

Abstract
Over the past several decades, metal-catalyzed cross-coupling has emerged as a very powerful strategy to functionalize carbon-based molecules. More recently, some of the cross-coupling methodologies have been adapted to inorganic compounds including boron-rich clusters. The development of this chemistry relies on the ability to synthesize halogenated boron-rich clusters which can serve as electrophilic cross-coupling partners with nucleophilic substrates in the presence of a metal catalyst. While the cross-coupling chemistry with boron-clusters is conceptually reminiscent to that of its hydrocarbon counterparts, several key aspects including the spheroidal bulk of clusters, and the distinct nature of boron-halogen / boron-heteroatom bonds make this chemistry unique. The utility of metal-catalyzed cross-coupling can be extended to several classes of polyhedral boranes including neutral and anionic carboranes, metallaboranes, and carbon-free boranes. Importantly, cross-coupling enables a suite of boron-heteroatom (C, N, O, P, S) couplings to prepare boron cluster-based systems that can be used for ligand design, medicinal chemistry, and materials applications.
Graphical Abstract
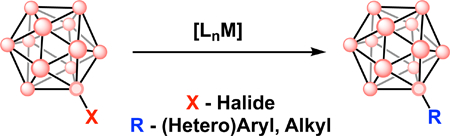
Introduction
Metal-catalyzed cross-coupling is a powerful method for constructing larger molecules by covalently bonding molecular fragments.1–7 These reactions serve an important role in organic synthesis due to the adaptability of metal catalysts to specific molecules including complex aromatic compounds and unsaturated hydrocarbons. Among such reactions, palladium-catalyzed cross-coupling between an electrophile and nucleophile has shown broad utility in synthesis. Pd-catalyzed cross-coupling generally features three elementary steps: oxidative addition of a electrophile, transmetallation of a nucleophile, and coupling of the electrophile and nucleophile by reductive elimination, Fig. 1.
Fig 1.
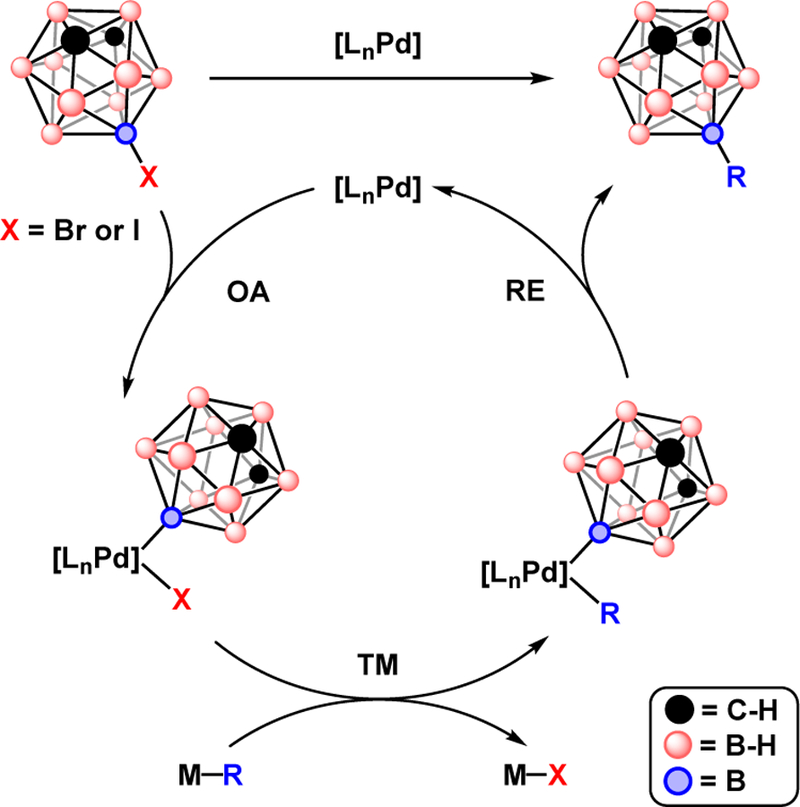
Generalized Pd-catalyzed cross-coupling of a representative halocarborane (halogen is attached on the B(9) position of a meta-carborane cluster). OA = oxidative addition, TM = transmetallation, RE = reductive elimination.
In the 1950s researchers discovered a new class of boron-rich molecules characterized by 3-center-2-electron bonds that stabilize polyhedral borane structures into cage-like (closo-) structures, Fig. 2.8 Formation of closo-polyhedral boranes results in a 3-dimensional (3D) σ-aromaticity that is considered a 3-dimensional analog of 2-dimensional aromatic molecules (e.g., benzene). At the onset of the discovery of polyhedral boranes, a fundamental interest pertaining to the reactivity of these clusters has emerged. Several reactions observed with aromatic hydrocarbons, such as electrophilic and nucleophilic substitution, have been subsequently applied to boron clusters. This similar reactivity lead to investigation into whether polyhedral boranes can undergo metal-mediated reactions like Pd-catalyzed cross-coupling reactions of aryl halides. Reactions of ortho/meta-carborane with Hg(CF3COO)2 in triflic acid yielded [Hg(CF3COO)(9-B-o/m-C2B10H11)] carboranyl compounds reminiscent to the early work by Heck with arylmercury species.9,10 Following these results, successful Pd-catalyzed cross-coupling of B-iodo-carboranes with Grignard reagents established an early precedent for using metal-catalyzed cross-coupling to derivatize polyhedral boranes.
Fig 2.
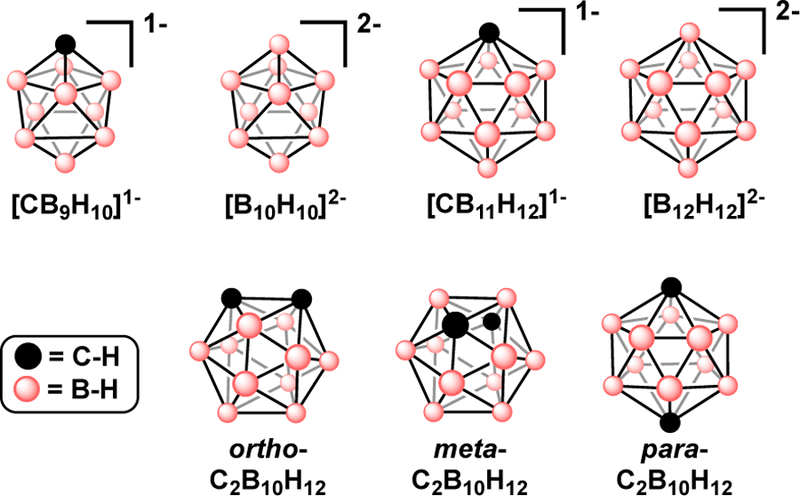
Polyhedral borane clusters that can serve as precursors to electrophilic substrates that can be subsequently used in metal-catalyzed cross-coupling.
This article will describe how metal-catalyzed cross-coupling has been developed, and deployed, to a broad subset of polyhedral boranes. Emphasis is placed on principles that are applicable to many compositions of polyhedral boranes. First, we will present how metal-catalyzed B-C coupling with various polyhedral boranes establishes the broad utility of cross-coupling for different polyhedral borane compositions. Then, the various metal-catalyzed B-N, B-O, and B-P couplings highlight the nuances of using polyhedral boranes as cross-coupling partners.
Polyhedral Boranes as Substrates
Ten- and 12-vertex closo-boranes have garnered attention due to their stability and synthetic accessibility. Their 3D aromaticity also imparts high thermal and chemical stability that makes these polyhedral boranes interesting for molecular building blocks and for scaffolds in supramolecular materials such metal-organic frameworks,11 macrocycles,12,13 dendrimers, and covalent nanoparticle cores.14 Ten- and 12-vertex closo-boranes exist as neutral, anionic and dianionic molecules depending on the composition of the boron-cluster core. The commonly encountered 10-vertex closo-boranes are [B10H10]2− and [CB9H10]1−; and the common 12-vertex closo-boranes are [B12H12]2−, [CB11H12]1−, and three isomers (ortho-, meta-, para-) of C2B10H12, Fig. 2. These 3D aromatic molecules are frequently compared to benzene, but they are more sterically similar to adamantane.15–17 For example, ortho-C2B10H12 has a calculated van der Waals volume of 148 Å3, whereas benzene has a volume of 79 Å3 and the non-aromatic adamantane has a volume of 136 Å3.15,18 The larger volume and σ-aromaticity of polyhedral boranes requires additional consideration when using them as replacements for phenyl or adamantly moieties.15
Polyhedral boranes also possess exohedral σ-bonds that allow covalent bonding between a boron cluster core and other molecular fragments.The electronic coupling between exohedral moieties and the boron cluster core can be engineered by the boron-cluster composition, this tunablility presents exciting opportunities for designing the electronic properties of polyhedral borane-based materials.
Controlled engineering of borane-based materials relies on precise ways of covalently attaching molecular fragments to the boron-cluster core. For polyhedral boranes that contain a cage heteroatom, such as a carbon vertex in closo-C2B10H= or [closo-CB11H12]1-, the C-H vertex proton is much more acidic compared to the hydridic B-H vertices.15 The higher acidity of the C-H proton allows it to be deprotonated with a strong base and the resulting C− vertex can undergo SN2 reactions with electrophiles to produce carbon substituted polyhedral boranes. Such SN2 reactions are used to selectively append functional groups to the carbon vertices of neutral and anionic carboranes. However, this functionalization approach is not useful for [B10H10]2− and [B12H12]2-, which have no C-H vertices, or when seeking to attach a functional group to a boron vertex.
Inductive effects can be transmitted through polyhedral boranes to exohedral functional groups.19 In terms of electronic effects, the more electronegative carbon vertices of carboranes (e. g. closo-C2B10H12, [closo-CB9H10]1− or [closo-CB11H12]1−) have an electron withdrawing effect, whereas bonding to the boron vertices produces an electron donating effect. The downside to the facile SN2 substitution of C-H vertices is that the carboranyl fragment always acts as an electron withdrawing group and the electron donating effects of the polyhedral boranes are not conferred to the exohedral moiety. Thus, a diverse set of methods that allow functionalization of the boron vertices are critical to fully capture the utility of polyhedral boranes in hierarchal designs.
B-H Functionalization
Direct B-H functionalization methods provide new synthons for studying 3D aromaticity in molecular systems. Among these methods, reactions of electrophiles (E+) with the electron-rich B-H vertices of polyhedral boranes form a variety of B-E bonds. Halogenated polyhedral boranes can be readily synthesized by reaction of elemental halogens with polyhedral boranes in the presence of a Lewis acid catalyst (AlCl3 or FeCl3), Fig. 3. These halogenation reactions also produce polyhalogenated boranes that serve as important entry-points to metal-catalyzed cross-coupling. Cage reconstruction is an additional approach to synthesizing halogenated boranes, it involves reacting an open (nido-) polyhedral borane with BX3 or BH2X (X = F, Cl, Br, or I) to install a B-X vertex and form a closo- structure, Fig. 3. Both, electrophilic and cage reconstruction methods can be applied to produce polyhalogenated compounds.
Fig 3.
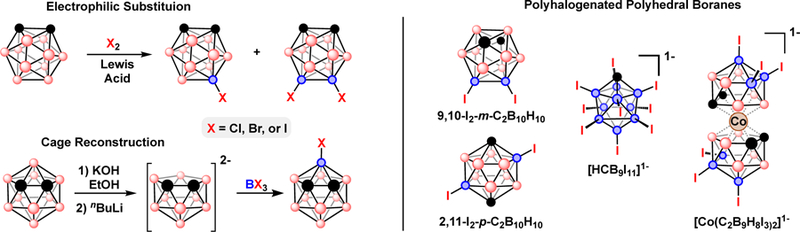
Left) Electrophilic halogenation of ortho-carborane and cage-reconstruction of ortho-carborane by deboronation and subsequent “recapitation”. Right) Examples of polyiodinated polyhedral boranes used in Pd-catalyzed cross-coupling.
Metal-catalyzed B-vertex Functionalization
Of the known polyhedral boranes, the 12-vertex carboranes have the largest number of cross-coupling reactions demonstrated. Carboranes containing two carbon vertices, C2B10H12, are neutral molecules with 3 isomers named by the distance (ortho-, meta-, para-) between the carbon atoms, Fig. 2.20 The anisotropic σ-aromaticity of carboranes is often leveraged in different metal-catalyzed functionalization routes to install various functional groups onto different boron vertices of polyhedral boranes.21
Two forms of metal-catalyzed functionalization prevail in the literature: 1) metal-catalyzed B-H activation,22–25 and 2) metal-catalyzed cross-coupling at B-X (X = Br or I) bonds. Metal-catalyzed B-H activation strategies often feature a directing group bound to the boron cluster which coordinates an electrophilic metal center near a B-H vertex and promotes B-H bond cleavage.24–33 In contrast, metal-catalyzed cross-coupling relies on electron-rich metals to perform oxidative addition into B-X bonds, Fig. 1. These two methods are complimentary because they target electronically different vertices. For example, metal-catalyzed B-H activation can be used to halogenate vertices that do not readily undergo electrophilic halogenation or to create nucleophilic cross-coupling partners for metal-catalyzed cross-coupling.
One advantage of metal-catalyzed cross-coupling is that it allows coupling wherever a halogen can be installed. This leverages the breadth of polyhedral borane halogenation reactions to create a vast library of potential substrates. Successful cross-coupling reactions with Grignard reagents were performed in the presence of Pd and Ni catalyst under conditions closely resembling those used for aryl iodides. Early on, a view emerged that carboranes are a 3-dimensional analog of benzene because of their aromaticity, susceptibility to electrophilic alkylation and halogenation, and metal-catalyzed cross-coupling. This lead to the development of functionalization reactions that closely resemble those of carbon-based aromatic molecules. However, further development of polyhedral borane cross-coupling calls for methods that account for their distinct electronic and steric properties.
Boron-Halogen Bond Activation
Most reports of polyhedral borane cross-coupling target the weakest boron-halogen bond which is a B-I bond, whereas other B-X (X = F, Cl, or Br) bonds were originally dismissed as too unreactive, Fig. 4.34–36 The lack of reactivity in B-bromo and B-chloro polyhedral boranes in cross-coupling reactions was attributed to the stronger B-X bonds, compared to aryl C-X bonds, which prevented oxidative addition of B-bromocarboranes. This difference in B-X bond strength allowed radiolabelling of carboranes with 76Br and 19F because the resulting B-bromo- and B-fluoro-carboranes do not undergo oxidative addition in the presence of commonly used Pd catalysts.35,36 A survey of the catalyst systems used in cross-coupling of carboranes reveals that the majority of Pd-catalyzed reactions use a small set of phosphine ligands, mainly triphenylphosphine (PPh3).
Fig. 4.
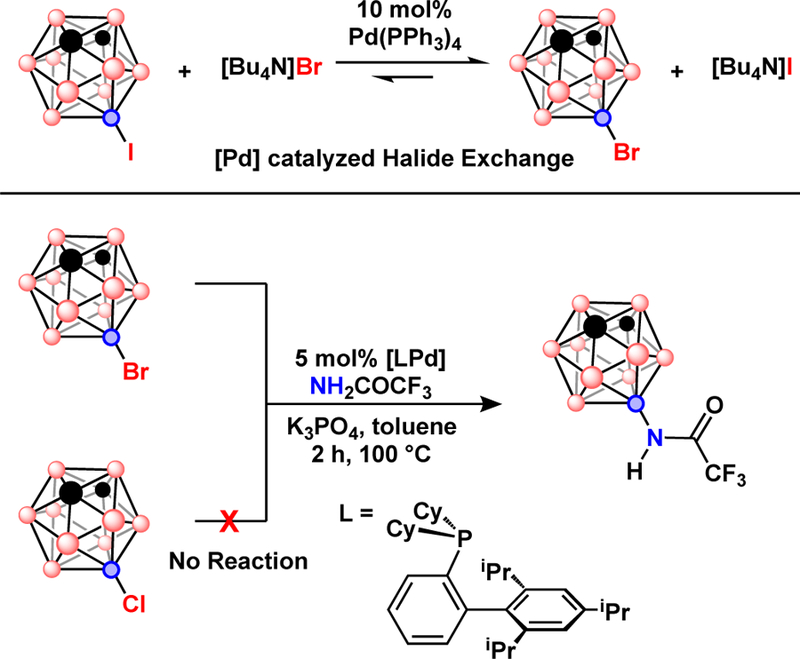
Top) Pd-catalyzed halide exchange with 9-I-m-C2B10H11, the equilibrium lies heavily to the right (ref 33). Bottom) Reactivity comparison between 9-Br-m-C2B10H11 and 9-Cl-m-C=B10H11 in a Pd-catalyzed amidation.
The reactivity of B-bromo-carboranes in Pd-catalyzed cross-coupling was finally overcome by using electron-rich dialkyl biaryl phosphines, which are known to activate C-Cl and C-OTf bonds of many carbon-based molecules, Fig. 4. Now, the difference in reactivity among B-X bonds presents heterofunctionalization pathways through catalyst-controlled sequential cross-coupling.37
Metal-Catalyzed B-C Bond Formation
Despite the narrow scope of catalysts examined for polyhedral borane cross-coupling, a variety of B-C coupling reaction types have been demonstrated. The first reports of polyhedral borane cross-coupling feature reactions of B-iodo-carboranes and Grignard reagents in the presence of Pd catalysts.38–41 Afterwards, Neigishi-type reactions were shown to produce B-Csp3 and B-Csp2 bonds with 5- and 6-member heterocycles.42 Sonogishira-, Heck-, and Suzuki-type cross-coupling reactions were also demonstrated on carboranes. Many of these reactions were adapted to polyhedral boranes with different sizes, compositions, and charge. Most importantly, the body of work on polyhedral borane B-C coupling reveals general rules and trends that can inform cross-coupling methods on several classes of polyhedral boranes.
Kumada- and Neigishi-type Coupling
Cross-coupling with Grignard reagents using Pd and Ni catalysts found [Ni(PPh3)4] to be extremely inactive compared to [Pd(PPh3)4].38 This type of Pd-catalyzed cross-coupling is often used to couple alkyl, aryl, vinyl, and alkynyl groups to B-iodo-boranes. Typical reactions use ethereal solvents such as Et2O and THF, and [Pd(PPh3)2Cl2] or [Pd(PPh3)4] as the catalyst Fig. 5.38,39,41 These reactions often require several hours or days, of refluxing depending on the Grignard reagent used. Cross-coupling of aryl, vinyl, and alkynyl substrates is more efficient than cross-coupling of alkyl groups. When using alkylmagnesium halides as the cross-coupling partner, reduction of B-iodo-carboranes (C2B10H11I) to their parent carborane (C2B10H12) was observed. Analogous, reduction of aryl halides in the presence of Pd catalysts has been observed.43–45 Importantly, reaction kinetics for this type of cross-coupling chemistry can be improved when precatalysts featuring biaryl ligands are used.46
Fig. 5.
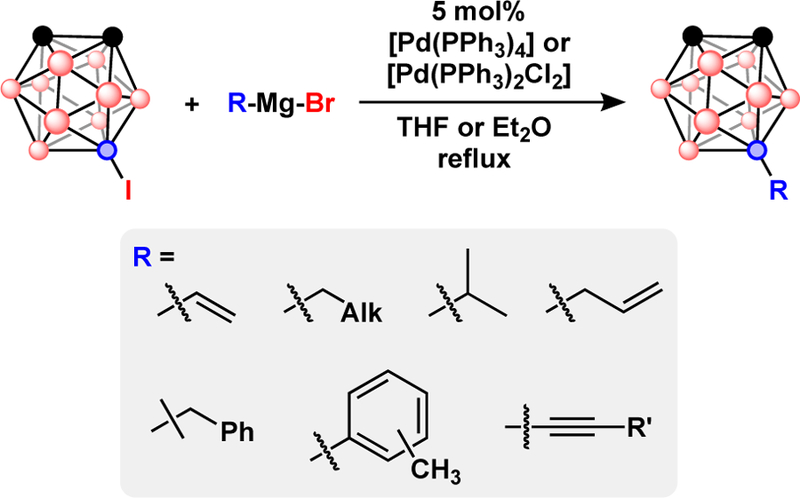
Pd-catalyzed Kumada cross-coupling with 9-I-o-C2B10H11.
The resulting low yield and long reaction times are attributed to palladium hydride complexes that may form by β-hydride elimination from alkylpalladium intermediates. Hawthorne and co-workers reported that addition of CuI as co-catalysts significantly improved reaction yields by reducing the propensity toward carborane reduction.40,47,48 Several types ofmetal-catalyzed cross-coupling reactions of B-iodo-carboranes with alkynes using Grignard reagents to form B-C bonds are known.39 Later, Hawthorne and co-workers demonstrated the adventitious role of a CuI co-catalyst when using alkynyl Grignard reagents.40,48 Importantly, these early cross-coupling methodology techniques were used to prepare rod-like carborane molecules in a step-wise iterative manner.49
Milder cross-coupling reactions using organozinc reagents demonstrated coupling of heterocycles to p- and m-carboranes using [Pd(PPh3)2Cl2], [Pd(PPh3)4], and [Pd(dppb)Cl2], Fig. 6.42 Notably, this allowed coupling of B-iodo-carboranes with an unprotected ethynyl steroid, 17-α-ethynylestradiol, to produce the B-C coupled product despite the presence of two alcohol groups that could have led to B-O coupled products.50 An additional advantage of using organozinc reagents is that they do not rely on a base to generate the nucleophilic cross-coupling partner. This allows the use of 9-I-o-C2B10H11 as a cross-coupling partner which is more prone to deboronation than 9-I-m-C2B10H11.51 Deboronation is a particular issue for ortho-carboranes because the resulting anionic nido-carboranes can act as chelating ligands that could sequester the metal catalyst from the reaction pool.
Fig. 6.
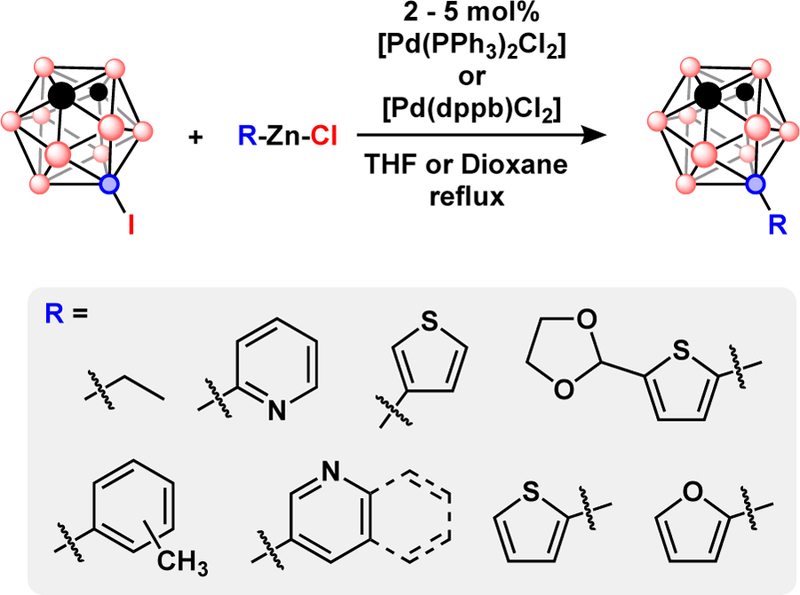
Pd-catalyzed Neigishi cross-coupling with 9-I-m-C2B10H11.
Sonogishira-, Heck-, and Suzuki-type Coupling
Beletskaya et al. reported Sonogishira-type reactivity when coupling B-iodo-carboranes and acetylene derivatives in the presence of CuI and [Pd(PPh3)2Cl2] as catalysts and pyrrolidine as the solvent, Fig. 7.42 The push to develop mild transmetallation reagents extended to B-C bond formation through Heck coupling of olefins, Fig. 7.52 However, 2-vinylpyridine, butyl acrylate and acrylonitrile did not react, presumably due to slow coordination of the olefins to Pd. This work features the use of a Pd(II) palladacycle as a precatalyst to generate the catalytically active P(0) species. This was found to greatly increase the product yield by extending the lifetime of the catalyst system and points to a long standing problem with catalyst stability during carborane cross-coupling.
Fig. 7.
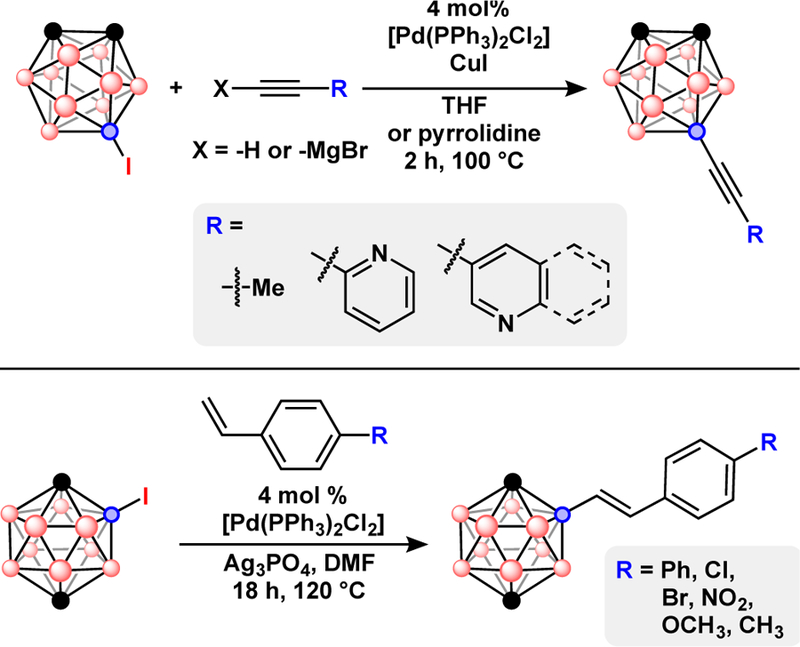
Top) Pd-catalyzed Sonogishira and Kumada cross-coupling of acetylenes with 9-I-o-C2B10H11. Bottom) Pd-catalyzed Heck cross-coupling of styrenes with 2-I-p-C2B10H11.
Interest in bench-stable transmetallation agents lead to the development of cross-coupling with aryl boronic acid nucleophiles. Cross-coupling of aryl boronic acids was first reported with 2-I-p-C2B10H11 in the presence of CsF and Pd2dba3/dppb, Fig. 8. Suzuki cross-coupling with 3-I-o-C2B10H11 was also reported using [Pd(PPh3)4] as the catalyst and K2CO3 as a base in a toluene:methanol mixture. Importantly, these cross-coupling reaction were demonstrated using arylboronic acids (4-NO2, 3-NO2, 3-CN, 3-CHO) that are not compatible with the previously mentioned Kumada and Neigishi reaction conditions.53 More recently, the Suzuki coupling of p-tolyl boronic acid with 9-Br-m-C2B10H11 produced 2-p-tolyl-m-C2B10H11 via the “cage-walking” isomerization mechanism, vide infra. These Suzuki cross-coupling reactions employ inorganic bases such as F− or OH− to facilitate the transmetallation step,54 but these bases are strong nucleophiles that can lead to deboronation of o- and m-C2B10H11 during cross-coupling.51,53
Fig. 8.
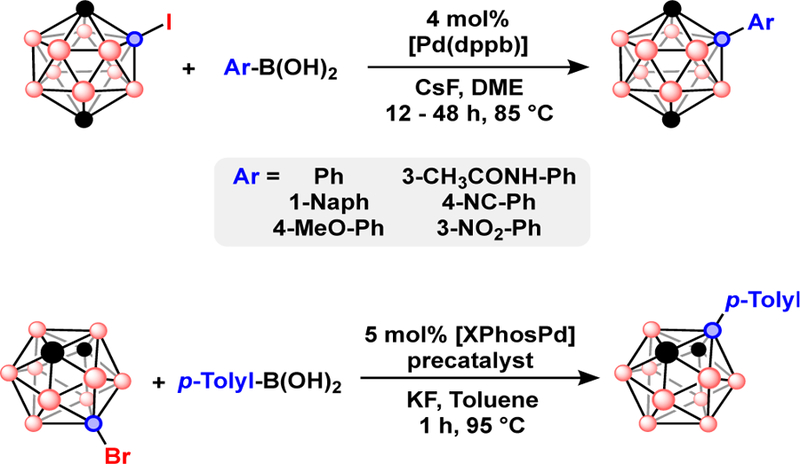
Top) Pd-catalyzed Suzuki cross-coupling of arylboronic acids with 2-I-p-C2B10H11. Bottom) Pd-catalyzed “cage-walking” and Suzuki cross-coupling of p-tolylboronic acid with 9-Br-m-C2B10H11 to produce 2-p-tolyl-m-C2B10H11.
Metal-Catalyzed B-C Coupling Reactions with Metallaboranes
In addition to carboranes, polyhedral boranes with transition metals as vertices are known to participate in halogenation reactions. Examples of metal-catalyzed cross-coupling also extend to various iodinated metallaboranes, heteroboranes, and anionic closo-borates. Many of these reactions feature cross-coupling of B-I bonds to form B-C bonds using methods similar to those reported for the neutral carboranes. Among the many metallacarboranes reported, the metal bis(dicarbollide)s have garnered the most attention due to their simple preparation, high stability and redox activity.55,56 Metal bis(dicarbollide)s can be prepared by deboronation of carborane and subsequent reaction of the resulting nido-carborane with a metal salt precursor. Metal-catalyzed cross-coupling can be applied in two ways to metal bis(dicarbollide) synthesis. First, B-iodo-carborane can be functionalized and deboronatated, then the resulting dicarbollides can be assembled from the functionalized nido-carboranes.11,57,58 Second, iodinated metal bis(dicarbollides) can be subjected to Kumada and Neigishi coupling to form B-C bonds, Fig. 9.59–62 Cross-coupling of 8-I-3–3’-Co(1,2-C2B9H10)(1’,2’-C2B9H11) with alkynes under Sonogishira conditions produced a tethered dicarbollide through an intramolecular B-H activation.60,62 A structurally similar metallaborane, the metallatricarbadecaboranyl, is also susceptible to Sonogishira coupling of trimethylsilyl, ester, and ferrocenyl acetylenes, Fig. 10.
Fig. 9.
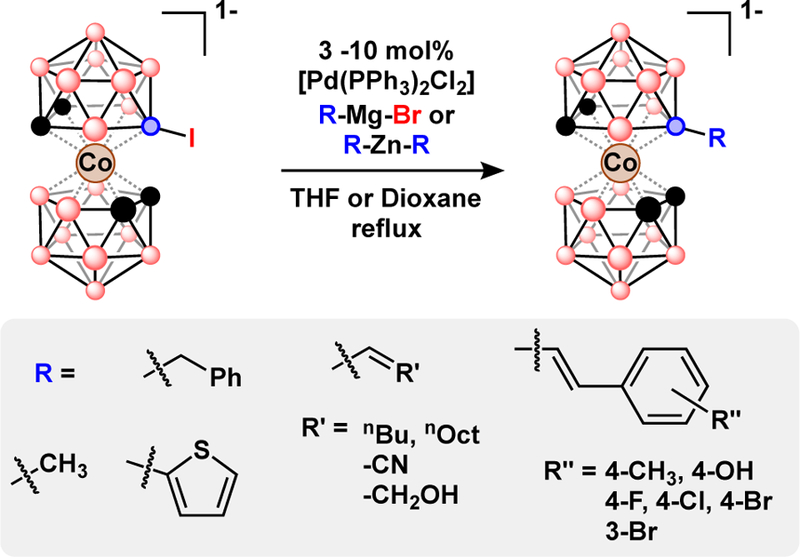
Pd-catalyzed Kumada and Negishi cross-coupling with B-iodo-cobalt bis(dicarbollide), [8-I-3,3’-Co(1,2-C2B9H10)(1’,2’-C2B9H11)]−.
Fig. 10.
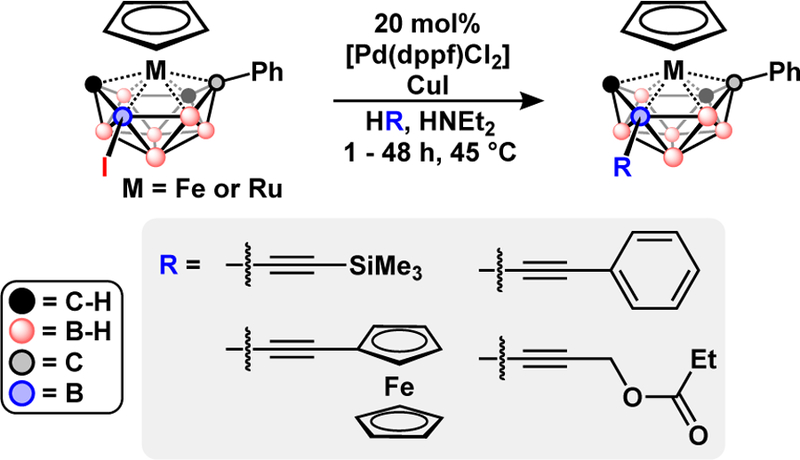
Pd-catalyzed Sonogishira cross-coupling of acetylenes with iodo-metallatricarbadecaborane.
Similar metal-catalyzed cross-coupling methods have been applied to less common metallaboranes. Neigishi coupling was demonstrated on the 6-vertex pentagonal-pyramidal nido-carboranes to attach ethynyl groups to the boron vertices.63,64 The resulting [Cp*Co(2,3-C2B4H3)] compounds were assembled into dimeric and trimeric multi-metallic compounds to study the electronic coupling between the [Cp*Co(2,3-C2B4H3)] metal-centers. Selective functionalization of different boron vertices of the (2,3-C2B4H3) ligands allowed subtle tuning of the electronic coupling between [Cp*Co(2,3-C2B4H3)] metal centers.64 In that work, two [Cp*Co(2,3-C2B4H3)] units were linked through a diethynyl bridge at two different boron vertices to form B(5)-B(5’) and B(7)-B(7’) bound dimers. Cyclic voltammetry revealed that the B(7)-B(7’) bound dimer has more Co-Co electronic communication than the B(5)-B(5’) bound dimer, further demonstrating the utility of precise B-vertex functionalization in the context of hierarchal design.
Metal-Catalyzed B-C Coupling Reactions with Polyhedral Borane Anions
Anionic polyhedral boranes are interesting compounds for applications in catalysis, as weakly coordinating anions, and as ionic materials such as ionic liquids, liquid crystals, or electrolytes. The anionic [CB11H12]1− and dianionic [B12H12]2− boranes are isoelectronic with the neutral C2B10H12 carboranes, and they exhibit higher reactivity towards electrophiles and halogens than the neutral carboranes.65 Anionic polyhedral boranes also undergo Kumada and Neigishi coupling of alkyl, aryl and alkynyl groups in the presence of [Pd(PPh3)2Cl2] and [Pd(PPh3)4] catalysts, Fig. 11.66–74 The reaction conditions used for cross-coupling with anionic polyhedral boranes closely resemble those reported for the neutral carborane cross-coupling, although the poor solubility of the anionic polyhedral boranes in organic solvents such as Et2O and toluene can lead to slower catalystic reactivity.
Fig 11.
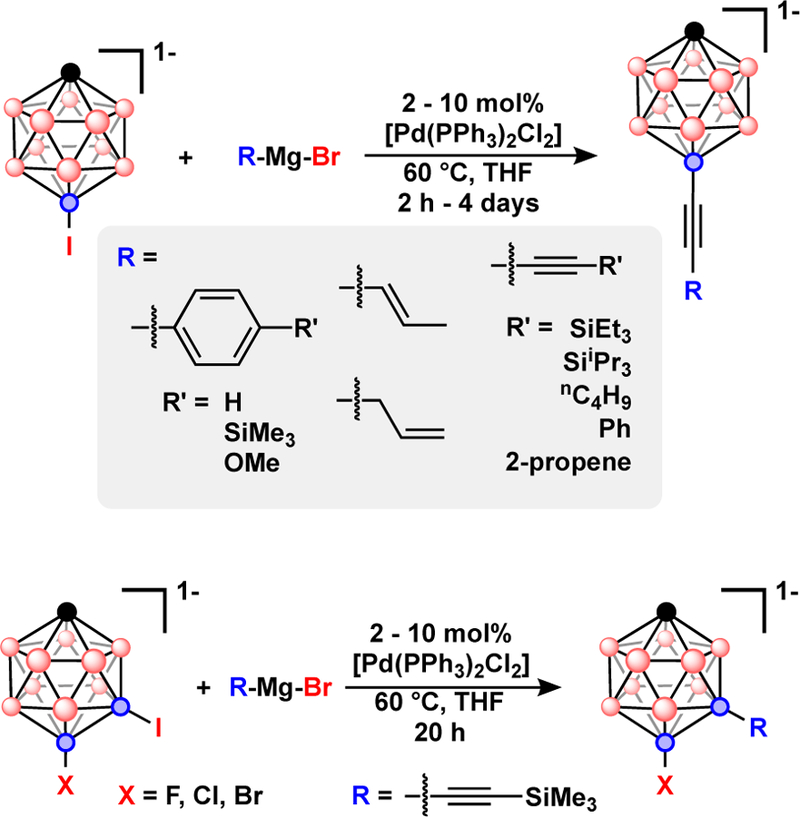
Top) Pd-catalyzed Kumada cross-coupling with [12-I-CB11H11]1−. Bottom) Halide selective Pd-catalyzed Kumada cross-coupling with [7-I-12-X-CB11H10]1− (X = F, Cl, or Br).
The 10- and 12-vertex monocarborane anions are also compatible with Grignard and organozinc reagents, which allows Pd-catalyzed cross-coupling of alkyl, alkenyl, alkynyl, and aryl groups. The 10-vertex [CB9H11]-1 carborane anions bearing amino-, diazo-, sulfonium-, and carboxylate groups tolerate Pd-catalyzed cross-coupling with alkyl groups using hexylmagnesium bromide and hexylzinc chloride as nucleophiles. In addition to B-C bond formation, of the Pd catalyst phosphine ligand to form B-P bonds was observed when the alkylzinc or alkylmagnesium halide was not present in excess.70,71
Kumada reactions also function with the dianionic [B10H9I]2 and [B12H11I]2− as the cross-coupling electrophiles to produce B-C bound alkyl and phenyl groups, Fig. 12.75–78 The metal-catalyzed B-C coupling reactions were adapted to the charge compensated 9-I-1,7-(Me2S)2-B12H9 with similar efficiency. Contrary to previous reports, a CuI co-catalyst was not necessary to achieve a high yield, however, the Grignard reagents used in this study did not possesses any β-hydrogens that can undergo β-hydride elimination as described previously.
Fig. 12.
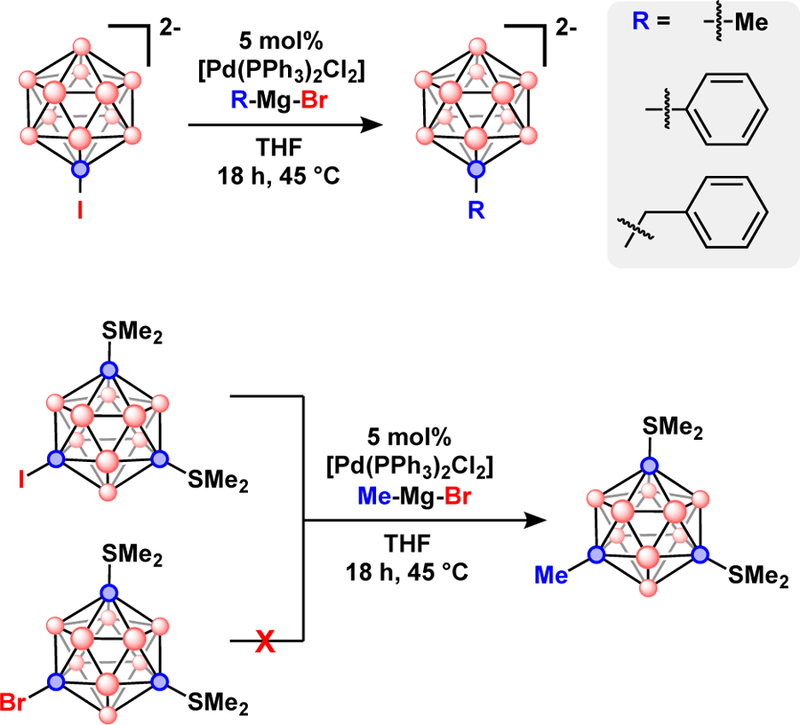
Top) Pd-catalyzed Kumada cross-coupling with [I-B12H11]2−. Bottom) Halide selective Pd-catalyzed Kumada cross-coupling with the charge compensate 9-X-1,7-(Me2S)2-B12H9 (X = Br, or I).
Interestingly, nido-4-I-7-Me3N-7-CB10H11 was successfully coupled with allylmagnesium bromide in the presence of [Pd(PPh3)4] to yield nido-4-CH2=CHCH2-7-Me3N-7-CB10H11.66 This represents a rare example of cross-coupling using a nido-carborane since they are known to coordinate Pd in an η5-fashion that can poison the metal catalyst.79–81
Lastly, cyanation of polyhedral boranes is of interest because the -CN group can be converted to other valuable synthons such as amides and amines. No direct electrophilic B-CN coupling methods have been reported, thus cyanation of polyhedral boranes proceeds via metal-catalyzed cross-coupling37,82,83 or nucleophilic displacement of phenyliodonium reagents.84 Notably, cyanation of [12-I-1-CB11H11]-1 with CuCN can proceed at 250 °C in the absence of a Pd catalyst, albeit use of Pd(PPh3)2Cl2 reduces the reaction time from 120 minutes to 15 min at 180 °C.85 Likewise, in the early studies by Trofimenko at DuPont, researchers showed that perhalogenated B12- and B10-based clusters can undergo partial substitution with cyanide when these compounds were irradiated with high-power UV light.86 No follow-up of this work has been reported since and the generality of photo-mediated polyhedral borate reactivity remain to be explored further.
Metal-Catalyzed B-N, B-O, B-S, B-P Bond Formation
The reports of B-C bond formation establish that Pd-catalyzed cross-coupling can be applied to several types of polyhedral boranes and many of the strategies used in aryl halide cross-coupling can be adapted to polyhedral boranes. Progress in metal-catalyzed boron-heteroatom coupling in polyhedral boranes closely tracks with the development of new catalyst systems for aryl halide cross-coupling. In the past two decades Buchwald-Hartwig amination reactions demonstrated a high substrate tolerance, and many effective ligand designs were developed.3,5 In comparison, most reports of Pd-catalyzed B-C coupling in polyhedral boranes employ triphenylphosphine or similar phosphines such as P(o-Tol)3 and Ph2P(CH2)nPPh2 {n = 1–3}. The recent emergence of metal-catalyzed B-N, B-O, and B-P coupling reactions has been driven by advances in catalyst ligand design. While these catalysts were originally developed for aryl-based systems, the selection of these catalysts for boron clusters is not straightforward, further underscoring the distinct electronic and steric properties of polyhedral boranes, Fig. 13.
Fig. 13.
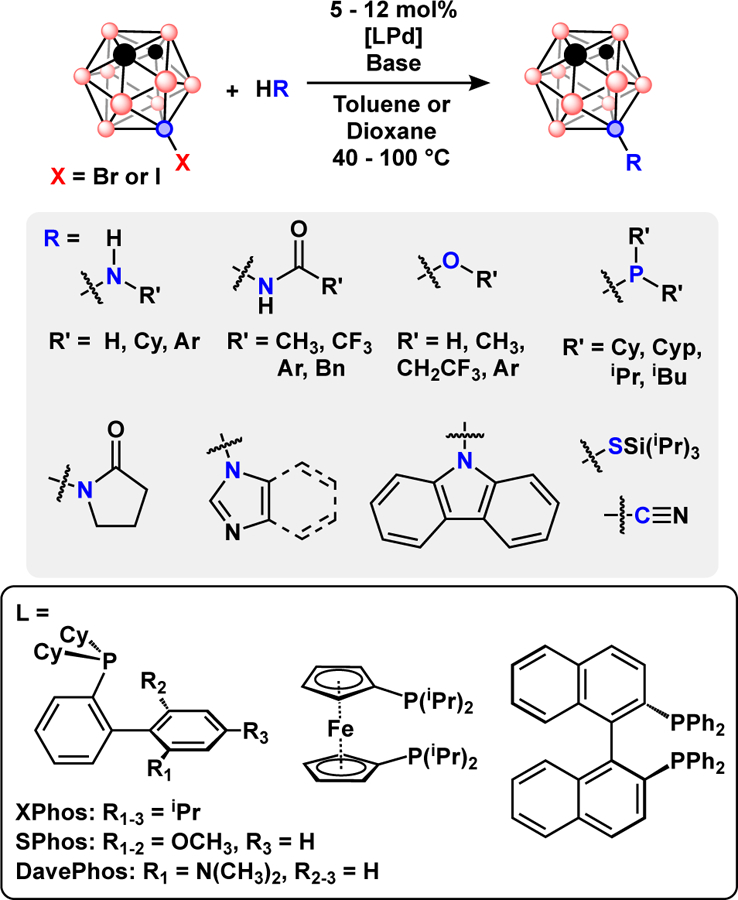
Top) Pd-catalyzed Buchwald-Hartwig with 9-I-m-C2B10H11, inset shows commonly used phosphine ligands for metal-catalyzed B-N, B-O, and B-P coupling.
Metal-Catalyzed B-N and B-O Coupling
The first report of catalytic carborane aminations was demonstrated on 2-I-p-C2B10H11 with imidazole, morpholine, and aromatic amines using Pd2dba3/BINAP as the catalyst and NatBuO as a base.87 Coincidently, B-OH coupling was observed as a significant by-product of these reactions which likely formed due to residual moisture present in the reagents (base or solvent). This report also examined the efficacy of other catalyst ligands such as PR3, IMes, and dialkyl biaryl phosphines and found that they were also capable of catalyzing B-N coupling, albeit with much lower yields. Later, the catalytic amidation of several B-iodo-C2B10H11 isomers was reported using the dialkyl biaryl phosphine (DavePhos) and K3PO= as the base.88
These works illuminate the critical role of the phosphine ligand in facilitating metal-catalyzed B-N coupling. When BINAP is used as a NaH base and day-long reaction times are required, whereas using DavePhos allows for a milder base (K3PO4) and only 2-hour reaction times. More importantly, a screening of dialkyl biaryl phosphines revealed that ditbutyl biaryl phosphines were particularly ineffective and it likely explains why the dialkyl biaryl phosphines examined by Beletskaya were not as effective as BINAP in amination reactions. Using the dicyclohexyl biaryl phosphines, SPhos and XPhos, allowed B-N and B-O coupling of B-bromo-carboranes with a diverse set of coupling partners.37,89 In order for B-bromo-carborane cross-coupling to occur it was important for the Pd precursor and phosphine ligand to be deployed as a precatalyst complex instead of using separate Pd(0) and phosphine sources.37 Similar dependence on catalyst precursor was observed in Heck coupling of styrenes with 2-I-p-C2B10H11, vide supra.52
Pd-catalyzed B-O coupling reactions are operationally similar to B-N amination and amidation reactions. When using the BINAP ligand strong base was required which limited the substrate scope to carboranes that are resistant to deboronation.90 In contrast, using SPhos allowed B-O coupling at the B(3) position (the position most susceptible to deboronation by strong nucleophiles) of 3-Br-o-C2B10H11.37 Moreover, the B-OH coupling reaction occurs in a 1:1 mixture of 1,4-dioxane:H2O, and these conditions can be further adapted to B-CN coupling using K4[Fe(CN)6] as a mild CN− source (vide supra).37
Metal-Catalyzed B-P and B-S Cross-Coupling
Forming boron-heteroatom bonds allowed the study of carboranes as tuneable functional groups. Although there are examples of B-P bond formation in polyhedral boranes, these routes require high temperatures or multi-step synthesis. First Pd-catalyzed B-P coupling was observed via B-H activation of [B10H10]2- and [B12H12]2− by [Pd(PMe2Ph)Cl2] which exchanged a B-H hydride for a PMe2Ph ligand.91 The resulting product was a mixture of mono- and disubstituted charge compensated phosphonium boranes, [1-PMe2-Ph-B12H11]−1 and 1,7-(PMe2Ph)2-B12H10. Similar reactions with [Pd(SMe2)Cl2] produced sulfonium boranes (1,7-(Me2S)2-B12H10).91
The first metal-catalyzed B-P cross-coupling in polyhedral boranes was observed as a by-product in Neigishi reactions when a 10-vertex B-iodo-carborane was coupled with a PCy3 ligand producing the zwitterionic 12-PCy3-1-COOH-1-CB9H8.70 Later, metal-catalyzed B-P cross-coupling with neutral carboranes was demonstrated between 9-I-m-C2B10H11 and diphenylphosphine using a Pd2dba3/DIPPF catalyst system.92 As with previous reports that use Pd2dba3 the reaction was sluggish with a 36-hour reaction time at 120 °C. Whether or not the resulting carboranyl phosphines further catalyzed the consumption of 9-I-m-C2B10H11 was undetermined. The B-bound carboranyl phosphines were much better electron donors than the C-bound isomers as determined from the decreased v[CO] in trans-Rh(PR3)2(CO)Cl, again showcasing the electron donating effects of bond through the boron vertices of carboranes. These studies established the effect of 9-meta-carboranyl substituent in the context of phosphine ligands and demonstrated that this substituent is more electron-donating than the majority of alkyl-based functional groups currently available.
B-S coupling via metal-catalyzed cross-coupling is a relatively new addition to the polyhedral borane chemists’ toolbox.93 This is because polyhedral borane B-H vertices can be converted to B-S groups through electrophilic routes.94 However, this limits the study of thio-carboranes to the vertices susceptible to electrophilic substitution. Pd-catalyzed cross-coupling of B-iodo-carboranes with HSSi(iPr)3 enables installation of a protected thiol group at electron-rich and electron-poor boron vertices.93 Self-assembled monolayers of 3-SH-o-C2B10H11 and 9-SH-o-C2B10H11 were deposited on gold and the static water contact angle of the SAMs was measured. The 3-SH-o-C2B10H11 SAM produced a contact angle of 80° versus the 9-SH-o-C2B10H11 SAM contact angle of 62°. This difference in contact angle is attributed to the orientation of the acidic C-H protons of 9-SH-o-carborane SAMs forming hydrogen bonds with the water droplet with leads to a smaller contact angle. There are a number of unexplored synthetic opportunities remaining for a broader utilization of metal-catalysed cross-coupling for the synthesis of boron clusters with appended thiol and other chalcogen units that would be valuable as building blocks for materials assembly.
Mechanistic Considerations of Metal-Catalyzed Polyhedral Borane Cross-Coupling
Choosing a Catalyst System
Clearly, new catalyst systems beyond Pd(PPh3)n {n = 1–3} were required to facilitate boron-heteroatom metal-catalyzed cross-coupling, and an effort was made to use phosphine ligands which can enforce a cis- coordination at the Pd-center. Phosphine ligands such as dppb, DIPPF, BINAP, and XantPhos were found to facilitate a variety of B-N, B-O, and B-P bond forming reactions.37,87,88,90,92,95 Notably, these works feature a broader catalyst screen which showed that electron-rich ligands such as dialkyl biaryl phosphines, PCy3, PtBu3, and IMes have some activity towards B-N and B-O bond formation.87 Furthermore, we demonstrated that, with an appropriately electron-rich ligand, B-bromo-carboranes can be excellent cross-coupling substrates.37 This advance is also attributed to the use of Pd(II) palladacycle precatalysts, which deploy the active catalyst species without releasing inhibitory ligands such as dba.96,97 The dba ligand is known to coordinate to palladium phosphine complexes thereby supressing the rate of oxidative addition to aryl halides. A similar effect is likely in play during cross-coupling chemistry when halo-carboranes are used as coupling partners and it would work against the presumably slow oxidative addition into B-Br bonds of B-bromo-carboranes.37
Oxidative Addition and Transmetallation
As mentioned before, most polyhedral borane cross-couplings use B-iodo-boranes, while B-bromo- and B-chloro-boranes until recently have been viewed as too unreactive. Attempts at isolating oxidative addition compounds of B-iodo-carborane with Pd complexes were unsuccessful, presumably due to the equilibrium strongly disfavouring oxidative addition.34 Surrogate oxidative addition complexes were prepared with Pt and a pseudo-oxidative addition Pd-Hg bimetallic provide clues about the possible nature of catalytically relevant intermediates98
The calculated Connolly volume of carboranes is ~30% larger than benzene and they are spherical, as opposed to planar aromatic molecules.15,18,99 This means that carboranes are a much more sterically demanding substrate at the catalyst site than their planar aromatic analogues. Several reports of Pd-catalyzed amination of carborane feature similar dialkylbiaryl phosphine ligands albeit with dramatically different results.37,87–89 Phosphines with -tBu groups showed poor conversion of B-I to B-N bonds,87 whereas phosphines with cyclohexyl groups gave complete conversion in a matter of hours instead of days.37,88,89
Although electron-rich dialkyl biaryl phosphines have been used as ligands in B-bromo-carborane Pd-catalyzed cross-coupling, these Pd catalysts have not been able to active the B-Cl bond of B-chloro-carboranes. Despite the inability to perform oxidative addition into B-Cl bonds, stoichiometric studies of the above mentioned Hg-Pd bimetallic complex show that B-chloro-carboranes can undergo Pd-catalyzed transmetallation to form B-N bonds.98 This is based on a proposed oxidative addition complex that could form upon extrusion of Hg from the Hg-Pd bimetallic complex. The proposed oxidative addition complex quickly reductively eliminates B-chloro-carborane. However, in the presence of excess nucleophile, transmetallation can occur resulting in the formation of a B-N bond. This suggests that the oxidative addition complex formed with B-chloro-carborane is unstable and quickly reductively eliminates the B-chloro-carborane unless it is trapped by a nucleophile.
Catalyst systems that can perform oxidative addition into C-Br and C-Cl bonds exhibit much higher metal-catalyzed cross-coupling efficiency than aryl iodides. It is believed that weaker Pd-Br and Pd-Cl bonds enable faster transmetallation at the expense of slower oxidative addition rates. The lack of isolable carborane oxidative addition complexes have made the study of carborane transmetallation rates challenging.34 Despite this, slow transmetallation with B-iodo-carboranes has been implicated in several cross-coupling reactions. As mentioned above, Heck coupling of styrenes bearing electron withdrawing groups with 2-I-p-C2B10H11 required much longer reaction times and some did not participate in the reaction at all.52
Isomerization via Cage-Walking
The use of dialkyl biaryl phosphine ligands for Pd-catalyzed cross-coupling of B-bromo-carboranes lead to the recent discovery of a unique isomerization mechanism, “cage-walking”, similar to the chain-walking isomerization observed in carbon-based molecules.89 The “cage-walking” isomerization enables metal-catalyzed cross-coupling to occur at vertices far from the initial location of B-Br bond activation, Fig. 15. This “off-cycle” mechanism is currently the only way to attach functional groups at the B(5) and B(4) positions of meta-carborane without relying on thermal isomerization processes. Whether the “cage-walking” mechanism was operative depended on the steric hindrance produced at the Pd metal center by the phosphine ligand. Using the bulkier XPhos ligand produced significantly more “cage-walking” regioisomers than the smaller DavePhos and SPhos ligands which showed cross-coupling mostly at the B(9) position.
Fig. 15.
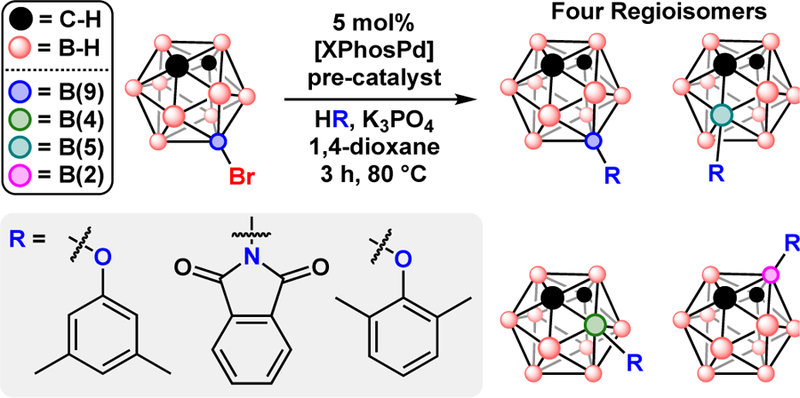
Four regioisomers of R-meta-carborane produced by Pd-catalyzed “cage-walking”.
Besides relying on ligand sterics, the “cage-walking” regioisomer distribution can be shifted from favouring B(9) to favouring B(2) substitution by changing the steric profile at the nucleophilic position of the cross-coupling partner. Between two phenols, 3,5-dimethylphenol and 2,6-dimethylphenol, the more sterically hindered 2,6-dimethylphenol yielded the B(2) isomer as the major product while the less hindered 3,5-dimethylphenol yielded an equal distribution of regioisomers.
The isomer distribution is believed to depend on the transmetallation step, where steric bulk at the Pd center impedes transmetallation and allows the “cage-walking” process to generate more reactive B-bromo-carborane isomers, Fig. 14. Since the carboranyl fragment has an anisotropic electron distribution, as the carborane “cage-walks” to electron deficient vertices, B(2) vs. B(9), it produces an increasingly cationic Pd metal center that is more likely to overcome the steric repulsion between the catalyst complex and nucleophilic cross-coupling partner.
Fig. 14.
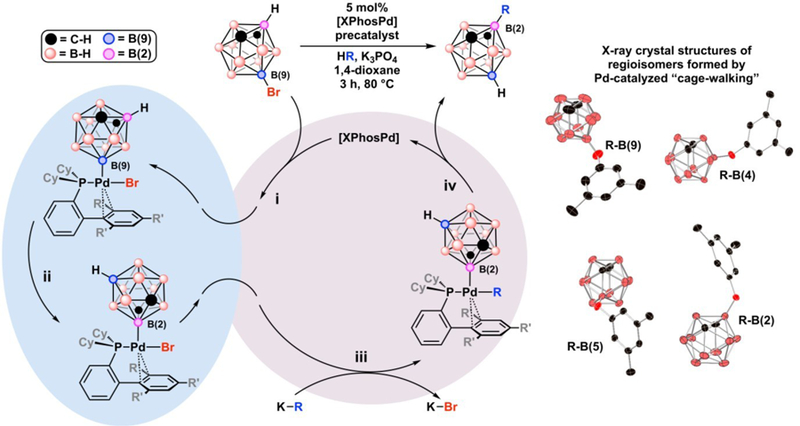
Proposed cross-coupling cycle for the formation of “cage-walking” products, Step i. oxidative addition, Step ii. “cage-walking” isomerization, Step iii. Transmetallation, Step iv. Reductive elimination. X-ray crystal structures of “cage-walking” regioisomers of B-3,5-dimethylphenoxy-m-C2B10H11.
Metal-Catalyzed Cross-Coupling of Polyhalogenated Boron Clusters
Aside from their unique 3D aromaticity and chemical stability, the spherical shape of polyhedral boranes presents a molecular scaffold for building three-dimensionally defined constructs. For example, functionalization of three vertices that share a common face creates a tripodal architecture that is not easily accessible with typical organic building blocks. Interest in creating polysubstituted polyhedral boranes lead to exploration of Pd-catalyzed cross-coupling methods for attaching several functional groups onto polyhalogenated boranes.
Pd-catalyzed cross-coupling of disubstituted carborane was first reported for the Kumada cross-coupling of B-diiodo-carboranes, I2-C2B10H 10,41,48 then extended to anionic69,72,73 and dianionic boranes.76,85 Later Hawthorne and co-workers reported the Pd-catalyzed methylation of hexaiodo cobalt bis(dicarbollide) with MeMgBr to produce hexamethyl cobalt bis(dicarbollide), Fig 16.59 The most metal-catalyzed cross-coupling reactions performed on a single polyhedral borane was reported on the [1-H-CB9I9]−1 anion where all nine B-I bonds were cross-coupling with MeMgBr in the presenc [Pd(PPh3)2Cl2] over the course of 10 days.100 These examples demonstrate the feasibility of functionalizing an entire face of a polyhedral borane using cross-coupling reactions. Such Pd-catalyzed polysubstitution was applied to B-tetraiodo-ortho-carborane (8,9,10,12-I4-o-C2B10H8) to synthesize 8,10-Ph2-9,12-I2-o-C2B10H8 and 8,9,10,12-(allyl)4-o-C2B10H8. Noteworthy was the selective arylation of the B(8) and B(10) vertices when using phenylmagnesium bromide as a cross-coupling partner.101 This selectivity was attributed to the steric bulk of the phenyl substituents which may have prevented functionalization of the more sterically hindered B(9) and B(12) vertices. Similar steric restrictions were observed for the Pd-catalyzed cross-coupling of the hexaiodo monocarborane anion, [1-Ph-7,8,9,10,11,12-I5-CB11H5]−, where cross-coupling occurred at the less sterically congested B(7–11) positions, but not at the B(12) position, to yield [1-Ph-7,8,9,10,11-(tolyl)5-12-I-CB11H5]−.102
Fig. 16.
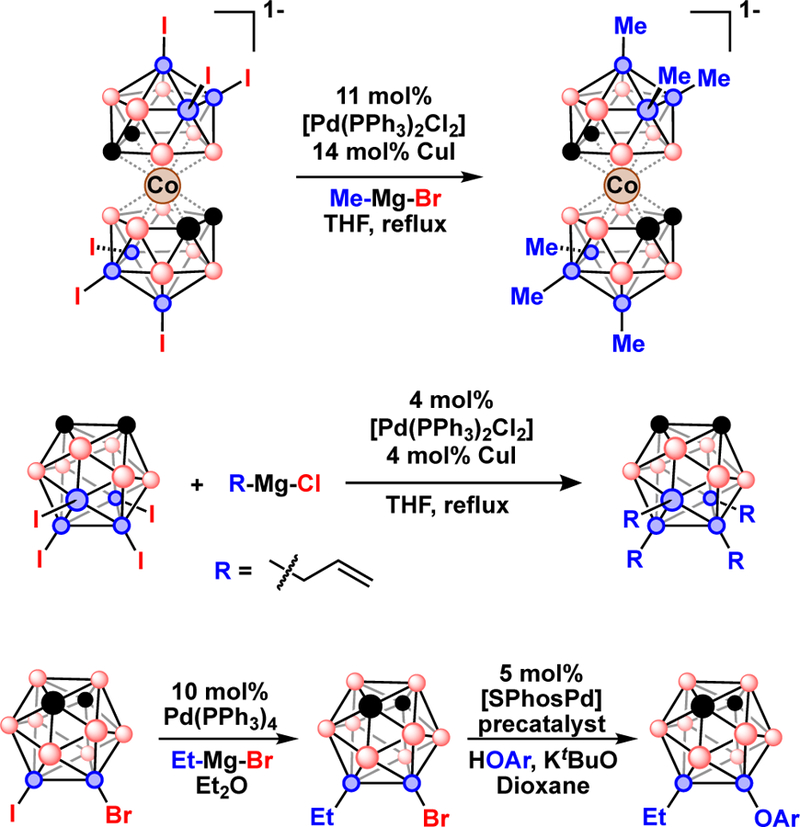
Top) Pd-catalyzed methylation of [3,3’-commo-(8,9,12-I3-CoC2B9H8)2]. Middle) Pd-catalyzed alkylation of 8,9,10,12-I4-o-C2B10H8 to produce 8,9,10,12-(allyl)4-o-C2B10H8. Bottom) Sequential Pd-catalyzed B-I then B-Br bond cross-coupling of 9-I-10-Br-m-C2B10H10.
Recently we demonstrated the first example of a sequential metal-catalyzed B-C coupling at a B-I vertex followed by B-O coupling at a B-Br vertex by using different phosphine ligands for Pd-catalyzed B-I and B-Br bond activation, Fig. 16.37 In this work [Pd(PPh3)2Cl2] was used to selectively perform Kumada B-C coupling at the B-I vertex of 9-I-10-B-m-C2B10H10 to produce 9-Et-10-Br-m-C2B10H10 which was then subjected to Buchwald-Hartwig B-O coupling at the B-Br vertex using [SPhosPd] to produce 9-Et-10-(O-3,5-dimethylphenyl)-m-C2B10H10.
An alternative approach to heterodifunctionalization may be realized by relying on a chelate effect to limit the number of functional groups that get installed. Such a chelate effect is believed to limit metal-catalyzed B-O coupling of OH- to a single B-Br vertex of 9,10-Br2-m-C2B10H10 to produce the monohydroxylated compound 9-Br-10-OH-m-C2B10H10 whereas coupling of 3,5-dimethylphenolate afforded the disubstituted product. This difference in reactivity may also be due to the steric bulk of the phenolate, which may reduce the propensity towards chelating the Pd center, and the possible deprotonation of the hydroxyl group of 9-Br-10-OH-m-C2B10H10 may lead to a stronger chelate effect. Alternatively, chelation of a metal catalyst to a polyhedral borane can allow functionalization of B-H vertices adjacent to the chelating moiety. This approach was used to install five aryl groups onto [1-COOH-CB11H11]1− at B-H vertices adjacent to the carboxylic acid directing group metal-catalyzed B-H activation.29 Similar metal-catalyzed B-H polyfunctionalization strategies were reported for 10- and 12-vertex polyhedral boranes.24,31,33
Conclusions and Outlook
Metal-catalyzed cross-coupling is emerging as a powerful method for installing functional groups at the boron vertices of polyhedral boranes. A broad body of work has demonstrated that cross-coupling strategies are applicable to several classes of polyhedral boranes.103 Subsequently, metal-catalyzed cross-coupling of polyhedral boranes enabled their use in materials science, organometallic chemistry, surface science, and medicinal chemistry, Fig. 17. For example, the vertex specificity of metal-catalyzed cross-coupling allowed the synthesis of a linear cobalt bis(dicarbollide) used to construct metal-organic frameworks with high methane uptake capacity.11 Additionally, the vertex specific cross-coupling was used to control the dipole moment orientation of surface adsorbed carborane thiols and carboxylic acids on Au and Ge surfaces, respectively.93,104 Using polyhedral boranes as surface ligands allowed nearly defect free self-assembly and tuning of surface properties such as work function and wettability. Similarly, the vertex specific cross-coupling of B-iodo-carboranes enabled synthesis of a panel of carborane-based anethetics (“boronicaines”) analogous to lidocaine to study their molecular docking characteristics relative to lidocaine and an adamanyl-based analog. Noteworthy was the ability to control the duration and efficacy of the boronicaine isomers which surpassed that of their phenyl- and adamantly-based analogues.105
Fig. 17.
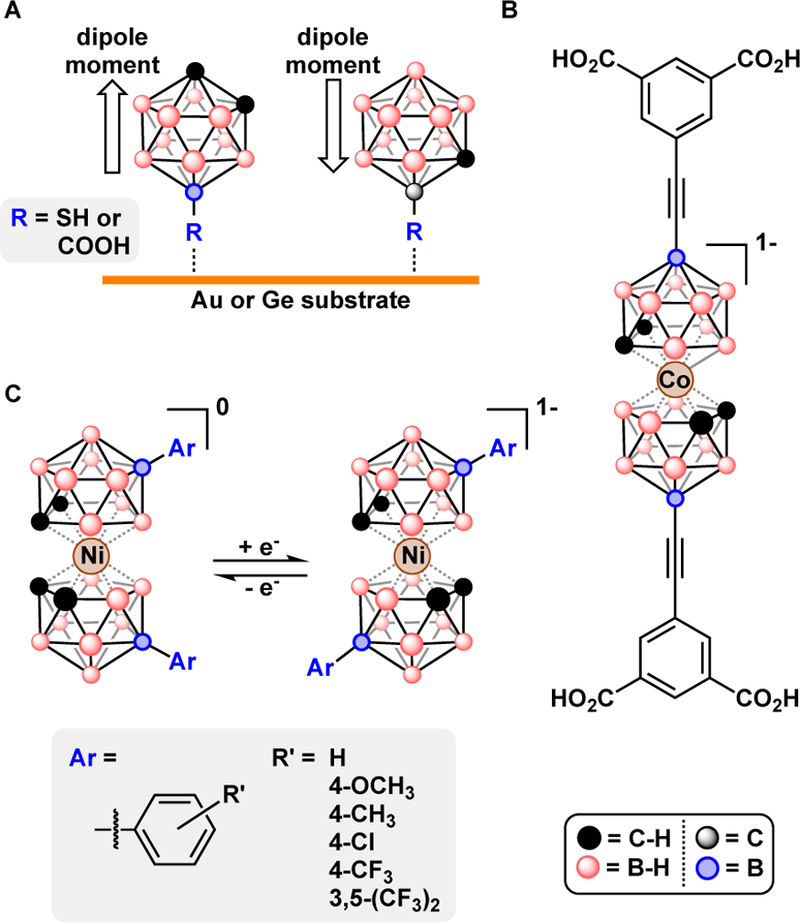
A) Functionalized o-carborane thiols and carboxylic acids used for self-assembled monolayers on gold and germanium surfaces, respectively. B) A linear cobalt bis(dicarbollide) used for constructing metal-organic frameworks. C) Arylated nickel bis(dicarbollide)s used as redox shuttles in dye-sensitized solar cells.
Furthermore, the broad functional group tolerance of metal-catalyzed cross-coupling allowed study of structure-activity relationships in arylated nickel bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Researchers showed that by tuning the inductive effects of the aryl groups attached to the cobalt bis(dicarbollide) cage can tune the electrochemical NiIII/IV redox couple and the resulting open circuit voltage of dye-sensitized solar cells.57 Recently, our group demonstrated control of the linkage isomerism of a bis(o-carborane) ligand to preferentially bind in a κ2-B,C-binding mode in phosphorescent Pt(II) coordination complexes by addition of ethyl groups to bis(o-carborane) to form B-tetraethyl-bis(o-carborane), 9,9’,12,12’-Et4-1,1’-bis(o-C2B10H9).106 Such differences in the κ 2 binding mode of bis(o-carborane) (κ 2-B,C- vs. κ2-C,C-bound) manifest as subtle differences in the electrochemical redox potential and luminescent properties of Pt(II) coordination complexes.107
Many of the new cross-coupling methods for polyhedral boranes leverage the growing number of aryl halide cross-coupling catalysts to generate B-C, B-N, B-O, B-S, and B-P bonds. These examples of metal-catalyzed boron-heteroatom coupling may provide clues in the future to generate B-B and B-Si bonds comparable to the C-B and C-Si bonds reported for aryl halides. Notably, while carborane species with exopolyhedral B-B bonds were recently synthesized via a B-H activation route,108 B-Si analogues have remained elusive. Progress in these topics opens opportunities for preparing polyhedral borane nucleophilic cross-coupling partners108 and the possibility of cross-coupling polyhedral boranes with each other providing access to a new class of linked polyboranes. Another outstanding challenge in polyhedral borane cross- coupling chemistry is the inactivity of B-Cl bonds and B-OTf bonds toward oxidative addition. And a new question arises, what type of catalyst system has the right electronic and steric features to activate B-Cl and B-OTf bonds? We note that Ni-based catalyst systems have been recently developed to activate strong bonds in organic substrates (e.g. C-N, C-O) and these and/or similar methods could be applied to solve this challenge.109–114 Furthermore, metal-mediated photoredox catalysis on boron cluster electrophiles has not been reported, albeit such reaction manifolds are well documented in 2D aromatic molecule literature.115
It is important to recognize that B-halo-carboranes are similar, but different, from their 2D aromatic aryl halides. In the context of metal-catalyzed cross-coupling the spheroidal steric bulk and stronger B-X (X = Cl, Br, I) bonds differentiate polyhedral borane cross-coupling from hydrocarbon cross-coupling. Although this difference in reactivity stymied polyhedral borane cross-coupling, it also gives rise to functionalization strategies that are not readily accessible in hydrocarbon chemistry. Namely the orthogonal reactivity toward B-X bonds with different catalysts and “cage-walking” isomerization open avenues to new boron cluster molecules and advance our ability to use polyhedral boranes as scaffolds for unique three-dimensionally defined building blocks.
Acknowledgements
A.M.S. thanks the UCLA Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, the Alfred P. Sloan Foundation for a Fellowship in Chemistry, Research Corporation for Science Advancement (RCSA) for a Cottrell Scholar Award, and the National Institutes of Health (NIH) for a Maximizing Investigators Research Award (MIRA, R35GM124746). R.M.D. is grateful for support from the Department of Defense through the National Defense Science and Engineering Graduate Fellowship. We thank all past and present co-workers at UCLA for contributing to the work highlighted in this article.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.de Meijere A and Diederich F, Eds., Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2004. [Google Scholar]
- 2.Johansson Seechurn CCC, Kitching MO, Colacot TJ and Snieckus V, Angew. Chemie Int. Ed, 2012, 51, 5062–5085. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Castillo P and Buchwald SL, Chem. Rev, 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biffis A, Centomo P, Del Zotto A and Zecca M, Chem. Rev, 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- 5.Hartwig JF, Acc. Chem. Res, 2008, 41, 1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Twilton J, Le CC, Zhang P, Shaw MH, Evans RW and MacMillan DWC, Nat. Rev. Chem, 2017, 1, 0052. [Google Scholar]
- 7.Beletskaya IP and Ananikov VP, Chem. Rev, 2011, 111, 1596–1636. [DOI] [PubMed] [Google Scholar]
- 8.King RB, Chem. Rev, 2001, 101, 1119–1152. [DOI] [PubMed] [Google Scholar]
- 9.Zakharkin LI and Pisareva IV, Izv. Akad. Nauk SSSR Ser. Khim, 1978, 252.
- 10.Heck RF, J. Am. Chem. Soc, 1968, 90, 5546–5548. [Google Scholar]
- 11.Kennedy RD, Clingerman DJ, Morris W, Wilmer CE, Sarjeant AA, Stern CL, O’Keeffe M, Snurr RQ, Hupp JT, Farha OK and Mirkin CA, Cryst. Growth Des, 2014, 14, 1324–1330. [Google Scholar]
- 12.Grimes RN, Angew. Chemie Int. Ed. English, 1993, 32, 1289–1290. [Google Scholar]
- 13.Badr IHA, Diaz M, Hawthorne MF and Bachas LG, Anal. Chem, 1999, 71, 1371–1377. [DOI] [PubMed] [Google Scholar]
- 14.Axtell JC, Saleh LMA, Qian EA, Wixtrom AI and Spokoyny AM, Inorg. Chem, 2018, 57, 2333–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholz M and Hey-Hawkins E, Chem. Rev, 2011, 111, 7035–7062. [DOI] [PubMed] [Google Scholar]
- 16.Issa F, Kassiou M and Rendina LM, Chem. Rev, 2011, 111, 5701–5722. [DOI] [PubMed] [Google Scholar]
- 17.Leśnikowski ZJ, J. Med. Chem, 2016, 59, 7738–7758. [DOI] [PubMed] [Google Scholar]
- 18.Ristori S, Oberdisse J, Grillo I, Donati A and Spalla O, Biophys. J, 2005, 88, 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otsuka M, Takita R, Kanazawa J, Miyamoto K, Muranaka A and Uchiyama M, J. Am. Chem. Soc, 2015, 137, 15082–15085. [DOI] [PubMed] [Google Scholar]
- 20.Bregadze VI, Chem. Rev, 1992, 92, 209–223. [Google Scholar]
- 21.Qiu Z, Tetrahedron Lett, 2015, 56, 963–971. [Google Scholar]
- 22.Hoel EL and Hawthorne MF, J. Am. Chem. Soc, 1974, 96, 6770–6771. [Google Scholar]
- 23.Mirabelli MGL and Sneddon LG, J. Am. Chem. Soc, 1988, 110, 449–453. [Google Scholar]
- 24.Quan Y, Qiu Z and Xie Z, Chem. - A Eur. J, 2018, 24, 2795–2805. [DOI] [PubMed] [Google Scholar]
- 25.Duttwyler S, Pure Appl. Chem, 2018, 90, 733–744. [Google Scholar]
- 26.Zhang Y, Sun Y, Lin F, Liu J and Duttwyler S, Angew. Chemie - Int. Ed, 2016, 55, 15609–15614. [DOI] [PubMed] [Google Scholar]
- 27.Lyu H, Quan Y and Xie Z, Chem. - A Eur. J, 2017, 23, 14866–14871. [DOI] [PubMed] [Google Scholar]
- 28.Cheng R, Li B, Wu J, Zhang J, Qiu Z, Tang W, You SL, Tang Y and Xie Z, J. Am. Chem. Soc, 2018, 140, 4508–4511. [DOI] [PubMed] [Google Scholar]
- 29.Lin F, Yu J-L, Shen Y, Zhang S-Q, Spingler B, Liu J, Hong X and Duttwyler S, J. Am. Chem. Soc, 2018, 140, 13798–13807. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Wang T, Wang L, Sun Y, Lin F, Liu J and Duttwyler S, Chem. - A Eur. J, 2018, 24, 15812–15817. [DOI] [PubMed] [Google Scholar]
- 31.Liang X, Shen Y, Zhang K, Liu J and Duttwyler S, Chem. Commun, 2018, 9–12. [DOI] [PubMed]
- 32.Eleazer BJ, Smith MD and Peryshkov DV, Organometallics, 2016, 35, 106–112. [Google Scholar]
- 33.Eleazer BJ, Smith MD and Peryshkov DV, J. Organomet. Chem, 2017, 829, 42–47. [Google Scholar]
- 34.Marshall WJ, Young RJ and Grushin VV, Organometallics, 2001, 20, 523–533. [Google Scholar]
- 35.Winberg KJ, Mume E, Tolmachev V and Sjöberg S, J. Label. Compd. Radiopharm, 2005, 48, 195–202. [Google Scholar]
- 36.Ishita K, Khalil A, Tiwari R, Gallucci J and Tjarks W, Eur. J. Inorg. Chem, 2018, 2018, 2821–2825. [Google Scholar]
- 37.Dziedzic RM, Saleh LMA, Axtell JC, Martin JL, Stevens SL, Royappa AT, Rheingold AL and Spokoyny AM, J. Am. Chem. Soc, 2016, 138, 9081–9084. [DOI] [PubMed] [Google Scholar]
- 38.Zakharkin LII, Kovredov AII, Ol’shevskaya VAA and Shaugumbekova ZSS, J. Organomet. Chem, 1982, 226, 217–222. [Google Scholar]
- 39.Zakharkin LI, Kovredov AI, Ol’shevskaya VA, Ol VA and Constants P, Bull. Acad. Sci. USSR, Div. Chem. Sci, 1985, 34, 809–812. [Google Scholar]
- 40.Zheng Z, Jiang W, Zinn AA, Knobler CB and Hawthorne MF, Inorg. Chem, 1995, 34, 2095–2100. [Google Scholar]
- 41.Li J, Logan CF and Jones M, Inorg. Chem, 1991, 30, 4866–4868. [Google Scholar]
- 42.Beletskaya IP, Bregadze VI, Ivushkin VA, Petrovskii PV, Sivaev IB, Sjöberg S and Zhigareva GG, J. Organomet. Chem, 2004, 689, 2920–2929. [Google Scholar]
- 43.Tamao K, Sumitani K, Kiso Y, Zembayashi M, Fujioka A, Kodama S, Nakajima I, Minato A and Kumada M, Bull. Chem. Soc. Jpn, 1976, 49, 1958–1969. [Google Scholar]
- 44.Zask A and Helquist P, J. Org. Chem, 1978, 43, 1619–1620. [Google Scholar]
- 45.Viciu MS, Grasa GA and Nolan SP, Organometallics, 2001, 20, 3607–3612. [Google Scholar]
- 46.Anderson KP, Mills HA, Mao C, Kirlikovali KO, Axtell JC, Rheingold AL and Spokoyny A, 10.26434/chemrxiv.7252586.v1. [DOI] [PMC free article] [PubMed]
- 47.Yang X, Knobler CB, Zheng Z and Hawthorne MF, J. Am. Chem. Soc, 1994, 116, 7142–7159. [Google Scholar]
- 48.Jiang W, Knobler CB, Curtis CE, Mortimer MD and Frederick Hawthorne M, Inorg. Chem, 1995, 34, 3491–3498. [Google Scholar]
- 49.Herzog A, Jalisatgi SS, Knobler CB, Wedge TJ and Hawthorne MF, Chem.--Eur. J, 2005, 11, 7155–7174. [DOI] [PubMed] [Google Scholar]
- 50.Beletskaya IP, Bregadze VI, Ivushkin VA, Zhigareva GG, Petrovskii PV and Sivaev IB, Russ. J. Org. Chem, 2005, 41, 1359–1366. [Google Scholar]
- 51.Fox MA and Wade K, J. Organomet. Chem, 1999, 573, 279–291. [Google Scholar]
- 52.Eriksson L, Winberg KJ, Claro RT and Sjöberg S, J. Org. Chem, 2003, 68, 3569–3573. [DOI] [PubMed] [Google Scholar]
- 53.Endo Y, Aizawa K and Ohta K, Heterocycles, 2010, 80, 369. [Google Scholar]
- 54.Lennox AJJ and Lloyd-Jones GC, Angew. Chemie - Int. Ed, 2013, 52, 7362–7370. [DOI] [PubMed] [Google Scholar]
- 55.Sivaev IB and Bregadze VI, Collect. Czechoslov. Chem. Commun, 1999, 64, 783–805. [Google Scholar]
- 56.Dash BP, Satapathy R, Swain BR, Mahanta CS, Jena BB and Hosmane NS, J. Organomet. Chem, 2017, 849–850, 170–194. [Google Scholar]
- 57.Spokoyny AM, Li TC, Farha OK, Machan CW, She C, Stern CL, Marks TJ, Hupp JT and Mirkin CA, Angew. Chemie - Int. Ed, 2010, 49, 5339–5343. [DOI] [PubMed] [Google Scholar]
- 58.Safronov AV, Sevryugina YV, Pichaandi KR, Jalisatgi SS and Hawthorne MF, Dalt. Trans, 2014, 43, 4969. [DOI] [PubMed] [Google Scholar]
- 59.Mortimer MD, Knobler CB and Hawthorne MF, Inorg. Chem, 1996, 35, 5750–5751. [DOI] [PubMed] [Google Scholar]
- 60.Rojo I, Teixidor F, Kivekäs R, Sillanpää R and Viñas C, J. Am. Chem. Soc, 2003, 125, 14720–14721. [DOI] [PubMed] [Google Scholar]
- 61.Rojo I, Teixidor F, Viñas C, Kivekäs R and Sillanpää R, Chem. - A Eur. J, 2003, 9, 4311–4323. [DOI] [PubMed] [Google Scholar]
- 62.Farràs P, Olid-Britos D, Viñas C and Teixidor F, Eur. J. Inorg. Chem, 2011, 2011, 2525–2532. [Google Scholar]
- 63.Russell JM, Sabat M and Grimes RN, Organometallics, 2002, 21, 4113–4128. [Google Scholar]
- 64.Yao H, Sabat M, Grimes RN, Zanello P and Fabrizi de Biani F, Organometallics, 2003, 22, 2581–2593. [Google Scholar]
- 65.Jelínek T, Baldwin P, Scheidt WR and Reed CA, Inorg. Chem, 1993, 32, 1982–1990. [Google Scholar]
- 66.Morris JH, Henderson W and Ol’shevskaya VA, J. Chem. Soc., Dalt. Trans, 1998, 1951–1959.
- 67.Grüner B, Janoušek Z, King BT, Woodford JN, Wang CH, Všetečka V and Michl J, J. Am. Chem. Soc, 1999, 121, 3122–3126. [Google Scholar]
- 68.Franken A, Kilner CA, Thornton-Pett M and Kennedy JD, J. Organomet. Chem, 2002, 657, 176–179. [Google Scholar]
- 69.Finze M, Inorg. Chem, 2008, 47, 11857–11867. [DOI] [PubMed] [Google Scholar]
- 70.Ringstrand B, Monobe H and Kaszynski P, J. Mater. Chem, 2009, 19, 4805. [Google Scholar]
- 71.Ringstrand B, Kaszynski P, Januszko A and Young VG, J. Mater. Chem, 2009, 19, 9204–9212. [Google Scholar]
- 72.Himmelspach A and Finze M, J. Organomet. Chem, 2010, 695, 1337–1345. [Google Scholar]
- 73.Himmelspach A, Reiss GJ and Finze M, Inorg. Chem, 2012, 51, 2679–2688. [DOI] [PubMed] [Google Scholar]
- 74.Hailmann M, Konieczka SZ, Himmelspach A, Löblein J, Reiss GJ and Finze M, Inorg. Chem, 2014, 53, 9385–9399. [DOI] [PubMed] [Google Scholar]
- 75.Peymann T, Knobler CB and Hawthorne MF, Inorg. Chem, 1998, 37, 1544–1548. [Google Scholar]
- 76.Kultyshev RG, Liu S, Leung HT, Liu J and Shore SG, Inorg. Chem, 2003, 42, 3199–3207. [DOI] [PubMed] [Google Scholar]
- 77.Bernard R, Cornu D, Luneau D, Naoufal D, Scharff JP and Miele P, J. Organomet. Chem, 2005, 690, 2745–2749. [Google Scholar]
- 78.Rzeszotarska E, Novozhilova I and Kaszyński P, Inorg. Chem, 2017, 56, 14351–14356. [DOI] [PubMed] [Google Scholar]
- 79.Warren L Jr and Hawthorne M, J. Am. Chem, 1968, 90, 4823–4828. [Google Scholar]
- 80.Jasper SA, Huffman JC and Todd LJ, Inorg. Chem, 1995, 34, 6430–6439. [DOI] [PubMed] [Google Scholar]
- 81.Jasper SA, Huffman JC and Todd LJ, Inorg. Chem, 1998, 37, 6060–6064. [DOI] [PubMed] [Google Scholar]
- 82.Rosenbaum AJ, Juers DH and Juhasz MA, Inorg. Chem, 2013, 52, 10717–10719. [DOI] [PubMed] [Google Scholar]
- 83.Wingen LM and Scholz MS, Inorg. Chem, 2016, 55, 8274–8276. [DOI] [PubMed] [Google Scholar]
- 84.Kaszyński P and Ringstrand B, Angew. Chemie - Int. Ed, 2015, 54, 6576–6581. [DOI] [PubMed] [Google Scholar]
- 85.Dwulet GE and Juhasz MA, Inorg. Chem. Commun, 2015, 51, 26–28. [Google Scholar]
- 86. U.S. Pat., 3,373,098, 1968.
- 87.Beletskaya IP, Bregadze VI, Kabytaev KZ, Zhigareva GG, Petrovskii PV, Glukhov IV and Starikova ZA, Organometallics, 2007, 26, 2340–2347. [Google Scholar]
- 88.Sevryugina Y, Julius RL and Hawthorne MF, Inorg. Chem, 2010, 49, 10627–10634. [DOI] [PubMed] [Google Scholar]
- 89.Dziedzic RM, Martin JL, Axtell JC, Saleh LMA, Ong TC, Yang YF, Messina MS, Rheingold AL, Houk KN and Spokoyny AM, J. Am. Chem. Soc, 2017, 139, 7729–7732. [DOI] [PubMed] [Google Scholar]
- 90.Kabytaev KZ, Mukhin SN, Glukhov IV, Starikova ZA, Bregadze VI and Beletskaya IP, Organometallics, 2009, 28, 4758–4763. [Google Scholar]
- 91.Jasper SA, Jones RB, Mattern J, Huffman JC and Todd LJ, Inorg. Chem, 1994, 33, 5620–5624. [Google Scholar]
- 92.Spokoyny AM, Lewis CD, Teverovskiy G and Buchwald SL, Organometallics, 2012, 31, 8478–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kabytaev KZ, Everett TA, Safronov AV, Sevryugina YV, Jalisatgi SS and Hawthorne MF, Eur. J. Inorg. Chem, 2013, 2488–2491.
- 94.Jain L, Jain VK, Kushwah N, Pal MK, Wadawale AP, Bregadze VI and Glazun SA, Coord. Chem. Rev, 2014, 258–259, 72–118. [Google Scholar]
- 95.Mukhin SN, Kabytaev KZ, Zhigareva GG, Glukhov IV, Starikova Z. a, Bregadze VI and Beletskaya IP, Organometallics, 2008, 27, 5937–5942. [Google Scholar]
- 96.Amatore C and Jutand A, Coord. Chem. Rev, 1998, 178–180, 511–528. [Google Scholar]
- 97.Fairlamb IJS, Org. Biomol. Chem, 2008, 6, 3645. [DOI] [PubMed] [Google Scholar]
- 98.Saleh LMA, Dziedzic RM, Khan SI and Spokoyny AM, Chem. - A Eur. J, 2016, 22, 8466–8470. [DOI] [PubMed] [Google Scholar]
- 99.Connolly ML, J. Am. Chem. Soc, 1985, 107, 1118–1124. [Google Scholar]
- 100.Tsang CW, Yang Q, Sze ETP, Mak TCW, Chan DTW and Xie Z, Inorg. Chem, 2000, 39, 3582–3589. [DOI] [PubMed] [Google Scholar]
- 101.Olid D, Viñas C and Teixidor F, Chem. - A Eur. J, 2012, 18, 12936–12940. [DOI] [PubMed] [Google Scholar]
- 102.Jelínek T, Plešek J, Heřmánek S and Štíbr B, Collect. Czechoslov. Chem. Commun, 1986, 51, 819–829. [Google Scholar]
- 103.Olid D, Núñez R, Viñas C and Teixidor F, Chem. Soc. Rev, 2013, 42, 3318. [DOI] [PubMed] [Google Scholar]
- 104.Serino AC, Anderson ME, Saleh LMA, Dziedzic RM, Mills H, Heidenreich LK, Spokoyny AM and Weiss PS, ACS Appl. Mater. Interfaces, 2017, 9, 34592–34596. [DOI] [PubMed] [Google Scholar]
- 105.Kracke GR, VanGordon MR, Sevryugina YV, Kueffer PJ, Kabytaev K, Jalisatgi SS and Hawthorne MF, ChemMedChem, 2015, 10, 62–67. [DOI] [PubMed] [Google Scholar]
- 106.Kirlikovali KO, Axtell JC, Gonzalez A, Phung AC, Khan S and Spokoyny AM, Chem. Sci, 2016, 7, 5132–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kirlikovali KO, Axtell JC, Anderson K, Djurovich PI, Rheingold AL and Spokoyny AM, Organometallics, 2018, 37, 3122–3131. [Google Scholar]
- 108.Cheng R, Qiu Z and Xie Z, Nat. Commun, 2017, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dander JE, Baker EL and Garg NK, Chem. Sci, 2017,8, 6433–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ibrahim AD, Tokmic K, Brennan MR, Kim D, Matson EM, Nilges MJ, Bertke JA and Fout AR, Dalt. Trans, 2016, 45, 9805–9811. [DOI] [PubMed] [Google Scholar]
- 111.Ichiishi N, Malapit CA, Woźniak A and Sanford MS, Org. Lett, 2018, 20, 44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scheuermann ML, Johnson EJ and Chirik PJ, Org. Lett, 2015, 17, 2716–2719. [DOI] [PubMed] [Google Scholar]
- 113.Wei J, Liu KM and Duan XF, J. Org. Chem, 2017, 82, 1291–1300. [DOI] [PubMed] [Google Scholar]
- 114.Mousseau JJ and Charette AB, Acc. Chem. Res, 2013, 46, 412–424. [DOI] [PubMed] [Google Scholar]
- 115.Shaw MH, Twilton J and MacMillan DWC, J. Org. Chem, 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]