants with certain prognostic features that could potentially effectively undo the effect of balancing prognostic factors between the intervention groups during randomisation. This may be of particular concern where allocation concealment was not ensured, leading to selection bias in the allocation of interventions where some participants but not others were using substances.
In the case of subgroup analysis in individual studies or secondary data analysis of subgroups in existing trials, we initially aimed to exclude all such studies as secondary analyses may have potentially been motivated by data‐driven post hoc hypothesis testing of non‐prespecified subgroups, leading to potentially spurious results, effectively selecting groups with specific prognostic features, interfering with randomisation and rendering such studies observational in nature (Sun 2014; Wang 2007).
Therefore, due to the paucity of studies in this area and because the implications of including such studies in reviews have not been empirically tested (it could be argued that randomisation still applies to participants in studies using subgroups and secondary analyses), we decided to include studies with a mixed population and subgroup and secondary analyses using a set of modified inclusion criteria.
To include RCTs reporting on outcomes where 100% of participants have BOTH a severe mental illness AND co‐occurring substance use disorder.
For studies that specifically examined the efficacy of risperidone in dual diagnosis, but that included mixed populations in randomisation: to include RCTs where ≥ 70% of participants had BOTH a severe mental illness AND co‐occurring substance use disorder. For these studies we used data from all participants as if they had a dual diagnosis.
In those RCTs where outcomes are reported for sub‐groups (i.e. secondary data analyses of existing data) with both severe mental illness and co‐occurring substance use, we reported on these and added only the relevant sub‐group into the meta‐analysis if possible.
In addition we will note the following.
In case RCTs randomising mixed populations, we highlighted whether such studies reported adequate allocation concealment procedures.
For secondary and subgroup analyses, we systematically recorded whether participants were randomised to risperidone or comparator following stratification into groups with and without co‐occurring substance use disorder either in the study or parent study.
In case of studies conducting subgroup analyses or secondary data analyses of subgroups, we recorded whether this analysis was pre‐specified in the original study protocol, and if not, rated these studies at potential high risk of selective outcome reporting by virtue of their design.
For data extraction of studies of mixed populations and subgroups or secondary analysis of subgroups, we extracted categorical or continuous data at study endpoint or change data within subgroups for risperidone and comparator drugs.
Should meta‐analyses have been possible, we would have examined the impact of excluding "lower quality studies" from the analysis.
Other minor changes
One minor change was the inclusion of one study that included a few participants younger than 18 years (age inclusion criteria 17 to 45 years).
We also included participants if they happened to be smokers, despite the protocol stating we would exclude nicotine dependence. We did, however, exclude studies where the substance use disorder under study was nicotine dependence or where investigators looked at the impact of antipsychotic treatments on smoking in particular.
As the review yielded only few studies with even lower numbers of studies providing data for different outcomes we were unable to conduct any planned funnel plots to detect publication bias, subgroup analyses, or sensitivity analyses.
Cochrane guidelines now recommend fewer primary outcomes and at least one primary outcome to be an adverse effect. We specified fewer mental state outcomes as primary outcomes and added incidence of adverse effect to be a primary outcome. We have reworded outcomes such as 'significant improvement' or 'significant response' to current wording of 'clinically important change'. The types of outcome specified as important have not changed.
Contributions of authors
Henk Temmingh: protocol development, searching, study selection, data extraction, data analysis, report writing.
Taryn Williams: protocol development, study selection, data extraction, comments and editing of final report.
Nandi Siegfried: protocol development, study selection, data extraction, comments and editing of final report.
Dan Stein: protocol development, comments and editing of final report.
Sources of support
Internal sources
-
South African Cochrane Centre, South Africa.
For their assistance and support during the protocol development
-
Department of Psychiatry and Mental Health, University of Cape Town, South Africa.
Dr Temmingh is a lecturer on joint University of Cape Town and Western Cape Department of Health conditions of employment. Dr Siegfried, Dr Amos and professor Stein are supported by the Department of Psychiatry and Mental Health, University of Cape Town.
-
Western Cape Department of Health, Valkenberg hospital, Cape Town, South Africa.
Dr Temmingh is on joint University of Cape Town and Department of Health conditions of employment and is salaried by the Western Cape Department of Health
-
Harry Crossley Foundation, South Africa.
Dr Temmingh is the recipient of the 2013 Harry Crossley Senior Clinical Research Fellowship. He received support from this fellowship for travel and subsistence during his sabbatical leave.
-
Goldman Trust, South Africa.
Dr Temmingh received a travel grant to attend the Cochrane workshop at the London School of Hygiene and Tropical medicine in 2011
External sources
No sources of support supplied
Declarations of interest
Henk Temmingh has received a speaker honorarium from Pharma Dynamics and was a recipient of the Harry Crossley Clinical Fellowship that provided support for travel and subsistence during his sabbatical.
Taryn Williams has no known conflicts of interest outside her employment by the MRC Unit on Anxiety and Stress Disorders.
Nandi Siegfried provides technical consultation on the efficacy of drugs at the request of a managed care organization, and receives an honorarium. Nandi has not appraised risperidone in this work.
Dan Stein has received research grants or consultancy honoraria (or both) from AstraZeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Orion, Pfizer, Pharmacia, Roche, Servier, Solvay, Sumitomo, and Wyeth.
Edited (no change to conclusions)
References
References to studies included in this review
Akerele 2007 {published data only (unpublished sought but not used)}
- Akerele E, Levin FR. Comparison of olanzapine to risperidone in substance‐abusing individuals with schizophrenia. American Journal on Addictions 2007;16(4):260‐8. [DOI] [PubMed] [Google Scholar]
- Akerele EO, Biderman L, Levin FR. A pilot study of olanzapine/risperidone for the treatment of cocaine/marijuana use disorder in individuals with schizophrenia. 64th Annual Scientific Meeting of the College on Problems of Drug Dependence;2002 Jun 8‐13, Quebec, Canada. 2002.
- Akerele EO, Gill KB, Vosburg SK, Levin FR. Efficacy of atypical neuroleptics in treatment of substance use among schizophrenic individuals. Drug and Alcohol Dependence 2002;66(Suppl 1):S4. [Google Scholar]
Brunette 2011 {published and unpublished data}
- Brunette M, Dawson R, O'Keefe CD, Noordsy DL, Green A. Clozapine versus other antipsychotics for schizophrenia and co‐occurring cannabis use disorder. Schizophrenia Bulletin 2011;37(suppl_1):297. [Google Scholar]
- Brunette MF, Dawson R, O'Keefe CD, Narasimhan M, Noordsy DL, Wojcik J, et al. A randomized trial of clozapine versus other antipsychotics for cannabis use disorder in patients with schizophrenia. Journal of Dual Diagnosis 2011;7(1‐2):50‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00149955. Cannabis and schizophrenia: effects of clozapine. www.ClinicalTrials.gov/ct/show/ (Accessed 06 January 2016).
Machielsen 2014 {published data only}
- Machielsen M, Haan L. Differential effect of clozapine and risperidone on craving related brain activity in patients with schizophrenia and cannabis use disorder. Schizophrenia Bulletin 2011;37(suppl_1):145. [Google Scholar]
- Machielsen MW, Veltman DJ, Brink W, Haan L. Comparing the effect of clozapine and risperidone on cue reactivity in male patients with schizophrenia and a cannabis use disorder: A randomized fMRI study. www.sciencedirect.com/science/article/pii/S0920996417301688 (accessed prior to 28 November 2017). [DOI] [PubMed]
- Machielsen MWJ, Veltman DJ, Brink W, Haan L. The effect of clozapine and risperidone on attentional bias in patients with schizophrenia and a cannabis use disorder: an fMRI study. Journal of Psychopharmacology 2014;28(7):633‐642. [DOI] [PubMed] [Google Scholar]
- Haan L, Machielsen MWJ. Effect of different antipsychotic medications on craving and craving related brain activity in patients with schizophrenia and cannabis abuse or dependence: a randomized controlled study comparing clozapine and risperidone. www.trialregister.nl/trialreg/index.asp (Accessed 06 January 2016).
Noordsy 2010 {published and unpublished data}
- NCT00573287. First episode schizophrenia and cannabis‐related disorder study. clinicaltrials.gov/ct2/show/NCT00573287 (Accessed 06 January 2016).
- Noordsy D. First episode schizophrenia and cannabis abuse: is there a role for clozapine?. Proceedings of the 15th Biennial Winter Workshop in Psychoses; 2009 Nov 15‐18; Barcelona, Spain. 2009.
- Noordsy D, Green JA. Clozapine versus risperidone for people with first episode schizophrenia and co‐occurring cannabis use disorder. Schizophrenia Research 2010;117(2‐3):165‐6. [Google Scholar]
- Noordsy DL. Personal Communication. Personal Communication 2012.
Sevy 2011 {published data only}
- Sevy S, Robinson DG, Napolitano B, Gallego J, Patel RC, Soto‐Perello JM, et al. Clinical and substance use outcomes of first‐episode schizophrenia patients with a lifetime diagnosis of cannabis use disorders randomly assigned to risperidone or olanzapine for 16 weeks. Schizophrenia bulletin 2009;35(suppl_1):355‐6. [Google Scholar]
- Sevy S, Robinson DG, Sunday S, Napolitano B, Miller R, McCormack J, et al. Olanzapine versus risperidone in patients with first‐episode schizophrenia and a lifetime history of cannabis use disorders: 16‐week clinical and substance use outcomes. Psychiatry Research 2011;188(3):310‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smelson 2006 {published data only}
- Smelson D, Nyhuis AW, Faries DE, Tunis SL. Impact of substance use on attrition and medication switching in an effectiveness trial for individuals with schizophrenia: implications for relapse prevention. The American Psychiatric Association, 158th Annual meeting, Psychosomatic medicine: Integrating psychiatry and medicine; 2005 May 21‐26; Atlanta, Georgia. Arlington, VA: American Psychiatric Association, 2005.
- Smelson D, Nyhuis AW, Faries DE, Tunis SL, Ascher‐Svanum H, Montgomery WS. Impact of substance use on attrition and medication switching in an effectiveness trial for individuals with schizophrenia. Australian and New Zealand Journal of Psychiatry 2005;39(2 Suppl):A60. [Google Scholar]
- Smelson DA, Tunis SL, Nyhuis AW, Faries DE, Kinon BJ, Ascher‐Svanum H. Antipsychotic treatment discontinuation among individuals with schizophrenia and co‐occurring substance use. Journal of Clinical Psychopharmacology 2006;26(6):666‐7. [DOI] [PubMed] [Google Scholar]
Swartz 2008 {published data only}
- Swartz MS, Wagner HR, Swanson JW, Stroup TS, McEvoy JP, Reimherr F, et al. The effectiveness of antipsychotic medications in patients who use or avoid illicit substances: results from the CATIE study. Schizophrenia Research 2008;100(1‐3):39‐52. [DOI] [PubMed] [Google Scholar]
van Nimwegen 2008 {published data only}
- Nimwegen L, Haan L, Beveren N, Laan W, Brink W, Linszen D. Subjective well‐being and craving for cannabis in first psychosis, a randomized double blind comparison of olanzapine versus risperidone. Schizophrenia Research 2006;81(supplement):141. [Google Scholar]
- Nimwegen LJ, Haan L, Beveren NJM, Helm M, Brink W, Linszen D. Effect of olanzapine and risperidone on subjective well‐being and craving for cannabis in patients with schizophrenia or related disorders: a double‐blind randomized controlled trial. Canadian Journal of Psychiatry. Revue Canadienne de Psychiatrie 2008;53(6):400‐5. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Blin 1996 {published data only}
- Blin O, Azorin JM, Bouhours P. Antipsychotic and anxiolytic properties of risperidone, haloperidol, and methotrimeprazine in schizophrenic patients. Journal of Clinical Psychopharmacology 1996;16(1):38‐44. [DOI] [PubMed] [Google Scholar]
Gaebel 2010 {published data only}
- Gaebel W, Schreiner A, Bergmans P, Arce R, Rouillon F, Cordes J, et al. Relapse prevention in schizophrenia and schizoaffective disorder with risperidone long‐acting injectable vs quetiapine: results of a long‐term, open‐label, randomized clinical trial [NCT00216476]. Neuropsychopharmacology 2010;35(12):2367‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Green 2001 {published data only}
- Green A. Cannabis and schizophrenia‐clozapine vs risperidone. www‐commons.cit.nih.gov/crisp/index.html (Accessed 19 February 2001).
Harvey 2007 {published data only}
- Harvey PD, Hassman H, Mao L, Gharabawi GM, Mahmoud RA, Engelhart LM. Cognitive functioning and acute sedative effects of risperidone and quetiapine in patients with stable bipolar I disorder: a randomized, double‐blind, crossover study. Journal of Clinical Psychiatry 2007;68(8):1186‐94. [DOI] [PubMed] [Google Scholar]
- Hassman H, Glass S, Krefetz D, Pinho M, Ridolfi E, Engelhardt L, et al. Cognitive function and acute sedative effects of risperidone and quetiapine in patients with stable Bipolar I Disorder: a randomized, double‐blind, crossover study. 46th Annual NCDEU (New Clinical Drug Evaluation Unit) Meeting; 2006 June 12‐15. Boca Raton, FL., 2006; Vol. 213.
- Hassman HA, Glass SJ, Engelhart L, Mao L, Rodriguez S, Gharabawi G, et al. Differences in cognitive function and acute sedative effects of risperidone and quetiapine among individuals with stable Bipolar I Disorder. Neuropsychopharmacology 2005;30(Suppl 1):S133‐4. [Google Scholar]
Ikuta 2014 {published data only}
- Ikuta T, Robinson DG, Gallego JA, Peters BD, Gruner P, Kane J, et al. Subcortical modulation of attentional control by second‐generation antipsychotics in first‐episode psychosis. Psychiatry Research 2014;221(2):127‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kerfoot 2011 {published data only}
- Kerfoot KE, Rosenheck RA, Petrakis IL, Swartz MS, Keefe RS, McEvoy JP, et al. Substance use and schizophrenia: adverse correlates in the CATIE study sample. Schizophrenia Research 2011;132(2‐3):177‐82. [DOI] [PubMed] [Google Scholar]
Liemburg 2011 {published data only}
- Liemburg E, Aleman A, Bous J, Hollander K, Knegtering H. An open randomized pilot trial on the differential effects of aripiprazole versus risperidone on anhedonia and subjective well‐being. Pharmacopsychiatry 2011;44(3):109‐13. [DOI] [PubMed] [Google Scholar]
Liu 2008 {published data only}
- Liu ZR, Zhang W, Lin H. A comparative study of risperidone and olanzapine in the treatment of mental disorders due to use of alcohol. Chinese Magazine of Drug Abuse Prevention and Treatment 2008;14(3):138‐9. [Google Scholar]
NCT00063349 {published data only}
- Green AI, Burgess ES, Dawson R, Zimmet SV, Strous RD. Alcohol and cannabis use in schizophrenia: effects of clozapine vs. risperidone. Schizophrenia Research 2003;60(1):81‐5. [DOI] [PubMed] [Google Scholar]
- NCT00063349. Clozapine, cannabis and first episode schizophrenia. www.ClinicalTrials.gov/ct/show/ (Accessed 06 January 2016).
NCT00130923 {published data only}
- Green AI, Brunette MF, Dawson R, Buckley P, Wallace AE, Hafez H, et al. Long‐acting injectable vs oral risperidone for schizophrenia and co‐occurring alcohol use disorder: a randomized trial. Journal of Clinical Psychiatry 2015;76(10):1359‐65. [DOI] [PubMed] [Google Scholar]
- NCT00130923. Risperidone long‐acting for alcohol and schizophrenia treatment (R‐LAST). www.ClinicalTrials.gov/ct/show/ (Accessed 06 January 2016).
NCT00169026 {unpublished data only}
- NCT00169026. Alcoholism and schizophrenia: effects of clozapine. www.ClinicalTrials.gov/ct/show/ (Accessed 06 January 2016).
NCT00498550 {published data only}
- NCT00498550. Cannabis and schizophrenia: effects of clozapine. www.ClinicalTrials.gov/ct/show/ (Accessed 06 January 2016).
Nejtek 2008 {published data only}
- Nejtek VA, Avila M, Chen LA, Zielinski T, Djokovic M, Podawiltz A, et al. Do atypical antipsychotics effectively treat co‐occurring bipolar disorder and stimulant dependence? A randomized, double‐blind trial. Journal of Clinical Psychiatry 2008;69(8):1257‐66. [DOI] [PubMed] [Google Scholar]
Perlis 2006 {published data only}
- Perlis RH, Baker RW, Zarate Jr CA, Brown EB, Schuh LM, Jamal HH, et al. Olanzapine versus risperidone in the treatment of manic or mixed states in bipolar I disorder: a randomized, double‐blind trial. Journal of Clinical Psychiatry 2006;67(11):1747‐53. [DOI] [PubMed] [Google Scholar]
Rezayat 2014 {published data only}
- Rezayat AA, Hebrani P, Behdani F, Salaran M, Marvast MN. Comparison the effectiveness of aripiprazole and risperidone for the treatment of acute bipolar mania. Journal of Research in Medical Sciences 2014;19(8):733‐8. [PMC free article] [PubMed] [Google Scholar]
Rubio 2006a {published data only}
- Rubio G, Marfinez‐Gras I, Ponce G, Lopez‐Munoz F, Alamo C, Jimenez‐Arriero MA, et al. Risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity: a long‐term controlled study. European Neuropsychopharmacology 2005;15(Suppl 3):S476. [Google Scholar]
- Rubio G, Martinez I, Ponce G, Jimenez‐Arriero MA, Lopez‐Munoz F, Alamo C. Long‐acting injectable risperidone compared with zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity. Canadian Journal of Psychiatry 2006;51(8):531‐9. [DOI] [PubMed] [Google Scholar]
Rubio 2006b {published data only}
- Rubio G, Martine I, Recio A, Ponce G, Lopez‐Munoz F, Alamo C, et al. Risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity: a long‐term randomized, controlled, crossover study. European Journal of Psychiatry 2006;20(3):133‐46. [Google Scholar]
- Rubio G, Martinez‐Gras I, Ponce G, Jimenez‐Arriero MA, Lopez‐Munoz F, Alamo C. Long‐acting injectable risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity. 159th Annual Meeting of the American Psychiatric Association; 2006, May 20‐25. Toronto, Canada, 2006. [DOI] [PubMed]
Sachs 2002 {published data only}
- Sachs GS, Grossman F, Ghaemi SN, Okamoto A, Bowden CL. Combination of a mood stabilizer with risperidone or haloperidol for treatment of acute mania: a double‐blind, placebo‐controlled comparison of efficacy and safety. American Journal of Psychiatry 2002;159(7):1146‐54. [DOI] [PubMed] [Google Scholar]
Sajatovic 2002 {published data only}
- Sajatovic M, Mullen JA, Sweitzer DE. Efficacy of quetiapine and risperidone against depressive symptoms in outpatients with psychosis. Journal of Clinical Psychiatry 2002;63(12):1156‐63. [DOI] [PubMed] [Google Scholar]
Smulevich 2005 {published data only}
- Smulevich AB, Khanna S, Eerdekens M, Karcher K, Kramer M, Grossman F. Acute and continuation risperidone monotherapy in bipolar mania: a 3‐week placebo‐controlled trial followed by a 9‐week double‐blind trial of risperidone and haloperidol [NCT00253162]. European Neuropsychopharmacology 2005;15(1):75‐84. [DOI] [PubMed] [Google Scholar]
van Nimwegen 2008a {published data only}
- Nimwegen L, Haan L, Beveren N, Laan W, Brink W, Linszen D. Obsessive‐compulsive symptoms in a randomized, double‐blind study with olanzapine or risperidone in young patients with early psychosis. Journal of Clinical Psychopharmacology 2008;28(2):214‐8. [DOI] [PubMed] [Google Scholar]
Yatham 2007 {published data only}
- Yatham LN, Fallu A, Binder CE. A 6‐month randomized open‐label comparison of continuation of oral atypical antipsychotic therapy or switch to long acting injectable risperidone in patients with bipolar disorder. Acta Psychiatrica Scandinavica 2007;434:50‐6. [DOI] [PubMed] [Google Scholar]
Zhangyue 2005 {published data only}
- Zhangyue B, Liu X. Aripiprazole and risperidone in the treatment of schizophrenia. Chinese Journal of Behavioral Medical Science 2005;14(10):923‐5. [Google Scholar]
References to studies awaiting assessment
Greenspan 2005 {published data only}
- Greenspan A, Kosik‐Gonzalez C, Bossie C, Zhu Y, McLemore J, Gharabawi G. Atypical antipsychotics in patients with schizophrenia and comorbid substance abuse. 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26; Atlanta, Georgia, USA. 2005:Nr279.
Johnsen 2010 {published data only}
- Johnsen E, Kroken RA, Wentzel‐Larsen T, Jorgensen HA. Effectiveness of second‐generation antipsychotics: a naturalistic, randomized comparison of olanzapine, quetiapine, risperidone, and ziprasidone [NCT00932529]. BMC Psychiatry 2010;10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjelby E, Jorgensen H A, Kroken R A, Loberg E M, Johnsen E. Anti‐depressive effectiveness of olanzapine, quetiapine, risperidone and ziprasidone: a pragmatic, randomized trial. BMC Psychiatry 2011;11:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00208143 {unpublished data only}
- NCT00208143. Seroquel (quetiapine) therapy for schizophrenia and schizoaffective disorders and comorbid cocaine and/or amphetamine abuse/dependence: a comparative study with risperidone. clinicaltrials.gov/show/NCT00208143 (Accessed 06 January 2016).
San 2012 {published data only}
- San L, Arranz B, Perez V, Safont G, Corripio I, Ramirez N, et al. One‐year, randomized, open trial comparing olanzapine, quetiapine, risperidone and ziprasidone effectiveness in antipsychotic‐naive patients with a first‐episode psychosis. Psychiatry Research 2012;200(2‐3):693‐701. [DOI] [PubMed] [Google Scholar]
Yatham 2003 {published data only}
- Yatham L N, Beaulieu S, Schaffer A, Kauer‐Sant'Anna M, Kapczinski F, Lafer B, et al. Optimal duration of risperidone or olanzapine adjunctive therapy to mood stabilizer following remission of a manic episode: A CANMAT randomized double‐blind trial. Mol Psychiatry 2016;21(8):1050‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatham L, et al. Atypical antipsychotics for continuation and maintenance treatment after an acute manic episode. clinicaltrials.gov/show/NCT01977300 2003.
References to ongoing studies
NCT01639872 {unpublished data only}
- NCT01639872. Clozapine for cannabis use in schizophrenia (CLOCS). clinicaltrials.gov/ct2/show/NCT01639872 (Accessed 06 January 2016).
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1982
- Andreasen NC. Negative symptoms in schizophrenia. Definition and reliability. Archive of General Psychiatry 1982;39(7):784‐8. [DOI] [PubMed] [Google Scholar]
Ascher‐Svanum 2006
- Ascher‐Svanum H, Zhu B, Faries D, Lacro JP, Dolder CR. A prospective study of risk factors for nonadherence with antipsychotic medication in the treatment of schizophrenia. Journal of Clinical Psychiatry 2006;67(7):1114‐23. [DOI] [PubMed] [Google Scholar]
Baker 2010
- Baker AL, Hides L, Lubman DI. Treatment of cannabis use among people with psychotic or depressive disorders: a systematic review. Journal of Clinical Psychiatry 2010;71(3):247‐54. [DOI] [PubMed] [Google Scholar]
Baker 2012
- Baker AL, Thornton LK, Hides L, Dunlop A. Treatment of cannabis use among people with psychotic disorders: a critical review of randomised controlled trials. Current Pharmaceutical Design 2012;18(32):4923‐37. [DOI] [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [PUBMED: 2574607] [DOI] [PubMed] [Google Scholar]
Barnett 2007
- Barnett JH, Werners U, Secher SM, Hill KE, Brazil R, Masson K, et al. Substance use in a population‐based clinic sample of people with first‐episode psychosis. British Journal of Psychiatry 2007;190:515‐20. [DOI] [PubMed] [Google Scholar]
Baxter 2010
- Baxter S, Killoran A, Kelly MP, Goyder E. Synthesizing diverse evidence: the use of primary qualitative data analysis methods and logic models in public health reviews. Public Health 2010;124(2):99‐106. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315(7108):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Broderick 2003
- Broderick PA, Rahni DN, Zhou Y. Acute and subacute effects of risperidone and cocaine on accumbens dopamine and serotonin release using in vivo microvoltammetry on line with open‐field behavior. Progress in Neuro‐psychopharmacology & Biological Psychiatry 2003;27(6):1037‐54. [DOI] [PubMed] [Google Scholar]
Buckley 2006
- Buckley PF. Prevalence and consequences of the dual diagnosis of substance abuse and severe mental illness. Journal of Clinical Psychiatry 2006;67(Suppl 7):5‐9. [PubMed] [Google Scholar]
de Haan 2000
- Haan L, Lavalaye J, Linszen D, Dingemans PM, Booij J. Subjective experience and striatal dopamine D(2) receptor occupancy in patients with schizophrenia stabilized by olanzapine or risperidone. American Journal of Psychiatry 2000;157(6):1019‐20. [DOI] [PubMed] [Google Scholar]
de Haan 2002
- Haan L, Weisfelt M, Dingemans P M, Linszen D H, Wouters L. Psychometric properties of the Subjective Well‐Being Under Neuroleptics scale and the Subjective Deficit Syndrome Scale. Psychopharmacology (Berl) 2002;162(1):24‐8. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium; 2000 Oct 25‐28. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG editor(s). Chapter 9: Analysing data and undertaking meta‐analyses. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011 Available from www.cochrane‐handbook.org.
Dekker 2012
- Dekker N, Koeter M, Brink W. Craving for cannabis in patients with psychotic disorder, their non‐affected siblings and healthy controls: psychometric analysis of the obsessive compulsive drug use scale. International Journal of Methods in Psychiatriatric Research 2012;21(4):286‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Demyttenaere 2004
- Demyttenaere K, Bruffaerts R, Posada‐Villa J, Gasquet I, Kovess V, Lepine JP, et al. Prevalence, severity, and unmet need for treatment of mental disorders in the World Health Organization World Mental Health Surveys. JAMA 2004;291(21):2581‐90. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Drevets 2001
- Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA, et al. Amphetamine‐induced dopamine release in human ventral striatum correlates with euphoria. Biological Psychiatry 2001;49(2):81‐96. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Endicott 1978
- Endicott J, Spitzer RL. A diagnostic interview: the schedule for affective disorders and schizophrenia. Archive of General Psychiatry 1978;35(7):837‐44. [DOI] [PubMed] [Google Scholar]
Fioritti 1997
- Fioritti A, Ferri S, Galassi L, Warner R. Substance use among the mentally ill: a comparison of Italian and American samples. Community Mental Health Journal 1997;33(5):429‐42. [DOI] [PubMed] [Google Scholar]
First 1994
- First MB, RL Spitzer, M Gibbon, JBW Williams. Structured Clinical Interview for DSM‐IV Axis I Disorders‐Patient Edition (SCID‐I‐P, version 2). American Psychiatric Press, 1994. [Google Scholar]
Fleischhacker 2003
- Fleischhacker WW, Eerdekens M, Karcher K, Remington G, Llorca PM, Chrzanowski W, et al. Treatment of schizophrenia with long‐acting injectable risperidone: a 12‐month open‐label trial of the first long‐acting second‐generation antipsychotic. Journal of Clinical Psychiatry 2003;64(10):1250‐7. [DOI] [PubMed] [Google Scholar]
Fowler 1998
- Fowler IL, Carr VJ, Carter NT, Lewin TJ. Patterns of current and lifetime substance use in schizophrenia. Schizophrenia Bulletin 1998;24(3):443‐55. [DOI] [PubMed] [Google Scholar]
Franken 2002
- Franken IH, Hendriksa VM, Brink W. Initial validation of two opiate craving questionnaires the obsessive compulsive drug use scale and the desires for drug questionnaire. Addictive Behaviors 2002;27(5):675‐85. [DOI] [PubMed] [Google Scholar]
Fukushiro 2007
- Fukushiro DF, Alvarez Jdo N, Tatsu JA, Castro JP, Chinen CC, Frussa‐Filho R. Haloperidol (but not ziprasidone) withdrawal enhances cocaine‐induced locomotor activation and conditioned place preference in mice. Progress in Neuro‐psychopharmacology & Biological Psychiatry 2007;31(4):867‐72. [DOI] [PubMed] [Google Scholar]
Fukushiro 2008
- Fukushiro DF, Carvalho Rde C, Ricardo VP, Alvarez Jdo N, Ribeiro LT, Frussa‐Filho R. Haloperidol (but not ziprasidone) withdrawal potentiates sensitization to the hyperlocomotor effect of cocaine in mice. Brain Research Bulletin 2008;77(2‐3):124‐8. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology‐Revised. Rockville, MD: US Department of Health and Human Services, 1976:534‐7. [DHHS Publ No ADM 91‐338] [Google Scholar]
Guyatt 2011
- Guyatt G, Oxman A D, Akl E A, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction‐GRADE evidence profiles and summary of findings tables. J Clin Epidemiol 2011;64:383‐94. [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hasan 2012
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World Journal of Biological Psychiatry 2012;13(5):318‐78. [DOI] [PubMed] [Google Scholar]
Hasan 2013
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long‐term treatment of schizophrenia and management of antipsychotic‐induced side effects. World Journal of Biological Psychiatry 2013;14(1):2‐44. [DOI] [PubMed] [Google Scholar]
Hauli 2011
- Hauli KA, Ndetei DM, Jande MB, Kabangila R. The prevalence of substance use among psychiatric patients: the case study of Bugando Medical centre, Mwanza (northern Tanzania). Substance Abuse 2011;32(4):238‐41. [DOI] [PubMed] [Google Scholar]
Heishman 2009
- Heishman SJ, Evans RJ, Singleton EG, Levin KH, Copersino ML, Gorelick DA. Reliability and validity of a short form of the Marijuana Craving Questionnaire. Drug and Alcohol Dependence 2009;102(1‐3):35‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Horacek 2006
- Horacek J, Bubenikova‐Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, et al. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs 2006;20(5):389‐409. [DOI] [PubMed] [Google Scholar]
Hunter 2003
- Hunter R, Kennedy E, Song F, Gadon L, Irving CB. Risperidone versus typical antipsychotic medication for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000440] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jablensky 2000
- Jablensky A, McGrath J, Herrman H, Castle D, Gureje O, Evans M, et al. Psychotic disorders in urban areas: an overview of the Study on Low Prevalence Disorders. Australian and New Zealand Journal of Psychiatry 2000;34(2):221‐36. [DOI] [PubMed] [Google Scholar]
Janssen 1988
- Janssen PA, Niemegeers CJ, Awouters F, Schellekens KH, Megens AA, Meert TF. Pharmacology of risperidone (R 64 766), a new antipsychotic with serotonin‐S2 and dopamine‐D2 antagonistic properties. Journal of Pharmacology and Experimental Therapeutics 1988;244(2):685‐93. [PubMed] [Google Scholar]
Kapur 1995
- Kapur S, Remington G, Zipursky RB, Wilson AA, Houle S. The D2 dopamine receptor occupancy of risperidone and its relationship to extrapyramidal symptoms: a PET study. Life Sciences 1995;57(10):L103‐7. [DOI] [PubMed] [Google Scholar]
Kapur 1996
- Kapur S. 5‐HT2 antagonism and EPS benefits: is there a causal connection?. Psychopharmacology 1996;124(1‐2):35‐9. [DOI] [PubMed] [Google Scholar]
Kapur 2001
- Kapur S, Seeman P. Does fast dissociation from the dopamine D(2) receptor explain the action of atypical antipsychotics?: a new hypothesis. American Journal of Psychiatry 2001;158(3):360‐9. [DOI] [PubMed] [Google Scholar]
Katz 2010
- Katz G, Durst R, Shufman E, Bar‐Hamburger R, Grunhaus L. Cannabis abuse and severity of psychotic and affective disorders in Israeli psychiatric inpatients. Comprehensive Psychiatry 2010;51(1):37‐41. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13(2):261‐76. [DOI] [PubMed] [Google Scholar]
Kelly 2012
- Kelly TM, Daley DC, Douaihy AB. Treatment of substance abusing patients with comorbid psychiatric disorders. Addictive Behaviors 2012;37(1):11‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kessler 2004
- Kessler RC. The epidemiology of dual diagnosis. Biological Psychiatry 2004;56(10):730‐7. [DOI] [PubMed] [Google Scholar]
Kuhar 1996
- Kuhar MJ, Pilotte NS. Neurochemical changes in cocaine withdrawal. Trends in Pharmacological Sciences 1996;17(7):260‐4. [DOI] [PubMed] [Google Scholar]
Kuroki 2008
- Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second‐generation antipsychotic drugs: a validity of the serotonin‐dopamine hypothesis. Progress in Brain Research 2008;172:199‐212. [DOI] [PubMed] [Google Scholar]
Lacro 2002
- Lacro JP, Dunn LB, Dolder CR, Leckband SG, Jeste DV. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. Journal of Clinical Psychiatry 2002;63(10):892‐909. [DOI] [PubMed] [Google Scholar]
Lambert 2005
- Lambert M, Conus P, Lubman DI, Wade D, Yuen H, Moritz S, et al. The impact of substance use disorders on clinical outcome in 643 patients with first‐episode psychosis. Acta Psychiatrica Scandinavica 2005;112(2):141‐8. [DOI] [PubMed] [Google Scholar]
Lazary 2012
- Lazary J. Psychopharmacological boundaries of schizophrenia with comorbid cannabis use disorder: a critical review. Current Pharmaceutical Design 2012;18(32):4890‐6. [DOI] [PubMed] [Google Scholar]
Lehman 2004
- Lehman AF, Lieberman JA, Dixon LB, McGlashan TH, Miller AL, Perkins DO, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. American Journal of Psychiatry 2004;161(2 Suppl):1‐56. [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Davis JM, Engel RR, Kissling W, Kane JM. Definitions of response and remission in schizophrenia: recommendations for their use and their presentation. Acta Psychiatrica Scandinavica 2009;438:7‐14. [DOI] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353(12):1209‐23. [DOI] [PubMed] [Google Scholar]
Lukoff 1986
- Lukoff D, Nuechterlein KH, Ventura J. Manual for expanded Brief Psychiatric Rating Scale. Schizophrenia Bulletin 1986;12:594–602. [Google Scholar]
Machielsen 2009
- Machielsen MW, Haan L. Differences in efficacy on substance abuse between risperidone and clozapine supports the importance of differential modulation of dopaminergic neurotransmission. Psychopharmacology Bulletin 2009;42:40‐52. [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Mattick 2014
- Mattick RP, Breen C, Kimber J, Davoli M. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD002207.pub4] [DOI] [Google Scholar]
McIntyre 2007
- McIntyre RS, Soczynska JK, Woldeyohannes HO, Alsuwaidan M, Konarski JZ. A preclinical and clinical rationale for quetiapine in mood syndromes. Expert Opinion on Pharmacotherapy 2007;8(9):1211‐9. [DOI] [PubMed] [Google Scholar]
McLellan 1980
- McLellan AT, Luborsky L, Woody GE, O'Brien CP. An improved diagnostic evaluation instrument for substance abuse patients. The Addiction Severity Index. Journal of Nervous and Mental Disease 1980;168:26‐33. [DOI] [PubMed] [Google Scholar]
McLellan 1992
- McLellan AT, Kushner H, Metzger D, Peters R, Smith I, Grissom G, et al. The Fifth Edition of the Addiction Severity Index. J.Subst.Abuse Treat. 1992;9(0740‐5472 (Print)):199‐213. [DOI] [PubMed] [Google Scholar]
McLoughlin 2014
- McLoughlin BC, Pushpa‐Rajah JA, Gillies D, Rathbone J, Variend H, Kalakouti E, et al. Cannabis and schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD004837.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Menezes 1996
- Menezes PR, Johnson S, Thornicroft G, Marshall J, Prosser D, Bebbington P, et al. Drug and alcohol problems among individuals with severe mental illness in south London. British Journal of Psychiatry 1996;168(5):612‐9. [DOI] [PubMed] [Google Scholar]
Modestin 1997
- Modestin J, Nussbaumer C, Angst K, Scheidegger P, Hell D. Use of potentially abusive psychotropic substances in psychiatric inpatients. European Archives of Psychiatry and Clinical Neuroscience 1997;247(3):146‐53. [DOI] [PubMed] [Google Scholar]
Moller 2005
- Moller HJ. Risperidone: a review. Expert Opinion on Pharmacotherapy 2005;6(5):803‐18. [DOI] [PubMed] [Google Scholar]
Moller 2007
- Moller HJ. Long‐acting injectable risperidone for the treatment of schizophrenia: clinical perspectives. Drugs 2007;67(11):1541‐66. [DOI] [PubMed] [Google Scholar]
Naber 1995
- Naber D. A self‐rating to measure subjective effects of neuroleptic drugs, relationships to objective psychopathology, quality of life, compliance and other clinical variables. International Clinical Psychopharmacology 1995;10(Suppl 3):133‐8. [PubMed] [Google Scholar]
National Institute of Mental Health 1988
- National Institute of Mental Health Psychopharmacology Research Branch. Abnormal Involuntary Movement Scale (AIMS). Psychopharmacology Bulletin 1988;24:781‐3. [PubMed] [Google Scholar]
Newcomer 2007
- Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. Journal of Clinical Psychiatry 2007;68(Suppl 1):20‐7. [PubMed] [Google Scholar]
NICE 2009
- National Collaborating Centre for Mental Health (UK). Schizophrenia: Core Interventions in the Treatment and Management of Schizophrenia in Primary and Secondary Care (Update). (NICE Clinical Guidelines, No. 82). Leicester: British Psychological Society, 2009. [PubMed] [Google Scholar]
NICE 2011
- National Collaborating Centre for Mental Health (UK). Psychosis with Coexisting Substance Misuse: Assessment and Management in Adults and Young People. (NICE Clinical Guidelines, No. 120). Leicester: British Psychological Society, 2011. [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Owen 1996
- Owen RR, Fischer EP, Booth BM, Cuffel BJ. Medication noncompliance and substance abuse among patients with schizophrenia. Psychiatric Services 1996;47(8):853‐8. [DOI] [PubMed] [Google Scholar]
Perkins 2002
- Perkins DO. Predictors of noncompliance in patients with schizophrenia. Journal of Clinical Psychiatry 2002;63(12):1121‐8. [DOI] [PubMed] [Google Scholar]
Perkins 2006
- Perkins DO, Johnson JL, Hamer RM, Zipursky RB, Keefe RS, Centorrhino F, et al. Predictors of antipsychotic medication adherence in patients recovering from a first psychotic episode. Schizophrenia Research 2006;83(1):53‐63. [DOI] [PubMed] [Google Scholar]
Potvin 2009
- Potvin S, Blanchet P, Stip E. Substance abuse is associated with increased extrapyramidal symptoms in schizophrenia: a meta‐analysis. Schizophrenia Research 2009;113(1573‐2509 (Electronic), 2‐3):181‐8. [DOI] [PubMed] [Google Scholar]
Regier 1990
- Regier DA, Farmer ME, Rae DS, Locke BZ, Keith SJ, Judd LL, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 1990;264(19):2511‐8. [PubMed] [Google Scholar]
Robins 1988
- Robins LN, Wing J, Wittchen HU, Helzer JE, Babor TF, Burke J, et al. The Composite International Diagnostic Interview. An epidemiologic Instrument suitable for use in conjunction with different diagnostic systems and in different cultures. Archives of general psychiatry 1988;45(12):1069‐77. [PUBMED: 2848472] [DOI] [PubMed] [Google Scholar]
Robinson 2006
- Robinson DG, Woerner MG, Napolitano B, Patel RC, Sevy SM, Gunduz‐Bruce H, et al. Randomized comparison of olanzapine versus risperidone for the treatment of first‐episode schizophrenia: 4‐month outcomes. American Journal of Psychiatry 2006;163(12):2096‐102. [DOI] [PubMed] [Google Scholar]
Rosenberg 2009
- Rosenberg H. Clinical and laboratory assessment of the subjective experience of drug craving. Clinical Psychology Review 2009;29(6):519‐34. [DOI] [PubMed] [Google Scholar]
Ruggeri 2000
- Ruggeri M, Leese M, Thornicroft G, Bisoffi G, Tansella M. Definition and prevalence of severe and persistent mental illness. British Journal of Psychiatry 2000;177:149‐55. [DOI] [PubMed] [Google Scholar]
Salyers 2001
- Salyers MP, Mueser KT. Social functioning, psychopathology, and medication side effects in relation to substance use and abuse in schizophrenia. Schizophrenia Research 2001;48(1):109‐23. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Seeman 2002
- Seeman P. Atypical antipsychotics: mechanism of action. Canadian Journal of Psychiatry 2002;47(1):27‐38. [PubMed] [Google Scholar]
Shokraneh 2017
- Shokraneh F, Adams CE. Study‐based registers of randomized controlled trials: Starting a systematic review with data extraction or meta‐analysis. BioImpacts 2017;InPress:InPress. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simpson 1970
- Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212(0065‐1591):11‐9. [DOI] [PubMed] [Google Scholar]
Sinha 2006
- Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. Stress‐induced cocaine craving and hypothalamic‐pituitary‐adrenal responses are predictive of cocaine relapse outcomes. Archives of General Psychiatry 2006;63(3):324‐31. [DOI] [PubMed] [Google Scholar]
Sinha 2011
- Sinha R. New findings on biological factors predicting addiction relapse vulnerability. Current Psychiatry Reports 2011;13(5):398‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Siris 1990
- Siris SG. Pharmacological treatment of substance‐abusing schizophrenic patients. Schizophrenia Bulletin 1990;16(1):111‐22. [DOI] [PubMed] [Google Scholar]
Smelson 1997
- Smelson DA, Roy A, Roy M. Risperidone diminishes cue‐elicited craving in withdrawn cocaine‐dependent patients. Canadian Journal of Psychiatry 1997;42(9):984. [DOI] [PubMed] [Google Scholar]
Smelson 2002
- Smelson DA, Losonczy MF, Davis CW, Kaune M, Williams J, Ziedonis D. Risperidone decreases craving and relapses in individuals with schizophrenia and cocaine dependence. Canadian Journal of Psychiatry 2002;47(7):671‐5. [DOI] [PubMed] [Google Scholar]
Sobell 1992
- Sobell LC, Sobell MB. Timeline follow‐back: a technique for assessing self‐reported ethanol consumption. In: J Allen RZ Litten editor(s). Measuring Alcohol Consuption: Psychosocial and Biological Methods. Totowa NJ: Humana Press, 1992:41‐72. [Google Scholar]
Soyka 1993
- Soyka M, Albus M, Kathmann N, Finelli A, Hofstetter S, Holzbach R, et al. Prevalence of alcohol and drug abuse in schizophrenic inpatients. European Archives of Psychiatry and Clinical Neuroscience 1993;242(6):362‐72. [DOI] [PubMed] [Google Scholar]
Stroup 2003
- Stroup TS, McEvoy JP, Swartz MS, Byerly MJ, Glick ID, Canive JM, et al. The National Institute of Mental Health clinical antipsychotic trials of intervention effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophrenia Bulletin 2003;29(1):15‐31. [DOI] [PubMed] [Google Scholar]
Stuyt 2006
- Stuyt EB, Sajbel TA, Allen MH. Differing effects of antipsychotic medications on substance abuse treatment patients with co‐occurring psychotic and substance abuse disorders. American Journal on Addictions 2006;15(2):166‐73. [DOI] [PubMed] [Google Scholar]
Sun 2007
- Sun SX, Liu GG, Christensen DB, Fu AZ. Review and analysis of hospitalization costs associated with antipsychotic nonadherence in the treatment of schizophrenia in the United States. Current Medical Research and Opinion 2007;23(10):2305‐12. [DOI] [PubMed] [Google Scholar]
Sun 2014
- Sun X, Ioannidis JP, Agoritsas T, Alba AC, Guyatt G. How to use a subgroup analysis: users' guide to the medical literature. JAMA 2014;311(4):405‐11. [DOI] [PubMed] [Google Scholar]
Swofford 1996
- Swofford CD, Kasckow JW, Scheller‐Gilkey G, Inderbitzin LB. Substance use: a powerful predictor of relapse in schizophrenia. Schizophrenia Research 1996;20(1‐2):145‐51. [DOI] [PubMed] [Google Scholar]
Tort 2006
- Tort AB, Souza DO, Lara DR. Theoretical insights into the mechanism of action of atypical antipsychotics. Progress in Neuro‐psychopharmacology & Biological Psychiatry 2006;30(4):541‐8. [DOI] [PubMed] [Google Scholar]
Tsibulsky 1998
- Tsibulsky VL, Grocki S, Dashevsky BA, Kehne JH, Schmidt CJ, Sorensen SM, et al. Mixed D2/5‐HT2A antagonism of cocaine‐induced facilitation of brain stimulation reward. Pharmacology, Biochemistry, and Behavior 1998;59(2):275‐80. [DOI] [PubMed] [Google Scholar]
Tunis 2006
- Tunis SL, Faries DE, Nyhuis AW, Kinon BJ, Ascher‐Svanum H, Aquila R. Cost‐effectiveness of olanzapine as first‐line treatment for schizophrenia: results from a randomized, open‐label, 1‐year trial. Value Health 2006;9(2):77‐89. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
van Mastrigt 2004
- Mastrigt S, Addington J, Addington D. Substance misuse at presentation to an early psychosis program. Social Psychiatry and Psychiatric Epidemiology 2004;39(1):69‐72. [DOI] [PubMed] [Google Scholar]
Volkow 1997
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R, et al. Decreased striatal dopaminergic responsiveness in detoxified cocaine‐dependent subjects. Nature 1997;386(6627):830‐3. [DOI] [PubMed] [Google Scholar]
Volkow 2006
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. Journal of Neuroscience 2006;26(24):6583‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Volkow 2007
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M, et al. Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement. Journal of Neuroscience 2007;27(46):12700‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Volkow 2011
- Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F. Addiction: beyond dopamine reward circuitry. Proceedings of the National Academy of Sciences of the United States of America 2011;108(37):15037‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vothknecht 2011
- Vothknecht S, Schoevers RA, Haan L. Subjective well‐being in schizophrenia as measured with the Subjective Well‐Being under Neuroleptic Treatment scale: a review. Australian and New Zealand Journal of Psychiatry 2011;45(3):182‐92. [DOI] [PubMed] [Google Scholar]
Wade 2006
- Wade D, Harrigan S, Edwards J, Burgess PM, Whelan G, McGorry PD. Substance misuse in first‐episode psychosis: 15‐month prospective follow‐up study. British Journal of Psychiatry 2006;189:229‐34. [DOI] [PubMed] [Google Scholar]
Wang 2007
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine ‐ reporting of subgroup analyses in clinical trials. New England Journal of Medicine 2007;357(21):2189‐94. [DOI] [PubMed] [Google Scholar]
Weddington 1990
- Weddington WW, Brown BS, Haertzen CA, Cone EJ, Dax EM, Herning RI, et al. Changes in mood, craving, and sleep during short‐term abstinence reported by male cocaine addicts. A controlled, residential study. Archives of General Psychiatry 1990;47(9):861‐8. [DOI] [PubMed] [Google Scholar]
Weich 2009
- Weich L, Pienaar W. Occurrence of comorbid substance use disorders among acute psychiatric inpatients at Stikland Hospital in the Western Cape, South Africa. African Journal of Psychiatry 2009;12(3):213‐7. [DOI] [PubMed] [Google Scholar]
Wobrock 2009
- Wobrock T, Soyka M. Pharmacotherapy of patients with schizophrenia and substance abuse. Expert Opinion in Pharmacology 2009;10:353‐67. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Yatham 2005
- Yatham LN, Goldstein JM, Vieta E, Bowden CL, Grunze H, Post RM, et al. Atypical antipsychotics in bipolar depression: potential mechanisms of action. Journal of Clinical Psychiatry 2005;66(Suppl 5):40‐8. [PubMed] [Google Scholar]