

Abstract
Throughout the design and development of supramolecular receptors for anion binding, many different non-covalent anion-binding motifs have been employed. One motif seen in many host-guest systems is the sometimes weaker, ‘non-traditional’ aryl CH hydrogen bond. From June Sutor’s discovery of the interaction and its subsequent dismissal by the field in the 1960s to today’s use of the aryl CH hydrogen bond in synthetic anion receptors, the path our lab took to begin studying this interaction has been influenced by many other researchers in the field. This feature article highlights the history and properties of the CH hydrogen bond, with a particular focus on aryl CH hydrogen bonds in anion recognition. We highlight select recent developments in the field of anion receptors utilizing aryl CH hydrogen bonds, with an emphasis on how this has influenced the evolution of our approach in designing fundamental studies on CH hydrogen bonding and exploiting this interaction in efforts aimed toward preferential anion binding.
Graphical Abstract
This Feature Article highlights recent approaches to anion recognition with a focus on aryl CH hydrogen bonds.

Introduction
Anionic species play diverse and complex roles in environmental, industrial, and biological systems, which necessitates chemical methods for detecting, sensing, sequestering, and selectively binding these negatively charged species to understand their fate, transport, and modes of action. As examples in the environment, anions are often found as natural and anthropogenic sources of pollution. Arsenate (AsO43−) contamination in Bangladeshi wells has caused one of the largest mass-poisonings in history, affecting an estimated 85 million people.1 Nitrate (NO3−) and dihydrogen phosphate (H2PO4−) are essential for plant growth and are used in fertilizers to increase crop yield; however, over-application of these anions can be extremely detrimental to the environment, reaching surrounding bodies of water through agricultural run-off and promoting eutrophication.2 As an example in industrial processes, anions such as sulfate (SO42−) also serve as major contaminants, and can thereby inhibit the effective vitrification of radioactive waste.3
In organisms, anions are essential for numerous biological processes. Chloride (Cl−) is used to regulate membrane transport and control nervous system function, and the misregulation of chloride is linked with serious diseases such as cystic fibrosis.4 The hydrosulfide anion (HS−) is currently being studied for its therapeutic potential as a signaling agent at low concentrations, but, at high concentrations, it is a deadly toxin and requires detailed monitoring in applications where exposure to the anion or its conjugate base (hydrogen sulfide, H2S) exists.5 Anions are even implicated in systems beyond our own planet. While perchlorate (ClO4−) serves as a rocket fuel additive and can lead to water contamination problems near terrestrial military bases (such as the Joint Base on Cape Cod, MA) and near flare manufacturing plants throughout California, perchlorate was also unexpectedly detected in soil on Mars.6,7 This finding perhaps hints at past microbial life on the Red Planet,7a and may suggest a future environmental cleanup challenge during terraforming by future humans seeking to populate other locations within the solar system.7b
To understand, and potentially to monitor, the complicated roles that anions play in these many systems, the complex modes of action between an anionic “guest” and a molecular “host” has received increasing attention. Anions present several challenges as targets for molecular/ion recognition, including: (i) Anions tend to be harder to bind by traditional electrostatic interactions because they are larger, more polarizable, and more diffuse than comparable cations. (ii) Anions exist in a diversity of molecular geometries, ranging from spherical (the halides) to planar (nitrate) to octahedral (SiF62−), among other forms.8a (iii) Anions typically serve as weak to moderate bases, so their speciation can be pH dependent. As a result, proton transfer might occur rather than, e.g., hydrogen bond formation during their interactions with a host. (iv) Anions tend to be highly solvated and particularly mobile, especially in polar protic solvents. Despite these challenges, supramolecular host-guest systems have emerged over the past few decades as a way to continuously monitor anions through reversible, non-covalent interactions.8 Molecular design and anion binding motifs can be used to modify and tailor host receptors for specific anion guests.9 Given the widespread use of hydrogen bonding in Nature, it is no surprise that a very popular approach that strongly mimics how proteins bind substrates is through the use of hydrogen bonding.9d,10
Our Native Oregonian and famous sister school Beaver, Linus Pauling, predicted the significance of the hydrogen bond well before confirmation of its influence on the structure of DNA or the folding of proteins.11–13 In fact, despite decades of debate on the hydrogen bond, much of Pauling’s quite simple description of the hydrogen bond in The Nature of the Chemical Bond still drives today’s more inclusive, lengthy formal definition.10,14 Pauling defines a hydrogen bond quite succinctly as occurring “under certain conditions [when] an atom of hydrogen is attracted by rather strong forces to two atoms, instead of only one, so that it may be considered to be acting as a bond between them”.14
Pauling’s definition reflects the traditional perspective of the hydrogen bond seen in structural biology, where the total interaction of the hydrogen bond is predominantly electrostatic and the distance between the donor and acceptor is less than the sum of the van der Waals radii.10,15 This classical definition of the hydrogen bond also reflects what many are taught in introductory chemistry courses: X–H···A reflects the strongly polar hydrogen bond donor groups X–H (X = O, N, or halogen) on one side and hydrogen bond acceptor atoms A (A = O, N, halogen, etc.) on the other (Fig. 1a).
Fig. 1.
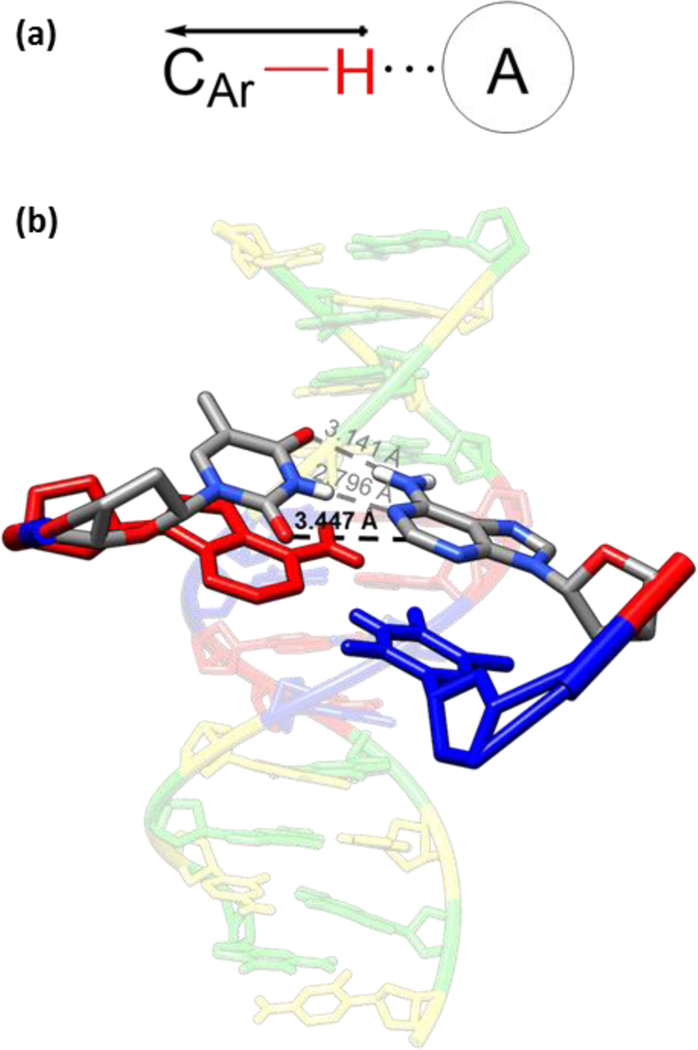
(a) A representation of a polarized aryl CH hydrogen bonding interaction with an anion and (b) a highlight of the adenine·thymine dimer with the traditional and non-traditional hydrogen bonding interactions highlighted.13 PDB ID: 4HLI60
The definition and classification of a hydrogen bond has evolved quite a bit since the early observations and predictions of this attractive interaction, and a knowledge of this evolving history is perhaps useful in understanding the relatively recent emergence of CH bonds as hydrogen bond donors in molecule and anion recognition.15 In fact, the fields of host-guest chemistry and anion recognition now regularly employ acidic CH hydrogens as H-bond donors, and the resultant interactions have often been deemed “weak” hydrogen bonds (irrespective of some stricter definitions we may learn in introductory organic chemistry courses).8,9,10,14 These and related emergent hydrogen bonding interactions are now well-recognized, in part due to an improved understanding of the interplay of the various attractive forces that comprise these interactions, including electrostatics, van der Waals forces, covalency, and degree of polarization.10,15,16
History and definition of the CH···X hydrogen bond
The first indication of the existence of a possible CH hydrogen bond (HB) appears to have occurred in 1935, around the same time as studies emerged about more traditional hydrogen bonds.11,12 However, these non-traditional CH hydrogen bonds were largely ignored until the early 1960s when D. June Sutor first published a systematic approach to define the existence of CH hydrogen-bonds in crystal structures.17 Her survey of crystal structures with “‘short’ intermolecular and intramolecular C···O contacts” was the first step toward defining this weak interaction but was limited in scope to molecules containing C‒H···O contacts.17
A few years later, Donohue challenged the Sutor definition of these short contacts as hydrogen bonds, in part suggesting the 2.6 Å contact was too long to be considered significant.18 With this dismissal—which appeared in a book celebrating the life and work of Linus Pauling and received almost no critical response—progress in the field halted until almost two decades later when Taylor and Kennard published a comprehensive survey of the neutron scattering data of 113 structures from the Cambridge Structural Database containing short C‒H···X contacts.19 In that work, they conclusively corroborated Sutor’s observations of the existence of C‒H···O hydrogen bond and systematically defined the properties of C‒H···X HBs. They also expanded the definition of these short contacts to include general C‒H···X interactions, where X = N, O, and Cl. They continued to postulate that “the C‒H···X hydrogen bond may be a significant factor in determining the minimum energy packing arrangements of small organic molecules that contain nitrogen”.19 A recent review by Schwalbe provides a wonderful analysis of Sutor’s role in the discovery, controversy, and ultimate vindication of the importance of the CH hydrogen bond.20 Shortly after her death in 1990, Desiraju dedicated “The C–H···O Hydrogen Bond: Structural Implications and Supramolecular Design” to Dr. Sutor’s memory.21
Nineteen years after the Taylor and Kennard work, Desiraju and Steiner published their book The Weak Hydrogen Bond: In Structure and Biology, wherein they further described the nature of the CH hydrogen bond.10 This weak hydrogen bond would then differ from classical “strong” hydrogen bonds defined as X–H···A, where A and X are assumed to be highly electronegative (e.g., O, N) and can approach each other closely, with the HBs observed between H2O molecules in crystalline ice serving as an example.15 Similarly, in defining the weak hydrogen bond, A and X are only of moderate electronegativity (e.g., CH hydrogen bonds where X is C). The definition and properties presented by Steiner et al. provided the following standard definition guiding current research in the field of supramolecular anion receptors:
“A X–H···A interaction is a hydrogen bond if i) it constitutes a local bond and ii) X–H acts as a proton donor to A: in the case of X–H + B: → X–H···:B. This definition implies a dipole-dipole interaction with a directional dependence.”10,15
While this clear definition of the hydrogen bond emerged in the late 1990s and the field of crystal engineering was transformed in the mid-1990s by the CH···X interaction, the field of supramolecular anion receptor chemistry did not begin to fully utilize or characterize this interaction in the solution-state until the mid-2000s.22,23 Recent work has shown that, when properly polarized by electron-withdrawing groups, CH HB donors can form hydrogen bonds similar in strength to those seen in more traditional HB donors.9d,24 These studies have also revealed several advantages in using CH HBs, including a greater resistance to proton transfer and pH-dependent host speciation, a greater affinity for softer anions in certain cases, and an overall additive effect to achieve strong binding (much like in the adenine·thymine base-pairs in the double helical backbone of DNA, Fig. 1b).13
Before we highlight current efforts in supramolecular anion receptors that utilize aryl CH hydrogen bonds as supporting interactions, we must first acknowledge the other, often competing and synergistic, supramolecular interactions at play in many such host-guest complexes. For this review, we focus on the interactions between an aromatic host and an anionic guest, but we will briefly touch on other competing forces, as well as solvent and entropic effects. Synthetic organic anion receptors commonly incorporate six main intermolecular and/or intramolecular interactions, alongside hydrophobic/solvophobic effects: ion pairing forces, dipole-anion forces, hydrogen bonding, halogen bonding, weak-σ interactions, and anion-π interactions (Fig. 2).8,9,25 All of these binding forces rely on an attractive force between two or more atoms of differing electrostatic potentials. Interestingly, aryl CH hydrogen bonds, halogen bonding, weak-σ interactions, and anion-π interactions are all dependent on electron-withdrawing functional groups to create a positive electrostatic potential within the molecule to “catch” the anion.9 In fact, the use of electron-withdrawing groups to flip the quadrupole moment in a phenyl ring to create a receptor capable of anion-π type interactions is how our group first stumbled into aryl CH hydrogen bonds.26
Fig. 2.
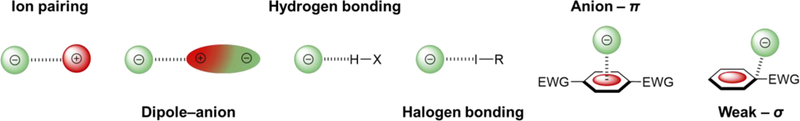
Depiction of common intrermolecular binding forces at play in host‒anionic guest systems
Aryl CH hydrogen bonding in anion receptors
In some of our early studies on anion recognition, published in 2008 with collaborator Ben Hay and then-doctoral student Orion Berryman, we designed a series of sterically-geared electronegative triaryl-substituted triethylbenzene receptors with different dinitro-substitution patterns on the aryl substituents (1 and 2, Fig. 3).26 In designing these receptors we sought to experimentally probe the continuum between weak-σ interactions and anion-π interactions in neutral aromatic hosts;27 at the time of this research, the anion-π literature was heavily weighted towards computational studies, with a few solution-state studies of receptors that often featured other competing binding forces (e.g., ion-pairing or hydrogen bonding).27c One key finding that fell out of these halide binding studies was not a direct measurement of the strength of the anion-π or the weak-σ interactions; rather, it was the surprising appearance of downfield shifts in 1H NMR spectroscopy titration studies that indicated the possibility of a different binding force at play: aryl CH hydrogen bonding. The substitution pattern of the dinitro groups allowed for discrimination between competing arene-anion and CH-anion binding interactions, since the 3,5-dinitro substituted receptor 2 was predicted to block aryl CH···X− hydrogen bonding sterically, while the 2,4-dinitro substituted receptor 1 allowed for two CH hydrogen bonds from the two weakly acidic ortho hydrogens within the anion binding pocket (Fig. 3).26
Fig. 3.
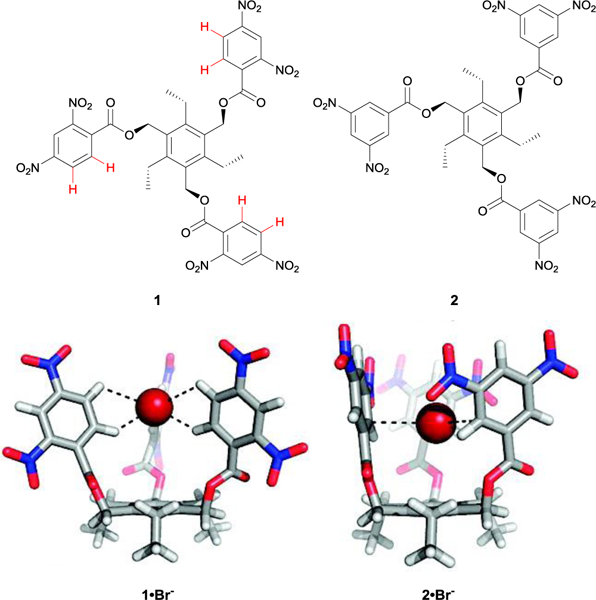
Optimized geometries of 1·Br− and 2·Br−. The 2,4-dinitro substituted triethylbenzene receptor 1 binds its guest via aryl CH hydrogen bonds versus the weak–σ binding mode depicted in the 3,5-dinitro substituted receptor 2. Figure adapted with permission of the American Chemical Society from ref. 26. Copyright 2008.
A key contemporary experimental investigation at the time also explored the aryl CH···X− interaction, as reported by Sessler, Hay, Lee, and coworkers in the context of a series of strapped calix[4]pyrroles.28 These systems—designed to bind chloride—contained either a phenyl, pyrrole, or furan moiety in the bridge/strap (Fig. 4). When compared to the unsubstituted calix[4]pyrrole, the phenyl and pyrrole straps (3 and 4, respectively) increased the affinity toward chloride by one and two orders of magnitude, respectively; however, the furan strapped system (5) showed an order of magnitude lower binding than the phenyl strapped system.28 This study was one of the first experimental examples to show the significance of the aryl CH···X− hydrogen bond as a supporting interaction for anion binding in synthetic hosts.
Fig. 4.
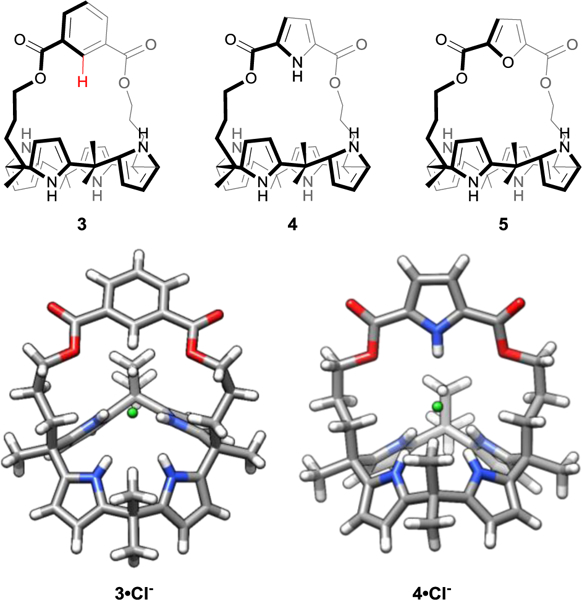
Series of strapped calix[4]pyrroles designed to bind chloride and the crystal X-ray diffraction structures of the Cl− complex. The key CH···Cl− interaction is clearly depicted in the 3·Cl− crystal structure (bottom). The added aryl CH hydrogen bond interaction available in the phenyl-strapped system 3 binds Cl− stronger than an unsubstituted calix[4]pyrrole and the furan-strapped system 5.
Shortly after these two studies were published, Colleti and Re performed high level computations to determine the strength of binding of the halide ions (F−, Cl−, Br−, and I−) to benzene.29 Their results suggested bifurcated aryl CH···X− hydrogen bonds of intermediate strength to be the preferred binding mode of F−, Cl−, Br−, and I− with benzene. However, a stronger, singular aryl CH···X− HB dominated the fluoride-benzene interaction. This study, in combination with the previous solution-state analyses, appeared to rekindle interest in using aryl CH···X− hydrogen bonding as an additional supporting interaction in complex host-guest systems.9
This series of earlier studies also helped inspire the longstanding collaboration between the Johnson and Haley labs at the University of Oregon in designing arylethynyl urea anion receptors, with a recent focus on phenylethynyl hosts (e.g. 6, Fig. 5a). This scaffold serves as a modified version of our original pyridylethynyl bis-urea and bis-sulfonamide receptors in which the core pyridine/pyridinium is replaced with a phenyl ring that is not subject to proton transfer (7, Fig. 5b).30 Our traditional pyridine and pyridinium-based receptors (8 and 9, Respectively, Fig. 5c) showed a pH dependency, limiting the scope of the anions we could bind and making studies at a physiological pH more challenging due to competing proton transfer processes between host, anions, and solvent.31 To overcome these limitations, we asked: did protonated pyridine need to be present in these receptor scaffolds? Graduate students at the time Calden Carroll, and later Blake Tresca, realized that the para-position on the aromatic core of the scaffold provided an easily functionalizable, fortuitous handle for polarizing the CH HB and studying substituent effects. In transitioning from a pyridine to a phenyl core, the opportunity to utilize aryl CH hydrogen bonding to bind anions was realized: by functionalizing the para-position on the core benzene ring with an electron-withdrawing substituent, the acidity of the aryl CH pointing into the binding pocket could be tuned as a hydrogen bond donor (Fig. 6, 10).32
Fig. 5.
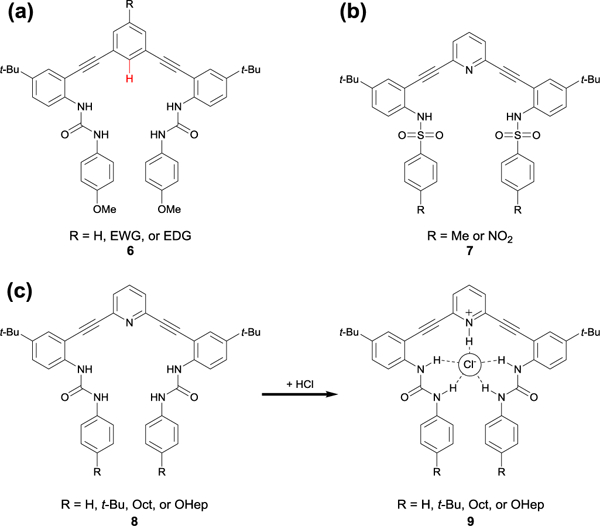
(a) Generic scaffold of the phenylethynyl bis-urea anion receptors our lab has used to investigate aryl CH hydrogen bonding; (b) bis-sulfonamide scaffold 7; and (c) original pH sensitive pyridine (8) to pyridinium (9) anion binding receptors.
Fig. 6.
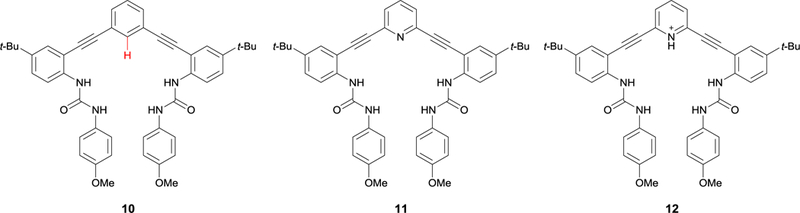
The three anion receptors that brought our lab into its current generation of aryl CH hydrogen bond studies. Tresca et al. compared the binding affinities of the phenyl- (10), pyridine- (11), and pyridinium-core (12) receptors to realize the potential of the supporting aryl CH HB in our scaffolds.
Fig. 10.
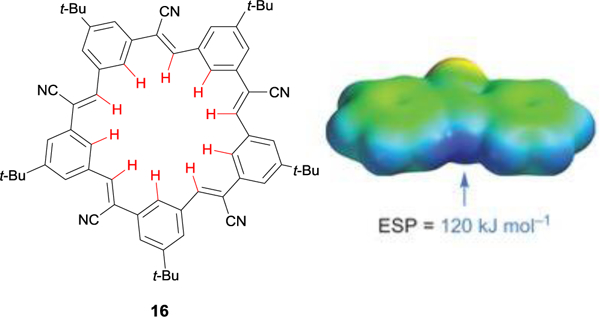
Cyanostilbene-based macrocycle “cyanostar” 16 obtained through the Knoevenagel condensation binds anions through 10 aryl CH HBs per host and a strongly electropositive binding pocket. Electrostatic potential surface map reproduced with permission of Springer Nature from ref. 45. Copyright 2013.
Similar to Sessler and coworkers’ research with strapped calix[4]pyrroles, Tresca et al. compared the affinity of halides with this phenyl-based receptor to those of the pyridine and pyridinium receptors (Fig. 6, 11 and 12).32 Receptor 12 showed the strongest binding for Cl−, attributed to the strong NH+ hydrogen bond combined with ion-pairing interactions.32 Phenyl-core receptor 10 featured an aryl CH HB in the binding pocket, which was further polarized by ortho-substituted alkynes. The resulting host-guest complex was not quite as stable as the complex with the pyridinium core when binding Cl−, as it showed a binding affinity an order of magnitude lower than 12. In comparison to receptor 11, scaffold 10 also showed an order of a magnitude stronger affinity toward Cl−, which was attributed to the repulsion of the nitrogen lone pair toward anions in the binding pocket of 11. 1H NMR titration studies showed a downfield shift of the internal proton resonance, indicating the participation of the aryl CH hydrogen bond in 10·Cl−, and a crystal structure analysis of 10·Cl− showed short contacts between the aryl CH and Cl− at 3.579(3) Å, demonstrating the clear participation of the aryl CH HB in this scaffold (Fig. 7).32
Fig. 11.
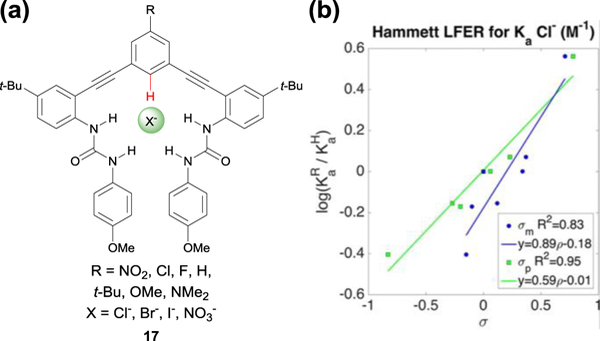
(a) Chemical structure of the differentially substituted phenylethynyl bis-urea receptors 17 implemented in the LFER study by Tresca et al. (b) Hammett plot of the binding constants of the various receptors with Cl−, indicating a σp relationship between the binding strength and aryl CH hydrogen bond donor. Hammett plot reproduced with permission of the American Chemical Society from ref. 44. Copyright 2015.
Fig. 12.
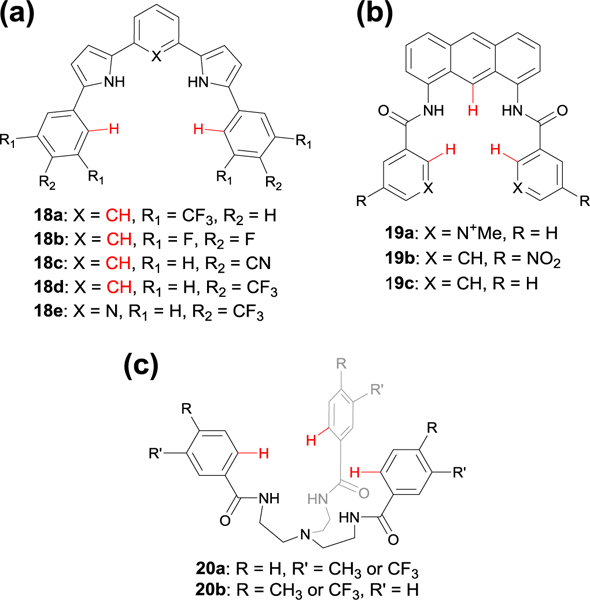
Chemical structures of various anion receptors used to probe the strength of the aryl CH hydrogen bond through differential substitution of (a) arylpyrrole oligomers 18, (b) anthracene-amide based receptors 19, and (c) TREN-based receptors 20.
Fig. 7.
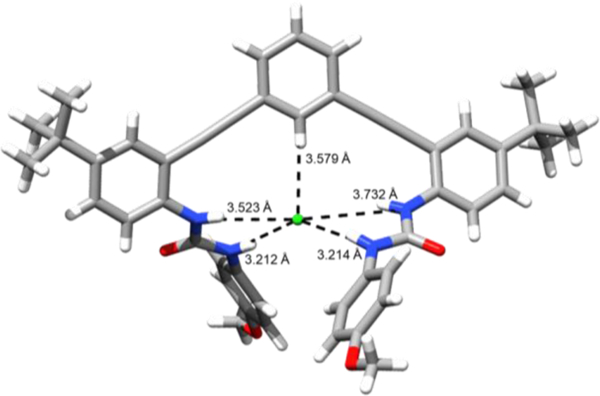
X-ray crystal structure of phenylethynyl bis-urea receptor 10 binding Cl‒ through urea NH and aryl CH hydrogen bonds.
The arylethynyl bis-urea scaffolds reported from our group are inherently flexible and capable of binding anions in a variety of conformations, with the aryl CH HBs acting as supporting anion binding motifs to traditional urea NH HBs or anion-π interactions.32,33 This is not an uncommon approach in designing anion receptors.8a As a related approach to these flexible hosts, many others have shown that moderate to strong anion binding is also possible with multiple aliphatic and/or aryl CH HBs in pre-organized macrocyclic receptors.34,9b-9d For example, triazolophane macrocycle receptors bind anions solely through aryl CH hydrogen bonds and serve as an early example of shape persistent hosts for anions, also reported in 2008.35 Flood et al. exploited the large diploe moment of 1,4-disubstituted 1,2,3-triazole groups linked by 1,3-disubstituted aromatic groups to pre-organize at least six acidic, polarized aryl CH groups pointing into the center of the macrocyclic ring (Fig. 8, 13). This series of neutral macrocycles showed selective binding toward halides utilizing only aryl CH hydrogen bonding, establishing the significance and strength of the aryl CH···X− interaction to bind anions in solution and the solid state.35
Fig. 8.
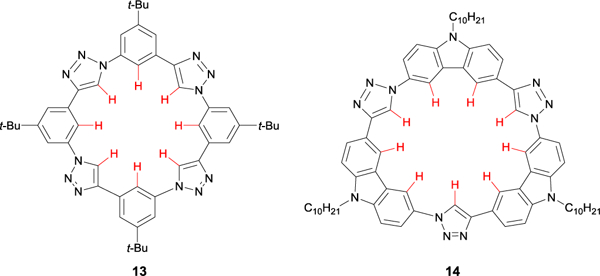
Triazolophane macrocycle 13 and related tricarbazolo triazolophane macrocycle 14 bind anionic guests solely through aryl CH hydrogen bonds.
Fig. 13.
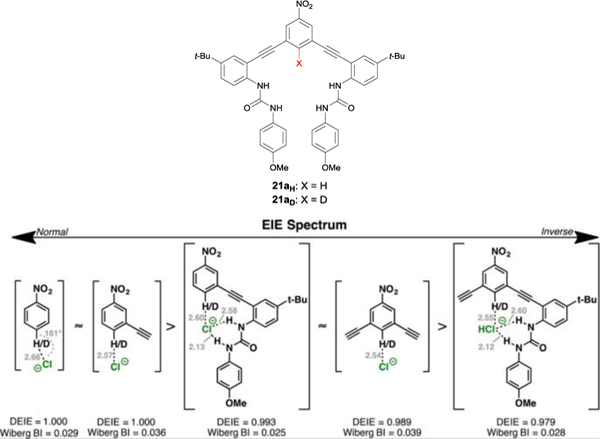
Deuterium labeled anion receptor 21 and subsequent computed EIE values involving the chloride complexes with fragments of receptor 20H/D. EIE spectrum reproduced with permission of the American Chemical Society from ref 51. Copyright 2017.
Since that first triazolophane publication in 2008, the Flood group has reported a multitude of elegant differentially-substituted triazolophanes and their anion-binding properties.36 In one spectacular example in 2016, Lee et al. replaced the phenyl linkers in the macrocycle with carbazole groups to create a rigid receptor (Fig. 8, 14) easily synthesized (in one pot) and in high yields (70% on an 8-gram scale). This tricarbazolo triazolophane structure showed highly cooperative binding with high affinities toward larger, more diffuse, and notoriously weakly-coordinating anions, such as SbF6– and PF6– in 20% MeOH in CHCl3 or the per-deutero equivalent. Strong π-stacking within this system also appeared to play a significant role in the self-assembly of this shape-persistent macrocycle into slip-stacked sandwiches in solution and at the liquid/solid interface, forming 2D crystalline honeycomb and flower polymorphs. Despite these supporting intermolecular interactions, host binding toward the anions results solely due to the activated aryl CH hydrogen bonds.36 The uniqueness of the triazole subunit – a conjugated ring that is easy to synthesize through “click” chemistry with a highly activated CH groups – has led to its incorporation into a variety of other anion-binding scaffolds, including foldamers,38 pyrrolyl-based triazolophane macrocycles,39 strapped calix[4]pyrroles,40 and anion-responsive self-assembled bis(triazole)benzamide receptors,41 among others.42
Fig. 14.
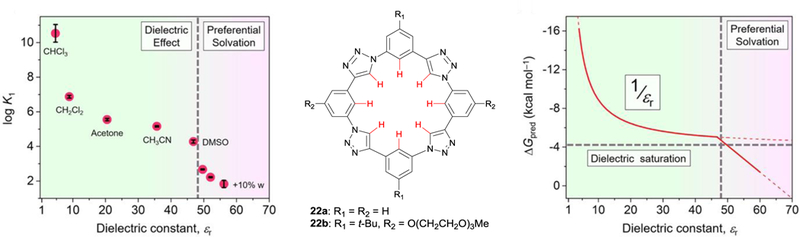
Understanding solvent effects on aryl CH···anion receptors is the next frontier in understanding the nature of this unique bond. Triazolophane macrocycles 22a and 22b showed a predictable 1/εr dependency in aprotic solvents but an unexpected linear decrease in anion association strength in protic solvents. Graphs reproduced with permission of Elsevier from ref. 52. Copyright 2017.
Another approach to incorporate aryl CH hydrogen bond donors lies in ring-strained hydrocarbon macrocycles featuring aryl CH groups directed into the strained macrocyclic cavity. In 2016, the Stępień group synthesized octulene 15, a structural homologue to kekulene, which has hyperbolic curvature with approximately 30 kcal mol–1 in strain energy.43 DFT geometries of the unsubstituted and methoxy-substituted ring showed a deep, saddle-like ring with eight aryl CH bonds pointing into the center of the large cavity (Fig. 9). The electrostatic potential (ESP) of these internal hydrogens was shown to be 23–24 kcal mol–1, making these aryl CH hydrogens about half as positive as the Flood triazolophane receptor (ESP = 41–55 kcal mol–1).37 This electrostatic potential is achieved through a neutral aromatic belt that lacks electron-withdrawing groups, but interestingly, is on par with the ESP of the aryl CH hydrogen bond donor in our most polarized, electropositive arylethynyl bis-urea receptors (ESP = 28.9 and 22.1 kcal mol–1 when the R group in Fig. 5 = NO2 and Cl, respectively).44
Fig. 9.
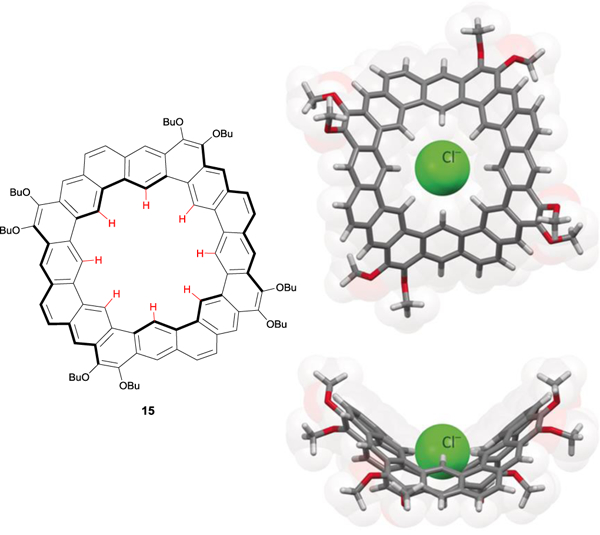
Chemical structure of aromatic belt octulene 15 and the gas-phase DFT geometries (level of theory: ωB97XD/6‐31G(d,p)) of the chloride-adduct showing the hyperbolic host pocket with eight aryl CH hydrogen bonds. Molecular models are reproduced with permission of Wiley from ref. 43. Copyright 2016.
With eight electropositive aryl CH hydrogen bond donors and a rigid, pre-organized binding cavity around the same size as that shown in triazolophane,35 Stępień and coworkers were able to bind Cl− with an association constant (Ka) of 2.2 × 104.43 This Ka, measured in 1% CD2Cl2 in C6D6, is particularly strong for a receptor that binds Cl− through only moderately-activated aryl CH hydrogen bonds. In comparison, the Flood triazolophanes, which feature many more electropositive aryl CH hydrogen bond donors, show a Ka of 1.3 × 105 in CH2Cl2.35 The strong association of octulene with Cl− shows the combined strength of these “weak” aryl CH hydrogen bond and may suggest that incorporating strain into a macrocylic host may be another strategy to increase the acidity and hydrogen bond donor strength of aryl CH hydrogen bonds.43
An even larger shape-persistent macrocyclic host was reported by Lee, Flood, and coworkers, again featuring all aryl CH hydrogen bonds oriented into the host cavity for anion recognition.45 This C5-symmetric penta-t-butyl-pentacyanopentabenzo[25]annulene macrocycle, aptly named “cyanostar”, was obtained through a one-pot Knoevenagel self-condensation (Fig. 10). It strongly binds large, weakly coordinating anions through polarized cyanostilbene aryl and olefinic CH hydrogen bonds. It is important to note that the cooperative π-stacking behavior of the cyanostars with large anions plays a role in creating a 2-to-1 host-to-guest “sandwich” complex.45 This electropositive binding pocket, combined with a total of 20 CH hydrogen bonds, resulted in an overall binding affinity of log β12 > 11 for weakly coordinating anions PF6−, ClO4−, and BF4− in 40% CD3OD in CD2Cl2. Since that initial report on cyanostars, the Flood group has continued to investigate the anion binding properties of differentially-substituted cyanostar macrocycles and has contributed significantly to the field of aryl CH···X− hydrogen bonding by investigating the nature of the contributions of the aryl CH···X− hydrogen bond in these host-guest complexes.46
Physical organic chemistry investigations into the nature of the aryl CH hydrogen bond
To employ aryl CH HBs as functional anion-binding motifs in supramolecular structures effectively—and perhaps still necessary in the recent past to convince skeptics that this attractive interaction rises to the level of inclusion as a hydrogen bond—detailed studies on the nature and contribution of these “non-traditional” hydrogen bond donors have been undertaken. Our lab became interested in using classical physical organic techniques to examine aryl CH HBs after receptor 10 showed moderate binding strength toward Cl−.32 We hypothesized that we could modulate the strength of the aryl CH HB in the binding pocket by installing various electron donating or withdrawing groups in the para position to the HB donor to study substituent effects. If these CH HBs were truly fundamentally related to their more traditional NH and OH counterparts, their HB binding energies should show linear free energy relationships to, e.g., traditional Hammett constants.
In 2016, Tresca and colleagues in our lab implemented a linear free energy relationship (LFER) study to probe the characteristics of these CH···X− interactions by modulating the HB strength through these varied para-substituents.44 The modular synthesis of our receptors allowed us to build a series of receptors (Fig. 11a, 17).44 We reported the association constants of the series of receptors with chloride, bromide, iodide, and nitrate in water-saturated CHCl3. We found that the binding energies were well described in LFER studies by using Hammett parameters, showing that the acidity of the aryl CH HB could be modulated by EWGs and EDGs in the para position, much like NH and OH HBs. Additionally, plotting the electrostatic potential of the aryl CH HB (calculated at the B3LYP/6–31+g(d) level of theory) against the ΔG of binding in solution revealed that the aryl CH HB was an important contributor to the overall binding energy, with the strongest aryl CH anion HB contributing up to ‒2.20 kcal mol‒1.44 In fact, the aryl CH HB amounted to as high as 47% of the total binding energy with I−.44
This research also highlighted differences in binding strength between the harder anions (Cl− and Br−) and the softer anions (I− and NO3−). Performing Hammett plots with σp or σm parameters for the different para substituents revealed subtle differences between the various anions.44 Binding energies with Cl− fit σp values best (Fig. 11b), while binding energies with I− better fit σm, suggesting that resonance contributions may play a more important role in binding the harder Cl− than when binding the softer I−. We also saw that NO3− fit both σp and σm equally well, perhaps due to the added geometric considerations of the larger, trigonal planar anion.44 Using the induction (F) and resonance (R) parameters developed by Swain and Lupton enabled determination of the inductive and resonance contributions of the receptors when binding the different anions. This analysis also revealed slightly higher resonance contributions for the harder anions than for the softer anions. These findings reinforce that linear free energy relationships can be a powerful tool in deciphering subtleties in non-covalent interactions, and potentially even provide approaches to achieving selectivity for different anions.44
Even without resorting to comprehensive LFER investigations, many other studies have explored the effect of polarizing CH bonds with electron withdrawing and donating groups. For example, in 2014 the Hill group reported an arylpyrrole oligomer possessing pyrrole NH and aryl CH hydrogen bonds for anion binding.47 These aryl CH hydrogen bonding motifs could be polarized through functional groups in the ortho-, meta-, and para-positions (Fig. 12a). When comparing five different receptors (18a-e) to six different anions (Cl−, HCO3−, AcO−, H2PO4−, NO3−, and Br−), the authors could not pinpoint a consistent trend across all host-guest pairs, with one exception: host 18e.47 This receptor, which combined a nitrogen lone pair pointing into the binding pocket with aryl CH HBs activated at the meta-position, bound all of the anions the weakest. This was likely due to the steric and/or electrostatic repulsion from the nitrogen lone pair pointing into the binding pocket. For the remaining host-guest pairs, the authors concluded that steric hindrance of the anion binding pocket was just as important to consider as the polarization of the aryl CH HB in host-guest interactions.47
The Kang group also reported on the effect of polarization of aryl CH hydrogen bonds on anion binding.48 Their receptors utilized an amide NH HB, a central anthracene CH HB, and an aryl CH polarized by an ortho pyridinium, a para-nitro group, or a control receptor without substituents (Fig. 12b, 19a-c, respectively). The unsubstituted receptor 19c showed no affinity toward a range of anions, while the slightly-more polarized receptor 19b only bound H2PO4−.48 Receptor 19a, however, which featured the most polarized aryl CH hydrogen bond, was able to bind all four anions studied (H2PO4−, HSO4−, Cl−, and Br−). In this case, the extent of polarization of this aryl CH HB, along with the favorable electrostatic interactions and possible N-methyl pyridinium CH HBs in 19a, were critical in creating a favorable host-guest interaction in solution.48
We previously collaborated with our colleagues in the lab of Michael Pluth at the University of Oregon to show that receptors of the type in Fig. 12c could bind the highly nucleophilic hydrosulfide anion (HS−, conjugate base of hydrogen sulfide, H2S).56 These studies revealed that a short CH···S contact contributed to the strong association of hydrosulfide in these complexes, and solution phase measurements supported the existence of this HB as well. In a continued attempt to determine the contribution of aryl CH hydrogen bonds in anion binding, the Pluth group published a series of tribenzamide TREN-based receptors (Fig. 12, 20).49 Within their series, two receptors were functionalized with CF3 electron withdrawing groups either in the meta (20a) or para (20b) position relative to the amide functional group. In the para position (20b), the CF3 group polarized the NH HB donor, making it more acidic, through both inductive and resonance effects. Likewise, in the meta position (20a), the CF3 group more greatly polarized the aryl CH HB donor. Titration of 20a and 20b (R = CF3) with TBASH revealed higher binding affinities than a receptor with an unfunctionalized aryl ring. Furthermore, they saw that the Ka for 20b was three times greater than for 20a, suggesting that the amide NH HB was more important in anion binding than the aryl CH HB.
To further explore the system, the Pluth group then installed methyl groups in both the para (20b, R = CH3) and meta (20a, R’ = CH3) position to the amide functional group, decreasing the acidity of both NH and CH HB. Through titration of the methyl- substituted 20a and 20b with TBASH, the authors saw lower binding affinities than the unsubstituted receptor but did not observe a significant difference in binding strengths between the two methyl-substituted receptors.49
Electrostatic potential surface (EPS) maps also serve as an efficient physical organic tool to visualize binding pockets and the extent of aryl CH hydrogen bond polarization without requiring the need to synthesize and study the anion-binding properties of a series of receptors. In their initial report on cyanostar macrocycles, Flood et al. attributed the strong binding toward weakly coordinating anions both to the electropositive cavity of the cyanostar and to the large size of the binding pocket (~4.5 A).45 To visualize this cavity, they used an electrostatic potential map of an intermediate building block to show that the nitrile group was able to polarize the vinylic and aryl CH bonds, thereby lining the inner cavity with an electropositive region (Fig. 10). Using calculations at the B3LYP/6–31G* level of theory, the authors calculated the EPS of the vinyl CH HB in the advanced intermediate at 29 kcal mol–1, which represents a highly polarized CH bond and thus a strong CH HB donor.45,50
Another way to study the contribution of a CH hydrogen bond to the overall anion binding in a receptor is through the use of deuterium equilibrium isotope effects (DEIEs); such studies are quite challenging to perform on traditional NH and OH donors due to proton exchange. We are fortunate to have a scaffold that presents a CH donor that is quite easy to label with deuterium (Fig. 13), and thus we embarked on a study with collaborator Paul Cheong’s lab to investigate EIEs in an aryl CH HB an anion receptor.51 In this investigation, receptors 21aH and 21aD were titrated with chloride in d6-DMSO and monitored through 13C NMR titrations. We reported a normal DEIE, with Ka21aH / Ka21aD = 1.019 +/− 0.010.51 We also reported the computed DEIEs of fragments of the receptors (Fig. 13).51 Interestingly, we saw that various fragments of the receptor showed an inverse DEIE. These results were surprising because they showed that the DEIE of the fragments would not be additive, as the inverse DEIEs would not sum to a positive DEIE, as was determined experimentally. Further analysis suggested that the origin of the different, normal DEIE of 21aH and 21aD was an emergent phenomenon resulting from combination of functional groups and binding geometries present in the host.51
Probing solvent effects in the aryl CH···X− interaction
We would be remiss if we did not emphasize that—especially with weak interactions—the binding forces alone do not always dominate binding structure, selectivity, strength, etc.; rather, solvent effects and entropy (through enthalpy-entropy compensation, preorganization, and cooperativity, among other factors) play their own critical, oftentimes ambiguous roles.50,51 Unfortunately, our understanding of solvent effects in general in synthetic host-guest complexes remains incomplete, and efforts to understand these effects in anion recognition are in their infancy. This is therefore a roadblock in understanding and predicting how receptors with any variety of binding motifs will interact with and select various anions in solution, particularly in water.51
Until recently, most of our understanding of solvent effects come from empirical reports of receptors examined in a few solvents. In 2017, the Flood group published a comprehensive study to untangle the forces that drive anion binding in macrocylic receptors, including electrostatics and solvent effects (Fig. 14).52 Experimental 1H NMR titrations with triazolophane receptor 22b and tetrabuylammonium chloride were conducted in solvents with a range in dieletric constant from εr = 4.7 (CHCl3) to εr = 56.2 (10% v/v H2O in DMSO).52 Additionally, DFT calculations were performed on receptor 22a to provide further insight into the binding events. From their experimental and computational results, the authors discovered a 1/εr dependence on anion affinity in aprotic solvents (Fig. 14). As the dielectric constant of the solvent decreased, the electrostatic forces of the receptor on the anion dominated the anion binding event and binding behavior became more and more similar to gas-phase calculations. As the dielectric constant increased, electrostatics gave way to other inter- and intramolecular forces, such as dispersion, induction, and exchange forces. With the switch from aprotic solvents to a mixture of DMSO and water, Flood et al. found a deviation from the 1/εr dependency: instead of plateauing, binding affinities began to decrease linearly in a fashion that was not predicted by computational binding models (Fig. 14).52
This unexpected trend in solvent influence on the strength of anion binding highlights how many forces are truly at play in these host-anion systems. While the strength of aryl CH hydrogen bonds can improve the overall association strength in a host-guest system, protect from proton transfer reactions, and even aid in anion binding selectivity, the role of dynamic electrostatic and solvent forces clearly warrants further scrutiny.53–55
Our future in the field of aryl CH···anion hydrogen bonding
Flood’s comprehensive approach to teasing apart solvent effects on anion binding is a notable contribution to the understanding of CH-anion recognition, and they make sure to highlight how much work remains in generalizing our understanding of solvent effects and moving theoretical models to shift from the gas phase into the more relevant solvent phase.52 We are inspired to continue thinking “beyond the electrostatic regime” in order to explain the deviation from the dielectric dependency upon moving into protic solvents, water-DMSO mixtures, and even neat water; to investigate solvent effects on our more flexible anion receptors; to explore the fundamental CH HB interactions and its role in driving anion binding selectivity; and to study the impact of solvent on hosts with binding geometries not perfectly designed for the guest.
The use of aryl CH hydrogen bonds and other anion binding approaches in the development of molecular probes and sensors for anions of biological relevance is another area that requires continued exploration. In one case, these pursuits led us to report the first examples of supramolecular receptors for the reversible binding of biologically-critical yet highly-reactive hydrosulfide (HS−) anion.56 Subsequent to these studies, new receptors targeting these types of biologically relevant anions through the use of aryl CH HBs have appeared.49,57 We are now further exploring the use of aryl CH hydrogen bonds to bind other reactive, yet biologically-relevant (hydro)chalcogenide anions, including hydroselenide and hydrogen sulfate.58
We also note that the studies on CH-anion HBs have focused on organic solvent mixtures predominantly, so there is still plenty of opportunity to study CH HBs in water to parallel other studies on anion recognition in water.53,55 We foresee combining the utility and tunability of the aryl CH···X– interaction with halogen bonding interactions to achieve strong and selective anion detection in water. These types of interaction motifs are now starting to appear in the design of organocatalysts and as bioisosteres in drug discovery. Finally, new generations of chemists continue to inspire us with the development of new binding motifs to consider for anion recognition, with a recent report showing the RCF2H group can serve as a HB donor that may mimic the function of ROH HB donors.59
Acknowledgements
Our work on CH hydrogen bonding has been primarily supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award number R01‐GM087398. Recent work on further applying CH hydrogen bonding to problems in the environment has also been supported by the National Science Foundation (CHE-1607214). This work was also supported by the Bradshaw and Holzapfel Research Professorship in Transformational Science and Mathematics to DWJ.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Hossain MF, Agric. Ecosyst. Environ, 2006, 113, 1–16. [Google Scholar]
- 2.(a) Fields S, Environ. Health. Perspect, 2004, 112, A556–A563; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) U.S. Environmental Protection Agency Nutrient Pollution: The Problem, https://www.epa.gov/nutrientpollution/problem, (accessed November 2018); [Google Scholar]; (c) Power JF and Schepers JS, Agric. Ecosyst. Environ, 1989, 26, 165–187; [Google Scholar]; (d) Strebel WHM, Wick K, Heumesser C and Schmid EJ, Environ. Manage, 2012, 111, 178–186; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mekonnen MM and Hoekstra AY, Water Resour. Res, 2018, 54, 345–358. [Google Scholar]
- 3.Moyer BA, Custelcean R, Hay BP, Sessler JL, Bowman-James K, Day VW and Kang S-O, Inorg. Chem, 2013, 52, 3473–3490. [DOI] [PubMed] [Google Scholar]
- 4.Gray MA, Winpenny JP, Verdon B, McAlroy H and Argent BE, Biosci. Rep. 1995, 15, 531–541. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Physiol. Rev, 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- 6.Coates JD and Achenbach LA, Nat. Rev. Microbiol, 2004, 2, 569–580. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ojha L, Wilhelm MB, Murchie SL, McEwen AS, Wray JJ, Hanley J, Massé M and Chojnacki M, Nat. Geosci. 2015, 8, 829–832; [Google Scholar]; (b) Kerr RA, Science, 2013, 340, 138; [DOI] [PubMed] [Google Scholar]; (c) While this comment is made with a bit of our tongues in our cheeks, we must acknowledge some inspiration from sci-fi extraterrestrial terraforming visions, such as that found in the Mars Trilogy by Kim Stanley Robinson. [Google Scholar]
- 8.(a) For an exhaustive review on approaches to anion recognition and an analysis of anion sizes, geometries, and interaction types, see: Molina P, Zapata Fand Caballero A, Chem. Rev‥ 2017, 117, 9907–9972; [DOI] [PubMed] [Google Scholar]; (b) Sessler JL, Gale PA and Cho W-S, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, 2006; [Google Scholar]; (c) Gale PA and Dehaen W, Anion Recognition in Supramolecular Chemistry, Springer, Berlin, 2010. [Google Scholar]
- 9.For more comprehensive reviews of anion recognition see: (a) Steed JW and Atwood JL, Supramol. Chem, John Wiley and Sons, 2nd edn, 2009; [Google Scholar]; (b) Gale PA, Howe ENW and Wu X, Chem. 2016, 1, 351–422; [Google Scholar]; (c) Zhao J, Yang D, Yang X-J and Wu B, Coord. Chem. Rev, 2019, 378, 415–444; [Google Scholar]; (d) For a thorough review of CH hydrogen bond donors in anion recognition, see: Cai J and Sessler JL, Chem. Soc. Rev, 2014, 43, 6198–6213. [DOI] [PubMed] [Google Scholar]
- 10.For a comprehensive history and description of the weak hydrogen bond see: Desiraju GRand Steiner T, The Weak Hydrogen Bond: In Structural Chemistry and Biology, Oxford University Press, Oxford, 1999. [Google Scholar]
- 11.(a) Pauling L, J. Am. Chem. Soc, 1935, 57, 2680–2684; [Google Scholar]; (b) Pauling L and Delbrück M, Science, 1940, 92, 77–79; [DOI] [PubMed] [Google Scholar]; (c) The University of Oregon (Ducks) and Oregon State University (Beavers) have a productive collaboration and competition in educating students in our state, and we have many research collaborations (for instance, the NSF Center for Sustainable Materials Chemistry is a longstanding collaboration between many faculty at both institutions, e.g., Fulton BL, Perkins CK, Mansergh RH, Jenkins MA, Gouliouk V, Jackson MN Jr., Ramos JC, Rogovoy NM, Gutierrez-Higgins MT, Boettcher SW, Conley JF, Keszler DA, Hutchison JE and Johnson DW, Chem. Mater. 2017, 29, 7760–7765). On the athletics fields the Ducks and Beavers are fierce, fairly friendly, rivals. [Google Scholar]
- 12.Around the same time as Pauling’s observations on the hydrogen bond, Kumler also noted its importance to dielectric constants, dipole moments, and hydrogen bonds in amides, among other functional groups. Of particular relevance to this Feature Article, Kumler likely offered the first observation of a CH HB between molecules of HCN; see: Kumler WD, J. Am. Chem. Soc, 1935, 57, 600–605. [Google Scholar]
- 13.(a) Quinn JR, Zimmerman SC, Del Bene JE and Shavitt I, J. Am. Chem. Soc, 2007, 129, 934–941; [DOI] [PubMed] [Google Scholar]; (b) Jeong KS, Tjivikua T, Muehldorf A, Deslongchamps G, Famulok M and Rebek J Jr., J. Am. Chem. Soc, 1991, 113, 201–209. [Google Scholar]
- 14.Pauling L, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 1939. [Google Scholar]
- 15.Desiraju GR, Angew. Chem. Int. Ed, 2011, 50, 52–59. [DOI] [PubMed] [Google Scholar]
- 16.(a) Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B and Nesbitt DJ, Pure Appl. Chem, 2011, 83, 1619–1636; [Google Scholar]; (b) Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B and Nesbitt DJ, Pure Appl. Chem, 2011, 83, 1637–1641. [Google Scholar]
- 17.(a) Sutor DJ, Acta. Cryst, 1958, 11, 453–458; [Google Scholar]; (b) Sutor DJ, Nature, 1962, 195, 68–69; [Google Scholar]; (c) Sutor DJ, J. Chem. Soc, 1963, 1105–1110. [Google Scholar]
- 18.J. Donohue in Structural Chemistry and Molecular Biology, ed. A. Rich and N. Davidson, W. H. Freeman, San Francisco, 1968, pp. 443–465.
- 19.Taylor R and Kennard O, J. Am. Chem. Soc, 1982, 104, 5063–5070. [Google Scholar]
- 20.Schawlbe CH, Crystallogr. Rev, 2012, 18, 191–206. [Google Scholar]
- 21.Desiraju GR, Acc. Chem. Res, 1996, 29, 441–449. [DOI] [PubMed] [Google Scholar]
- 22.The first account of a solution-state quantification of the C–H···X– interaction was in fact published in 1990 but the next publication was over a decade later. See: Farnham WB, Roe DC, Dixon DA, Calabrese JC, Harlow RL, J. Am. Chem. Soc, 1990, 112, 7707–7718. [Google Scholar]
- 23.Some examples of solution-state quantifications of the CH hydrogen bond: (a) Ilioudis CA, Tocher DAand Steed JW, J. Am. Chem. Soc, 2004, 126, 12395–12402; [DOI] [PubMed] [Google Scholar]; (b) Maeda H and Kusunose Y, Chem. Eur. J, 2005, 11, 5661–5666; [DOI] [PubMed] [Google Scholar]; (c) Vega IED, Gale PA, Light ME and Loeb SJ, Chem. Commun, 2005, 4913–4915; [DOI] [PubMed] [Google Scholar]; (d) Chimielewski MJ and Jurczak J, Chem. Eur. J, 2006, 12, 7652–7667; [DOI] [PubMed] [Google Scholar]; (e) Adcock JL and Zhang H, J. Org. Chem, 1995, 60, 1999–2002. [Google Scholar]
- 24.A selection of recent work not highlighted herein: (a) McDonald KP, Ramabhadran RO, Lee S, Raghavachari Kand Flood AH, Org. Lett, 2011, 13, 6260–6263; [DOI] [PubMed] [Google Scholar]; (b) Gilday LC, White NG and Beer PD, Dalton Trans, 2012, 41, 7092–7097; [DOI] [PubMed] [Google Scholar]; (c) Massena CJ, Riel AMS, Neuhaus GF, Decato DA and Berryman OB, Chem. Commun, 2015, 51, 1417–1420; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hua Y, Ramabhadran RO, Uduehi EO, Karty JA, Raghavachari K and Flood A, Chem. Eur. J, 2011, 17, 312–321. [DOI] [PubMed] [Google Scholar]
- 25.b/>Bianchi A, Bowman-James K and Garcia-Espana E, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997. [Google Scholar]
- 26.Berryman OB, Sather AC, Hay BP, Meisner JS and Johnson DW, J. Am. Chem. Soc, 2008, 130, 10895–10897. [DOI] [PubMed] [Google Scholar]
- 27.(a) Hay BP and Bryanstev VS, Chem. Commun, 2008, 2417–2428; [DOI] [PubMed] [Google Scholar]; (b) Berryman OB, Bryanstev VS, Stay DP, Johnson DW and Hay BP, J. Am. Chem. Soc, 2007, 129, 48–58; [DOI] [PubMed] [Google Scholar]; (c) For an overview of related work, a snapshot of research at the time, and pictures of much younger versions of two of the guilty parties, see our earlier Feature Article: Berryman OB, Johnson DW, Chem. Commun. 2009, 3143–3153. [DOI] [PubMed] [Google Scholar]
- 28.Yoon D-W, Gross DE, Lynch VM, Sessler JL, Hay BP and Lee C-H, Angew. Chem. Int. Ed, 2008, 47, 5038–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coletti C and Re N, J. Phys. Chem. A, 2009, 113, 1578–1585. [DOI] [PubMed] [Google Scholar]
- 30.Berryman OB, Johnson CA II, Zakharov LN, Haley MM and Johnson DW, Angew. Chem. Int. Ed, 2008, 47, 117–120. [DOI] [PubMed] [Google Scholar]
- 31.Carroll CN, Berryman OB, Johnson CA II, Zakharov LN, Haley MMand Johnson DW, Chem. Commun, 2009, 2520–2522. [DOI] [PubMed] [Google Scholar]
- 32.Tresca BW, Zakharov LN, Carroll CN, Johnson DW and Haley MM, Chem. Commun, 2013, 49, 7240–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Carroll CN, Coombs BA, McClintock SP, Johnson CA II, Berryman OB, Johnson DW and Haley MM, Chem. Commun, 2011, 47, 5539–5541; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle JM, Lakshminarayanan PS, Carroll CN, Zakharov LN, Haley MM and Johnson DW, Cryst. Growth Des, 2011, 11, 5144–5152; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Engle JM, Carroll CN, Johnson DW and Haley MM, Chem. Sci, 2012, 3, 1105–1110; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Watt MM, Zakharov LN, Haley MM and Johnson DW, Angew. Chem. Int. Ed, 2013, 52, 10275–10280; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gavette JV, Mills NS, Zakharov LN, Johnson CA II, Johnson DW and Haley MM, Angew. Chem. Int. Ed, 2013, 52, 10270–10274; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gavette JV, Evoniuk CJ, Zakharov LN, Carnes ME, Haley MM and Johnson DW, Chem. Sci, 2014, 5, 2899–2905; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Eytel LM, Brueckner AC, Lohrman JA, Haley MM, Cheong PH-Y and Johnson DW, Chem. Commun, 2018, 54, 13208–13211. [DOI] [PubMed] [Google Scholar]
- 34.For the sake of this review, we focus on receptors implementing aryl CH hydrogen bonds. For a comprehensive review on combined interactions, see ref. 8a.
- 35.Li Y and Flood AH, Angew. Chem. Int. Ed, 2008, 47, 2649–2652. [DOI] [PubMed] [Google Scholar]
- 36.A selection of triazolophane papers: (a) McDonald KP, Hua Y, Lee S and Flood A, Chem. Commun, 2012, 48, 5065–5075; [DOI] [PubMed] [Google Scholar]; (b) Li Y and Flood AH, J. Am. Chem. Soc, 2008, 130, 12111–12122; [DOI] [PubMed] [Google Scholar]; (c) Ramabhadran RO, Hua Y, Flood AH and Raghavachari K, Chem. Eur. J. 2011, 17, 9123–9129; [DOI] [PubMed] [Google Scholar]; (d) Bandyopadhyay I, Raghavachari K and Flood AH, Chem. Phys. Chem, 2009, 10, 2535–2540; [DOI] [PubMed] [Google Scholar]; (e) Zahran EM, Hua Y, Li Y, Flood AH and Bachas LG, Anal. Chem, 2010, 82, 368–375. [DOI] [PubMed] [Google Scholar]
- 37.Lee S, Hirsch BE, Liu Y, Dobscha JR, Burke DW, Tait SL and Flood AH, Chem. Eur. J, 2016, 22, 560–569. [DOI] [PubMed] [Google Scholar]
- 38.(a) Meudtner RM and Hecht S, Angew. Chem. Int. Ed, 2008, 47, 4926–4930; [DOI] [PubMed] [Google Scholar]; (b) Lee S, Hua Y and Flood AH, J. Org. Chem, 2014, 79, 8383–8396; [DOI] [PubMed] [Google Scholar]; (c) Hua Y and Flood AH, J. Am. Chem. Soc, 2010, 132, 12838–12840; [DOI] [PubMed] [Google Scholar]; (d) Juwarker H, Lenhardt JM, Pham DM and Craig SL, Angew. Chem. Int. Ed, 2008, 47, 3740–3743. [DOI] [PubMed] [Google Scholar]
- 39.Sessler JL, Cai J, Gong H-Y, Yang X, Arambula JF and Hay BP, J. Am. Chem. Soc, 2010, 132, 14058–14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher MG, Gale PA, Hiscock JR, Hursthouse MB, Light ME, Schmidtchen FP and Tong CC, Chem. Commun, 2009, 3017–3019. [DOI] [PubMed] [Google Scholar]
- 41.(a) Curiel D, Espinosa A, Más-Montoya M, Sánchez G, Tárraga A and Molina P, Chem. Commun, 2009, 7539–7541; [DOI] [PubMed] [Google Scholar]; (b) García F, Aragó J, Viruela R, Ortí E and Sánchez L, Org. Biomol. Chem, 2013, 11, 765–772. [DOI] [PubMed] [Google Scholar]
- 42.(a) Hua Y and Flood AH, Chem. Soc. Rev, 2010, 39, 1262–1271; [DOI] [PubMed] [Google Scholar]; (b) Schulze B and Schulbert US, Chem. Soc. Rev, 2014, 43, 2522–2571. [DOI] [PubMed] [Google Scholar]
- 43.Majewski MA, Hong Y, Lis T, Gregoliński J, Chmielewski PJ, Cybińska J, Kim D and Stępień M, Angew. Chem. Int. Ed, 2016, 55, 14072–14076. [DOI] [PubMed] [Google Scholar]
- 44.Tresca BW, Hansen RJ, Chau CV, Hay BP, Zakharov LN, Haley MM and Johnson DW, J. Am. Chem. Soc, 2015, 137, 14959–14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S, Chen C-H and Flood AH, Nat. Chem, 2013, 5, 704–710. [DOI] [PubMed] [Google Scholar]
- 46.A selection of cyanostar-related publications: (a) Qiao B, Anderson JR, Pink Pand Flood AH, Chem. Commun, 2016, 52, 8683–8686; [DOI] [PubMed] [Google Scholar]; (b) Fatila EM, Twum EB, Sengupta A, Pink M, Karty JA, Raghavachari K and Flood AH, Angew. Chem. Int. Ed, 2016, 55, 14057–14062; [DOI] [PubMed] [Google Scholar]; (c) Bensen CR, Fatila EM, Lee S, Marzo MG, Pink M, Mills MB, Preuss KE and Flood AH, J. Am. Chem. Soc, 2016, 138, 15057–15065; [DOI] [PubMed] [Google Scholar]; (d) Fatila EM, Twum EB, Karty JA and Flood AH, Chem. Eur. J, 2017, 23, 10652–10662. [DOI] [PubMed] [Google Scholar]
- 47.Rossom WV, Terentyeva TG, Sodeyama K, Matsushita Y, Tateyama Y, Ariga K and Hill JP, Org. Biomol. Chem, 2014, 12, 5492–5499. [DOI] [PubMed] [Google Scholar]
- 48.Choi Y, Kim T, Jang S and Kang J, New. J. Chem, 2016, 40, 794–802. [Google Scholar]
- 49.Lau N, Zakharov LN and Pluth MD, Chem. Commun, 2018, 54, 2337–2340. [DOI] [PubMed] [Google Scholar]
- 50.Based on Pauling’s electronegativity scale, the electrostatic difference between carbon and hydrogen is 0.35.
- 51.Tresca BW, Brueckner AC, Haley MM, Cheong PH-Y and Johnson DW, J. Am. Chem. Soc, 2017, 139, 3962–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Sengupta A, Raghavachari K and Flood AH, Chem, 2017, 3, 411–427. [Google Scholar]
- 53.Cremer PS, Flood AH, Gibb BC and Mobley DL, Nat. Chem, 2018, 10, 8–16. [DOI] [PubMed] [Google Scholar]
- 54.Sengupta A, Liu Y, Flood AH and Raghavachari K, Chem. Eur. J, 2018, 24, 14409–14417. [DOI] [PubMed] [Google Scholar]
- 55.These solvation effects are well-recognized in the field of anion binding in general. For selected recent reviews on the thermodynamics of anion binding and discussions on solvation effects, see: (a) Gibb BC, Isr. J. Chem, 2011, 51, 798–806; [Google Scholar]; (b) Hillyer MB and Gibb BC, Annu. Rev. Phys. Chem, 2016, 67, 307–329; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Okur HI, Hladíková J, Rembert KB, Cho Y, Heyda J, Dzubiella J, Cremer PS and Jungwirth P, J. Phys. Chem. B, 2017, 121, 1997–2014. [DOI] [PubMed] [Google Scholar]
- 56.Hartle MD, Hansen RJ, Tresca BW, Prakel SS, Zakharov LN, Haley MM, Pluth MD and Johnson DW, Angew. Chem. Int. Ed, 2016, 55, 11480–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vázquez J and Sindelar V, Chem. Commun, 2018, 54, 5859–5862. [DOI] [PubMed] [Google Scholar]
- 58.(a) Fargher HA, Lau N, Zakharov LN, Haley MM, Johnson DW and Pluth MD, Chem. Sci, 2019, 10, 67–72; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deng C-L, Bard JP, Lohrman JA, Barker JE, Zakharov LN, Johnson DW, Haley MM, Angew. Chem. Int. Ed. 2019, Accepted Article, 10.1002/anie.201814431. [DOI] [PubMed] [Google Scholar]
- 59.Sessler CD, Rahm M, Becker S, Goldberg JM, Wang F and Lippard SJ, J. Am. Chem. Soc, 2017, 139, 9325–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reniciuk D, Blacque O, Vorlickova M and Spingler B, Nucl. Acids Res, 2013, 41, 9891–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]