

Abstract
Background
Peripheral arterial occlusive disease (PAOD) is a common cause of morbidity and mortality due to cardiovascular disease in the general population. Although numerous treatments have been adopted for patients at different disease stages, no option other than amputation is available for patients presenting with critical limb ischaemia (CLI) unsuitable for rescue or reconstructive intervention. In this regard, prostanoids have been proposed as a therapeutic alternative, with the aim of increasing blood supply to the limb with occluded arteries through their vasodilatory, antithrombotic, and anti‐inflammatory effects. This is an update of a review first published in 2010.
Objectives
To determine the effectiveness and safety of prostanoids in patients with CLI unsuitable for rescue or reconstructive intervention.
Search methods
For this update, the Cochrane Vascular Information Specialist searched the Specialised Register (January 2017) and the Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 1). In addition, we searched trials registries (January 2017) and contacted pharmaceutical manufacturers, in our efforts to identify unpublished data and ongoing trials.
Selection criteria
Randomised controlled trials describing the efficacy and safety of prostanoids compared with placebo or other pharmacological control treatments for patients presenting with CLI without chance of rescue or reconstructive intervention.
Data collection and analysis
Two review authors independently selected trials, assessed trials for eligibility and methodological quality, and extracted data. We resolved disagreements by consensus or by consultation with a third review author.
Main results
For this update, 15 additional studies fulfilled selection criteria. We included in this review 33 randomised controlled trials with 4477 participants; 21 compared different prostanoids versus placebo, seven compared prostanoids versus other agents, and five conducted head‐to‐head comparisons using two different prostanoids.
We found low‐quality evidence that suggests no clear difference in the incidence of cardiovascular mortality between patients receiving prostanoids and those given placebo (risk ratio (RR) 0.81, 95% confidence interval (CI) 0.41 to 1.58). We found high‐quality evidence showing that prostanoids have no effect on the incidence of total amputations when compared with placebo (RR 0.97, 95% CI 0.86 to 1.09). Adverse events were more frequent with prostanoids than with placebo (RR 2.11, 95% CI 1.79 to 2.50; moderate‐quality evidence). The most commonly reported adverse events were headache, nausea, vomiting, diarrhoea, flushing, and hypotension. We found moderate‐quality evidence showing that prostanoids reduced rest‐pain (RR 1.30, 95% CI 1.06 to 1.59) and promoted ulcer healing (RR 1.24, 95% CI 1.04 to 1.48) when compared with placebo, although these small beneficial effects were diluted when we performed a sensitivity analysis that excluded studies at high risk of bias. Additionally, we found evidence of low to very low quality suggesting the effects of prostanoids versus other active agents or versus other prostanoids because studies conducting these comparisons were few and we judged them to be at high risk of bias. None of the included studies assessed quality of life.
Authors' conclusions
We found high‐quality evidence showing that prostanoids have no effect on the incidence of total amputations when compared against placebo. Moderate‐quality evidence showed small beneficial effects of prostanoids for rest‐pain relief and ulcer healing when compared with placebo. Additionally, moderate‐quality evidence showed a greater incidence of adverse effects with the use of prostanoids, and low‐quality evidence suggests that prostanoids have no effect on cardiovascular mortality when compared with placebo. None of the included studies reported quality of life measurements. The balance between benefits and harms associated with use of prostanoids in patients with critical limb ischaemia with no chance of reconstructive intervention is uncertain; therefore careful assessment of therapeutic alternatives should be considered. Main reasons for downgrading the quality of evidence were high risk of attrition bias and imprecision of effect estimates.
Plain language summary
Prostanoids for people with severely blocked arteries of the leg
Background
People with severely blocked arteries of the leg suffer from pain, ulcers (areas showing loss of skin that do not heal easily), or gangrene (areas showing dead tissues resulting from loss of blood supply). This condition is usually associated with several risk factors, such as diabetes, smoking, high cholesterol, high blood pressure, obesity, and unhealthy lifestyle. The main treatments for people with this condition are surgical procedures performed to unblock the arteries. However, in some situations, surgical unblocking is not possible and amputation of part of the leg is required.
Prostanoids make up a family of medicines that could increase blood supply to the legs when taken orally or by injection. Prostanoids expand and open up small blood vessels and reduce the activity of inflammatory cells and platelets, preventing blood clots. We wanted to discover the benefits and harms of prostanoids for people whose leg arteries are severely blocked with no chance for surgical unblocking.
Review question
In this review, we studied the effect of prostanoids in people with severely blocked leg arteries who are not able to undergo any surgical unblocking procedure.
Study characteristics
We searched published and unpublished studies up to January 2017. We found 33 clinical trials with a total of 4477 participants; most were published in the 1980s and 1990s and were carried out in European countries. Eleven out of 33 studies received funding from pharmaceutical companies. Most studies included patients over 60 years old who had severe blocking of arteries of the leg; many also had diabetes. Follow‐up was usually less than 1 year.
Key results
We found that, when compared with placebo, prostanoids provided a small beneficial effect by alleviating pain in the leg at rest and improving ulcer healing. Prostanoids did not reduce deaths or the need for an amputation. We found that no studies evaluated the quality of life of people with this condition. We found insufficient evidence to compare effects of prostanoids against those of other medications or other prostanoids.
Our findings suggest that taking prostanoids does cause harm. When 1000 patients are treated with prostanoids, on average 674 (572 to 798) will experience adverse events, compared with 319 given placebo. Adverse events usually include nausea, vomiting, diarrhoea, headache, dizziness, and flushing. More severe adverse events include low blood pressure, chest pain, and abnormalities in heart rhythm.
Quality of the evidence
When evaluating effects of prostanoids on rest‐pain, ulcer healing, and adverse events, researchers provided moderate‐quality evidence; review authors downgraded this in most cases because of loss of participants to follow‐up. Evaluating cardiovascular mortality yielded evidence of low quality related to loss of participants to follow‐up and small numbers of reported events. On the other hand, the quality of evidence on risk of amputation was high.
Summary of findings
Background
Description of the condition
The term 'critical limb ischaemia' (CLI) should be used for all cases of chronic ischaemic rest‐pain, ulcers, or gangrene attributable to objectively proven arterial occlusive disease (Gerhard‐Herman 2016). CLI stands at the end of the most severe spectrum of peripheral arterial obstructive disease (PAOD). This multi‐factorial condition occurs secondary to the combination of endothelial dysfunction, dyslipidaemia, inflammatory and immunological factors, plaque rupture, and tobacco use. Unlike individuals with intermittent claudication (IC), patients with CLI have poor arterial blood flow to the lower limbs (resting perfusion), which is inadequate to sustain viability in the distal tissue bed. The European Working Group on CLI specifically defined this illness as the presence of ischaemic rest‐pain requiring analgesia for longer than two weeks, or ulceration, or gangrene of the lower extremity with ankle systolic blood pressure < 50 mmHg and/or toe systolic pressure < 30 mmHg (Anonymous 1991). Although the definition of CLI has evolved over time (Becker 2011), a recent multi‐disciplinary consensus ‐ the Peripheral Academic Research Consortium ‐ maintained those classic haemodynamic thresholds for patients with ischaemic rest‐pain, but defined CLI haemodynamic thresholds for patients with tissue loss as ankle systolic blood pressure < 70 mmHg and/or toe systolic pressure < 50 mmHg. This consensus definition also considers alternative haemodynamic parameters, such as transcutaneous oxygen (TcPO2) (< 20 mmHg for patients with ischaemic rest‐pain and < 40 mmHg for patients with tissue loss) and skin perfusion pressure (< 40 mmHg or < 30 mmHg, respectively) (Patel 2015).
Peripheral arterial disease (PAD) affects approximately 12% of adults increasing to 20% in people aged 70 years or older (Dua 2016). CLI is the initial clinical presentation in only 1% to 2% of cases of PAD, whereas 40% to 50% of those affected begin with atypical leg pain, and 10% to 35% with IC; 20% to 50% are asymptomatic. After five years of progressive functional impairment, a further 1% to 2% of PAD cases will result in CLI and eventual amputation (Hirsch 2006). However, a more recent meta‐analysis estimated the deterioration rate to be as high as 21% (95% confidence interval (CI) 12% to 29%) over five years (Sigvant 2016). Published epidemiological data on this condition are remarkably variable owing to use of different definitions of CLI over time; this represents the main cause of difficulty for proper identification of arterial insufficiency causing signs and symptoms in a large number of patients for epidemiological studies (Becker 2011).
Besides age (Hylton 2014; Ostchega 2007), the most important clinical predictors for CLI progression are smoking and diabetes (Howard 2015). The risk associated with smoking applies to all ages and increases with the number of cigarettes smoked. Major PAD deterioration occurs in people with claudication who are heavy smokers (Aquino 2001). Diabetes is associated with greater severity of PAOD in the lower limbs, especially in the arteries below the knee (Jude 2001). People with claudication and diabetes are also at higher risk of amputation and mortality than those with IC but without diabetes (Aquino 2001; Jude 2001). However, it is difficult to determine the true impact of this condition on the prognosis of patients with CLI for two reasons: (1) the proportion of patients with diabetes is variable in different studies assessing the natural history of CLI; and (2) diagnosis of CLI in patients with diabetes is challenging owing to frequent comorbid neuropathy and infectious complications, which may lead to tissue lesions (Becker 2011).
The prognosis for limb and patient survival is variable in chronic CLI. The most important risk factors for amputation are those involved in progression to CLI (Howard 2015), as well as an ankle brachial index (ABI) below 0.5 (Jelnes 1986). Within a six‐month period, 20% of patients die, 35% live but require amputation, and the remaining 45% live with no immediate need for amputation (Dormandy 2000). Meta‐analysis of 10 case series and non‐interventional or placebo arms of randomised controlled trials revealed a mortality rate of 22% (95% CI 12% to 32%) and an amputation rate of 30% (95% CI 19% to 42%) after 12 to 18 months' follow‐up (Abu 2015). Even though the strength of evidence on pooled estimates was low owing to high risk of bias, heterogeneity, and imprecision, these results are consistent with previous observations (Dormandy 2000; Hirsch 2006; Norgren 2007) and with the findings of a more recent retrospective study, which reported a mortality rate of 15% among patients with CLI after one year, and 24% after two years' follow‐up (Melillo 2016).
The prognosis after amputation is even worse. According to the Second European Consensus Document on chronic CLI (Anonymous 1991), perioperative mortality varies between 5% and 10% for below‐knee amputation, and between 15% and 20% for above‐knee amputation, respectively. This information is consistent with that provided in more recent reports, which estimated in‐hospital mortality rates for minor and major amputees as 3.6% to 4.6% and 16.8% to 19.8%, respectively (Malyar 2016; Moxey 2010). A second amputation is required in 30% of cases (Norgren 2007), and full mobility is achieved in only 56% of patients who have a minor amputation and in 17% of those who have a major amputation (above‐ and below‐knee) one year after the procedure (Suckow 2012). Furthermore, it is well known that patients with PAD have elevated risks of myocardial infarction, congestive heart failure, stroke, transient ischaemic attack, and cardiovascular death (Criqui 2015).
Quality of life in patients with CLI is significantly impaired compared with that in the general population and in patients with less severe PAOD (Sprengers 2010). Psychological testing of these patients has typically disclosed quality of life indices similar to those of patients with cancer at critical or even terminal phases (Albers 1992). Most people with CLI who experienced amputations have reported mobility limitations, rest‐pain (amputation‐related pain or ischaemic‐related pain in the affected or contralateral limb), and depression as prevalent domains undermining their quality of life (Suckow 2015). Therefore, because of its negative impact on quality of life and poor prognosis in terms of both limb and patient survival, CLI is a critical health issue.
First‐line therapeutic options for CLI are limited to percutaneous transluminal angioplasty or surgical revascularisation. Unfortunately, many patients with CLI are poor candidates for either procedure because of comorbidities or vascular anatomy (lack of conduit). These patients have only medical treatment as a therapeutic alternative, and amputation (when necessary) as the last chance to survive.
Angiogenic factors have suggested favour for neovascularisation by increasing collateral circulation and enhancing blood flow to ischaemic limbs. Despite promising results of early studies assessing the efficacy of this innovative treatment, systematic reviews and meta‐analyses examining patients with PAD and CLI (Hammer 2013; Miao 2014) showed no significant differences between treatment and control groups in terms of amputation, mortality at one year, or wound healing at six months' follow‐up.
Other therapeutic strategies for non‐surgical patients, such as autologous implantation of bone marrow mononuclear cells and spinal cord stimulation, have proved modestly effective in decreasing the amputation rate and reducing pain during variable periods of follow‐up (Moazzami 2014; Ubbink 2013). However, the quality of evidence regarding these strategies is considered moderate.
Description of the intervention
Medical therapies that decrease pain, promote healing of skin lesions, and reduce the risk of amputation would be attractive alternatives for patients with CLI non‐suitable for revascularisation procedures. Several drugs have been used at this stage (e.g. cilostazol, pentoxifylline, naftidrofuryl) and have led to no significant benefit. Prostanoids have been used for treatment of individuals with PAD for longer than two decades because some trials have recommended their use (Balzer 1991; Brock 1990; ICAI Group 1999; Norgren 1990; Trubestein 1989; Verstraete 1994). This family of drugs consists of the following: prostaglandin E1 (also referred to as PGE1, or alprostadil, generally by intravenous or intra‐arterial administration for 21 days); prostacyclin (also referred to as PGI2, or epoprostenol, by intravenous administration for four to seven days, or by intra‐arterial administration for 72 hours); iloprost (by intravenous administration for 14 to 28 days, oral for 28 days up to one year); lipo‐ecraprost (by intravenous administration for 50 days); and ciprostene (by intravenous administration for seven days).
How the intervention might work
Prostanoids have pharmacological activities on endothelial cells, vascular smooth muscle cells, and platelets that could favourably alter the otherwise inexorable downhill course of CLI. These include inhibition of platelet activation, adhesion, and aggregation; vasodilatation; vascular endothelial cytoprotection; and inhibition of leucocyte activation (Balzer 1991; Brock 1990; ICAI Group 1999; Norgren 1990; Robertson 2013; Trubestein 1989).
Why it is important to do this review
A few meta‐analyses such as Creutzig 2004 and Loosemore 1994 and reviews such as Dormandy 1996 have been published, but they did not include all types of prostanoids and all routes of administration. The first version of this Cochrane review, published in 2010, took into account new approaches regarding this therapeutic option to find conclusive evidence about the effectiveness and safety of the whole family of prostanoids for patients with CLI, including only randomised controlled trials (RCTs) considered to have low or moderate risk of bias (Ruffolo 2010).
Even though other review authors have performed updated systematic reviews on this topic, their approaches were narrower in terms of interventions evaluated (Brodszky 2011), or broader in terms of study populations, including patients with PAOD of any severity (Vitale 2016).
Moreover, since publication of the first version of this Cochrane systematic review, methodological standards have changed substantially. Currently, the preferred tool for appraisal of internal validity of RCTs is Cochrane's tool for assessing risk of bias (Higgins 2011b); use of any selection criterion based on a threshold of global quality assessment is discouraged. For these reasons, it is necessary to update this systematic review with the goal of reassessing eligibility of previously excluded studies and strength of evidence regarding effectiveness and safety of this pharmacological therapy.
Objectives
To determine the effectiveness and safety of prostanoids in patients with CLI unsuitable for rescue or reconstructive intervention.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials.
Types of participants
People irrespective of age or gender, presenting with critical limb ischaemia of atherosclerotic origin, without chance of rescue or reconstructive intervention. We did not include studies with patients given a diagnosis of thromboangiitis obliterans, also known as Buerger's disease, unless more than 80% of participants fulfilled inclusion criteria, or disaggregated data were available for the group of participants with CLI of atherosclerotic origin.
Types of interventions
Prostaglandin E1 (PGE1, alprostadil), prostacyclin (PGI2, epoprostenol), iloprost, beraprost, cisaprost, ciprostene, clinprost, ecraprost, or taprostene compared with placebo or other pharmacological control treatments (e.g. pentoxifylline, cilostazol, naftidrofuryl, angiogenic therapy, other prostanoids). We did not include surgical treatments or other medical non‐pharmacological treatments as comparators.
Types of outcome measures
Primary outcomes
Cardiovascular mortality (e.g. due to myocardial infarction, stroke, arrhythmia or variation form the normal rhythm of the heartbeat, sudden death)
Total amputations (major plus minor)
Quality of life (measured according to a validated quality of life questionnaire)
Adverse events of treatment
Secondary outcomes
Evaluation of rest‐pain and/or use of analgesic drugs (measured according to a validated pain scale and a validated questionnaire, respectively)
Evolution of tissue lesions (healing or non‐healing ulcers, according to surface area increase or decrease, and presence or absence of granulation tissue)
Major amputations (above or below knee)
Minor amputations (partial feet or fingers)
Ankle brachial index (ABI)
All‐cause mortality
Search methods for identification of studies
We did not restrict language of publication.
Electronic searches
For this update, the Cochrane Vascular Information Specialist (CIS) searched the following databases for relevant trials.
Cochrane Vascular Specialised Register (January 2017).
Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 1) via the Cochrane Register of Studies Online.
See Appendix 1 for details of the search strategy used to search CENTRAL.
The Cochrane Vascular Specialised Register is maintained by the CIS and is constructed from weekly electronic searches of MEDLINE Ovid, Embase Ovid, the Cumulative Index to Nursing and Allied Health Literature (CINAHL), and the Allied and Complementary Medicine Database (AMED), and through handsearching of relevant journals. A full list of databases, journals, and conference proceedings that have been searched, as well as the search strategies used, is presented in the Specialised Register section of the Cochrane Vascular module in the Cochrane Library (www.cochranelibrary.com).
The CIS and the review authors separately searched the following trial registries for details of ongoing and unpublished studies (January 2017).
ClinicalTrials.gov (www.clinicaltrials.gov).
World Health Organization International Clinical Trials Registry Platform (www.who.int/trialsearch).
International Standard Randomised Controlled Trials Number (ISRCTN) Registry (www.isrctn.com/).
See Appendix 2 for details of the search strategies.
Searching other resources
We identified additional articles by reviewing reference lists of both papers identified by electronic searches and systematic reviews. We contacted pharmaceutical companies to ask about additional studies (Actelion Pharmaceuticals, Bayer‐Schering, Italfarmaco, Mitsubishi Pharma America, Pfizer, UCB Pharma, United Therapeutics).
Data collection and analysis
Selection of studies
For this update, two review authors (VV, JVAF or DC) independently checked titles, abstracts, and keywords of all references retrieved. We retrieved the full text of all studies considered potentially relevant, and each review author assessed these studies independently using a Study Eligibility Form. We resolved disagreements between the two review authors by consensus or, finally, by consultation with a third review author (AC).
Data extraction and management
For this update, two review authors (VV, JVAF, VS or DC) independently collected the following data from each included study using a Data Extraction Form, provided by Cochrane Vascular.
Publication type and source, including language of publication, year of publication, and method of retrieval of the report.
Sources of support.
Trial design, including methods of generation and concealment of allocation sequences, along with type of control intervention.
Setting, including country, and level of care.
Participants, including selection criteria used and numbers of withdrawals and dropouts per group.
Interventions, including dose, route of administration, and duration of treatment.
Outcome measures, including modalities and schedules of assessment, adverse events, and overall mortality and details of its causes.
Analysis, including whether analysis was done according to the intention‐to‐treat principle.
Results, including averages and variations in individual outcome assessments and different comparisons, test statistics, and P values for comparison within and between groups.
We resolved disagreements by consensus or by consultation with a third review author (AC), if required. We contacted primary study authors by email to obtain additional information. We collected all data in original units; therefore, transformation for comparisons was not necessary.
Assessment of risk of bias in included studies
For this update, two review authors (VV, JVAF, VS, or DC) independently assessed potential risks of bias for all included RCTs using the Cochrane tool for assessing risk of bias (Higgins 2011b). This tool assesses bias in six different domains.
Random sequence generation (selection bias).
Allocation concealment (selection bias).
Blinding of participants and personnel (performance bias).
Blinding of outcome assessors (detection bias).
Incomplete outcome data (attrition bias).
Selective outcome reporting (reporting bias).
Other sources of bias.
Each domain received a score of high, low, or unclear risk of bias depending on each review author's judgement. We resolved disagreements by consensus and, if necessary, asked review author AC to act as arbitrator. We contacted study authors for clarification when doubt arose as to potential risk of bias.
Measures of treatment effect
Even though clinical heterogeneity was significant, we did perform an overall meta‐analysis of prostanoids versus placebo, including subgroup analysis by type of prostanoid. We also completed analyses of prostanoids versus other active agents by describing results of individual studies or performing meta‐analyses when more than one study reported the same outcome (prostanoids compared with pentoxifylline, naftidrofuryl, adenosine triphosphate (ATP), or inositol niacinate) and we considered results to show clinical homogeneity. In addition, we completed analyses of prostanoids versus other prostanoids for the specific comparisons described in the included studies (iloprost vs PGE1, clinprost vs lipo‐PGE1). Finally, we prepared a qualitative synthesis of reported adverse events by type of prostanoid.
We analysed dichotomous data by using risk ratios (RRs) with a confidence interval (CI) of 95%. We analysed continuous data by using standardised mean differences (SMDs) with a CI of 95%.
Unit of analysis issues
For this update, we considered the individual participant as the unit of analysis. In the event that outcome information was reported as numbers of events instead of numbers of participants with at least one event, we did not include the study in the meta‐analysis.
Dealing with missing data
In the event that data were missing from the full reports, we planned to move the study to Studies awaiting classification until we could obtain further information from the study authors. However, in four cases (potentially relevant studies published as abstracts), authors did not have the data, or did not reply, so we excluded their reports (Fonseca 1991; Menzoian 1995; Mingardi 1993; Schwarz 1995). We excluded another seven studies because trial authors reported only outcome measures combining participants with non‐critical limb ischaemia or with chance of reconstructive intervention and did not reply to our request for disaggregate data (Bertele 1999; Feng 2009; Guan 2003; Heidrich 1991; Ohtake 2014).
Assessment of heterogeneity
To test for statistical heterogeneity, although of limited power, we used the Chi² test with significance level set at P < 0.1 and the I² test. We interpreted the I² test as indicated in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: represents considerable heterogeneity.
For robustness of results, and to perform a conservative analysis, if I² was 0% to 40%, suggesting not important heterogeneity, we reported results using a fixed‐effect model; if moderate heterogeneity was suggested by an I² between 30% and 60%, we reported results using a random‐effects model; and if I² was > 50%, suggesting substantial heterogeneity, we did not perform meta‐analyses.
Assessment of reporting biases
We attempted to obtain study protocols to assess for selective outcome reporting. When study protocols were unavailable, we compared methods and results sections of the report.
We used funnel plots to assess small‐study effects when 10 or more studies were available for outcomes (Higgins 2011a). Several explanations can be offered for asymmetry in a funnel plot, including true heterogeneity of effect with respect to trial size, poor methodological design (and hence bias of small trials), and publication bias. We interpreted these results carefully.
Data synthesis
We performed random‐effects meta‐analysis when we found little evidence of clinical heterogeneity across studies. Additionally, when we found good evidence of homogeneous effects across studies, we performed fixed‐effect meta‐analysis (see Assessment of heterogeneity). We interpreted random‐effects meta‐analyses with due consideration of the whole distribution of effects. In addition, we performed statistical analyses according to the statistical guidelines provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). For dichotomous outcomes, we used the Mantel‐Haenszel method, and for continuous outcomes, we used the inverse variance method. We used Review Manager 5 (RevMan 2014) software to perform analyses.
Subgroup analysis and investigation of heterogeneity
When sufficient studies were available, we conducted subgroup analyses by type of prostanoid. We used the test for subgroup differences in Review Manager 5 (RevMan 2014) to compare subgroups.
Sensitivity analysis
We performed sensitivity analyses to explore the influence of risk of bias on treatment effect sizes by excluding studies with high risk of bias in the following domains: allocation concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessors (detection bias), and incomplete outcome data (attrition bias).
'Summary of findings' table
We presented the overall quality of evidence for each outcome according to the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach (Guyatt 2008), which takes into account five criteria.
Risk of bias.
Inconsistency.
Imprecision.
Publication bias.
Directness of results.
For each comparison, two review authors (VV and JVAF) independently rated the quality of evidence for each outcome as 'high', 'moderate', 'low', or 'very low' using GRADEpro GDT. We resolved discrepancies by consensus, or, if needed, by arbitration provided by a third review author (AC). For each comparison presented in the review, we presented a summary of evidence for main outcomes in a 'Summary of findings' table, which provides key information about the best estimate of the magnitude of effect in relative terms and absolute differences for each relevant comparison of alternative management strategies; numbers of participants and studies addressing each important outcome; and the rating of overall confidence in effect estimates for each outcome (Guyatt 2011; Schünemann 2011). When meta‐analysis was not possible, we presented results in a narrative 'Summary of findings' table.
We included in the 'Summary of findings' table data for all participants with critical limb ischaemia for the following outcomes.
Cardiovascular mortality.
Amputations (total: major and minor).
Quality of life.
Adverse events.
Rest‐pain relief.
Ulcer healing.
Results
Description of studies
Results of the search
See Figure 1.
1.
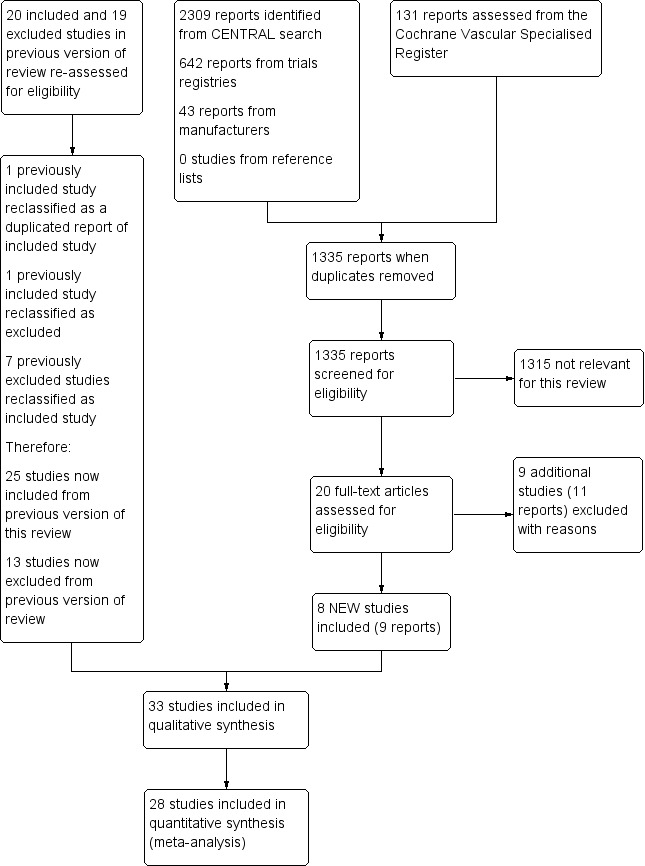
Study flow diagram.
*Out of 22 excluded studies, nine studies evaluated an inappropriate intervention for control group, seven included participants not fulfilling inclusion criteria with no available disaggregated outcome data, four were abstracts with no available full text, one had a quasi‐randomised design, and another was unpublished and had no available results.
For this update, we reassessed all 39 previously included (20) and excluded (19) studies for eligibility according to changes in selection criteria. We reclassified one previously included study as a duplicate report (Diehm 1987), reclassified another previously included study as excluded owing to inappropriate intervention for the control group (Beischer 1998), and reclassified seven previously excluded studies as included studies (Alstaedt 1993; Bandiera 1995; Böhme 1994; Cronenwett 1986; Diehm 1989; Karnik 1986; Trübestein 1989). We made no change to the other studies.
Included studies
We have listed details of the included studies below and in the Characteristics of included studies table.
For this update, we reassessed seven previously excluded studies for inclusion (Alstaedt 1993; Bandiera 1995; Böhme 1994; Cronenwett 1986; Diehm 1989; Karnik 1986; Trübestein 1989), and we identified eight new included studies (Belch 2011; Castagno 2000; Esato 1995; Jogestrand 1985; NCT00596752; Reisin 1997; Sakaguchi 1984; Schuler 1984). We reclassified one previously included study (Diehm 1987) as a preliminary report of another included study (Diehm 1988), based on information provided by personal contact with study authors. We reclassified another previously included study as excluded owing to inappropriate intervention for the control group (Beischer 1998).
In total, we included in the review 33 trials with a total of 4477 randomised participants (Alstaedt 1993; Balzer 1991; Bandiera 1995; Belch 1983; Belch 2011; Böhme 1989; Böhme 1994; Brass 2006; Brock 1990; Castagno 2000; Cronenwett 1986; Diehm 1988; Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Esato 1995; Guilmot 1991; Hossmann 1983; Jogestrand 1985; Karnik 1986; Linet 1991; NCT00596752; Negus 1987; Norgren 1990; Reisin 1997; Sakaguchi 1984; Schellong 2004; Schuler 1984; Stiegler 1992; Telles 1984; Trubestein 1987; Trübestein 1989).
Most studies were published in English, but nine were published in German (Böhme 1989; Böhme 1994; Brock 1990; Diehm 1988; Diehm 1989; Hossmann 1983; Karnik 1986; Stiegler 1992; Trubestein 1987), one in Japanese (Esato 1995), and one in Italian (Bandiera 1995). We obtained information from the ClinicalTrials.gov database on the unpublished clinical trial NCT00596752.
We included 21 studies that compared a prostanoid versus placebo intravenously administered: seven studies on PGE1 (Diehm 1988; Jogestrand 1985; NCT00596752; Reisin 1997; Schuler 1984; Stiegler 1992; Telles 1984), three on PGI2 (Belch 1983; Cronenwett 1986; Hossmann 1983), six on iloprost (Balzer 1991; Brock 1990; Diehm 1989; Dormandy 1991; Guilmot 1991; Norgren 1990), and three single studies on lipo‐ecraprost (Brass 2006), taprostene (Belch 2011), or ciprostene (Linet 1991). Two additional studies compared high‐dose and low‐dose oral iloprost versus placebo (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B).
Five studies compared PGE1 against other active drugs. One study compared PGE1 infused intravenously (iv) versus pentoxifylline (Trübestein 1989), and another compared the same prostanoid versus naftidrofuryl (Böhme 1994). Another two studies compared intra‐arterial (ia) administered PGE1 versus adenosine triphosphate (ATP) (Böhme 1989; Trubestein 1987), and a third compared high‐dose and low‐dose PGE1 infused ia versus oral inositol niacinate (Sakaguchi 1984).
Two studies compared PGI2 versus naftidrofuryl: One used the ia route of administration (through a 21 spring wire guide (SWG) catheter inserted into the common femoral artery) (Negus 1987), and the other used the iv route (Karnik 1986).
Finally, five studies compared different prostanoids: Four studies evaluated iv iloprost versus PGE1 (Alstaedt 1993; Bandiera 1995; Castagno 2000; Schellong 2004), and another compared TTC‐909 (clinprost incorporated in lipid microspheres) versus lipo‐PGE1 (Esato 1995), both administered iv.
No studies evaluating beraprost or cisaprost fulfilled the inclusion criteria.
Length of treatment ranged from three days to four weeks, except for one cross‐over study, for which length of treatment was three hours with a washout period of one day between interventions (Schellong 2004). Length of participant follow‐up ranged from two weeks to six months, except for two studies, in which follow‐up lasted three years and four years (Sakaguchi 1984 and NCT00596752, respectively).
Excluded studies
We have listed details of excluded studies in the Characteristics of excluded studies table.
For this update, we identified nine new excluded studies (Esato 1997; Feng 2009; NCT00059644; Nizankowski 1985; Ohtake 2014; Sakaguchi‐Shukichi 1990; Sert 2008; Weiss 1991; Ylitalo 1990). We reclassified one previously included study as excluded (Beischer 1998). We also reclassified and included seven studies that were excluded in the previous review owing to high risk of bias (see Included studies), making a total of 22 excluded studies.
Among the 22 excluded studies, the most frequent criteria for exclusion were inappropriate or no intervention for the control group (Arosio 1998; Beischer 1998; Breuer 1995; Ceriello 1998; Di Paolo 2005; Esato 1997; Petronella 2004; Sert 2008; Weiss 1991) and participants not fulfilling inclusion criteria with no available disaggregated outcome data (Bertele 1999; Feng 2009; Guan 2003; Heidrich 1991; Nizankowski 1985; Ohtake 2014; Ylitalo 1990). Four out of 22 studies were abstracts for which we were unable to obtain full text in spite of contacting trial authors for further information (Fonseca 1991; Menzoian 1995; Mingardi 1993; Schwarz 1995). Finally, one study was non‐randomised (Sakaguchi‐Shukichi 1990), and another was unpublished and no results were available (NCT00059644).
Risk of bias in included studies
None of the included studies presented low risk of bias in all domains, and most studies presented at least one domain with high risk of bias (see Characteristics of included studies; Figure 2; and Figure 3).
2.
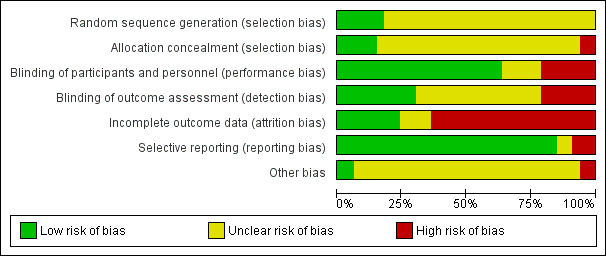
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
3.
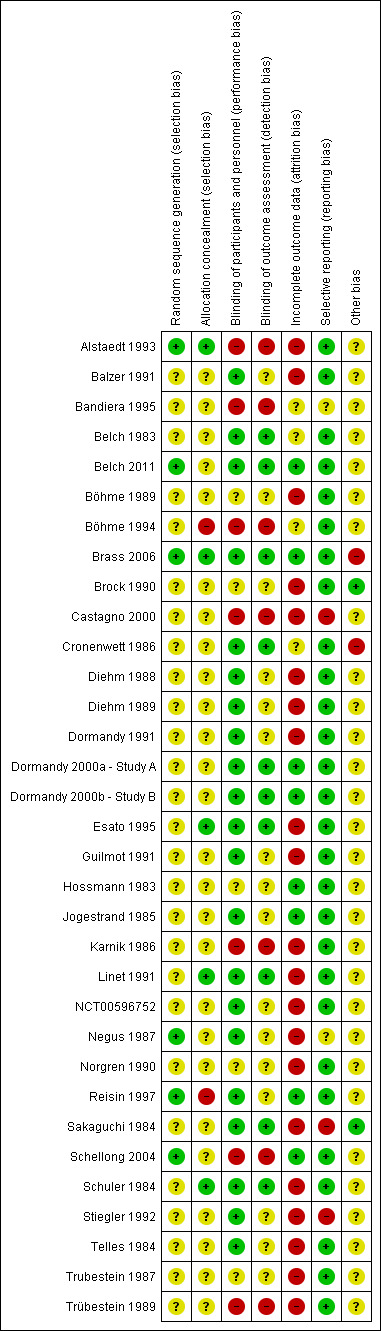
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
Of the 33 included studies, six (18%) described the method used for the randomisation process (see Characteristics of included studies). One study used a random number table (Alstaedt 1993), three used a computer‐generated randomisation list (Belch 2011; Brass 2006; Schellong 2004), one study reported use of random numbers (Negus 1987), and another stated that investigators used a randomised, permuted block design (Reisin 1997). The remaining studies did not provide sufficient information, and we deemed that they had unclear risk of bias.
Seven studies (21%) provided an explanation of the allocation concealment process. Five studies described use of an adequate method for allocation concealment, which included central allocation (Alstaedt 1993; Brass 2006), a sealed packaging method (Esato 1995), and a code of drug assignment supplied by the study sponsor (Linet 1991; Schuler 1984). Two studies reported that researchers used a randomisation or allocation list (Böhme 1994; Reisin 1997); we therefore considered them to have high risk of bias. The remaining studies did not provide sufficient information, and we deemed that they had unclear risk of bias.
Blinding
Of the 33 included studies, 21 (64%) reported a double‐blind design, and we considered them to be at low risk of performance bias (see Characteristics of included studies). Seven studies (21%) were open‐label studies; we therefore considered them to have high risk of bias owing to the subjective nature of reported outcomes (Alstaedt 1993; Bandiera 1995; Böhme 1994; Castagno 2000; Karnik 1986; Schellong 2004; Trübestein 1989). Five studies (15%) provided no statement or information about blinding (Böhme 1989; Brock 1990; Hossmann 1983; Norgren 1990; Trubestein 1987), and we deemed that they were at unclear risk of bias.
Among the 21 studies described by trial authors as double‐blind, 10 stated that not only participants and study personnel, but also outcome assessors, were blinded (Belch 1983; Belch 2011; Brass 2006; Cronenwett 1986; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Esato 1995; Linet 1991; Sakaguchi 1984; Schuler 1984); we therefore considered them to be at low risk of detection bias. The remaining 11 studies (Balzer 1991; Diehm 1988; Diehm 1989; Dormandy 1991; Guilmot 1991; Jogestrand 1985; NCT00596752; Negus 1987; Reisin 1997; Stiegler 1992; Telles 1984) did not mention whether outcome assessors were blinded; therefore we considered them to be at unclear risk of detection bias. We considered the seven open‐label studies to be at high risk of bias in this domain as they did not include a blinded outcome assessor (Alstaedt 1993; Bandiera 1995; Böhme 1994; Castagno 2000; Karnik 1986; Schellong 2004; Trübestein 1989). Five studies (15%) provided no statement or information about blinding of outcome assessment (Böhme 1989; Brock 1990; Hossmann 1983; Norgren 1990; Trubestein 1987); we deemed that these studies had unclear risk of bias.
Incomplete outcome data
Of the 33 studies included, 30 (90%) reported participant withdrawals (see Characteristics of included studies). Twenty‐one (64%) studies had high risk of bias in this domain: 16 described an attrition rate greater than 10% (Alstaedt 1993; Balzer 1991; Böhme 1989; Brock 1990; Diehm 1989; Dormandy 1991; Esato 1995; Guilmot 1991; Karnik 1986; Linet 1991; NCT00596752; Negus 1987; Norgren 1990; Sakaguchi 1984; Schuler 1984; Stiegler 1992), one reported unbalanced withdrawals in the control group (Castagno 2000), and four did not perform intention‐to‐treat (ITT) analysis (Diehm 1988; Telles 1984; Trubestein 1987; Trübestein 1989). Four studies did not provide sufficient information, and we deemed that they were at unclear risk of bias in this domain (Bandiera 1995; Belch 1983; Böhme 1994; Cronenwett 1986); we considered the remaining eight studies to have low risk of attrition bias owing to complete follow‐up or attrition rate less than 10% and ITT analysis (Belch 2011; Brass 2006; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Hossmann 1983; Jogestrand 1985; Reisin 1997; Schellong 2004).
Selective reporting
Of the 33 studies included, 28 (85%) reported all outcomes specified in the methods section (see Characteristics of included studies). We considered three studies as having high risk of selective reporting bias because we noted differences between outcomes reported in the methods and results sections (Stiegler 1992); because only the composite efficacy outcome was reported (Castagno 2000); or because reported outcomes were subjective and were not clearly defined (Sakaguchi 1984). Two studies did not provide prespecified outcomes in the methods section (Bandiera 1995; Negus 1987); we deemed that they had unclear risk of bias.
One of the included studies is a report of adverse events of a larger efficacy study, which we could not retrieve by applying search strategies and contacting trial authors (Bandiera 1995).
Regarding publication bias, we did not detect important asymmetries in the following outcomes from funnel plots performed for meta‐analyses, including at least 10 studies that compared prostanoids versus placebo: total amputations (Figure 4), all‐cause mortality, rest‐pain relief (dichotomous variable), and ulcer healing.
4.
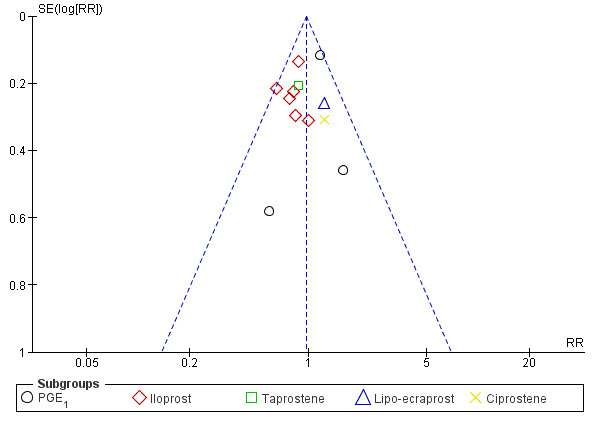
Funnel plot of comparison: 1 Prostanoids versus placebo, outcome: 1.2 Total amputations.
Other potential sources of bias
We considered two studies to have low risk of bias because trial authors provided a complete description of baseline characteristics of included participants and identical care programmes for both groups (Brock 1990; Sakaguchi 1984). One study presented unbalanced baseline characteristics and co‐interventions between groups (Cronenwett 1986), and another was terminated early because of futility (Brass 2006); therefore we considered these studies to have high risk of bias.
The remaining 29 studies presented unclear risk of bias in this domain as the result of incomplete description of care programmes and baseline risk of included participants (Alstaedt 1993; Balzer 1991; Bandiera 1995; Belch 1983; Belch 2011; Böhme 1989; Böhme 1994; Castagno 2000; Diehm 1988; Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Esato 1995; Guilmot 1991; Hossmann 1983; Jogestrand 1985; Karnik 1986; Linet 1991; NCT00596752; Negus 1987; Norgren 1990; Reisin 1997; Schellong 2004; Schuler 1984; Stiegler 1992; Telles 1984; Trubestein 1987; Trübestein 1989).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
Summary of findings for the main comparison. Prostanoids compared with placebo for critical limb ischaemia.
| Prostanoids compared with placebo for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: prostanoids Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with prostanoids | |||||
| Cardiovascular mortality Follow‐up: range 2 months to 7 months | Study population | RR 0.81 (0.41 to 1.58) | 1170 (3 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 32 per 1000 | 26 per 1000 (13 to 51) | |||||
| Total amputations Follow‐up: range 1 month to 12 months | Study population | RR 0.97 (0.86 to 1.09) | 2825 (12 RCTs) | ⊕⊕⊕⊕ HIGHc | ||
| 269 per 1000 | 261 per 1000 (231 to 293) | |||||
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included trials reported this outcome. | |
| Adverse events (participants) Follow‐up: range 3 weeks to 7 months | Study population | RR 2.11 (1.79 to 2.50) | 655 (8 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 319 per 1000 | 674 per 1000 (572 to 798) | |||||
| Rest‐pain relief (dichotomous) Follow‐up: range 1 month to 12 months | Study population | RR 1.30 (1.06 to 1.59) | 1179 (10 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 228 per 1000 | 296 per 1000 (241 to 362) | |||||
| Ulcer healing Follow‐up: range 3 weeks to 6 months | Study population | RR 1.24 (1.04 to 1.48) | 1719 (11 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 358 per 1000 | 444 per 1000 (373 to 530) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level because most studies were at high risk of bias (attrition bias). bDowngraded by one level for not meeting optimal information size (OIS) (for a 25% relative risk ratio (RRR), alpha = 0.05 and beta = 0.20, OIS = 13342). The 95% confidence interval includes both appreciable benefit and harm. cEven though eight trials were at high risk of attrition bias, a sensitivity analysis excluding those studies did not modify the effects of prostanoids (RR 0.91, 95% CI 0.75 to 1.10).
Summary of findings 2. Prostanoids compared with pentoxifylline for critical limb ischaemia.
| Prostanoids compared with pentoxifylline for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: prostanoids Comparison: pentoxifylline | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with pentoxifylline | Risk with prostanoids | |||||
| Cardiovascular mortality | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Total amputations Follow‐up: range 1 month to 6 months | Study population | RR 0.50 (0.05 to 5.27) | 70 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | ||
| 57 per 1000 | 29 per 1000 (3 to 301) | |||||
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse events (participants) Follow‐up: range 1 month to 6 months | Study population | RR 0.60 (0.24 to 1.47) | 70 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | ||
| 286 per 1000 | 171 per 1000 (69 to 420) | |||||
| Rest‐pain relief | Study population | RR 1.40 (0.72 to 2.72) | 70 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | ||
| 286 per 1000 | 400 per 1000 (206 to 777) | |||||
| Ulcer healing | Study population | RR 1.61 (1.13 to 2.30) | 70 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | ||
| 514 per 1000 | 828 per 1000 (581 to 1000) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by two levels owing to high risk of performance bias (open‐label study) and attrition bias, and owing to imbalance in baseline characteristics of participants with no mention of methods used for random sequence generation or allocation concealment. bDowngraded by one level owing to wide confidence interval.
Summary of findings 3. Prostanoids compared with naftidrofuryl for critical limb ischaemia.
| Prostanoids compared with naftidrofuryl for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: prostanoids Comparison: naftidrofuryl | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with naftidrofuryl | Risk with prostanoids | |||||
| Cardiovascular mortality | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Total amputations Follow‐up: range 6 months to 4 years | Study population | RR 0.96 (0.57 to 1.64) | 29 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 667 per 1000 | 640 per 1000 (380 to 1000) | |||||
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse events (participants) Follow‐up: range 3 days to 2 weeks | Study population | RR 3.29 (0.27 to 40.19) | 55 (2 RCTs) | ⊕⊕⊝⊝ LOWb,c | ||
| 36 per 1000 | 118 per 1000 (10 to 1000) | |||||
| Rest‐pain relief Follow‐up: range 3 weeks to 4 weeks | Study population | RR 1.07 (0.83 to 1.39) | 75 (3 RCTs) | ⊕⊕⊝⊝ LOWb,d | ||
| 605 per 1000 | 648 per 1000 (502 to 841) | |||||
| Ulcer healing Follow‐up: range 3 weeks to 4 weeks | Study population | RR 0.97 (0.46 to 2.03) | 39 (2 RCTs) | ⊕⊕⊝⊝ LOWb,c | ||
| 429 per 1000 | 416 per 1000 (197 to 870) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level owing to high risk of attrition bias. bDowngraded by one level owing to imprecision issues (low rate of events). cDowngraded by one level owing to high risk of bias (one trial presented high risk of performance and detection bias, and the other high risk of attrition bias). dDowngraded by one level owing to high risk of bias (most studies presented high risk of performance and detection bias or performance bias).
Summary of findings 4. PGE1 compared with ATP for critical limb ischaemia.
| PGE1 compared with ATP for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: PGE1 Comparison: ATP | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with ATP | Risk with PGE1 | |||||
| Cardiovascular mortality | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Total amputations Follow‐up: range 3 weeks to 12 months | Study population | RR 0.26 (0.09 to 0.74) | 91 (2 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 310 per 1000 | 80 per 1000 (28 to 229) | |||||
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse event (participants) Follow‐up: range 3 weeks to 12 months | Study population | RR 2.78 (1.41 to 5.48) | 91 (2 RCTs) | ⊕⊕⊝⊝ LOWa,b | ||
| 190 per 1000 | 530 per 1000 (269 to 1000) | |||||
| Rest‐pain relief | See comments. | ‐ | 34 (1 RCT) | ‐ | One study reported rest‐pain as a continuous outcome. Reanalysis was not possible. | |
| Ulcer healing (complete) Follow‐up: median 3 weeks | Study population | RR 0.72 (0.17 to 3.03) | 31 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 231 per 1000 | 166 per 1000 (39 to 699) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ATP: adenosine triphosphate; CI: confidence interval; PG: prostaglandin; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level owing to high risk of attrition bias. bDowngraded by one level owing to a wide confidence interval resulting from a low rate of events.
Summary of findings 5. PGE1 compared with inositol niacinate for critical limb ischaemia.
| PGE1 compared with inositol niacinate for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: PGE1 Comparison: inositol niacinate | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with inositol niacinate | Risk with PGE1 | |||||
| Cardiovascular mortality | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Total amputations | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse events (participants) Follow‐up: range 2 weeks to 6 weeks | Study population | RR 1.60 (0.79 to 3.23) | 65 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 333 per 1000 | 533 per 1000 (263 to 1000) | |||||
| Rest‐pain relief Follow‐up: range 2 weeks to 6 weeks | Study population | RR 1.45 (0.88 to 2.37) | 65 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 500 per 1000 | 725 per 1000 (440 to 1000) | |||||
| Ulcer healing Follow‐up: range 2 weeks to 6 weeks | Study population | RR 3.06 (0.41 to 22.79) | 65 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 56 per 1000 | 170 per 1000 (23 to 1000) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; PG: prostaglandin; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level owing to high risk of attrition bias. bDowngraded by one level owing to a low rate of events resulting in wide confidence intervals.
Summary of findings 6. Iloprost compared with PGE1 for critical limb ischaemia.
| Iloprost compared with PGE1 for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: iloprost Comparison: PGE1 | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with PGE1 | Risk with iloprost | |||||
| Cardiovascular mortality | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Total amputations Follow‐up: range 4 weeks to 6 months | Study population | RR 0.89 (0.66 to 1.21) | 209 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 466 per 1000 | 415 per 1000 (308 to 564) | |||||
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse events (participants) Follow‐up: range 2 days to 4 weeks | Study population | RR 2.35 (1.82 to 3.04) | 511 (4 RCTs) | ⊕⊕⊕⊝ MODERATEa | ||
| 202 per 1000 | 476 per 1000 (368 to 615) | |||||
| Rest‐pain relief Follow‐up: range 21 days to 28 days | Study population | RR 1.15 (0.90 to 1.47) | 228 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 491 per 1000 | 565 per 1000 (442 to 722) | |||||
| Ulcer healing Follow‐up: range 21 days to 28 days | Study population | RR 1.24 (0.95 to 1.63) | 228 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 536 per 1000 | 664 per 1000 (509 to 873) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; PG: prostaglandin; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level owing to high risk of bias (performance, detection, and attrition bias). bDowngraded by one level owing to a low rate of events resulting in a wide confidence interval.
Summary of findings 7. Clinprost compared with lipo‐PGE1 for critical limb ischaemia.
| Clinprost compared with lipo‐PGE1 for critical limb ischaemia | ||||||
| Patient or population: people with critical limb ischaemia Setting: hospital Intervention: clinprost Comparison: lipo‐PGE1 | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with lipo‐PGE1 | Risk with clinprost | |||||
| Cardiovascular mortality | See comments. | ‐ | (1 RCT) | ‐ | The trial authors mentioned the incidence of 1 death due to acute heart failure during the trial in the clinprost group. They did not discriminate whether this happened in the CLI from atherosclerotic origin group or in the thromboangiitis obliterans (TAO) group. | |
| Total amputations | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Quality of life | See comments. | ‐ | ‐ | ‐ | None of the included studies reported this outcome. | |
| Adverse events Follow‐up: median 28 days | Study population | RR 1.83 (0.56 to 5.96) | 135 (1 RCT) | ⊕⊕⊕⊝ MODERATEa | The trial authors did not discriminate whether adverse events happened in the CLI from atherosclerotic origin group or in the TAO group. | |
| 58 per 1000 | 106 per 1000 (32 to 346) | |||||
| Rest‐pain relief Follow‐up: median 28 days | Study population | RR 1.05 (0.58 to 1.87) | 67 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 394 per 1000 | 414 per 1000 (228 to 737) | |||||
| Ulcer healing Follow‐up: median 28 days | Study population | RR 1.18 (0.74 to 1.89) | 72 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | ||
| 457 per 1000 | 539 per 1000 (338 to 864) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CLI: critical limb ischaemia; PG: prostaglandin; RCT: randomised controlled trial; RR: risk ratio; TAO: thromboangiitis obliterans. | ||||||
| GRADE Working Group grades of evidence. High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level owing to few events, resulting in a wide confidence interval that includes both harms and benefits. bDowngraded by one level owing to high risk of bias (attrition rate > 10%).
Prostanoids versus placebo
As we could obtain neither concrete nor homogeneous definitions of some of the predefined outcomes, we considered the following outcomes as a dichotomous outcome: evaluation of rest‐pain relief when it was reported as the number or proportion of participants who experienced any improvement on a pain scale (considered no relief if pain was the same or worse); and ulcer healing when it was reported as the number or proportion of participants who had any decrease in surface area and/or presence of granulation tissue (considered no healing if surface area was similar or bigger and/or granulation tissue was absent).
Although we noted some clinical heterogeneity (study designs, types of prostanoid, and routes of administration differed significantly among included studies), we completed a global meta‐analysis of any type of prostanoid given via any route versus placebo. The aim was to allow a meaningful meta‐analysis of the same class of drug with the same expected biochemical action.
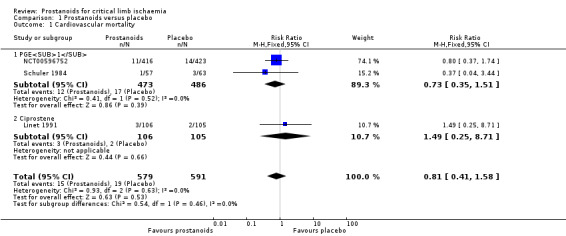
Cardiovascular mortality
Three studies reported cardiovascular mortality (two on PGE1 (NCT00596752; Schuler 1984), and one on ciprostene (Linet 1991)), with a similar incidence in both groups (RR 0.81, 95% CI 0.41 to 1.58; 1170 participants; I² = 0%; Analysis 1.1). We used a fixed‐effect model because statistical heterogeneity was not important.
1.1. Analysis.
Comparison 1 Prostanoids versus placebo, Outcome 1 Cardiovascular mortality.
Subgroup analysis by type of prostanoid revealed no differences for cardiovascular mortality (test for subgroup differences: Chi² = 0.54, df = 1 (P = 0.46), I² = 0%).
The quality of this evidence is low given that all studies had high risk of attrition bias and the number of events in each group was small, resulting in a wide confidence interval that includes both appreciable benefit and harm (see Table 1).
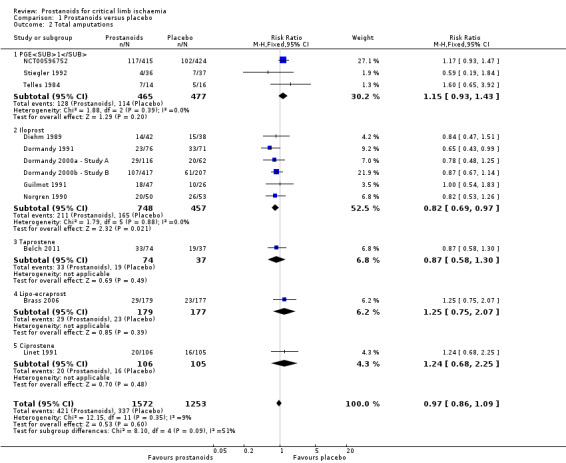
Total amputations
Twelve studies reported amputations: One study reported major, minor, and total amputations (Norgren 1990); three reported major and minor amputations (Belch 2011; Guilmot 1991; NCT00596752); four reported major amputations (Brass 2006; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B); and the remaining four reported the total number of amputations, including major and minor (Diehm 1989; Linet 1991; Stiegler 1992; Telles 1984).
We pooled results for total (major and minor) amputations across the 12 studies (three on PGE1 (NCT00596752; Stiegler 1992; Telles 1984), six on iloprost (Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991; Norgren 1990), one on taprostene (Belch 2011), one on lipo‐ecraprost (Brass 2006), and another on ciprostene (Linet 1991)). We found that 421/1572 events were reported in the prostanoids group and 337/1253 in the placebo group (RR 0.97, 95% CI 0.86 to 1.09; I² = 9%; Analysis 1.2). We used a fixed‐effect model owing to lack of important statistical heterogeneity.
1.2. Analysis.
Comparison 1 Prostanoids versus placebo, Outcome 2 Total amputations.
Subgroup analysis by type of prostanoid revealed differences for total amputations (test for subgroup differences: Chi² = 8.10, df = 4 (P = 0.09), I² = 50.6%) owing to better results with iloprost against placebo (RR 0.82, 95% CI 0.69 to 0.97) than with the other prostanoids, which showed minimal or no effect.
Even though several trials were at high risk of attrition bias, when we performed a sensitivity analysis excluding those studies (Diehm 1989; Dormandy 1991; Guilmot 1991; Linet 1991; NCT00596752; Norgren 1990; Stiegler 1992; Telles 1984), effects of prostanoids with regards to the incidence of total amputations were not modified (RR 0.91, 95% CI 0.75 to 1.10; 1269 participants; I² = 0%; Figure 5). For this reason, we consider the quality of this evidence to be high (see Table 1).
5.
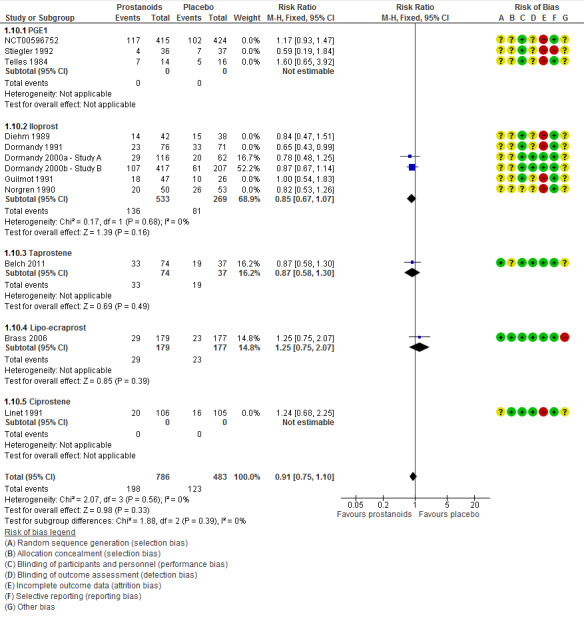
Forest plot of comparison: 1 Prostanoids versus placebo, outcome: 1.10 Total amputations. Sensitivity analysis excluding studies at high risk of attrition bias.
Quality of life
None of the included studies reported this outcome.
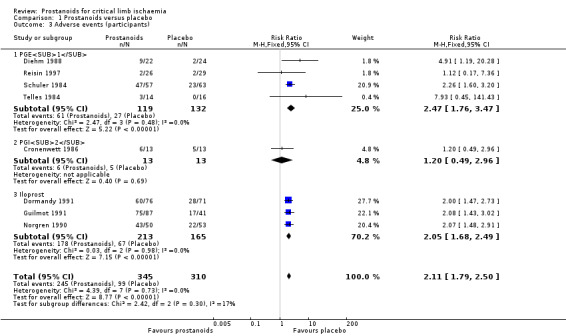
Adverse events
Fourteen studies reported adverse events. We excluded six studies from the meta‐analysis because they reported total numbers of adverse events instead of numbers of participants with at least one event (Belch 1983; Belch 2011; Linet 1991; NCT00059644), or they reported tolerability outcomes entailing clinical heterogeneity in relation to the definition of adverse event (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B).
Eight studies reported extractable information about numbers of participants with adverse events, and we included them in the meta‐analysis (four studies on PGE1 (Diehm 1988; Reisin 1997; Schuler 1984; Telles 1984), one on PGI2 (Cronenwett 1986), and three on iloprost (Dormandy 1991; Guilmot 1991; Norgren 1990)). Pooled results showed that 245/345 participants in the prostanoids group had adverse effects compared with 99/310 in the placebo group (RR 2.11, 95% CI 1.79 to 2.50; I² = 0%; Analysis 1.3). We used a fixed‐effect model because statistical heterogeneity was unimportant.
1.3. Analysis.
Comparison 1 Prostanoids versus placebo, Outcome 3 Adverse events (participants).
Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 2.42, df = 2 (P = 0.30), I² = 17.4%).
A sensitivity analysis excluding trials at high risk of selection bias did not affect the pooled estimate (RR 2.13, 95% CI 1.80 to 2.52; 600 participants; I² = 0%) (Reisin 1997). Nevertheless, the quality of evidence regarding this outcome was moderate owing to high risk of attrition bias in six out of eight studies (Diehm 1988; Dormandy 1991; Guilmot 1991; Norgren 1990; Schuler 1984; Telles 1984); exclusion of these studies from the sensitivity analysis influenced the overall effect (RR 1.18, 95% CI 0.51 to 2.71; 81 participants; I² = 0%), resulting in a wider confidence interval that included both appreciable benefit and harm (see Table 1).
Rest‐pain relief
We found 18 studies that reported rest‐pain relief. We excluded four studies from the meta‐analysis owing to unavailability of extractable or enough data for an appropriate analysis (Belch 2011; Hossmann 1983; Jogestrand 1985; Stiegler 1992).
We included 10 studies in a fixed‐effect meta‐analysis for dichotomous assessment of rest‐pain relief (one on PGE1 (Diehm 1988), two on PGI2 (Belch 1983; Cronenwett 1986), six on iloprost (Balzer 1991; Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991), and one on lipo‐ecraprost (Brass 2006)). As subgroup analysis revealed no differences between high‐dose and low‐dose iloprost against placebo for this outcome (test for subgroup differences: Chi² = 0.82, df = 1 (P = 0.37), I² = 0%) (RR 1.13, 95% CI 0.88 to 1.46; 739 participants; 10 studies; I² = 24%), we presented data from the two studies combined (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B). Pooled results indicate that 204/665 participants who received prostanoids and 117/514 who received placebo achieved rest‐pain relief (RR 1.30, 95% CI 1.06 to 1.59; I² = 18%; Analysis 1.4). Subgroup analysis revealed no differences between different types of prostanoids (test for subgroup differences: Chi² = 5.35, df = 3 (P = 0.15), I² = 43.9%).
1.4. Analysis.
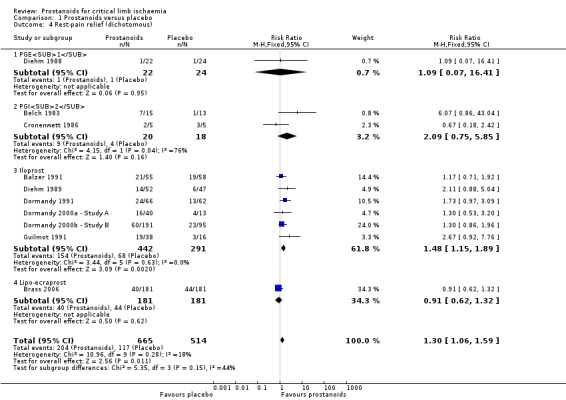
Comparison 1 Prostanoids versus placebo, Outcome 4 Rest‐pain relief (dichotomous).
The quality of this evidence was moderate owing to high risk of attrition bias in five studies (see Table 1). A sensitivity analysis excluding those studies (Balzer 1991; Diehm 1988; Diehm 1989; Dormandy 1991; Guilmot 1991) yielded a similar incidence of rest‐pain relief in the group that received prostanoids compared with the group given placebo (RR 1.13, 95% CI 0.88 to 1.46; 739 participants; I² = 24%; Analysis 1.5). However, estimates lost precision and resulted in a wider confidence interval that included both appreciable benefit and harm.
1.5. Analysis.
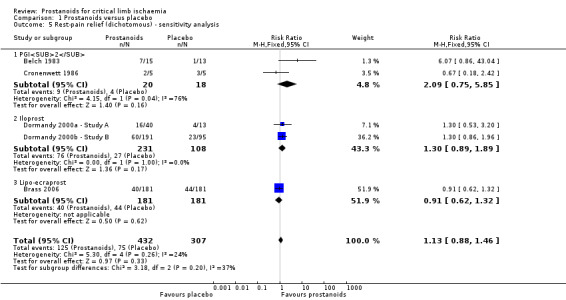
Comparison 1 Prostanoids versus placebo, Outcome 5 Rest‐pain relief (dichotomous) ‐ sensitivity analysis.
We included three studies with 984 participants in a meta‐analysis for continuous measurement of rest‐pain relief (two on PGE1 (NCT00596752; Schuler 1984) and one on PGI2 (Cronenwett 1986)). We summarised rest‐pain scores in a random‐effects model meta‐analysis for standardised mean differences (SMDs), obtaining 0.16 (95% CI ‐0.10 to 0.42; I² = 42%; Analysis 1.6). Confidence intervals of the prostanoids subgroups overlapped, and studies were too few to permit conclusions about whether one type of prostanoid produced more rest‐pain relief than another. The quality of this evidence is low owing to high risk of bias and inconsistency across included studies.
1.6. Analysis.
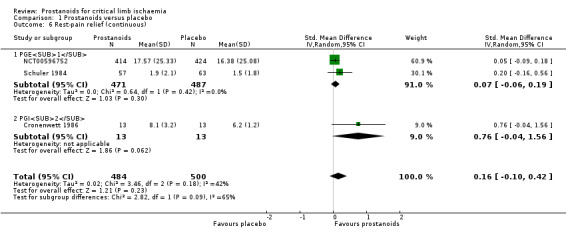
Comparison 1 Prostanoids versus placebo, Outcome 6 Rest‐pain relief (continuous).
Two other studies reported results regarding rest‐pain relief qualitatively, stating no significant differences between treatment and control groups (Linet 1991; Telles 1984).
Reduction in analgesic consumption
We found five studies that reported analgesics consumption. We excluded three studies from meta‐analysis: two because extractable data were unavailable (Brock 1990; NCT00596752), and another because investigators reported a continuous outcome (Belch 1983).
We included two studies in a fixed‐effect meta‐analysis for dichotomous assessment of reduction in analgesics consumption (one on PGE1 (Diehm 1988), and one on iloprost (Balzer 1991)). Pooled results showed that 47/64 participants receiving prostanoids and 38/71 receiving placebo achieved a reduction in analgesic consumption (RR 1.40, 95% CI 1.08 to 1.82; 135 participants; I² = 0%; Analysis 1.7). Subgroup analysis by type of prostanoid showed no differences for this outcome (test for subgroup differences: Chi² = 0.35, df = 1 (P = 0.56), I² = 0%).
1.7. Analysis.
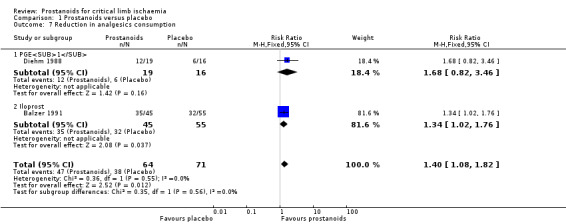
Comparison 1 Prostanoids versus placebo, Outcome 7 Reduction in analgesics consumption.
Belch 1983 reported a significant reduction in mean daily analgesic consumption compared with baseline in both groups of participants, but the mean difference between groups was not statistically significant neither during the infusion (mean difference (MD) ‐0.20, 95% CI ‐1.06 to 0.66) nor after four days' follow‐up (MD 0.40, 95% CI ‐0.53 to 1.33).
Two studies described results regarding analgesics consumption qualitatively, indicating no significant differences between treatment and control groups (Belch 2011; Telles 1984).
The quality of the evidence regarding this outcome is low owing to high risk of bias and imprecision, and a small number of events did not reach the optimal information size.
Ulcer healing
We found 13 studies that reported ulcer healing. We excluded two studies from meta‐analysis: one because it included only diabetic patients with a significant proportion of neuropathic and microangiopathic ulcers and therefore represented a clinically different population from other studies (Brock 1990), and the other because investigators measured ulcer healing as a continuous outcome (Belch 2011).
We included the remaining 11 studies in a random‐effects meta‐analysis (five studies on PGE1 (Diehm 1989; NCT00596752; Schuler 1984; Stiegler 1992; Telles 1984), one on PGI2 (Cronenwett 1986), four on iloprost (Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991), and another on ciprostene (Linet 1991)). As subgroup analysis revealed no differences between high‐dose and low‐dose iloprost against placebo for this outcome (test for subgroup differences: Chi² = 0.07, df = 1 (P = 0.80), I² = 0%), we presented data from the two studies combined (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B). Pooled results show that 381/946 participants in the prostanoid group and 277/773 in the placebo group had some degree of ulcer healing (RR 1.24, 95% CI 1.04 to 1.48; 11 studies; I² = 39%; Analysis 1.8). Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 2.59, df = 3 (P = 0.46), I² = 0%).
1.8. Analysis.
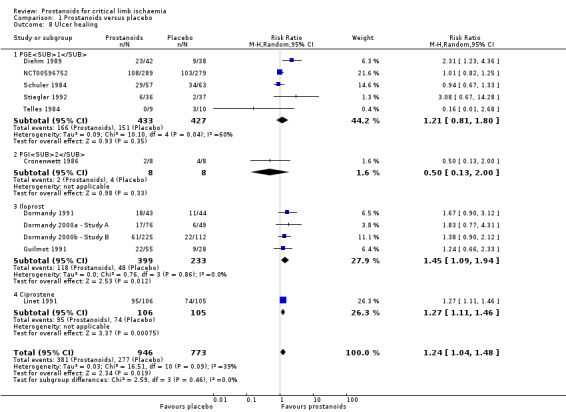
Comparison 1 Prostanoids versus placebo, Outcome 8 Ulcer healing.
We performed sensitivity analysis excluding eight studies at high risk of attrition bias (Diehm 1989; Dormandy 1991; Guilmot 1991; Linet 1991; NCT00596752; Schuler 1984; Stiegler 1992; Telles 1984), which showed an influence on overall effect (RR 1.33, 95% CI 0.83 to 2.13; 478 participants; I² = 19%). Estimates lost precision, resulting in a wider confidence interval that included both appreciable benefit and harm. Therefore, the quality of this evidence is moderate (see Table 1).
Belch 2011 reported that the group of participants receiving taprostene experienced no significant decrease in median ulcer size (161 to 177 mm²), whereas participants in the placebo group experienced an increase in median ulcer size (201 to 292 mm²) by the end of the infusion period.
Major amputation
Eight studies reported major amputations (one on PGE1 (NCT00596752), five on iloprost (Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991; Norgren 1990), one on taprostene (Belch 2011), and one on lipo‐ecraprost (Brass 2006)). As subgroup analysis revealed no differences between high‐dose and low‐dose iloprost against placebo for this outcome (test for subgroup differences: Chi² = 0.01, df = 1 (P = 0.93), I² = 0%), we presented data from the two studies combined (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B).
A fixed‐effect meta‐analysis showed that 294/1373 participants in the prostanoid group and 247/1057 in the placebo group experienced this event (RR 0.84, 95% CI 0.73 to 0.98; I² = 0%; Analysis 1.10). Subgroup analysis by type of prostanoid revealed no differences for major amputations (test for subgroup differences: Chi² = 2.98, df = 3 (P = 0.40), I² = 0%).
1.10. Analysis.
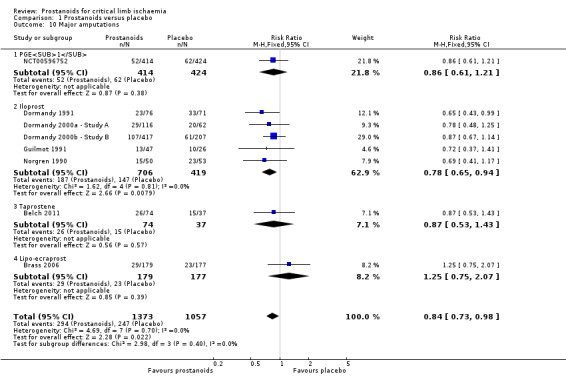
Comparison 1 Prostanoids versus placebo, Outcome 10 Major amputations.
We conducted a sensitivity analysis excluding studies with high risk of attrition bias (Dormandy 1991; Guilmot 1991; NCT00596752; Norgren 1990), thus diluting the beneficial effect of prostanoids for major amputations (RR 0.91, 95% CI 0.75 to 1.11; 1269 participants; I² = 0%). Therefore, the quality of evidence regarding this outcome is moderate.
Minor amputation
Four studies reported the incidence of minor amputations (one on PGE1 (NCT00596752), two on iloprost (Guilmot 1991; Norgren 1990), and another on taprostene (Belch 2011)). We observed this event in 81/485 participants in the prostanoid group and in 46/413 in the placebo group (RR 1.55, 95% CI 1.11 to 2.16; I² = 0%; Analysis 1.11). We used a fixed‐effect model owing to lack of important statistical heterogeneity. Subgroup analysis by type of prostanoid revealed no differences for minor amputations (test for subgroup differences: Chi² = 1.85, df = 2 (P = 0.40), I² = 0%).
1.11. Analysis.
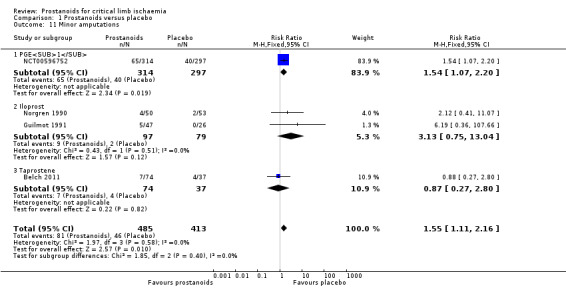
Comparison 1 Prostanoids versus placebo, Outcome 11 Minor amputations.
The quality of this evidence is low owing to high risk of attrition bias and imprecision. If we exclude studies at high risk of bias, only one study is left (Belch 2011). This study reported a similar incidence of minor amputations in those who received taprostene and those who received placebo (RR 0.87, 95% CI 0.27 to 2.80).
Ankle brachial index
Two studies reported ankle brachial index (ABI) (one on PGE1 (Telles 1984), and another on ciprostene (Linet 1991)). These studies presented data differently; therefore we could not perform a meta‐analysis.
Telles 1984 reported no significant improvement in ABI for both groups of participants at 24 hours (PGE1 group, median 0.43, range 0 to 1.15; placebo group, median 0.50, range 0 to 1.0) or at one month after infusion (measurements of effect not reported).
Linet 1991 reported no differences for this outcome between participants who received ciprostene (average range 0.60 ± 0.038 to 0.67 ± 0.052) and those given placebo (0.60 ± 0.047 to 0.69 ± .047) during the study.
The quality of this evidence is moderate owing to high risk of attrition bias.
All‐cause mortality
Eleven studies reported all‐cause mortality (two on PGE1 (NCT00596752; Schuler 1984), one on PGI2 (Belch 1983), five on iloprost (Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Norgren 1990), one on taprostene (Belch 2011), one on lipo‐ecraprost (Brass 2006), and another on ciprostene (Linet 1991)). As subgroup analysis revealed no differences between high‐dose and low‐dose iloprost against placebo for this outcome (test for subgroup differences: Chi² = 0.02, df = 1 (P = 0.89), I² = 0%), we presented data from the two studies combined (Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B).
Pooled results indicate a mortality incidence of 142/1548 in the prostanoid group and 95/1249 in the placebo group (RR 1.04, 95% CI 0.81 to 1.33; I² = 3%). Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 6.78, df = 5 (P = 0.24), I² = 26.3%). When excluding studies with high risk of attrition bias (Diehm 1989; Dormandy 1991; Linet 1991; NCT00596752; Norgren 1990; Schuler 1984), we noted no influence on the overall effect of prostanoids on all‐cause mortality (RR 1.06, 95% CI 0.78 to 1.43; 1297 participants; I² = 27%). However, owing to loss of precision and widening of the confidence interval, the quality of this evidence is moderate.
Prostanoids versus other active agents
Owing to the presence of significant clinical heterogeneity (types of experimental and control drugs differed significantly among included studies), we described results obtained upon various comparisons of prostanoids versus other active agents and performed meta‐analysis only for comparisons considering the same control intervention, when possible.
Prostanoids versus pentoxifylline
A single study with 70 participants compared intravenous infusion of PGE1 versus pentoxifylline (Trübestein 1989).
Cardiovascular mortality
This study provided no information regarding this outcome.
Total amputations
This trial reported a "borderline amputation" (an unclear type of amputation) in 1/35 participants who received PGE1 and in 2/35 participants given pentoxifylline during the treatment phase (RR 0.50, 95% CI 0.05 to 5.27). The quality of this evidence is very low owing to high risk of bias and imprecision, with a small number of events not reaching the optimal information size (see Table 2).
Quality of life
This included study did not report this outcome.
Adverse events
Trial authors reported adverse events in 6/35 participants who received placebo and in 10/35 who received pentoxifylline (RR 0.60, 95% CI 0.24 to 1.47). Premature treatment discontinuation occurred only in 4/35 participants in the pentoxifylline group and in no participants in the PGE1 group. The quality of this evidence is very low owing to high risk of bias and imprecision, with a small number of events not reaching the optimal information size (see Table 2).
Rest‐pain relief and consumption of analgesics
Data show some degree of rest‐pain relief for 14/35 participants in the PGE1 group and for 10/35 participants in the pentoxifylline group (RR 1.40, 95% CI 0.72 to 2.72), while in 16/35 participants in the PGE1 group and for 14/35 participants in the pentoxifylline group, this symptom disappeared (RR 1.11, 95% CI 0.65 to 1.91) after four weeks' follow‐up. Data show no differences regarding consumption of analgesic drugs: 22/30 participants in the PGE1 group and 17/29 in the pentoxifylline group reduced or discontinued consumption of analgesics by completion of treatment (RR 1.25, 95% CI 0.86 to 1.82). The quality of this evidence is very low owing to high risk of bias and imprecision issues (see Table 2).
Ulcer healing
In the PGE1 group, 29/35 participants experienced complete or partial ulcer healing according to a score system that included area, depth, floor, and edges of lesions as well as maintenance of a functioning extremity, compared with 18/35 participants in the pentoxifylline group who achieved this endpoint (RR 1.61, 95% CI 1.13 to 2.30). The quality of evidence provided by this randomised trial is very low owing to high risk of bias and imprecision issues (see Table 2).
Major or minor amputations
This study reported the incidence of an unclear type of amputation, which we could not classify as major and could not classify as minor.
Ankle brachial index
The included study did not report ABI as an outcome.
All‐cause mortality
This included study did not report information regarding this outcome.
Prostanoids versus naftidrofuryl
Three studies with 75 participants compared a prostanoid with naftidrofuryl (one on intravenously administered PGE1 (Böhme 1994), another on intravenous PGI2 (Karnik 1986), and a third on intra‐arterial PGI2 (Negus 1987)).
Cardiovascular mortality
None of these studies reported the incidence of cardiovascular mortality.
Total amputations
A single study reported amputation events, which presented in 9/14 participants in the PGI2 group and in 10/15 participants in the naftidrofuryl group (RR 0.96, 95% CI 0.57 to 1.64; Analysis 2.1) (Negus 1987). The quality of this evidence was low owing to high risk of attrition bias and imprecision issues.
2.1. Analysis.
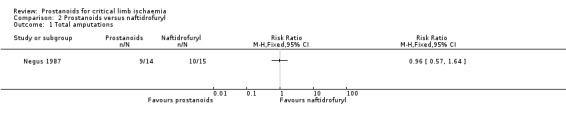
Comparison 2 Prostanoids versus naftidrofuryl, Outcome 1 Total amputations.
Quality of life
None of the included studies reported this outcome.
Adverse events
We included two studies in a random‐effects meta‐analysis of adverse events (Böhme 1994; Negus 1987). We excluded Karnik 1986 from meta‐analysis because investigators stated numbers of adverse events in each group, instead of numbers of participants with at least one adverse event. We observed that 6/27 participants who received prostanoids and 1/28 who received naftidrofuryl presented adverse events during the infusion (RR 3.29, 95% CI 0.27 to 40.19; I² = 40%; Analysis 2.2). Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 1.56, df = 1 (P = 0.21), I² = 35.7%). The quality of evidence is low owing to high risk of bias and imprecision issues (see Table 3).
2.2. Analysis.
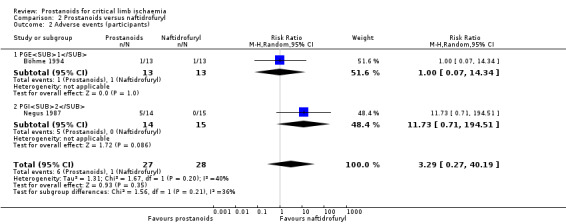
Comparison 2 Prostanoids versus naftidrofuryl, Outcome 2 Adverse events (participants).
Rest‐pain relief and consumption of analgesics
The included studies reported rest‐pain relief as a dichotomous outcome, defined as any improvement in a pain scale at the end of treatment, which ranged between 3 and 21 days (Böhme 1994; Karnik 1986; Negus 1987). A fixed‐effect meta‐analysis showed that 24/37 participants who received prostanoids and 23/38 who received naftidrofuryl achieved some degree of rest‐pain relief (RR 1.07, 95% CI 0.83 to 1.39; I² = 22%; Analysis 2.3). Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 1.60, df = 2 (P = 0.45), I² = 0%).
2.3. Analysis.
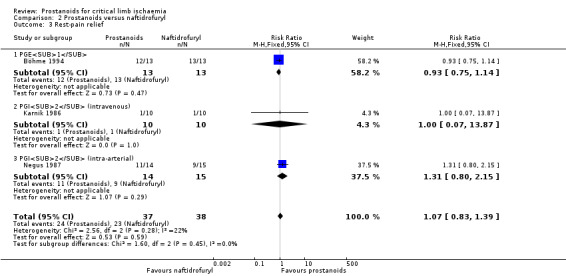
Comparison 2 Prostanoids versus naftidrofuryl, Outcome 3 Rest‐pain relief.
Sensitivity analysis excluding one study at high risk of selection bias (Böhme 1994) showed no influence on overall effect (RR 1.28, 95% CI 0.77 to 2.13; 49 participants; I² = 0%). Similarly, neither a sensitivity analysis excluding two studies at high risk of performance and detection bias (open‐label trials) (Böhme 1994; Karnik 1986), nor another excluding one study at high risk of attrition bias (Negus 1987), showed any influence on the estimated effect of prostanoids for this outcome (RR 1.31, 95% CI 0.80 to 2.15; and RR 0.93, 95% CI 0.75 to 1.14, respectively). Nevertheless, we consider the quality of this evidence as low owing to high risk of bias in critical domains for this subjective outcome and imprecision issues, including not reaching optimal information size (see Table 3).
Negus 1987 reported that participants experiencing relief of rest‐pain reduced their consumption of analgesics (11/14 participants in PGI2 group and 9/15 in naftidrofuryl group; RR 1.31, 95% CI 0.80 to 2.15). No other included studies reported this outcome. The quality of this evidence is low owing to high risk of attrition bias and imprecision issues.
Ulcer healing
A meta‐analysis of two studies that reported evolution of tissue lesions showed that 7/18 participants receiving a prostanoid and 9/21 receiving naftidrofuryl experienced partial or complete ulcer healing (RR 0.97, 95% CI 0.46 to 2.03; I² = 0%; Analysis 2.4) (Böhme 1994; Karnik 1986). Subgroup analysis by type of prostanoid revealed no differences for this outcome (test for subgroup differences: Chi² = 0.52, df = 1 (P = 0.47), I² = 0%).
2.4. Analysis.
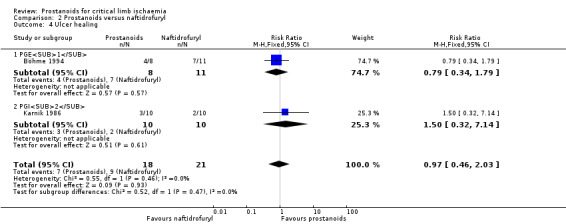
Comparison 2 Prostanoids versus naftidrofuryl, Outcome 4 Ulcer healing.
The quality of the evidence is very low owing to high risk of bias and imprecision issues (see Table 3).
Major or minor amputations
One study reported the incidence of minor amputations in 4/14 participants in the PGI2 group and in 3/15 in the naftidrofuryl group (RR 1.43, 95% CI 0.39 to 5.28), with occurrence of major amputation in 5/14 participants in the PGI2 group and in 7/15 in the naftidrofuryl group (RR 0.77, 95% CI 0.32 to 1.86) (Negus 1987). The quality of this evidence is low owing to high risk of attrition bias and the low rate of events not reaching the optimal information size.
Ankle brachial index
Negus 1987 reported pretreatment ABI stratified by severity of disease at long‐term follow‐up, with no mention of post‐treatment measurements for this outcome. No other included studies reported this outcome.
All‐cause mortality
None of the included studies reported this outcome.
PGE1 versus ATP
Cardiovascular mortality
None of the included studies comparing PGE1 and ATP reported the incidence of cardiovascular deaths.
Total amputations
Two studies with 91 participants evaluated effects of PGE1 compared with ATP (Böhme 1989; Trubestein 1987). Fixed‐effect meta‐analyses showed that 4/49 participants in the PGE1 group underwent amputations compared with 13/42 in the ATP group (RR 0.26, 95% CI 0.09 to 0.74; I² = 0%; Analysis 3.1). The quality of this evidence is low owing to high risk of attrition bias and a low rate of events, resulting in a wide confidence interval (see Table 4).
3.1. Analysis.
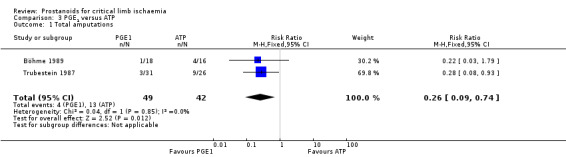
Comparison 3 PGE1 versus ATP, Outcome 1 Total amputations.
Quality of life
None of the included studies comparing PGE1 and ATP reported this outcome.
Adverse events
Two studies with 91 participants evaluated effects of PGE1 compared with ATP (Böhme 1989; Trubestein 1987). Fixed‐effect meta‐analyses showed that 26/49 participants in the PGE1 group and 8/42 in the ATP group experienced adverse events (RR 2.78, 95% CI 1.41 to 5.48; I² = 14%; Analysis 3.2). The quality of this evidence is low owing to high risk of attrition bias and imprecision issues (see Table 4).
3.2. Analysis.
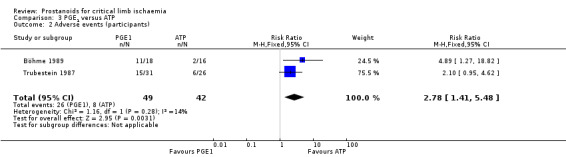
Comparison 3 PGE1 versus ATP, Outcome 2 Adverse events (participants).
Rest‐pain relief
None of the included studies comparing PGE1 and ATP reported rest‐pain relief as a dichotomous outcome. Trubestein 1987 reported that both groups of participants experienced significant rest‐pain relief as a continuous outcome measured on a ten‐point analogue scale, changing from an initial mean of 6 to a mean of 3 at three weeks in the PGE1 group, and from an initial mean of 6 to a mean of 3.5 in the ATP group at three weeks. This study did not provide standard deviation; therefore reanalysis was not possible.
Böhme 1989 reported rest‐pain relief as part of a composite outcome along with consumption of analgesics and ulcer healing, and did not provide enough information to show how many participants experienced some degree of improvement for this outcome.
Reduction in analgesics consumption
Trubestein 1987 reported that consumption of analgesics was reduced or discontinued in 14/31 participants who received PGE1 and in 10/26 who received ATP (RR 1.17, 95% CI 0.63 to 2.19). We considered the quality of this evidence as low owing to high risk of attrition bias and imprecision issues.
Böhme 1989 reported this outcome as part of a composite outcome, along with rest‐pain relief and ulcer healing, not providing enough information to show how many participants experienced some degree of reduction in consumption of analgesics.
Ulcer healing
Trubestein 1987 reported that all participants in both groups experienced some degree of ulcer healing. Additionally, 3/18 in the PGE1 group and 3/13 in the ATP group presented complete healing of trophic lesions (RR 0.72, 95% CI 0.17 to 3.03). We considered the quality of this evidence as low owing to high risk of attrition bias and imprecision issues (see Table 4).
Böhme 1989 reported healing of necrosis as a composite outcome, along with pain relief and consumption of analgesics, not providing enough information to indicate how many participants experienced ulcer healing as a separate outcome.
Major or minor amputations
Information was insufficient to further classify amputation events reported by Böhme 1989 and Trubestein 1987 into major or minor amputations.
Ankle brachial index
None of the included studies comparing PGE1 and ATP reported this outcome.
All‐cause mortality
Böhme 1989 reported that 1/18 participants in the PGE1 group and 1/16 in the ATP group died during follow‐up (RR 0.89, 95% CI 0.06 to 13.08). The quality of this evidence is low owing to high risk of attrition bias and a low rate of events, resulting in a wide confidence interval.
PGE1 versus inositol niacinate
A single study with 76 participants compared high‐dose (0.15 ng/kg/min) and low‐dose (0.05 ng/kg/min) intra‐arterial infusion of PGE1 versus orally administered inositol niacinate (Sakaguchi 1984).
Cardiovascular mortality
The included study did not report this outcome.
Total amputations
Sakaguchi 1984 reported that most participants in the three groups that experienced no improvement in ulcer healing underwent surgical treatment, including amputation, and did not specify the incidence of these events.
Quality of life
The included study did not report this outcome.
Adverse events
Sakaguchi 1984 reported adverse events in 16/22, 9/25, and 6/18 participants in high‐dose PGE1, low‐dose PGE1 and inositol niacinate groups, respectively. As reanalysis of these data showed no statistical differences in the incidence of adverse events between high‐dose and low‐dose PGE1 groups compared with inositol niacinate, we present data for the two PGE1 groups combined (RR 1.60, 95% CI 0.79 to 3.23). The quality of this evidence is low owing to high risk of attrition bias and a low rate of events, resulting in a wide confidence interval (see Table 5).
Rest‐pain relief
Sakaguchi 1984 reported that 18/22 participants who received high‐dose PGE1, 16/25 who received low‐dose PGE1 and 9/18 given inositol niacinate experienced some degree of rest‐pain relief. As reanalysis of this data showed no statistical differences in the incidence of rest‐pain relief between high‐dose and low‐dose PGE1 groups compared with inositol niacinate, we present data for the two PGE1 groups combined (RR 1.45, 95% CI 0.88 to 2.37). The quality of this evidence is low owing to high risk of attrition bias and imprecision issues (see Table 5).
Reduction in analgesics consumption
The included study did not report this outcome.
Ulcer healing
Sakaguchi 1984 reported that 6/22 participants in the high‐dose PGE1 group, 2/25 in the low‐dose PGE1 group, and 1/18 in the inositol niacinate group achieved ulcer healing after treatment. Reanalysis of these data revealed no statistical differences in the incidence of this dichotomous outcome between high‐dose and low‐dose PGE1 groups compared with inositol niacinate. For this reason, we present data for the two PGE1 groups combined (RR 3.06, 95% CI 0.41 to 22.79). The quality of this evidence is low owing to high risk of attrition bias and imprecision issues resulting from a low rate of events (see Table 5).
Major or minor amputations
The included study did not report this outcome.
Ankle‐brachial index
The included study did not report this outcome.
All‐cause mortality
Sakaguchi 1984 reported that one death was recorded in each group of participants within one year after completion of therapy. As reanalysis of these data showed no statistical differences in the incidence of this dichotomous outcome between high‐dose and low‐dose PGE1 groups compared with inositol niacinate, we present data for the two PGE1 groups combined (RR 0.77, 95% CI 0.07 to 7.94) .The quality of this evidence is low owing to high risk of attrition bias and imprecision issues.
Prostanoids versus other prostanoids
Owing to the presence of significant clinical heterogeneity between the two comparisons among different prostanoids (study designs and route of administration differed significantly among included studies), we described results obtained from various comparisons of prostanoids versus other prostanoids. We performed meta‐analysis only for comparisons considering the same control intervention when more than one study reported the same outcome.
Iloprost versus PGE1
Four studies with 511 participants compared iloprost versus PGE1 (Alstaedt 1993; Bandiera 1995; Castagno 2000; Schellong 2004).
Cardiovascular mortality
None of the included studies comparing iloprost versus PGE1 reported the incidence of this event.
Total amputations
Alstaedt 1993 reported that 44/106 participants in the iloprost group and 48/103 in the PGE1 group underwent amputation procedures (RR 0.89, 95% CI 0.66 to 1.21). No other included studies comparing iloprost versus PGE1 reported this outcome. The quality of this evidence is low owing to high risk of bias and imprecision issues (see Table 6).
Quality of life
None of the included studies comparing iloprost versus PGE1 reported this outcome.
Adverse events
Four studies with 511 participants comparing iloprost versus PGE1 reported this outcome (Alstaedt 1993; Bandiera 1995; Castagno 2000; Schellong 2004). Fixed‐effect meta‐analysis showed that 127/259 participants who received iloprost and 51/252 who received PGE1 experienced adverse events (RR 2.35, 95% CI 1.82 to 3.04; I² = 0%; Analysis 4.1). Sensitivity analysis excluding studies with high risk of attrition bias revealed influence on the overall effect (RR 1.91, 95% CI 0.99 to 3.69; 172 participants; 2 studies; I² = 0%). The quality of this evidence is moderate owing to high risk of bias.
4.1. Analysis.
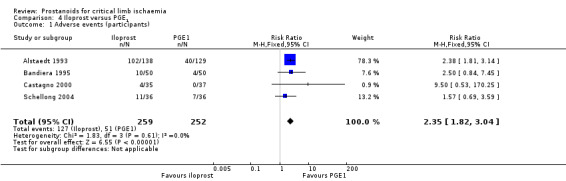
Comparison 4 Iloprost versus PGE1, Outcome 1 Adverse events (participants).
Rest‐pain relief
A single study reported this outcome (Alstaedt 1993), stating that 55/87 participants who received iloprost and 50/91 who received PGE1 experienced some degree of pain relief (RR 1.15, 95% CI 0.90 to 1.47) by the end of treatment. The quality of this evidence is low owing to high risk of bias and imprecision issues (see Table 6).
Reduction in analgesics consumption
None of the included studies comparing iloprost versus PGE1 reported this outcome.
Ulcer healing
Alstaedt 1993 reported that 60/112 participants in the iloprost group and 50/116 in the PGE1 group achieved some degree of improvement or healing of trophic lesions (RR 1.24, 95% CI 0.95 to 1.63) by the end of treatment. No other included studies comparing iloprost versus PGE1 reported this outcome.The quality of this evidence is low owing to high risk of bias and imprecision issues (see Table 6).
Major or minor amputations
Alstaedt 1993 reported that minor amputations were performed in 19/106 participants receiving iloprost and in 19/103 receiving PGE1 (RR 0.97, 95% CI 0.55 to 1.73), and 34/106 participants in the iloprost group and 28/103 in the PGE1 group had major amputations (RR 1.18, 95% CI 0.78 to 1.80) after six months' follow‐up. No other included studies comparing iloprost versus PGE1 reported amputation events. We consider this evidence as low quality owing to high risk of bias and imprecision issues.
Ankle brachial index
None of the included studies comparing iloprost versus PGE1 reported this outcome.
All‐cause mortality
A single study reported this outcome (Alstaedt 1993). Death occurred in 8/106 participants in the iloprost group and in 15/103 in the PGE1 group (RR 0.52, 95% CI 0.23 to 1.17) by six months' follow‐up. The quality of this evidence is low owing to high risk of attrition bias and a low rate of events, resulting in a wide confidence interval.
Clinprost versus lipo‐PGE1
A single study with 135 participants compared clinprost incorporated in lipid microspheres versus lipo‐PGE1 (Esato 1995). We present results for the subgroup of 72 participants with CLI of atherosclerotic origin, excluding those for the subgroup with a diagnosis of thromboangiitis obliterans (TAO; 63 participants). For the outcomes "cardiovascular mortality", "adverse events", and "total mortality", we could not retrieve disaggregated data.
Cardiovascular mortality
Authors of the single study included for this comparison mentioned the incidence of one death due to acute heart failure in the clinprost group during the trial (Esato 1995). This report does not specify whether this happened in the CLI of atherosclerotic origin group or in the TAO group.
Total amputations
None of the included studies comparing clinprost versus lipo‐PGE1 reported this outcome.
Quality of life
None of the included studies comparing clinprost versus lipo‐PGE1 reported this outcome.
Adverse events
This study reported an incidence of adverse events of 7/66 participants in the clinprost group and 4/69 in the lipo‐PGE1 group (RR 1.83, 96% CI 0.56 to 5.96), although trial authors did not report adverse events separately for participants with PAOD or TAO. The quality of this evidence is moderate owing to imprecision issues because events were few, resulting in a wide confidence interval that included both harms and benefits (see Table 7).
Rest‐pain relief
Esato 1995 reported that 14/34 participants in the clinprost group and 13/33 in the lipo‐PGE1 group achieved some degree of rest‐pain relief (RR 1.05, 95% CI 0.58 to 1.87). The quality of this evidence is low owing to high risk of attrition bias and imprecision due to a small number of events (see Table 7).
Reduction in analgesics consumption
None of the included studies comparing clinprost versus lipo‐PGE1 reported this outcome.
Ulcer healing
Esato 1995 stated that 20/37 participants in the clinprost group and 16/35 in the lipo‐PGE1 group achieved some degree of ulcer healing (RR 1.18, 95% CI 0.74 to 1.89). The quality of this evidence is low owing to high risk of attrition bias and imprecision due to a small number of events (see Table 7).
Major or minor amputations
None of the included studies comparing clinprost versus lipo‐PGE1 reported this outcome.
Ankle brachial index
None of the included studies comparing clinprost versus lipo‐PGE1 reported this outcome.
All‐cause mortality
As mentioned above for "cardiovascular mortality", authors of the single study included for this comparison mentioned the incidence of one death due to acute heart failure in the clinprost group during the trial (Esato 1995). This report does not specify whether this happened in the CLI of atherosclerotic origin group or in the TAO group.
Adverse events analysed by type of prostanoid
PGE1
PGE1 was the experimental intervention in 16 studies: seven versus placebo (Diehm 1988; Jogestrand 1985; NCT00596752; Reisin 1997; Schuler 1984; Stiegler 1992; Telles 1984), four versus iloprost (Alstaedt 1993; Bandiera 1995; Castagno 2000; Schellong 2004), one versus pentoxifylline (Trübestein 1989), two versus ATP (Böhme 1989; Trubestein 1987), one versus naftidrofuryl (Böhme 1994), and another versus inositol niacinate (Sakaguchi 1984). One study compared lipo‐PGE1, a preparation of PGE1 incorporated in lipid microspheres, versus clinprost (Esato 1995).
Taken together, participants treated with a prostanoid reported the following as the most common adverse events: phlebitis, thrombophlebitis or pain at the infusion site or both, headache, nausea, flushing, and hypotension. During intra‐arterial infusion, few participants presented swelling, dull pain, redness, and/or fever (Sakaguchi 1984; Trubestein 1987). Cardiovascular events, such as atrial fibrillation, chest pain, dyspnoea, and severe changes in blood pressure, were occasionally reported (NCT00596752; Reisin 1997; Schuler 1984; Stiegler 1992). Five studies stated that symptoms improved with a decrease in infusion rate (Böhme 1994; Diehm 1988; Sakaguchi 1984; Schuler 1984; Telles 1984).
PGI2
Even though PGI2 was the experimental drug in five studies (Belch 1983; Cronenwett 1986; Hossmann 1983; Karnik 1986; Negus 1987), only three of them reported adverse events: two versus placebo (Belch 1983; Cronenwett 1986), and one versus naftidrofuryl (Karnik 1986). The most commonly reported adverse events were flushing, headache, nausea and vomiting, painful skin inflammation, diarrhoea, and hypotension. One patient presented exanthema (Karnik 1986).
Iloprost
Iloprost was the experimental intervention in 12 studies: 11 versus placebo (Alstaedt 1993; Balzer 1991; Bandiera 1995; Brock 1990; Castagno 2000; Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991; Norgren 1990), and one versus PGE1 (Schellong 2004).
The most commonly reported adverse events were headache, nausea, and flushing (Alstaedt 1993; Balzer 1991; Brock 1990; Dormandy 1991; Guilmot 1991; Norgren 1990). Two studies reported phlebitis or adverse reaction at the injection site, or both (Diehm 1989; Norgren 1990). Seven studies reported abdominal pain, diarrhoea, vomiting, and diaphoresis (Alstaedt 1993; Bandiera 1995; Brock 1990; Diehm 1989; Dormandy 1991; Guilmot 1991; Norgren 1990). Participants in eight studies presented changes in blood pressure ‐ both severe hypotension and hypertension (Alstaedt 1993; Bandiera 1995; Brock 1990; Castagno 2000; Diehm 1989; Dormandy 1991; Guilmot 1991; Norgren 1990). Four studies alluded to cardiac adverse events, such as arrhythmia, chest pain, pulmonary oedema, and myocardial infarction, among sporadic participants in four studies (Balzer 1991; Brock 1990; Dormandy 1991; Norgren 1990). One study reported an altered state of consciousness (confusion) (Castagno 2000), another reported a vasovagal syncope (Dormandy 1991), and two studies reported that two stroke events occurred in the treatment group (Diehm 1989; Norgren 1990).
In most cases, trial authors reported that symptoms were ameliorated when infusion speed was decreased (Bandiera 1995; Brock 1990; Diehm 1989; Dormandy 1991; Dormandy 2000a ‐ Study A; Dormandy 2000b ‐ Study B; Guilmot 1991).
Taprostene
One study compared the experimental drug taprostene versus placebo (Belch 2011). Trial authors described flushing and headache as the most common adverse events.
Ciprostene
A single study provided ciprostene as the experimental intervention versus placebo (Linet 1991). Trial authors reported that the most common adverse events were headache, nausea, and flushing. Hypotension, vomiting, jaw pain, and restlessness were less common. Five participants in the treatment group died as the result of heart failure, myocardial infarction, cardiac arrest, chronic obstructive pulmonary disease, and sepsis, respectively.
Lipo‐ecraprost
Lipo‐ecraprost was the experimental drug in one study (Brass 2006). Commonly reported adverse reactions secondary to infusion were headache, pain, hypotension, tachycardia, vasodilation, diarrhoea, nausea, and vomiting. Investigators also reported 11 cases of congestive heart failure and 14 myocardial infarctions.
Clinprost
One single study provided clinprost incorporated in lipid microspheres as the experimental intervention for comparison with lipo‐PGE1 (Esato 1995). Adverse events described in the clinprost group included nausea, anorexia, diarrhoea, dizziness, liver disorder, tinnitus, and frizzy hair, although trial authors did not report adverse events separately for participants with PAOD or thromboangitis obliterans.
Discussion
Summary of main results
We updated this systematic review including 33 studies that evaluated the effects and safety of prostanoids, a family of drugs proposed to increase blood supply, for patients with critical limb ischaemia, a severe arterial obstruction affecting the lower limbs that causes rest‐pain and necrosis of soft tissues, who are unable to receive any revascularisation procedure.
Prostanoids versus placebo
Moderate‐quality evidence shows small effects of prostanoids (see Table 1) in terms of rest‐pain relief, reduction of analgesics consumption, and ulcer healing compared with placebo. However, high‐quality evidence shows no effect on the incidence of total amputations. Nevertheless, subgroup analysis revealed a small reduction in the incidence of total amputations for iloprost. Regarding major and minor amputations, effects derived from moderate‐quality evidence showed opposite directions ‐ favouring prostanoids for major amputations, but favouring placebo for minor amputations. Selective outcome reporting across studies could have affected the direction of these results. Moderate‐quality evidence shows no differences in all‐cause mortality between placebo and prostanoids groups. Low‐quality evidence suggests no differences in cardiovascular mortality between placebo and prostanoid groups. None of the included studies reported measurements of quality of life. The incidence of adverse effects was consistently greater in those treated with prostanoids. The most frequently reported adverse events were headache, nausea, vomiting, diarrhoea, flushing, and hypotension.
Results from the meta‐analysis were not statistically heterogeneous. However, they should be appraised with caution because of the presence of clinical heterogeneity and the potential influence of bias. Effects of prostanoids on rest‐pain relief, major amputations, and adverse events were diluted with exclusion of studies at high risk of bias. All‐cause mortality and total amputations were not affected by the sensitivity analysis.
Prostanoids versus other agents
A single study comparing a prostanoid (prostaglandin E1 (PGE1)) versus pentoxifylline found no clear differences in effects on amputation, rest‐pain relief, reduction in analgesic consumption, or adverse events, although more participants who received PGE1 achieved some degree of ulcer healing (very low‐quality evidence; see Table 2). When compared with naftidrofuryl (see Table 3), prostanoids had no significant effect on rest‐pain relief, ulcer healing, amputations, or adverse events (low‐quality evidence). In two studies comparing PGE1 versus adenosine triphosphate (ATP) (see Table 4), PGE1 had a significant effect on reduction of total amputations, with an increased incidence of adverse events; the remaining outcome measures were not combined owing to different definitions for each study, but study authors separately reported no differences regarding the incidence of rest‐pain relief, reduction in consumption of analgesics, or ulcer healing (low‐quality evidence). A single study compared intra‐arterial PGE1 versus oral inositol niacinate, reporting no differences in the incidence of rest‐pain relief, reduction in consumption of analgesics, or ulcer healing (low‐quality evidence; see Table 5). Studies comparing prostanoids versus other agents did not report cardiovascular mortality or quality of life.
Head‐to‐head comparisons between prostanoids
We found two comparisons of different prostanoids: iloprost versus PGE1, and clinprost incorporated in lipid microspheres versus lipo‐PGE1. Participants treated with iloprost had a greater incidence of adverse events compared with those given PGE1 (moderate‐quality evidence; see Table 6). A single study reported no differences in the incidence of amputations, mortality, rest‐pain relief, and ulcer healing for the same comparison. Data show no differences for ulcer healing or rest‐pain relief in the subgroup of participants with peripheral arterial occlusive disease (PAOD)/critical limb ischaemia as reported by a single study on clinprost compared with lipo‐PGE1 (low‐quality evidence; see Table 7).
Overall completeness and applicability of evidence
The relevance of evidence gathered for this update on the efficacy of prostanoids for patients with CLI is uncertain for several reasons. Even though most of the included studies described consistent selection criteria used for the study population, the concept of critical limb ischaemia without a chance of rescue or reconstructive intervention is a subjective definition. Thus, the diagnosis for a particular participant could have varied across studies according to centre characteristics, availability of endovascular procedures, and physician judgement. Most included studies recruited participants from countries in Europe, and this could limit transferability of results to other populations. Moreover, given that most identified studies were developed during the 1980s and 1990s, and that during past decades, both standard of care for patients with chronic PAOD and endovascular technologies for revascularisation have significantly improved (Teraa 2016), we have reason to believe that a higher degree of severity of disease is currently needed if a patient will be considered as having no chance of reconstructive intervention.
Given the chronic nature of the condition of patients with CLI, and that most included studies provided short‐term results (one to six months), we could not provide conclusions regarding long‐term (> 1 year) efficacy and safety of prostanoids. Similarly, owing to different doses and routes of administration used by primary studies, and even though we performed subgroup analysis by type of prostanoid, we are unable to determine which was the optimal posology. Additionally, none of the included randomised controlled trials (RCTs) assessed the variable quality of life, which we consider to be a very important patient‐centred outcome.
Quality of the evidence
For this update, we included 33 RCTs with a total of 4477 participants. We present assessment of the risk of bias in Figure 2 and Figure 3. When considering selection bias, we noted that 27/33 and 26/33 studies had unclear risk of bias for random sequence generation and allocation concealment, respectively. Only 6/33 and 5/33 had low risk of bias for the aforementioned domains. Most studies (21/33) had low risk of performance bias, and 5/33 and 7/33 had uncertain and high risk of bias, respectively, for this domain. We rated 10/33 studies as having low risk of detection bias, and 16/33 and 7/33 as having unclear and high risk of detection bias, respectively. Finally, regarding incomplete outcome data, 21/33 studies had high risk of bias, and only 8/33 and 16/33 studies had low and unclear risk of bias, respectively. Attrition bias was a significant contributor to downgrading of the body of evidence for most outcomes.
For the comparison prostanoids versus placebo (see Table 1), the overall quality of evidence for most outcomes was moderate owing to high risk of attrition bias. Overall beneficial effects of prostanoids on the incidence of major amputations, ulcer healing, and pain relief were diluted with exclusion of studies with high risk of attrition bias. The same phenomenon was evident with the incidence of adverse events. Attrition bias did not influence overall effects of prostanoids on amputation while precision is maintained; therefore, we rated the quality of the evidence as high. Attrition bias did not influence overall effects of prostanoids on mortality and cardiovascular mortality. In this case, however, precision was affected; therefore, the quality of the evidence was low.
Unfortunately we could not perform sensitivity analysis for all outcomes and comparisons because in some cases, all studies included in the meta‐analysis had high risk of bias, and we therefore could not assess the influence of bias on these outcomes (cardiovascular mortality, rest‐pain relief as a continuous outcome, and reduction in analgesic consumption with prostanoids vs placebo; pain relief, ulcer healing, and adverse events with prostanoids vs naftidrofuryl; total amputations and adverse events with PGE1 vs ATP; and adverse events with iloprost vs PGE1).
For comparisons of prostanoids versus other agents (see Table 2; Table 3; Table 4; and Table 5), we included studies at high risk of bias that had a low incidence of events and a small sample size. Therefore we rated the quality of evidence as low or very low owing to imprecision and high risk of bias.
For comparisons between prostanoids (see Table 6; Table 7), we also included studies at high risk of bias that had a low incidence of events and a small sample size. Therefore we rated the quality of evidence as low or very low owing to imprecision and high risk of bias.
Potential biases in the review process
As described in the Excluded studies section, for this update we reclassified excluded studies from the original review as the result of changes in exclusion criteria regarding the quality of studies. We preferred this approach to minimising selection bias in the review process. Nevertheless, we had to exclude four studies owing to inability to retrieve a full text, and seven studies because they did not provide disaggregated data for the population of interest. Additionally, even though statistical analysis and funnel plot representation (Figure 4) did not introduce suspicion of publication bias, we were unable to find the larger trial mentioned by Bandiera 1995, and we could find neither published nor unpublished data from the study NCT00059644. Selective or inadequate outcome reporting was frequent, and only two trial authors replied when we attempted to contact them to obtain clarification (Brass 2006; Diehm 1988). In most cases, author contact information was neither available nor current (many studies were reported in the 1980s). We tried to obtain new contact information (email addresses), but most attempts were unsuccessful. Assumptions made when meta‐analysis was conducted are a potential source of bias. For rest‐pain relief and ulcer healing, we did not restrict the definition of improvement at any specific cut‐off point. Minimal and non‐clinically relevant improvements in pain and ulcer healing could lead to overestimation of effects. Additionally, even though most meta‐analyses had low statistical heterogeneity, for these outcomes most studies included heterogeneous measurements or non‐validated scales/scores.
Agreements and disagreements with other studies or reviews
Some published meta‐analyses stated the beneficial effects of iloprost (Loosemore 1994), PGE1 (Creutzig 2004), or both (Brodszky 2011), for patients with critical limb ischaemia (CLI) who are unsuitable for rescue or reconstructive intervention. This updated systematic review analysed all studies included in those publications and identified additional studies; this contributed to a more comprehensive synthesis of the body of evidence on efficacy and safety of the whole family of prostanoids.
Brodszky 2011 has highlighted that the previous version of this review pooled incorrect data for the rest‐pain relief outcome (Ruffolo 2010). For this update, we found considerable clinical heterogeneity regarding rest‐pain relief measurement across studies. Nevertheless, our criterion for pooling data was that the last measurement of rest‐pain assessment during the follow‐up period would be used as input, since this would be the most clinically relevant information for evaluating effects of prostanoids on this outcome beyond the immediate infusion period.
A recent systematic review assessed the efficacy of prostanoids for patients with PAOD of any severity, with a treatment phase of at least 24 hours and a minimal follow‐up period of four weeks (Vitale 2016). Although the direction of results regarding amputation outcomes presented by Vitale 2016 is consistent with that estimated by this update, differences in the ulcer healing pooled estimate could be explained by the fact that this update achieved greater statistical power by including additional studies.
Authors' conclusions
Implications for practice.
We found low‐quality evidence suggesting that prostanoids had no effect on the incidence of cardiovascular mortality when compared against placebo, and high‐quality evidence showing that prostanoids had no effect on the incidence of total amputations for the same comparison. Moderate‐quality evidence showed beneficial effects of prostanoids for rest‐pain relief and ulcer healing when compared against placebo; however, effects on rest‐pain relief were diluted when studies with high risk of bias were excluded. Additionally, we found moderate‐quality evidence showing a greater incidence of adverse effects with the use of prostanoids. None of the included studies reported quality of life measurements. The main reasons for downgrading the quality of evidence were high risk of attrition bias and imprecision of the included studies. We found evidence of low to very low quality on effects of prostanoids versus other active agents or versus other prostanoids because a small number of studies evaluated the same comparisons and had high risk of bias.
Current evidence regarding effects and safety of prostanoids for patients with critical limb ischaemia unsuitable for rescue or reconstructive interventions is insufficient to ascertain the balance of benefits and harms of this therapeutic strategy. Therefore, careful assessment of therapeutic alternatives should be considered.
Implications for research.
Results of this comprehensive systematic review suggest that planning of further research on this topic should address weaknesses identified in the included studies, with the aim of improving the quality of evidence. As the main reasons for downgrading evidence were high risk of attrition bias and imprecision issues, future randomised clinical trialists should increase efforts to obtain optimal sample sizes and to prevent losses to follow‐up, with the goal of improving the precision of effect estimates.
Additionally, future research should consider measurement of important patient‐centred outcomes such as quality of life, which was not addressed by any of the included studies. Use of validated instruments for measurement of objective outcomes such as ulcer healing and rest‐pain relief is imperative for preventing bias and increasing clinical consistency across studies. Finally, registration of protocols could prevent selective outcome reporting and risk of publication bias.
What's new
| Date | Event | Description |
|---|---|---|
| 10 January 2018 | Amended | Acknowledgements section amended with correct name of referee |
History
Protocol first published: Issue 2, 2007 Review first published: Issue 1, 2010
| Date | Event | Description |
|---|---|---|
| 11 May 2017 | New search has been performed | Searches rerun. Eight new studies included, nine new studies excluded. |
| 11 May 2017 | New citation required but conclusions have not changed | Searches rerun. Eight new studies included, nine new studies excluded. Review updated using current Cochrane standards including risk of bias and summary of findings. Review taken over by new authors. Conclusions not changed. |
| 16 February 2010 | Amended | CENTRAL search strategy amended |
| 29 April 2008 | Amended | Converted to new review format |
Acknowledgements
Cochrane Vascular, University of Edinburgh (Scotland), specially to Cathryn Broderick, Marlene Stewart, Karen Welch, and Elsbeth Hamilton, for support provided during development of this update.
Cochrane Central Executive, specially to Hayley Hassan, who kindly helped us contact other members of the Cochrane community for assessment of non‐English language studies.
Cochrane Germany, specially to Ingrid Töws, Valérie Labonté, and Karin Rau, for translation of reports published in the German language.
Cochrane Japan Branch, specially to Shinobu Kobayashi, for help with translation of reports prepared in the Japanese language.
Cochrane Hungary Branch, specially to Szimonetta Lohner and Klarissza Pál, for help in translation of a full text published in the Hungarian language.
IECS Cochrane Collaborating Centre (Argentina).
Instituto Universitario Hospital Italiano Cochrane Collaborating Centre (Argentina), specially to Virginia Garrote, who developed the search strategies for clinical trials registers and helped with full‐text retrieval.
Family and Community Medicine Division, Hospital Italiano de Buenos Aires, specially to Santiago Esteban, who helped with full‐text retrieval, and Haydee Giraudi for translation of a report prepared in the Italian language.
Study authors Eric Brass and Bruno Simini, for responding kindly when we requested additional information on studies.
UCB Pharma, for helping with full‐text retrieval and identification of published studies.
Antonio Ruffolo and Marina Romano, who wrote the protocol and the first version of the review, and their collaborators ‐ Anette Bluemle, Juan Gutierrez, Daniel Comande, and Valeria Amatto, whose previous work constituted foundations for the current update.
Lorna Watson, Catriona Keerie, Indrani Sen, Martin Teraa, and Shunjie Chua, for editorial and peer‐review input.
Appendices
Appendix 1. CENTRAL search strategy
| Search run on Tue Jan 31 2017 | ||
| #1 | MESH DESCRIPTOR Arteriosclerosis | 868 |
| #2 | MESH DESCRIPTOR Arteriolosclerosis EXPLODE ALL TREES | 0 |
| #3 | MESH DESCRIPTOR Arteriosclerosis Obliterans | 71 |
| #4 | MESH DESCRIPTOR Atherosclerosis | 619 |
| #5 | MESH DESCRIPTOR Arterial Occlusive Diseases | 724 |
| #6 | MESH DESCRIPTOR Intermittent Claudication | 712 |
| #7 | MESH DESCRIPTOR Ischemia | 789 |
| #8 | MESH DESCRIPTOR Peripheral Vascular Diseases EXPLODE ALL TREES | 2201 |
| #9 | (atherosclero* or arteriosclero* or PVD or PAOD or PAD ):TI,AB,KY | 9119 |
| #10 | ((arter* or vascular or vein* or veno* or peripher*) near3 (occlus* or reocclus* or re‐occlus* or steno* or restenos* or obstruct* or lesio* or block* or harden* or stiffen* or obliter*) ):TI,AB,KY | 7966 |
| #11 | (peripheral near3 dis*):TI,AB,KY | 3371 |
| #12 | (claudic* or IC):TI,AB,KY | 3063 |
| #13 | (isch* or CLI):TI,AB,KY | 23713 |
| #14 | arteriopathic or leriche*:TI,AB,KY | 61 |
| #15 | dysvascular*:TI,AB,KY | 10 |
| #16 | (leg near3 (occlus* or reocclus* or re‐occlus* or steno* or restenos* or obstruct* or lesio* or block* or harden* or stiffen* or obliter*) ):TI,AB,KY | 95 |
| #17 | (limb near3 (occlus* or reocclus* or re‐occlus* or steno* or restenos* or obstruct* or lesio* or block* or harden* or stiffen* or obliter*) ):TI,AB,KY | 145 |
| #18 | ((lower near3 extrem*) near3 (occlus* or reocclus* or re‐occlus* or steno* or restenos* or obstruct* or lesio* or block* or harden* or stiffen* or obliter*) ):TI,AB,KY | 77 |
| #19 | MESH DESCRIPTOR Leg EXPLODE ALL TREES WITH QUALIFIERS BS | 1107 |
| #20 | MESH DESCRIPTOR Iliac Artery | 144 |
| #21 | MESH DESCRIPTOR Popliteal Artery | 278 |
| #22 | MESH DESCRIPTOR Femoral Artery | 810 |
| #23 | MESH DESCRIPTOR Tibial Arteries | 33 |
| #24 | (((femor* or iliac or popliteal or fempop* or crural or poplite* or infrapopliteal or femdist* or inguinal or infrainquinal or tibial) near3 (occlus* or reocclus* or re‐occlus* or steno* or restenos* or obstruct* or lesio* or block* or harden* or stiffen* or obliter*) )):TI,AB,KY | 1157 |
| #25 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 or #24 | 43842 |
| #26 | MESH DESCRIPTOR Prostaglandins EXPLODE ALL TREES | 4727 |
| #27 | (*prost*):TI,AB,KY | 31745 |
| #28 | PGE*:TI,AB,KY | 1819 |
| #29 | PGI*:TI,AB,KY | 719 |
| #30 | (AS‐013 or ventavis or TTC‐909):TI,AB,KY | 8 |
| #31 | #26 OR #27 OR #28 OR #29 OR #30 | 32358 |
| #32 | #25 AND #31 | 2309 |
Appendix 2. Trials registries searches
Searches carried out by the CIS: 13 records
ClinicalTrials.gov
13 studies found for: ischemia or ischemic or peripheral arterial | prostanoid OR prostanoids OR prostaglandin OR prostaglandins
World Health Organization International Clinical Trials Registry Platform (WHO‐ICTRP)
ISRCTN Register
No results found for "(Condition: ischemia or ischemic or peripheral arterial AND Interventions: prostanoid OR prostanoids OR prostaglandin OR prostaglandins)"
Searches carried out by the review authors: 642 records
ClinicalTrials.gov: (prostanoid OR prostanoids OR prostaglandin OR prostaglandins)
WHO‐ICTRP: (prostanoid OR prostanoids OR prostagland*)
Data and analyses
Comparison 1. Prostanoids versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cardiovascular mortality | 3 | 1170 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.41, 1.58] |
| 1.1 PGE1 | 2 | 959 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.35, 1.51] |
| 1.2 Ciprostene | 1 | 211 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.25, 8.71] |
| 2 Total amputations | 12 | 2825 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.86, 1.09] |
| 2.1 PGE1 | 3 | 942 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.93, 1.43] |
| 2.2 Iloprost | 6 | 1205 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.69, 0.97] |
| 2.3 Taprostene | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.58, 1.30] |
| 2.4 Lipo‐ecraprost | 1 | 356 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.75, 2.07] |
| 2.5 Ciprostene | 1 | 211 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.68, 2.25] |
| 3 Adverse events (participants) | 8 | 655 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.11 [1.79, 2.50] |
| 3.1 PGE1 | 4 | 251 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [1.76, 3.47] |
| 3.2 PGI2 | 1 | 26 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.49, 2.96] |
| 3.3 Iloprost | 3 | 378 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [1.68, 2.49] |
| 4 Rest‐pain relief (dichotomous) | 10 | 1179 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [1.06, 1.59] |
| 4.1 PGE1 | 1 | 46 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.07, 16.41] |
| 4.2 PGI2 | 2 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.09 [0.75, 5.85] |
| 4.3 Iloprost | 6 | 733 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [1.15, 1.89] |
| 4.4 Lipo‐ecraprost | 1 | 362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.62, 1.32] |
| 5 Rest‐pain relief (dichotomous) ‐ sensitivity analysis | 5 | 739 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.88, 1.46] |
| 5.1 PGI2 | 2 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.09 [0.75, 5.85] |
| 5.2 Iloprost | 2 | 339 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.89, 1.89] |
| 5.3 Lipo‐ecraprost | 1 | 362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.62, 1.32] |
| 6 Rest‐pain relief (continuous) | 3 | 984 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.10, 0.42] |
| 6.1 PGE1 | 2 | 958 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.06, 0.19] |
| 6.2 PGI2 | 1 | 26 | Std. Mean Difference (IV, Random, 95% CI) | 0.76 [‐0.04, 1.56] |
| 7 Reduction in analgesics consumption | 2 | 135 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [1.08, 1.82] |
| 7.1 PGE1 | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.68 [0.82, 3.46] |
| 7.2 Iloprost | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.02, 1.76] |
| 8 Ulcer healing | 11 | 1719 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [1.04, 1.48] |
| 8.1 PGE1 | 5 | 860 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.81, 1.80] |
| 8.2 PGI2 | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.13, 2.00] |
| 8.3 Iloprost | 4 | 632 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.09, 1.94] |
| 8.4 Ciprostene | 1 | 211 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.11, 1.46] |
| 9 Ulcer healing ‐ sensitivity analysis | 3 | 478 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.83, 2.13] |
| 9.1 PGI2 | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.13, 2.00] |
| 9.2 Iloprost | 2 | 462 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.99, 2.15] |
| 10 Major amputations | 8 | 2430 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.73, 0.98] |
| 10.1 PGE1 | 1 | 838 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.61, 1.21] |
| 10.2 Iloprost | 5 | 1125 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.65, 0.94] |
| 10.3 Taprostene | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.53, 1.43] |
| 10.4 Lipo‐ecraprost | 1 | 356 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.75, 2.07] |
| 11 Minor amputations | 4 | 898 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [1.11, 2.16] |
| 11.1 PGE1 | 1 | 611 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [1.07, 2.20] |
| 11.2 Iloprost | 2 | 176 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.13 [0.75, 13.04] |
| 11.3 Taprostene | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.27, 2.80] |
| 12 All‐cause mortality | 11 | 2797 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.81, 1.33] |
| 12.1 PGE1 | 2 | 959 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.72, 2.41] |
| 12.2 PGI2 | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.03, 2.45] |
| 12.3 Iloprost | 5 | 1132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.60, 1.16] |
| 12.4 Taprostene | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.52, 2.53] |
| 12.5 Lipo‐ecraprost | 1 | 356 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [0.85, 3.75] |
| 12.6 Ciprostene | 1 | 211 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.48 [0.49, 12.48] |
1.9. Analysis.
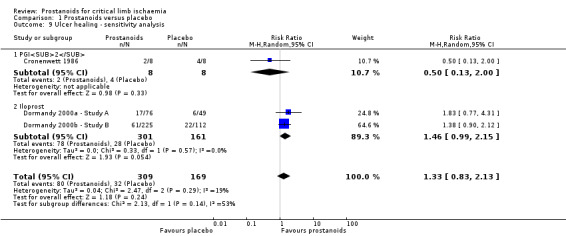
Comparison 1 Prostanoids versus placebo, Outcome 9 Ulcer healing ‐ sensitivity analysis.
1.12. Analysis.
Comparison 1 Prostanoids versus placebo, Outcome 12 All‐cause mortality.
Comparison 2. Prostanoids versus naftidrofuryl.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total amputations | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse events (participants) | 2 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 3.29 [0.27, 40.19] |
| 2.1 PGE1 | 1 | 26 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.34] |
| 2.2 PGI2 | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 11.73 [0.71, 194.51] |
| 3 Rest‐pain relief | 3 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.83, 1.39] |
| 3.1 PGE1 | 1 | 26 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.75, 1.14] |
| 3.2 PGI2 (intravenous) | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 13.87] |
| 3.3 PGI2 (intra‐arterial) | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.80, 2.15] |
| 4 Ulcer healing | 2 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.46, 2.03] |
| 4.1 PGE1 | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.34, 1.79] |
| 4.2 PGI2 | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.32, 7.14] |
Comparison 3. PGE1 versus ATP.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total amputations | 2 | 91 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.09, 0.74] |
| 2 Adverse events (participants) | 2 | 91 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.78 [1.41, 5.48] |
Comparison 4. Iloprost versus PGE1.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse events (participants) | 4 | 511 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.82, 3.04] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alstaedt 1993.
| Methods | Study design: multi‐centre, randomised, open‐label, controlled trial | |
| Participants |
Country: Germany Number of study centres: not stated Setting: hospital Number of participants: 267; iloprost group 138; PGE1 group 129 Age (median) years: iloprost group 70.5; PGE1 group 70.9 Gender ratio (M/F): iloprost group 78/60; PGE1 group 73/56 Inclusion criteria: PAOD stage IV secondary to obliterating arteriosclerosis or diabetic angiopathy with stable symptoms for at least 21 days. The diagnosis of PAOD was based on case history, clinical examination, angiography, and, for non‐diabetic participants, Doppler ankle pressures. Exclusion criteria: "Patients who were suitable for revascularization or who would probably require a major amputation within the first week of treatment". Other exclusion criteria included pregnancy or lactation period, type 1 diabetes mellitus, amputation within the previous 4 weeks, sympathectomy within the previous 3 months, bypass operation within the previous 6 weeks, prostaglandin treatment within the previous 2 months, unstable angina pectoris, heart failure (NYHA III, IV), myocardial infarction within the previous 3 months, osteomyelitis, platelets < 100,000 or > 500,000 per mL, haematocrit > 48%, haemoglobin < 10 g/dL, leucocytes < 3000/mL and serum creatinine > 40 mg%. |
|
| Interventions |
Experimental (1): intravenous infusion of iloprost over 6 hours Experimental (2): intravenous infusion of prostavasin (= 40 µg dry‐substance alprostadil) over 2 hours Duration: 28 days, follow‐up 6 months |
|
| Outcomes | Healing of or improvement in trophic lesions, relief of rest pain, overall clinical status, amputation (minor and major), mortality, side effects | |
| Notes | Source of funding: not stated ‐ probably Schering | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Low risk | Central allocation. "After giving their informed consent, patients were randomly assigned to receive either iloprost or PGE1 by the investigator after telephoning a central statistics institute, where the randomisation list was kept". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. "A double‐blind study design would have been desirable, but was not possible for reasons of practicality considering the different recommended regimens for the two drugs, the different formulations, the different dilution requirements and the different pharmaceutical stabilities of the two drugs". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals due to adverse effects occurred in 23/138 participants in the iloprost group, and in 4/129 in the PGE1 group. 4/138 and 2/129 participants in the iloprost and PGE1 groups who were amputated within the first week of treatment were excluded from primary analysis because they were considered "desperate cases" that should not have been entered into the study. 22/138 and 11/129 participants in the iloprost and PGE1 groups received less than 21 days of treatment. They were excluded from the primary analysis. Attrition rate was > 10% in both groups. 18/138 and 32/138 participants in the iloprost group were lost to follow‐up at 3 and 6 months. 15/138 and 26/138 participants in the PGE1 group were lost to follow‐up at 3 and 6 months. ITT analysis was performed at 6 months over a population of 209 participants, from 267 entered, considering participants with no assessment as non‐responders. |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the methods section were reported. |
| Other bias | Unclear risk | Imbalance in baseline population characteristics. Diabetics: 70.3% in iloprost group, 60.5% in PGE1 group. Mean ABI was lower in the iloprost group (0.23 vs 0.30). Identical care programmes were not clearly stated. |
Balzer 1991.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: Germany Number of study centres: 13 Setting: hospital Number of participants: 113; experimental group 55; control group 58 Age (median) years: experimental group 69; control group 67 Gender ratio (M/F): experimental group 33/22; control group 41/17 Inclusion criteria: nocturnal ischaemic pain at rest for at least 2 weeks, clinical signs of advanced PAOD (stage III or IV) Exclusion criteria: "patients with trophic lesions" (no further details) |
|
| Interventions |
Experimental: intravenous infusion of iloprost over 6 hours Control: placebo Duration: 14 days, follow‐up 4 weeks |
|
| Outcomes | Rest‐pain relief, use of analgesics, tolerability | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided besides stratification by diabetes mellitus |
| Allocation concealment (selection bias) | Unclear risk | "Patients were randomised within each centre". No additional information was provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was described as double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 10/55 patients in the iloprost group and 3/58 patients in the placebo group discontinued treatment prematurely. 11 patients withdrew from the study before completion; 102 patients included in the final analysis. ITT analysis not performed. |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoints reported as mentioned in the methods section. |
| Other bias | Unclear risk | Comparable groups at baseline as shown in Table 1. Identical care programmes not stated. |
Bandiera 1995.
| Methods | Study design: single‐centre, randomised, open‐label, controlled trial | |
| Participants |
Country: Italy Number of study centres: 1 Setting: hospital Number of participants: 100; alprostadil group 50; iloprost group 50 Age (mean) years: alprostadil group 68; iloprost group 66 Gender ratio (M/F): not stated Inclusion criteria: "patients suffering from critical ischaemia of the lower limbs" Exclusion criteria: not stated |
|
| Interventions |
Experimental (1): intravenous infusion of iloprost starting from 0.5 ng/kg/min up to a maximum of 2 ng/kg/min over 6 hours Experimental (2): intravenous infusion of alprostadil over 2 to 3 hours Duration: 3 to 4 weeks |
|
| Outcomes | Adverse events | |
| Notes | This is a report of adverse events in the context of an efficacy study for prostanoids in patients with CLI, not retrieved by search strategies. Trial author did not respond. Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. Subjective outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Complete follow‐up or ITT analysis not described |
| Selective reporting (reporting bias) | Unclear risk | Prespecified outcomes not specified in the methods section |
| Other bias | Unclear risk | Comparability between groups of participants was mentioned, but no information on characteristics of included patients was provided. Identical care programmes were not stated |
Belch 1983.
| Methods | Study design: single‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: UK Number of study centres: 1 Setting: hospital Number of participants: 28; experimental group 15; control group 13 Age years (mean ± SD): experimental group 67 ± 12; control group 69 ± 7 Gender ratio (M/F): experimental group 9/6; control group 10/3 Inclusion criteria: ischaemic rest pain Exclusion criteria: > 80 years of age; had diabetes, infection, or necrosis > 1 cm². |
|
| Interventions |
Experimental: intravenous infusion of epoprostenol (prostacyclin ‐ PGI2) Control: placebo Duration: 4 days |
|
| Outcomes | Rest‐pain relief; analgesic consumption; side effects 24 days, 1 month, and 6 months post treatment; mortality | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were unaware of their treatment group, as were the assessing surgeon and all staff on the ward, except for the physician who administered all treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The assessing surgeon and all staff on the ward, except for the physician who administered all treatment, were unaware of treatment groups. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No statement is provided regarding the number of randomised participants. Follow‐up for participants who received treatment was complete in both groups. |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoints were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1 Identical care programmes not stated |
Belch 2011.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Countries: UK, Finland, Sweden
Number of study centres: 9 Setting: hospital Number of participants: 111; experimental group 74; control group 37 Age years (mean ± SD): experimental group 72 ± 9; control group 75 ± 7 Gender ratio (M/F): experimental group 38/36; control group 23/14 Inclusion criteria: patients with critical limb ischaemia with symptoms in 1 leg, whose highest ankle systolic pressure measured by Doppler ultrasound in the affected leg was < 60 mmHg (non‐diabetic), or who had an ankle brachial systolic pressure index < 0.6. For diabetic patients, toe pressure < 40 mmHg or a TcPO2 < 20 mmHg Exclusion criteria:General exclusions: participating in other clinical trials or having received an investigational drug within the past 30 days; intention to administer any trial medication other than taprostene; previous admission to this study; inability to fulfil requirements of the study protocol (e.g. poor compliance); alcohol, medicine, or drug addicts and patients with psychiatric disorders; women with reproductive potential and inadequate contraception. Medical exclusions: difference > 30% in treadmill exercise test results during the run‐in period; iliac artery occlusions; successful angioplasty or vascular surgery therapy within the past 6 months; known hypersensitivity to taprostene; pathological ECG stress test (at onset of claudication pain), which limits walking distance or renders exercise training/treadmill inappropriate; MI within the past 3 months or acute ischaemic changes on resting ECG; heart failure NYHA III or IV; uncontrolled hypertension; stroke within the past 3 months; severe renal insufficiency (max creatinine 200 µmol/L); severe liver disease; polycythaemia; leucocytosis; thrombocytosis or thrombocytopaenia; history of bleeding diathesis; history of hyperviscosity syndromes; severe allergy; acute and chronic illness, which, according to investigator could prejudice the course of the study; concomitant or previous medications such as vasoactive substances for PAD if intake was not stopped at least 14 days before randomisation; treatment with prostanoids for PAD within the past 3 months |
|
| Interventions |
Experimental: intravenous infusion of taprostene twice a day over 2 hours, in different doses depending on the participant's weight Regimen A: Participants weighing < 65 kg received 200 µg of taprostene in 250 mL of sodium chloride. Regimen B: Participants weighing 66 to 100 kg received 400 µg of taprostene in 250 mL of sodium chloride. Regimen C: Participants weighing < 100 kg received 600 µg of taprostene in 250 mL of sodium chloride. Control: placebo (matching volume of sodium chloride) Duration: 4 weeks |
|
| Outcomes | Early response (combined outcome: complete pain relief with no requirement for analgesic therapy in participants without ulceration, or a decrease in ulcer size > 30%), amputation, death after 6 months' follow‐up, adverse events | |
| Notes | Run‐in period lasted 2 weeks. Source of funding: Wellcome Research Laboratories provided prostacyclin and glycine buffer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A computer‐generated randomization list was used for each stratum. Equal numbers of patients fulfilling the inclusion criteria were allocated to each stratum in accordance with their recruitment to regimens A, B or C". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. Trial author did not respond. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were unaware of their treatment group, as were the assessing surgeon and all staff on the ward, except for the physician who administered all treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The integrity of the blinding of the trial was maintained by ensuring that ulcer size was assessed by clinical staff who were not directly responsible for supervising the infusions". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Statistical analysis of endpoints was performed according to an ITT basis. All "dropouts", whether due to adverse events or failure to respond to treatment, were assessed as non‐responders. Apart from those who died, all participants completed the 12‐month follow‐up. |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoints were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1 Identical care programmes not stated |
Brass 2006.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: USA, UK, Japan Number of study centres: 5 Setting: hospital Number of participants: 560 planned; 383 randomised; ITT population 379: 190 placebo group, 189 lipo‐ecraprost group Age (mean) (range) years: (ITT population) experimental group 69.7 (43.7 to 99.4); control group 69.7 (42.7 to 96.4) Gender ratio (M/F): (ITT population) experimental group 127/62; control group 130/60 Inclusion criteria: atherosclerotic CLI, aged > 40 years, without revascularisation option, stratification by diabetic status Exclusion criteria: previous major amputation or if major amputation would be required; recent revascularisation; receiving antihypertensive therapy; clinical evidence of sepsis; ESRD; recent MI |
|
| Interventions |
Experimental: intravenous infusion of lipo‐ecraprost (ecraprost 60 µg) Control: placebo Duration: 8 weeks |
|
| Outcomes | Major amputation/death at 180 days, all‐cause mortality, cardiovascular adverse events, ulcer healing, rest‐pain relief, other adverse events. Follow‐up at 6 and 12 months | |
| Notes | The study terminated early by recommendation of the DSMB, after interim analysis: 383 randomised participants Source of funding: Mitsubishi Pharma Corporation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computed generated (data provided via personal communication with trial author) |
| Allocation concealment (selection bias) | Low risk | Centralised (data provided via personal communication with trial author) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Investigators and participants were blinded. Prepackaged study drug, active and placebo, indistinguishable with ID number (data provided via personal communication with trial author) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded (data provided via personal communication with trial author). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | At 6 months, withdrawals < 10% of the study population. "After 6 months, 13/190 patients in the placebo arm and 10/189 patients in the lipo‐ecraprost arm were unavailable for end‐point assessment". ITT analysis of 379 participants who received at least 1 dose of study medication (modified ITT analysis) |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoints reported as stated in the methods section |
| Other bias | High risk | Comparability of groups shown in Table 1 Identical care programmes not stated Early termination based on DSMB recommendation after a protocol prespecified interim analysis for futility |
Brock 1990.
| Methods | Study design: multi‐centre, randomised, placebo‐controlled trial | |
| Participants |
Country: Germany Number of study centres: 11 Setting: hospital Number of participants: 109; experimental group 56; control group 53 Age years: ≤ 40 to 80 Gender ratio (M/F): 61/48 Inclusion criteria: diabetic patients with ischaemic and/or neuropathic lesions Exclusion criteria: unstable diabetes; acute venous thrombosis or venous ulcers; indication for amputation; osteomyelitis, recent sympathectomy; unstable angina pectoris or miocardial infarction |
|
| Interventions |
Experimental: intravenous iloprost 2 ng/kg/min over 6 hours/d (in 40 to 50 mL saline) Control: placebo (40 to 50 mL saline) Duration: 28 days |
|
| Outcomes | Complete healing of tissue lesions, pain relief, analgesics consumption, tolerability | |
| Notes | Diabetic patients: 42 participants (75%) in iloprost group and 36 (68%) in placebo group had macroangiopathy with or without neuropathic lesions; the remaining participants presented lesions of microangiopathic, neuropathic, or undetermined origin. Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not clearly stated. Randomisation at study‐centre level |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not clearly described. Placebo and experimental groups received infusion with identical characteristics. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10% of study population, unbalanced between groups. ITT analysis was performed. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Low risk | Comparable groups as shown in Table 1 and Table 2 Identical care programmes |
Böhme 1989.
| Methods | Study design: 2‐centre, randomised, controlled trial | |
| Participants |
Country: Germany, Switzerland Number of study centres: 2 Setting: hospital Number of participants: 42 randomised; 34 analysed Age (range) years: 69 (33 to 85) Gender ratio (M/F): experimental group 11/7; control group 13/3 Inclusion criteria: PAOD stage III or IV Exclusion criteria: not stated |
|
| Interventions |
Experimental: intra‐arterial infusion of prostaglandin E1 (prostavasin) 10 to 20 µg over 60 minutes Control: adenosine triphosphate (ATP) 30 mg Duration: 23 days, follow‐up 12 months |
|
| Outcomes | Rest‐pain relief, healing of necrosis, consumption of analgesics, adverse effects (infusion pain or irritation), amputation, death | |
| Notes | Source of funding: not stated ‐ probably Schwartz‐Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10 % of study population. ITT analysis was not performed; only 34 of 42 randomised participants were analysed. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods section were reported. |
| Other bias | Unclear risk | Comparable groups as shown in Table 2 Identical care programmes not stated |
Böhme 1994.
| Methods | Study design: randomised controlled trial | |
| Participants |
Country: Germany Number of study centres: not stated Setting: hospital Number of participants: 30 randomised; 30 analysed. 15 experimental group; 15 control group Age (mean ± SD) years: experimental group 72 ± 11; control group 70 ± 9 Gender ratio (M/F): not stated Inclusion criteria: PAOD stage III or IV with symptoms lasting longer than 4 weeks, without chance of vascular surgery, catheter techniques, or fibrinolysis Exclusion criteria: decompensated heart failure, poorly controlled hypertension (systolic blood pressure > 200 mmHg), unstable coronary heart disease, renal failure (creatinine > 21.2 mg/dL) |
|
| Interventions |
Experimental: intravenous infusion of prostaglandin E1 (alprostadil) 40 µg twice a day over 2 hours Control: intravenous infusion of naftidrofuryl 400 mg twice a day over 2 hours Duration: 2 weeks, follow‐up 3 weeks |
|
| Outcomes | Rest pain, healing of necroses, response to treatment (combined outcome) | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | High risk | Participants were assigned to treatment groups according to a randomisation list. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Complete follow‐up at 21 days. ITT analysis not mentioned |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparability of groups shown in Tables 1 and 2 Identical care programmes not stated |
Castagno 2000.
| Methods | Study design: randomised controlled trial | |
| Participants |
Country: Italy Number of study centres: 1 Setting: hospital Number of participants: 93 randomised; 79 analysed. 49 experimental (1) group; 47 experimental (2) group; excluding stage II PAOD and connective tissue disorders, 35 in experimental (1) group and 37 in experimental (2) group Age (mean ± SD) years: experimental (1) group 69 ± 11; experimental (2) group 65 ± 11 (calculated while excluding aforementioned participants) Gender ratio (M/F): 51/21 (calculated while excluding aforementioned participants) Inclusion criteria: not stated. "The treatment regarded only patients in which a surgical revascularisation or reoperation was not proposable for matters related to the clinical picture of the patients". Exclusion criteria: not stated |
|
| Interventions |
Experimental (1): intravenous infusion of iloprost (half an ampoule, 0.50 mg/mL) in 250 mL of physiological solution once a day over 6 hours daily Experimental (2): intravenous infusion of alprostadil (60 µg) in 250 mL of physiological solution once a day over 2 hours Duration: experimental (1) 4 weeks; experimental (2) 3 weeks |
|
| Outcomes | Effectiveness (combined outcome), side effects (that could not be eliminated with adaptation of infusion speed and involved suspension of treatment) | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The assignment to iloprost or alprostadil has been randomised". |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not stated, probably an open‐label study owing to different duration of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not stated, probably an open‐label study owing to different duration of interventions |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals: 4 participants in the alprostadil group interrupted treatment (unbalanced attrition). ITT analysis was performed by review authors using individual participant data. |
| Selective reporting (reporting bias) | High risk | Only composite efficacy outcome reported; only severe adverse events reported |
| Other bias | Unclear risk | Identical care programmes not stated |
Cronenwett 1986.
| Methods | Study design: single‐centre, double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Country: USA Number of study centres: 1 Setting: hospital Number of participants: 26 (13 in each branch) Age (mean ± SD) years: experimental 66 ± 8, control 71 ± 10 Gender ratio (M/F): experimental 4/13, control 10/13 Inclusion criteria: extensive PAOD manifested by ischaemic ulcers or rest‐pain (stable or deteriorating for at least 2 months), surgically unreconstructable; fewer than 10 ulcers, area < 25 cm², not involving tendon, joint space, or bone Exclusion criteria: premenopausal women; patients with severe systemic illness |
|
| Interventions |
Experimental: intravenous infusion of PGI2 in glycine buffer, stable infusion of 6 ng/kg/min over 6 hours Control: placebo Duration: 3 days, monthly follow‐up during 6 months |
|
| Outcomes | Ulcer size, rest pain (considering frequency, severity, potency of analgesics consumed), "vascular laboratory testing" (composite outcome considering ankle, thigh, and toe brachial indexes, bidirectional Doppler analysis, reactive hyperaemia, and time to recover after reactive hyperaemia); "treatment success" (combined outcome: pain or ulcer reduction) | |
| Notes | Source of funding: University of Michigan Clinical Research Unit | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated "double‐blind fashion" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study was described as double‐blind. Ulcer size was measured by computerised planimetry (objective outcome). Pain was assessed by a "blind observer". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One participant died (myocardial infarction); no losses to follow‐up were reported. ITT analysis was not mentioned. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods section were reported. |
| Other bias | High risk | Baseline characteristics were unbalanced between groups (higher proportion of males in control group). Participants who did not respond (in ulcer size or pain reduction) were offered a second non‐blinded PGI2 infusion. Five out of 6 participants who received a second PGI2 infusion were "non‐responders" from the experimental group, and only one was from the control group (unbalanced co‐intervention). Identical care programmes not stated |
Diehm 1988.
| Methods | Study design: single‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: Germany Number of study centres: 1 Setting: hospital Number of participants: 50; 46 evaluated; experimental group 22; control group 24 Age (average) years: experimental group 65; control group 66 Gender ratio (M/F): experimental group 18/4; control group 16/8 Inclusion criteria: PAOD stage III, clinically in a steady state for 14 days before treatment start; < 70 years Exclusion criteria: blood vessel surgery; pregnancy; heart failure; MI within the previous 6 months; thrombocytosis > 4,000,000/µL; liver or kidney disease; uncontrolled diabetes mellitus |
|
| Interventions |
Experimental: intravenous infusion of prostavasin (60 µg PGE1) over 4 hours Control: placebo Duration: 3 weeks |
|
| Outcomes | Rest‐pain relief, analgesic consumption, side effects | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided regarding the method used for random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals < 10% of the study population (4/50). ITT analysis not performed |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported according to the methods section. |
| Other bias | Unclear risk | No information was provided regarding comparability of groups or care programmes. |
Diehm 1989.
| Methods | Study design: multi‐centre, randomised, double‐blind, parallel, placebo‐controlled trial | |
| Participants |
Country: Germany Number of study centres: 13 Setting: hospital Number of participants: 101 randomised; iloprost group 53; placebo group 48 Age (range) years: 40 to 80 Gender ratio (M/F): experimental group 34/19; control group 33/15 Inclusion criteria: age between 40 and 80 years, with diagnosis of PAOD stage IV, with stable symptoms, double‐sonographic ankle arterial blood pressure ≤ 50 mmHg; in patients with hypertension, difference between upper arm and ankle arteries ≤ 80 mmHg Exclusion criteria: diabetes mellitus, inflammatory arteriopathy, acute venous thrombosis, venous ulcers, peripheral neuropathy, possibility for lumen inaugurating process, attempt at revascularisation in the preceding 4 weeks, profunda‐plastic or sympathectomy within the past 3 months, gangrene and/or osteomyelitis that needs amputation, refractory hypertension, haematocrit > 0.50, bleeding diathesis, unstable angina or myocardial infarction, stroke or major cerebral circulation disorders within the past 6 months, acute or congestive heart failure, liver or kidney disease, other consuming or serious comorbidities, permanent anticoagulation in the past 3 months, long‐term therapy with acetylsalicylic acid within the past 2 weeks |
|
| Interventions |
Experimental: intravenous iloprost over 6 hours Control: placebo Duration: 28 days, follow‐up 6 months |
|
| Outcomes | Rest‐pain improvement, ulcer healing, improvement in arterial occlusion (peripheral ankle arterial pressure) | |
| Notes | Source of funding: Schering AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided regarding the method used for random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind. “Due to methodological problems in the quantification of the healing of ulcers, the main outcome, the study was conducted under double blind conditions”. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10%. ITT not performed |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Sparse information regarding comparability of groups of participants at baseline. Two unbalanced co‐interventions: 36% of participants in the iloprost group received antihypertensive therapy compared with 19% in the placebo group; 29% of participants in the iloprost group received non‐specified ointment‐based treatment compared with 13% in the placebo group. |
Dormandy 1991.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: UK Number of study centres: 14 Setting: hospital Number of participants: 151; experimental group 80; control group 71 Age (mean ± SD) (range) years: experimental group 73 ± 9.9 (33 to 89); control group 73 ± 9.7 (37 to 89) Gender ratio (M/F): experimental group 52/28; control group 36/35 Inclusion criteria: PAOD stage III or IV, unsuitable for surgical or catheter procedures Exclusion criteria: inflammatory arteriopathies; venous ulcers; frank peripheral neuropathy; other serious concomitant disease |
|
| Interventions |
Experimental: intravenous infusion of iloprost up to 2 ng/kg/min over 6 hours Control: placebo Duration: 28 days (participants with ulcer); 14 days (participants with rest‐pain) |
|
| Outcomes | Ulcer healing, rest‐pain relief, major amputation, death, side effects at end of treatment and at 6 months | |
| Notes | Source of funding: Schering Healthcare Ltd. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided regarding the method used for random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind. "Patients were randomly assigned to receive either iloprost or placebo (solvent without iloprost) which were identically packed". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | At 6 months, information concerning major amputations and deaths was available for 76/80 iloprost participants and 71/71 placebo participants. ITT analysis was performed. Follow‐up information concerning ulcer healing and pain relief at end of treatment was available for 67/80 iloprost participants and 70/71 placebo participants; but after 6 months for only 59/80 iloprost participants and 57/71 placebo participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the methods section were reported. |
| Other bias | Unclear risk | Imbalance in some baseline characteristics: "The groups were well matched for age, sex distribution, number of smokers, the pattern of arterial disease and ankle Doppler pressures, but there were more diabetics (49 vs. 31%) in the placebo group and more patients with only rest pain (46 vs. 38%) in the iloprost group". Identical care programmes not clearly stated |
Dormandy 2000a ‐ Study A.
| Methods | Study design: multi‐centre, randomised, dose‐ranging, double‐blind, placebo‐controlled trial | |
| Participants |
Country: France, Germany, Italy, Norway, Poland, Sweden, UK Number of centres: 35 in 7 countries Setting: hospital Number of participants: 178; experimental group (1) 58; experimental group (2) 58; control group 62 Age (mean) years (M/F): experimental group (1) 71/78; experimental group (2) 67/73; control group 69/ 73 Gender ratio (M/F): experimental group (1) 34/24; experimental group (2) 42/16; control group 37/25 Inclusion criteria: trophic skin lesions (ulcers or gangrene), rest‐pain due to severe arterial disease Exclusion criteria: acute onset or rapid deterioration of ischaemia; revascularisation procedure in previous 2 weeks; rapidly spreading cellulitis; regular treatment with antiplatelet other than aspirin; planned major amputation in the next 2 weeks |
|
| Interventions |
Experimental: oral iloprost; (1) low dose (100 µg twice daily); (2) high dose (200 µg twice daily) Control: placebo Duration: 4 weeks |
|
| Outcomes | Tolerability of doses, safety, death, major amputation, healing of trophic lesions, relief of rest‐pain at 6 months | |
| Notes | Source of funding: Schering AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded, as they were the attending physicians. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Information on only 1 participant at end of treatment and at 6‐month follow‐up was missing. ITT analysis was performed to analyse efficacy results at end of follow‐up. |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the methods section of this article were reported. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1a Identical care programmes not clearly stated |
Dormandy 2000b ‐ Study B.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: Finland, France, Germany, Hungary, Italy, Norway, Poland, Portugal, Sweden, UK Number of centres: 37 in 10 countries Setting: hospital Number of participants: 624; experimental group (1) 210; experimental group (2) 207; control group 207 Age (mean) years (M/F): experimental group (1) 66/74; experimental group (2) 65/74; control group 65/75 Gender ratio (M/F): not listed Inclusion criteria: trophic skin lesions (ulcers or gangrene), rest‐pain due to severe arterial disease Exclusion criteria: acute onset or rapid deterioration of ischaemia; revascularisation procedure in previous 2 weeks; rapidly spreading cellulitis; regular treatment with antiplatelet other than aspirin; planned major amputation in the next 2 weeks |
|
| Interventions |
Experimental: oral iloprost low dose (50 µg twice daily), high dose (150 µg twice daily) Control: placebo Duration: 1 year |
|
| Outcomes | Death, major amputation, healing of trophic lesions, rest‐pain relief at 1 year | |
| Notes | Source of funding: Schering AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded, as they were the attending physicians. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Information at end of treatment and at 12‐month follow‐up was missing for only 3 participants. ITT analysis was performed to analyse efficacy results at end of follow‐up. |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the methods section of the article were reported. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1b. Identical care programmes not clearly stated |
Esato 1995.
| Methods | Study design: multi‐centre, randomised, double‐blind, parallel, controlled trial | |
| Participants |
Country: Japan Number of centres: 113 Setting: hospital Number of participants: 135; experimental (1) group 66; experimental (2) group 69 Age (mean, range) years: experimental group 61.5 (22 to 89); control group 66.1 (32 to 91) Gender ratio (M/F): experimental group (2) 50/16; experimental (2) group 52/17 Inclusion criteria: chronic arterial occlusive disease (thromboangiitis obliterans or arteriosclerosis obliterans), with ischaemic ulcer and stable clinical condition Exclusion criteria: surgery to improve circulation within the previous 4 weeks; under 20 years of age; weight < 35 kg; hepatic, kidney, or heart failure; low blood pressure and glaucoma; pregnancy or breastfeeding; difficult determination of ischaemic ulcer; necrosis, exteriorisation of bone or tendon; ulcer present for longer than a year; ulcer due to neuropathic disorder |
|
| Interventions |
Experimental (1): intravenous infusion of TTC‐909 (clinprost incorporated in lipid microspheres) Experimental (2): intravenous infusion of lipo‐PGE1 Duration: 28 days |
|
| Outcomes | Rest‐pain relief, improvement in ulcer, adverse events, usefulness (comprehensive assessment of final improvement, overall safety, and natural repair process) | |
| Notes | TAO/ASO ratio: 17/37 experimental (1) group, 23/35 experimental (2) group. Improvement in rest‐pain and ulcer healing was reported separately for ASO participants, other outcomes were reported for participants with TAO and ASO combined. Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Low risk | Sealed packaging (apparently indistinguishable) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study: Participants and outcome assessors were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | For overall safety, 135/135 cases were analysed. For efficacy, 72/89 participants with ASO were analysed. ITT analysis was not performed. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods section were reported. |
| Other bias | Unclear risk | Comparable groups (for overall safety) as shown in Table 3 Identical care programmes not clearly stated |
Guilmot 1991.
| Methods | Study design: multi‐centre, randomised, placebo‐controlled trial | |
| Participants |
Country: France Number of study centres: 13 Setting: hospital Number of participants: 128; experimental group 87; control group 41 Age years: experimental group 68 ± 11; control group 68 ± 12 Gender ratio (M/F): experimental group 55/32; control group 30/11 Inclusion criteria: hospitalised men or postmenopausal women aged 40 to 85 years with PAOD due to atherosclerosis with and without diabetes mellitus. Furthermore, patients had to have 1 or more ulcers or areas of necrosis of arterial origin, or had to suffer from resting pain requiring regular analgesic treatment (PAOD stage III or IV). Ischaemia had to be confirmed by at least 2 of the following measurements: no pulsatility during vasodilatation measured with plethysmography; transcutaneous oxygen pressure (TcPO2) < 30 mmHg in the dependent foot, or systolic blood pressure in the first toe < 40 mmHg. In non‐diabetic patients, ankle systolic blood pressure index < 0.35 was an alternative indicator of ischaemia. Exclusion criteria: suitable for reconstructive vascular surgery or likely to require amputation in the near future; inflammatory arteriopathy such as Buerger's disease, venous ulcers, failure of reconstructive vascular surgery during the previous week, sympathectomy during the past 2 months, haematocrit > 55% or haemoglobin > 11 mmol/L (or 175 g/L), platelets < 100,000 or > 400,000/mm³, serum creatinine > 260 µmol/L, aminotransferase > 35 lU/L, continuous treatment with aspirin during the previous 2 weeks, congestive heart failure (III and IV NYHA stages), permanent hypertension (> 180 mmHg for systolic pressure and > 100 mmHg for diastolic pressure), cerebral ischaemic attack during the past 6 months |
|
| Interventions |
Experimental: intravenous infusion of iloprost over 6 hours Control: placebo Duration: 21 days |
|
| Outcomes | Healing and surface area of ulcers; rest‐pain relief; amputation; ankle systolic index; TcPO2 plethysmography; global evaluation (composite outcome carried out by investigators considering quantified and non‐quantified parameters); tolerability at days 21, 28, 60, and 120; side effects | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided regarding the method used for random sequence generation, with exception of stratification by history of diabetes mellitus |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10% of the study population. ITT analysis not performed |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1 Identical care programmes not stated |
Hossmann 1983.
| Methods | Study design: single‐centre, randomised, placebo‐controlled, cross‐over trial | |
| Participants |
Country: Germany Number of study centres: 1 Setting: hospital Number of participants: 10 Age (range) years: 33 to 77 years Gender ratio (M/F): 9/1 Inclusion criteria: PAOD stage III and IV Exclusion criteria: not stated |
|
| Interventions |
Experimental: intravenous infusion of prostacyclin PGI2 5 ng/kg/min Control: placebo Duration: 7 days (7 days between treatments) |
|
| Outcomes | Blood pressure, pain perception on a VAS | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not clearly stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study not described as blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study not described as blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Complete follow‐up |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Cross‐over design appears to be suitable for the outcomes assessed. Nevertheless, we may not exclude the presence of carry‐over effect. |
Jogestrand 1985.
| Methods | Study design: single‐centre, randomised, double‐blind, placebo‐controlled, cross‐over trial | |
| Participants |
Country: Sweden Number of study centres: 1 Setting: hospital Number of participants: 16 Age (mean ± SD) years: 67.18 ± 9.84 Gender ratio (M/F): 11/5. Inclusion criteria: PAOD stages III and IV. Exclusion criteria: not stated |
|
| Interventions |
Experimental: continuous intravenous infusion of prostacyclin PGE1 (15 µg/h) Control: placebo Duration: 72 hours (1 month between treatments) |
|
| Outcomes | Effects on peripheral blood flow (venous occlusion plethysmography, skin temperature, digital pulse plethysmography, systolic toe blood pressure, dynamic fluorescein angiography, vital capillary microscopy), rest‐pain | |
| Notes | Source of funding: grants from Tore Nilsons Fond för Medicinsk Forskning and the Swedish Medical Research Council 19x‐204 Cross‐over design |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomization was performed in groups of four patients and half of the subjects received saline first" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Each morning new PGE1‐solution was prepared in the pharmacy". "Nobody in the ward knew if a patient received PGE1 or saline". "At the dosage used, flush has never been observed. This fact assured the study blindness". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals < 10%. Two participants out of 16 were excluded from the study (1 because of myocardial infarction after the first treatment, 1 because of lack of co‐operation). A third participant was excluded from the evaluation of subjective outcomes because of atypical pain with peripheral neuropathy, discovered after admittance to hospital. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported according to the methods section. |
| Other bias | Unclear risk | Cross‐over design seems to be suitable for the outcomes assessed. Nevertheless, we may not exclude the presence of carry‐over effect. |
Karnik 1986.
| Methods | Study design: single‐centre, randomised, controlled trial. Cross‐over to alternative in 6 cases of primary treatment failure | |
| Participants |
Country: Germany Number of study centres: 1 Setting: hospital Number of participants: 20; prostacyclin 10; naftidrofuryl 10 Age (mean ± SD) years: 73.6 ± 10.7 prostacyclin group; 67.1 ± 10.1 naftidrofuryl group Gender ratio (M/F): 13/7; 6/4 in prostacyclin group, 7/3 in naftidrofuryl group Inclusion criteria: diagnosed peripheral arterial occlusion at stages III and IV, rest‐pain and/or ulcers, longer than 4 weeks Exclusion criteria: severe stenosis or occlusion of the iliac artery documented by angiography |
|
| Interventions |
Experimental: intravenous infusion of prostacyclin PGI2 5 ng/kg/min over 10 hours, on 5 successive days Control: intravenous infusion of naftidrofuryl 600 mg over 6 hours, on 7 successive days Duration: 4 weeks |
|
| Outcomes | Rest‐pain, evolution of tissue lesions (healing of ulcers and gangrenes), adverse events | |
| Notes | Funding source not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals ≥ 10% of the study population. At follow‐up after 4 weeks, 6 participants were included in the prostacyclin group and 5 in the naftidrofuryl group. ITT analysis was not performed. |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1 Identical care programmes not clearly stated |
Linet 1991.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Country: Australia, Austria, Belgium, Canada, Italy, France, Germany (2 centres), Mexico, Switzerland, UK (3 centres), USA (9 centres) Number of study centres: 22 Setting: hospital Number of participants: 211; ciprostene 106, placebo 105 Age (mean ± SD) years: experimental group 67.5 ± 1.36; control group 67.7 ± 1.31 Gender ratio (M/F): 114/97 Inclusion criteria: aged > 18 years with atherosclerotic PVD manifested by Ischaemic ulcers of the lower extremities (for 3 weeks or longer); not at a stage requiring amputation Exclusion criteria: infection/gangrene or exposed tendons or bones; CVD or MI within the previous 2 months; coagulation disorders; uncontrolled diabetes; hypertension; cancer; ARI; unstable angina |
|
| Interventions |
Experimental: intravenous infusion by mechanical pump of ciprostene up to 120 ng/kg/min over 8 hours daily Control: placebo Duration: 7 days |
|
| Outcomes | Healing of ulcers, rest‐pain relief, mortality, ABI, global functional status, amputation, adverse events, safety. Follow‐up of 4 months | |
| Notes | Source of funding: Upjohn Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Low risk | Code of drug assignment supplied by sponsor. Pharmacist followed code supplied by sponsor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Solutions of ciprostene and vehicle (placebo) were identical (clear fluid) in appearance. All study personnel with the exception of the pharmacist were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All study personnel with the exception of the pharmacist were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10% of the study population. Only 48 (45%) and 58 (55%) participants in the ciprostene and placebo groups completed the study, respectively. ITT analysis not mentioned |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Baseline imbalance between groups: hypertension history in 52% participants in the ciprostene group vs 35% in the placebo group Identical care programmes not stated |
NCT00596752.
| Methods | Study design: multi‐centre, randomised, double‐blind, phase 4, placebo‐controlled trial | |
| Participants |
Country: Czech Republic (2 centres), Germany, Mexico (3 centres), Poland (16 centres), Russian Federation (39 centres), Ukraine (17 centres) Number of study centres: 77 Setting: not stated Number of participants: 839; experimental group 415; control group 424 Age (mean ± SD) years: experimental group 66.8 ± 8.5; control group 66.4 ± 9.3 Gender ratio (M/F): alprostadil 123/293, placebo 117/306 Inclusion criteria: aged > 45 years with macro‐angiopathy; proven PAOD stage IV (with up to 2 ischaemic skin lesions for longer than 2 weeks) documented by complete angiography of pelvis, thigh, and calf within 1 month of inclusion; not in the position to be primarily revascularised or refuses surgery; systolic ankle pressure ≤ 70 mmHg in individuals without medial sclerosis of the lower limb artery, or systolic big toe pressure ≤ 50 mmHg in diabetic patients with medial sclerosis of the lower limb artery Exclusion criteria: imminent or foreseeable amputation; major amputation of the affected extremity; history of chronic alcohol or drug abuse; more than 2 ischaemic ulcerations; 1 ulcer ≥ 6 cm², both ulcers ≤ 1 cm² or at least 1 ulcer affecting the bone or tendons; acute ischaemia and peripheral vascular disorders of inflammatory or immunological origin; neuropathic or venous ulcers; Buerger's disease; septic gangrene; use of vasoactive medication or prostaglandins; treatment with prostanoids within 3 months before inclusion; surgical or interventional measures performed on the affected extremity within 3 months before start of study drug treatment |
|
| Interventions |
Experimental: intravenous infusion of alprostadil (prostavasin) 40 μg twice daily over 2 hours in 50 to 150 mL isotonic sodium chloride solution Control: intravenous infusion of placebo twice daily over 2 hours in 50 to 150 mL isotonic sodium chloride solution Duration: 4 weeks |
|
| Outcomes | Rate of complete healing of ulcerations after 12 weeks and 24 weeks; amputations (major and minor); intensity of rest‐pain induced by ischaemic lesions; increase/decrease in ulcer area ≥ 50% systolic pressure at ankle level; revascularisation procedures at 24 weeks; analgesic consumption; all‐cause mortality; cardiovascular mortality; cardiovascular morbidity (MI or stroke) during the course of the study (up to 196 days); adverse events | |
| Notes | Source of funding: UCB Pharma (UCB BIOSCIENCES GmbH) Collaborators: Aptiv Solutions |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated double‐blind study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 126/415 participants in alprostadil group and 143/424 in control group did not complete the study. "Subjects were analysed according to the actual treatment received. 4 placebo subjects were treated with Alprostadil, 3 Alprostadil subjects were treated with placebo. 1 placebo subject withdrew prior to start of study treatment". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported. |
| Other bias | Unclear risk | Comparability of groups according to randomisation not clearly stated Identical care programmes not stated |
Negus 1987.
| Methods | Study design: single‐centre, randomised, double‐blind, controlled trial | |
| Participants |
Country: UK Number of study centres: 1 Setting: hospital Number of participants: 29; experimental group 14; control group 15 Age (mean ± SD) years: experimental group 70.3 ± 11.9; control group 68.3 ± 10.1 Gender ratio (M/F): experimental group 9/5; control group 9/6 Inclusion criteria: ischaemic rest‐pain requiring analgesics; atherosclerotic lower limb arteries unsuitable for reconstructive surgery Exclusion criteria: not stated |
|
| Interventions |
Experimental: intra‐arterial infusion of prostacyclin (PGI2, epoprostenol) at a dose of 8 ng/kg/min Control: naftidrofuryl at a dose of 0.02 mg/kg/min Duration: 72 hours |
|
| Outcomes | Relief of rest‐pain and analgesic consumption after treatment, digital or forefoot amputation or major amputation up to 4 years, ABI | |
| Notes | Wellcome Research Laboratories Ltd. and Lipha Pharmaceuticals Ltd. helped in purchasing equipment. Source of financial assistance: SE Thames Regional Health Authority |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The choice of drug was by means of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | No information provided regarding the method used for allocation concealment ‐ probably done; "... those responsible for patient selection, pro forma completion and subsequent evaluation having no knowledge of which agent was delivered". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Four‐year follow‐up in only 5/10 participants in experimental group and 3/8 in control group (long‐term follow‐up of participants for whom proximal amputation was avoided initially). ITT analysis not mentioned |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information in methods section of the article; protocol not available |
| Other bias | Unclear risk | Comparable groups described in Table 1 Identical care programmes not stated |
Norgren 1990.
| Methods | Study design: multi‐centre, randomised, placebo‐controlled trial | |
| Participants |
Country: Finland (2 study centres), Poland (1 study centre), Sweden (6 study centres) Number of study centres: 9 Setting: hospital Number of participants: 103; experimental group 50; control group 53 Age (mean) (range) years: 70 (41 to 85) Gender ratio (M/F): 57/46 Inclusion criteria: 1 or more ischaemic ulcer of measurable size, for which a careful investigation including angiography showed that the case was unsuitable for reconstructive vascular surgery or interventional radiology Exclusion criteria: failed attempts at reconstruction in the past month, ≥ 85 years of age |
|
| Interventions |
Experimental: intravenous infusion of iloprost up to 2 ng/kg/min over 6 hours daily Control: placebo Duration: 14 days |
|
| Outcomes | Response to treatment, amputation, adverse events | |
| Notes | Source of funding: not stated (probably Schering AG) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided, besides stratification for centre and diabetes |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Twenty five patients in the treatment group and 35 patients in the control group did not reach the end of the follow‐up period" (n = 50 and 53, respectively). ITT analysis not performed. "As a result, data on clinical efficacy was limited to 46 of 50 patients in the iloprost group and to 52 of 53 patients in the placebo group". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Imbalance in baseline participant characteristics: ankle pressure 44 ± 34 mmHg in iloprost group, 57 ± 33 mmHg in placebo group (P < 0.05) Identical care programmes not clearly stated |
Reisin 1997.
| Methods | Study design: multi‐centre, randomised, placebo‐controlled trial | |
| Participants |
Country: Israel Number of study centres: 8 Setting: hospital Number of participants: 55; experimental group 26; control group 29 Age (mean ± SD) years: experimental group 66.07 ± 8.14; control group 66.41 ± 7.9 Gender ratio (M/F): experimental group 20/6; control group 22/7 Inclusion criteria: aged 35 to 80 years with both Fontaine class III or IV PAOD and NYHA functional Class II or III CHF that was chronic, compensated, and stable on digitalis, diuretics, or vasodilators Exclusion criteria: unstable angina pectoris; acute MI within 8 weeks of study entry; cardiomyopathy not associated with coronary artery disease; haemodynamically severe cardiac valvular abnormality; atrial or ventricular dysrhythmia requiring long‐term treatment; untreated hypertension; persistent hypotension; renal, endocrine, hematological, or hepatic disease sufficiently severe to affect the likelihood of survival |
|
| Interventions |
Experimental: intravenous infusion of PGE1 60 µg in 100 mL normal saline over 2 hours daily Control: placebo (100 mL normal saline) Duration: 14 days |
|
| Outcomes | Adverse events, incidence of clinical heart failure events | |
| Notes | Participants with CHF All participants received placebo infusion on day ‐1 and active drug on day 0. Active drug was given to all participants who chose to continue participation from days 15 to 28 (open‐label treatment phase). Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised, permuted block design, with block size of 4 (2 PGE1 and 2 placebo assignments within each block) to enable equivalent randomisation at each participating centre |
| Allocation concealment (selection bias) | High risk | Allocation list |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Three out of 53 participants withdrew because of adverse events during the randomised phase of the study: 2 in the placebo group (owing to abnormal liver enzymes and unstable angina pectoris), 1 in the PGE1 group (owing to blurred vision episode). ITT analysis performed |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups at baseline shown in Table 2 Identical care programmes not clearly stated |
Sakaguchi 1984.
| Methods | Study design: multi‐centre, randomised, controlled trial | |
| Participants |
Country: Japan Number of study centres: 10 Setting: hospital Number of participants: 76 randomised; 65 analysed. Experimental (1) group 22; experimental (2) group 25; control group 18 Age: not stated Gender ratio (M/F): not stated Inclusion criteria: ischaemic ulcer of the extremities due to chronic arterial occlusion in which surgical treatment such as arterial reconstructive surgery had not been indicated or other medical treatment had not been effective Exclusion criteria: not stated |
|
| Interventions | Participants were randomly allocated to 1 of the following therapies in which both an injection and oral capsules were given according to the double‐dummy method. Experimental (1): intra‐arterial infusion (through a catheter inserted into the femoral or brachial artery and a portable infusion pump) of PGE1 0.05 ng/kg/min (low dose) Experimental (2): intra‐arterial infusion (through a catheter inserted into the femoral or brachial artery and a portable infusion pump) of PGE1 0.15 ng/kg/min (high dose) Control: oral capsule containing inositol niacinate 200 mg Duration: 2 to 6 weeks |
|
| Outcomes | Early response to treatment (subjective), healing of ischaemic ulcer, improvement in rest‐pain, adverse events, recurrence of ischaemic ulcer (1 to 3 years' follow‐up) | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to the randomisation arm via the double‐dummy method (both an injection and oral capsules were given for 2 to 6 weeks). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The effect of PGE1 therapy was assessed by each physicians and the Evaluation Committee, which consisted of selected members. Discrepancies in assessments between the physicians and the Committee were subjected to the discussion to give a final judgement. All these assessments were carried out under blind conditions". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10%. ITT analysis not performed |
| Selective reporting (reporting bias) | High risk | No mention of a study protocol nor prespecified outcomes were provided in the methods section. Subjective endpoints not clearly defined. Additional analysis after pooling of low‐dose PGE1 and control groups to compare the results with those of the high‐dose PGE1 group on the basis that there was no difference between the first 2 groups |
| Other bias | Low risk | "In the background features of 65 patients subjected to the final evaluation, no serious bias between the groups were recognized". Identical care programmes |
Schellong 2004.
| Methods | Study design: multi‐centre, randomised, single‐blind, controlled, cross‐over trial | |
| Participants |
Country: Germany Number of study centres: 5 Setting: hospital Number of participants: 36 Age (mean ± SD) years: 70.3 ± 12.2 Gender ratio (M/F): 22/14 Inclusion criteria: PAOD stage III or IV that had been diagnosed on the basis of clinical criteria and confirmed by duplex ultrasonography or angiography; systolic arterial ankle pressure ≤ 70 mmHg; TcPO2 ≤ 20 mmHg, as measured in the supine patient at an electrode temperature of 44°C on the dorsum of the forefoot of the ischaemic extremity Exclusion criteria: known diabetes mellitus; history of sensitivity to PGE1 or iloprost; decompensated heart failure; MI within previous 6 months; suspected pulmonary oedema; pregnancy or lactation; severe CHD; unstable angina pectoris; simultaneous participation in another clinical trial; drug or alcohol addiction, as well as any serious disorder likely to jeopardise the participation |
|
| Interventions |
Experimental (1): intravenous infusion of PGE1 Experimental (2): intravenous infusion of iloprost Duration: 3 hours; 1 day of washout between treatments |
|
| Outcomes | Tolerability, adverse events | |
| Notes | Source of funding: Schwartz Pharma GmbH | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random plan in blocks of 4 per study site "Patients randomly assigned to consecutive therapy with the two prostaglandins using a computer generated random plan at blocks of four per study site" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participant‐blind study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcome assessors were not blinded. Most outcomes were subjective. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study (3 days). All randomised participants (36) were included in the ITT analysis. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Cross‐over design seems to be suitable for the outcomes assessed. Nevertheless, we may not exclude the presence of carry‐over effect. |
Schuler 1984.
| Methods | Study design: multi‐centre, randomised, double‐blind, placebo‐controlled, clinical trial | |
| Participants |
Country: USA Number of study centres: 22 Setting: hospital Number of participants: 123 randomised; 120 analysed: 57 participants PGE1 group, 63 participants placebo group Age (mean ± SD) years: not stated Gender ratio (M/F): 49/8 PGE1 group, 48/15 placebo group Inclusion criteria: male or postmenopausal female patients with 1 to 3 ischaemic ulcers of the lower extremities with greatest diameters ≤ 4 cm or greatest depth 0.5 cm that had been present for at least 3 weeks and did not show clinical signs of healing with conventional care. Patients with ischaemic ulcers were entered into the study if in the opinion of the principal investigator, surgical procedures were unsuitable, had previously failed or produced limited success, were not indicated, were refused by the patient, or could without undue risk of further tissue loss be judiciously postponed and performed at a later date if necessary. Exclusion criteria: cancer; premenopausal women; less than 18 years of age; unable to understand and grant truly informed consent; participating in other experimental drug studies; recent or unstable angina pectoris, pericardial effusion, idiopathic subaortic stenosis, congestive heart failure, recent myocardial infarction, or severe hypertension (diastolic blood pressure > 115 mmHg); chronic obstructive pulmonary disease or acute respiratory infection; poorly controlled diabetes mellitus; active hepatic, biliary, or peptic ulcer disease or ulcerative colitis or regional enteritis; any hematological or coagulation disorder; leg ulcers with associated chronic venous insufficiency; an ulcer that had tendon, periosteum, or a joint space in its base because of its location; ulcers overlying the calcaneus; ulcers associated with gross infection or gangrene in other areas |
|
| Interventions |
Experimental: intravenous infusion of PGE1, diluted with 0.9% saline solution to a concentration of 2.5 µgm/mL Control: placebo Duration: one or three 72‐hour infusions at 2‐week intervals |
|
| Outcomes | Rest‐pain, ulcer healing or significant change in size, adverse events | |
| Notes | Both interventions were administered by a central venous catheter, inserted through subclavian or internal jugular vein. Source of funding: The Upjohn Company |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided regarding the method used for random sequence generation, with the exception of stratification by centre. "Randomization was performed on a per center basis to eliminate the possibility of some centers treating a majority of patients with drugs while other centers treated a majority of patients with placebos". |
| Allocation concealment (selection bias) | Low risk | "On completion of the preliminary assessment, the obtaining of informed consents, and the decision that protocol requirements had been met, patients were assigned identification numbers; the pharmacist at each participating center followed a random code of drug assignment supplied by the Upjohn Project Coordinator". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The patient, the principal investigator, and all other personnel involved in patient care were blinded as to the nature of the infusion received. Only the pharmacist charged with preparing the infusion solution was aware of whether the patient was receiving PGE1 or a placebo, and the pharmacist was not aware of the patients' status at presentation or the subsequent clinical course". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Trial authors stated that all personnel involved in patient care, with the exception of the pharmacist, were blinded as to the nature of the infusion received (see above). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10% of the study population. Only 28/57 participants (49%) in the PGE1 group and 34/63 (54%) in the placebo group completed the study. Loss to follow‐up and withdrawals due to side effects that were drug related or intercurrent medical events, or due to technical difficulties with catheter placement and maintenance, or at the participants' request, were balanced. ITT analysis was not mentioned. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups at baseline. "There was no significant difference between the PGE1‐treated group and the placebo group with regard to the number of infusions received per patient". Identical care programmes not clearly stated |
Stiegler 1992.
| Methods | Study design: single‐centre, double‐blind, placebo‐controlled trial | |
| Participants |
Country: Germany Number of study centres: 1 Setting: hospital Number of participants: 117 recruited; 73 completed the study; experimental group 36; control group 37 Age (mean) (range) years: 69 (44 to 86) Gender ratio (M/F): 37/36 (completed) Inclusion criteria: individuals with type 2 diabetes with ulcers in the forefoot area (for at least 14 days) due to an arterial occlusive disease Exclusion criteria: not stated in the full text |
|
| Interventions |
Experimental: intravenous infusion of PGE1 (2 ampoules of 40 µg in NaCl 250 mL) Control: placebo Duration: 3 to 4 weeks |
|
| Outcomes | Ulcer sum‐score, rest pain, amputation rate, adverse events | |
| Notes | Diabetic patients Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals > 10% of study population. 32 participants had to be excluded afterwards because they did not meet inclusion‐exclusion criteria; 12 additional participants did not complete the study. ITT analysis not performed |
| Selective reporting (reporting bias) | High risk | Not every outcome is reported as stated in the methods section (analgesics consumption). |
| Other bias | Unclear risk | Comparability of groups sparsely described Identical care programmes not stated |
Telles 1984.
| Methods | Study design: 2‐centre, randomised, placebo‐controlled, double‐blind trial | |
| Participants |
Country: UK Number of study centres: 2 Setting: hospital Number of participants: 30; experimental group 14; control group 16 Age (mean) (range) years: 68.5 (40 to 84) Gender ratio (M/F): 20/10 Inclusion criteria: presenting rest‐pain alone, ischaemic ulceration, or both; unsuitable for reconstructive arterial surgery after angiographic assessment Exclusion criteria: not stated |
|
| Interventions |
Experimental: intravenous infusion of prostaglandin E1 (PGE1) up to 10 ng/kg/min in 0.9% saline Control: placebo Duration: over 72 hours |
|
| Outcomes | Rest‐pain relief, analgesic consumption, ulcer healing, ABI, amputation, blood pressure, heart rate, side effects. Follow‐up at 24 hours, 2 and 4 weeks | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not clearly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No mention regarding withdrawals or completeness of follow‐up. ITT analysis not performed |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups as shown in Table 1 Identical care programmes not stated |
Trubestein 1987.
| Methods | Study design: multi‐centre, randomised, controlled trial | |
| Participants |
Country: Germany Number of study centres: 4 Setting: hospital Number of participants: 57; experimental group 31; control group 26 Age (mean) years: experimental group 68; control group 63 Gender ratio (M/F): not stated Inclusion criteria: PAOD stages III and IV for at least 1 year of evolution; aged between 50 and 70 years. Arterial ulcers had to have been present for at least 14 days. Angiographically demonstrated stenoses and occlusions had to be localised in the femoral and lower leg arteries. Exclusion criteria: manifested heart insufficiency or vascular surgery within the previous 6 months |
|
| Interventions |
Experimental: intra‐arterial infusion of prostaglandin E1 (PGE1) 20 µg over 60 minutes daily in 50 mL saline solution Control: ATP 30 mg over 60 minutes daily in 50 mL saline solution Duration: 3 weeks |
|
| Outcomes | Relief of rest‐pain, use of analgesics, healing of or improvement in ulcers, change in clinical stage, maintenance of a functional extremity where amputation had been necessary, amputation, adverse events | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not mentioned |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not mentioned |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No mention regarding withdrawals or completeness of follow‐up. ITT analysis not performed |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Comparable groups regarding risk factors (hypertension, smoking, diabetes mellitus, and hyperlipoproteinaemia) Identical care programmes not stated |
Trübestein 1989.
| Methods | Study design: multi‐centre, randomised, controlled trial | |
| Participants |
Country: Germany Number of study centres: not stated Setting: hospital Number of participants: 85 entered; 70 analysed. Experimental group 35; control group 35 Age (mean) years: experimental group 75; control group 75 Gender ratio (M/F): not stated Inclusion criteria: PAOD stage IV for at least 1 year, arterial ulceration or necrosis for at least 14 days, and rest‐pain. Angiographically proven stenoses and occlusions had to be localised in the iliac, femoral, and lower leg arteries. Exclusion criteria: absolute indications for vascular reconstructive surgery or amputation;thromboangiitis obliterans and diabetes mellitus in combination with neuropathy, micro‐angiopathy, and osteolysis; myocardial infarction within the past 6 months; congestive heart failure; renal failure; pathological liver findings in laboratory tests |
|
| Interventions |
Experimental: intravenous infusion of prostaglandin E₁ (prostavasin) 40 µg in 250 mL saline solution twice a day, over 2 hours Control: intravenous infusion of pentoxifylline 300 mg in 250 mL saline solution twice a day, over 2 hours Duration: 4 weeks, follow‐up 6 months |
|
| Outcomes | Rest‐pain, analgesic consumption, ulcer/necrosis healing, amputation rate, adverse events | |
| Notes | Source of funding: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No mention regarding withdrawals or completeness of follow‐up. ITT analysis not performed |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as stated in the methods section. |
| Other bias | Unclear risk | Imbalance in baseline characteristics of participants: More participants in the control group had diabetes mellitus (7/35 vs 2/35). Longer duration of arterial occlusive disease in the experimental group (4 years vs 2.3 years) Identical care programmes not stated |
ABI: ankle brachial index. ARI: acute respiratory infection. ASO: arteriosclerosis obliterans. ATP: adenosine triphosphate. CHD: coronary heart disease. CHF: chronic heart failure. CLI: critical limb ischaemia. CVD: cardiovascular disease. DSMB: Data and Safety Monitoring Board. ECG: electrocardiogram. ESRD: end‐stage renal disease. ITT: intention‐to‐treat. µg: microgram. MI: myocardial infarction. ng: nanogram. NYHA: New York Heart Association functional classification. PAD: peripheral arterial disease. PAOD: peripheral arterial occlusive disease. PG: prostaglandin. PVD: peripheral vascular disease. SD: standard deviation. TAO: thromboangiitis obliterans. TcPO2: transcutaneous oxygen pressure. UK: United Kingdom. USA: United States of America. VAS: visual analogue score.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arosio 1998 | Inappropriate intervention for the control group. Study compared intravenous infusion of iloprost administered for 7 vs 28 days. |
| Beischer 1998 | Inappropriate intervention for the control group. Study compared different doses of the same prostanoid (iloprost). |
| Bertele 1999 | Most patients did not fulfil inclusion criteria: "Among patients not undergoing revascularization (total, 983 [63.0%]); 45.0% (44.0% in the alprostadil group and 45.9% in the control group) had no immediate indication for the intervention; 37.3% (37.2% in the alprostadil group and 37.4% in the control group) were considered ineligible for surgery because of the peripheral vascular condition (18.5% and 20.3%, respectively), the general clinical condition (9.4% and 9.5%, respectively), or both (9.4% and 7.5%, respectively)." We contacted the trial author for disaggregated outcome data for participants without chance of rescue or reconstructive intervention. The trial author did not respond. |
| Breuer 1995 | Inappropriate intervention for the control group. Study compared intravenous infusion of iloprost as an adjuvant of local surgical conservative therapy vs local surgical conservative therapy alone. |
| Ceriello 1998 | Inappropriate intervention for the control group. Study compared intravenous infusion of iloprost administered for 7 vs 28 days. |
| Di Paolo 2005 | Inappropriate intervention for the control group: extracorporeal blood oxygenation and ozonation (not pharmacological treatment) |
| Esato 1997 | Inappropriate intervention for the control group. Study compared different doses of the same prostanoid (clinprost). |
| Feng 2009 | Outcomes were reported as combined for all stages of PAOD. We contacted the trial author for disaggregated data for stages III and IV. The trial author did not respond. |
| Fonseca 1991 | Abstract. Full text not available. Trial author does not have it. Sponsor did not respond. |
| Guan 2003 | Outcomes were reported as combined for all stages of PAOD. We contacted the trial author for disaggregated data for stages III and IV. The trial author did not respond. |
| Heidrich 1991 | Outcomes were reported as combined for all stages of PAOD. Design: open ‐ uncontrolled follow‐up |
| Menzoian 1995 | Abstract. Full text not available. Trial author does not have a copy. Sponsor did not respond. |
| Mingardi 1993 | Abstract. Full text not available. Trial author did not respond. Sponsor does not have it. |
| NCT00059644 | No results available. Sponsor did not respond |
| Nizankowski 1985 | Outcomes were reported as combined for all participants including TAO, which represented 50% of the study population. We contacted the trial author for disaggregated data. Trial author did not respond. |
| Ohtake 2014 | Outcomes were reported combined for all stages of PAOD. We contacted the trial author for disaggregated data for stages III and IV. Trial author did not respond. |
| Petronella 2004 | Inappropriate intervention for the control group: surgical treatment (lumbar sympathectomy) |
| Sakaguchi‐Shukichi 1990 | Study design: not a randomised controlled trial |
| Schwarz 1995 | Abstract. Full text not available |
| Sert 2008 | Inappropriate intervention for the control group. Study compared intravenous infusion of iloprost as an adjuvant of routine treatment strategies vs routine treatment strategies alone. |
| Weiss 1991 | Inappropriate intervention for the control group. Study compared intravenous vs intra‐arterial infusion of the same prostanoid (iloprost). |
| Ylitalo 1990 | Most included participants (10/13) had PAOD stage II or TAO. |
PAOD: peripheral arterial obstructive disease. TAO: thromboangiitis obliterans.
Differences between protocol and review
We updated the introduction and its references for this version of the review.
For this update, we amended the objective to clarify the aim of the review, which is to assess the effectiveness and safety of prostanoids in patients with critical limb ischaemia (CLI) unsuitable for rescue or revascularisation procedures, as was stated in the abstract and mentioned under Types of participants.
We did not include studies of patients with a diagnosis of thromboangiitis obliterans, also known as Buerger's disease, to avoid overlap with a recently published review that focussed on this type of participant (Cacione 2016), and as a result of different subjacent pathophysiological mechanisms of this autoimmune disease.
We excluded surgical treatments and other medical non‐pharmacological treatments as comparators; we did not explicitly state this in the methods section, but they are not pharmacological treatments, and the effectiveness of these interventions has been addressed by other Cochrane systematic reviews (Ubbink 2013) and protocols (Sen 2011).
For this update, we rearranged selected outcomes to simplify understanding of main results in the 'Summary of findings' table. We removed "cardiovascular morbidity" as an outcome because it was poorly defined.
The previous version of this review specified study quality and risk of bias as exclusion criteria for eligible studies. According to current practice in Cochrane Reviews, we removed this restriction and analysed effects of bias and study quality in sensitivity analyses. For this purpose, two review authors (VV, JVAF or DC) independently assessed potential risks of bias for all included randomised controlled trials (RCTs) using Cochrane's tool for assessing risk of bias (Higgins 2011b).
For this update, we stated the thresholds for interpretation of heterogeneity as evaluated by results of the I² test, as indicated in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). As thresholds previously used were based on guidelines provided in the Cochrane Handbook for Systematic Reviews of Interventions, this did not affect the meta‐analyses performed for this review.
For this update, and as a result of changes to the aforementioned selection criteria, in some cases we reclassified both previously included and excluded studies (see Included studies and Excluded studies).
For this update, we extended the search for potentially relevant studies to clinical trials registers (see Search methods for identification of studies).
Software used in the protocol was RevMan 4.2, which was updated to RevMan 5.0 for the original review and to RevMan 5.3 for this update.
For this update, we restructured the results section. We included the subheadings "Prostanoids versus placebo", "Prostanoids versus other agents", and "Prostanoids versus other prostanoids", to provide a clearer report for readers. We included subgroup analyses of different prostanoids versus placebo in the analysis tree, which allowed us to perform and report statistical tests for subgroup differences. Following the same rationale, we eliminated the high‐dose and low‐dose iloprost versus placebo subgroup analysis performed by previous review authors, after considering the lack of statistical differences for subgroups or additional studies that evaluated a similar posology.
Contributions of authors
VV: served as leading review author, co‐ordinated the contributions of coauthors, and participated at all stages of this review update: study selection, contact with trial authors/manufacturers, risk of bias assessment, analysis of data, and writing of the manuscript. JVAF: participated at all stages of the review update: study selection, risk of bias assessment, data extraction, analysis, and writing of the manuscript. VS: contributed to translation of German language reports, risk of bias assessment, data extraction, analysis, and writing of the manuscript. DC: contributed to study selection, risk of bias assessment, and data extraction. JC: as a cardiovascular surgeon, contributed to interpretation of data from a clinical perspective. AC: provided methodological supervision for preparation of the original review and this update. He resolved disagreements regarding selection of studies, assessment of study quality, and data extraction.
Sources of support
Internal sources
-
Family and Community Medicine Service, Hospital Italiano de Buenos Aires, Argentina.
Provided financial support through time protection of the main author for completing the review and training
-
Instituto Universitario Hospital Italiano de Buenos Aires, Argentina.
Provided financial support through time protection of JVAF, as a fellow researcher in systematic reviews, and VV, as the Cochrane Associate Centre director
External sources
-
Chief Scientist Office, Scottish Government Health Directorates, Scottish Government, UK.
The Cochrane Vascular editorial base is supported by the Chief Scientist Office.
Declarations of interest
VV: none known. JVAF: none known. VS: none known. DC: none known. JC: none known. AC: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Alstaedt 1993 {published data only}
- Altstaedt HO, Berzewski B, Breddin HK, Brockhaus W, Bruhn HD, Cachovan M, et al. Treatment of patients with peripheral arterial occlusive disease Fontaine stage IV with intravenous iloprost and PGE1: a randomized open controlled study. Prostaglandins Leukotrienes Essential Fatty Acids 1993;49(2):573‐8. [DOI] [PubMed] [Google Scholar]
Balzer 1991 {published data only}
- Balzer K, Bechara G, Bisler H, Clevert HD, Diehm C, Heisig G, et al. Reduction of ischaemic rest pain in advanced peripheral arterial occlusive disease. A double blind placebo controlled trial with iloprost. International Angiology 1991;10(4):229‐32. [PubMed] [Google Scholar]
Bandiera 1995 {published data only}
- Bandiera G, Crisci D, Camilli S. Analysis of side effects of prostanoid‐based treatment in patients suffering from critical ischaemia of the lower limbs. Chronica Dermatologica 1995;5(2):249‐60. [Google Scholar]
Belch 1983 {published data only}
- Belch JJ, McKay A, McArdle B, Leiberman P, Pollock JG, Lowe GD, et al. Epoprostenol (prostacyclin) and severe arterial disease. A double‐blind trial. Lancet 1983;1(8320):315‐7. [DOI] [PubMed] [Google Scholar]
Belch 2011 {published data only}
- Belch JJ, Ray S, Rajput‐Ray M, Engeset J, Fagrell B, Lepantalo M, et al. The Scottish‐Finnish‐Swedish PARTNER study of taprostene versus placebo treatment in patients with critical limb ischemia. International Angiology 2011;30(2):150‐5. [PUBMED: 21427652] [PubMed] [Google Scholar]
Böhme 1989 {published data only}
- Böhme H, Brulisauer M, Hartel U, Bollinger A. Peripheral arterial occlusive disease stage III and IV. Medizinische Welt 1989;40(51‐52):1501‐3. [Google Scholar]
Böhme 1994 {published data only}
- Böhme H, Hartel U, Walter H. Naftidrofuryl in comparison to alprostadil in peripheral arterial occlusive disease stage III and IV. Medizinische Welt 1994;45(5):209‐13. [Google Scholar]
Brass 2006 {published and unpublished data}
- Brass EP, Anthony R, Dormandy J, Hiatt WR, Jiao J, Nakanishi A, et al. (Circulase investigators). Parenteral therapy with lipo‐ecraprost, a lipid‐based formulation of a PGE1 analog, does not alter six‐month outcomes in patients with critical leg ischemia. Journal of Vascular Surgery 2006;43(4):752‐9. [DOI] [PubMed] [Google Scholar]
Brock 1990 {published data only}
- Brock FE, Abri O, Baitsch G, Bechara G, Beck K, Corovic D, et al. Iloprost in the treatment of ischemic tissue lesions in diabetics: results of a placebo‐controlled multicenter study with a stable prostacyclin derivative. Schweizerische Medizinische Wochenschrift 1990;120(40):1477‐82. [PubMed] [Google Scholar]
Castagno 2000 {published data only}
- Castagno PL, Molfetta L, Merlo M, Barile G, Violato F, Buzzacchino A, et al. Prospects of prostanoid therapy. Preliminary results. Minerva Cardioangiologica 2000; Vol. 48, issue 1‐2:9‐18. [CN‐00278141] [PubMed]
Cronenwett 1986 {published data only}
- Cronenwett JL, Zelenock GB, Whitehouse WM Jr, Lindenauer SM, Graham LM, Stanley JC. Prostacyclin treatment of ischemic ulcers and rest pain in unreconstructable peripheral arterial occlusive disease. Surgery 1986;100(2):369‐75. [PubMed] [Google Scholar]
Diehm 1988 {published data only}
- Diehm C, Hubsch‐Muller C, Stammler F. Intravenous prostaglandin E1 therapy in patients with stage III peripheral arterial disease ‐ a double‐blind placebo controlled study. Prostaglandin E1 Wirkungen und Therapeutische Wirksamkeit. Berlin/Heidelberg/New York: Springer, 1988:133‐43. [Google Scholar]
- Diehm C, Stammler F, Hubsch C, Wilhelm C, Eckstein HH. Treatment of rest pain in peripheral arterial occlusive disease (PVVK) with intravenous prostaglandin infusion. Vasa Supplementum 1987;20:204‐5. [Google Scholar]
- Diehm C, Stammler F, Hubsch‐Muller C, Eckstein HH, Simini B. Clinical effects of intravenously administered prostaglandin E1 in patients with rest pain due to peripheral obliterative arterial disease (POAD) ‐ a preliminary report on a placebo‐controlled double‐blind study. Vasa Supplementum 1987;17:52‐6. [PubMed] [Google Scholar]
Diehm 1989 {published data only}
- Diehm C, Abri O, Baitsch G, Bechara G, Beck K, Breddin HK, et al. Iloprost, a stable prostacyclin derivative, in stage IV arterial disease. A placebo‐controlled multicentre study. Deutsche Medizinische Wochenschrift 1989;114(20):783‐8. [DOI] [PubMed] [Google Scholar]
Dormandy 1991 {published data only}
- Dormandy JA, U.K. Severe Limb Ischaemia Study Group. Treatment of limb threatening ischaemia with intravenous iloprost: a randomised double‐blind placebo controlled study. European Journal of Vascular Surgery 1991;5(5):511‐6. [DOI] [PubMed] [Google Scholar]
Dormandy 2000a ‐ Study A {published data only}
- Dormandy J, Belcher G, Boccalon H, Braga A, Breddin K, Catalano M, et al. Controlled randomized study of an oral prostacyclin analogue (iloprost) in patients with inoperable stage III and IV leg ischaemia. British Journal of Surgery 1996;83(4):558‐9. [Google Scholar]
- Dormandy JA. Two randomised and placebo‐controlled studies of an oral prostacyclin analogue (Iloprost) in severe leg ischaemia. Study A. European Journal of Vascular and Endovascular Surgery 2000;20(4):358‐62. [DOI] [PubMed] [Google Scholar]
Dormandy 2000b ‐ Study B {published data only}
- Dormandy JA. Two randomised and placebo‐controlled studies of an oral prostacyclin analogue (Iloprost) in severe leg ischaemia. Study B. European Journal of Vascular and Endovascular Surgery 2000;20(4):358‐62. [DOI] [PubMed] [Google Scholar]
Esato 1995 {published data only}
- Esato K, Yasuda K, Abe T, Hoshino S, Ishimaru S, Hori G, et al. Studies on the clinical efficacy and safety of TTC‐909 in the treatment of peripheral arterial occlusive disease: a multi‐center double‐blind comparison with Alprostadil incorporated in lipid microspheres [TTC‐909の慢性動脈閉塞症に対する臨床的有効性と安全性の検討 Alprostadil含有脂肪乳剤を対照薬とした多施設二重盲検群間比較試験]. Rinsho Iyaku 1995; Vol. 11, issue 10:2111‐41. [CN‐00543666]
Guilmot 1991 {published data only}
- Guilmot J‐L, Diot E, French Iloprost Study Group. Treatment of lower limb ischaemia due to atherosclerosis in diabetic and nondiabetic patients with iloprost, a stable analogue of prostacyclin. Results of a French Multicentre Trial. Drug Investigation 1991;3(5):351‐9. [Google Scholar]
Hossmann 1983 {published data only}
- Hossmann V, Auel H, Rucker W, Schror K. Long‐term infusion of epoprostenol (prostacyclin) in patients with occlusive arteriopathy stage III‐IV. Verhandlungen der Deutschen Gesellschaft fur Innere Medizin 1983;89:540‐4. [Google Scholar]
Jogestrand 1985 {published data only}
Karnik 1986 {published data only}
- Karnik R, Valentin A, Slany J. A randomized study on patients with peripheral arterial occlusive disease (PAOD) stage III and IV. Herz Kreislauf 1986;18(12):644‐7. [Google Scholar]
Linet 1991 {published data only}
- Linet OI, Eckert SM, Huang DC. The effect of ciprostene in patients with peripheral vascular disease (PVD) characterized by ischemic ulcers. Journal of Clinical Pharmacology 1991;31(1):81‐7. [DOI] [PubMed] [Google Scholar]
NCT00596752 {unpublished data only}
- Alprostadil in Peripheral Arterial Occlusive Disease (PAOD) Stage IV (ESPECIAL) [Multinational, prospective, randomized, double blind, placebo controlled, parallel groups study to assess the efficacy and safety of prostaglandin E1 in subjects with critical limb ischemia (Fontaine Stage IV)]. clinicaltrials.gov/ct2/show/NCT00596752. Date first received: 21 December 2007. [2005‐001970‐29 ( EudraCT Number); SP0777 (Sponsor ID)]
Negus 1987 {published data only}
- Negus D, Irving JD, Friedgood A. Intra‐arterial prostacyclin compared to praxilene in the management of severe lower limb ischaemia: a double blind trial. Journal of Cardiovascular Surgery (Torino) 1987;28(2):196‐9. [PubMed] [Google Scholar]
Norgren 1990 {published data only}
- Norgren L, Alwmark A, Angqvist KA, Hedberg B, Bergqvist D, Takolander R, et al. A stable prostacyclin analogue (Iloprost) in the treatment of ischaemic ulcers of the lower limb. European Journal of Vascular Surgery 1990;4(5):463‐7. [DOI] [PubMed] [Google Scholar]
Reisin 1997 {published data only}
Sakaguchi 1984 {published data only}
- Sakaguchi S. Prostaglandin E1 intra‐arterial infusion therapy in patients with ischemic ulcer of the extremities. International Angiology 1084;3:39‐41. [Google Scholar]
Schellong 2004 {published data only}
- Schellong S, Altmann E, Bilderling P, Rudofsky G, Waldhausen P, Rogatti W. Microcirculation and tolerability following i.v. infusion of PGE1 and iloprost: a randomized cross‐over study in patients with critical limb ischemia. Prostaglandins Leukotrienes and Essential Fatty Acids 2004;70(6):503‐9. [DOI] [PubMed] [Google Scholar]
Schuler 1984 {published data only}
- Rhodes RS, Heard SE. Detrimental effect of high‐dose prostaglandin Esub 1 in the treatment of ischemic ulcers. Surgery 1983; Vol. 93, issue 6:839‐42. [CN‐00177644] [PubMed]
- Schuler JJ, Flanigan DP, Holcroft JW, Ursprung JJ, Mohrland JS, Pyke J. Efficacy of prostaglandin E1 in the treatment of lower extremity ischemic ulcers secondary to peripheral vascular occlusive disease. Results of a prospective randomized, double‐blind, multicenter clinical trial. Journal of Vascular Surgery 1984;1(1):160‐70. [PUBMED: 6384558] [DOI] [PubMed] [Google Scholar]
Stiegler 1992 {published data only}
- Stiegler H, Diehm C, Grom E, Martin M, Morl H, Rudofsky G, et al. Placebo controlled, double‐blind study of the effectiveness of i.v. prostaglandin E1 in diabetic patients with stage IV arterial occlusive disease [Placebokontrollierte, doppelblinde Studie zur Wirksamkeit von i.v. Prostaglandin E1 bei Diabetikern mit AVK im Stadium IV]. VASA 1992;35 (Suppl):164‐6. [PubMed] [Google Scholar]
Telles 1984 {published data only}
- Telles GS, Campbell WB, Wood RFM, Collin J, Baird RN, Morris PJ, et al. Prostaglandin E1 in severe lower limb ischaemia: a double‐blind controlled trial. British Journal of Surgery 1984;71(7):506‐8. [DOI] [PubMed] [Google Scholar]
Trubestein 1987 {published data only}
- Trubestein G, Diehm C, Gruss JD, Horsch S. Prostaglandin E1 in chronic arterial disease ‐ a multicenter study. VASA Supplementum 1987;17:39‐43. [PUBMED: 3470965] [PubMed] [Google Scholar]
- Trubestein G, Ludwig M, Diehm C, Gruss JD, Horsch S. Prostaglandin E1 in stage III and IV arterial occlusive disease: results of a multi‐center trial. Deutsche Medizinische Wochenschrift 1987;112(24):955‐9. [DOI] [PubMed] [Google Scholar]
Trübestein 1989 {published data only}
- Trubestein G, Bary S, Breddin K, Diehm C, Gruss JD, Heinrich H, et al. Intravenous prostaglandin E1 versus pentoxifylline therapy in chronic arterial occlusive disease ‐ a controlled randomised multicenter study. VASA Supplementum 1989;28:44‐9. [PubMed] [Google Scholar]
References to studies excluded from this review
Arosio 1998 {published data only}
- Arosio E, Sardina M, Prior M, Marchi S, Zannoni M, Bianchini C. Clinical and circulatory effects of Iloprost either administered for 1 week or 4 weeks in patients with peripheral obstructive arterial disease at Leriche‐Fontaine stage III. European Review for Medical and Pharmacological Sciences 1998;2(2):53‐9. [PubMed] [Google Scholar]
Beischer 1998 {published data only}
- Beischer W, Dembski JC, Gruss JD, Hofgartner F, Horsch A, Horsch S, et al. Low‐dose iloprost infusions compared to the standard dose in patients with peripheral arterial occlusive disease Fontaine stage IV. DAWID Study Group. VASA. Zeitschrift fur Gefasskrankheiten. Journal for Vascular Diseases 1998;27(1):15‐9. [PubMed] [Google Scholar]
Bertele 1999 {published data only}
- Bertele V. Prostanoids for chronic critical leg ischemia. A randomized, controlled, open‐label trial with prostaglandin E1. Annals of Internal Medicine 1999;130(5):412‐21. [PubMed] [Google Scholar]
Breuer 1995 {published data only}
- Breuer C, Abri O. Prostacyclin (iloprost) as an adjuvant in local surgical therapy of stage IV arterial occlusive disease ‐ is quantification of the therapeutic effect possible with tcPO2 measurements?. VASA. Zeitschrift fur Gefasskrankheiten. Journal for Vascular Diseases 1995;24(1):62‐71. [PubMed] [Google Scholar]
Ceriello 1998 {published data only}
- Ceriello A, Sardina M, Motz E, Lizzio S, Tuniz D, Bianchini C. Effects of iloprost, either administered for 1 week or 4 weeks, on systemic hemodynamics, peripheral blood flow, and exercise tolerance in patients with peripheral occlusive arterial disease at Leriche‐Fontaine stage III. International Journal of Angiology 1998;7(1):28‐33. [Google Scholar]
Di Paolo 2005 {published data only}
- Paolo N, Bocci V, Salvo DP, Palasciano G, Biagioli M, Meini S, et al. Extracorporeal blood oxygenation and ozonation (EBOO): a controlled trial in patients with peripheral artery disease. International Journal of Artificial Organs 2005;28(10):1039‐50. [DOI] [PubMed] [Google Scholar]
Esato 1997 {published data only}
- Esato K, Yasuda K, Abe T, Hoshino S, Ishimaru S, Hori G, et al. Clinical study of TTC‐909 (Prostacyclin derivative, clinprost incorporated in lipid microspheres) in patients with peripheral arterial occlusive disease: establishment of optimal dose in multi‐center double‐blind comparative study. Rinsho Iyaku (Journal of Clinical Therapeutics and Medicines) 1995; Vol. 11, issue 10:2083‐110. [CN‐00543531]
- Esato K, Yasuda K, Abe T, Hoshino S, Ishimaru S, Hori G, et al. Clinical study of TTC‐909 (Prostacyclin derivative, clinprost incorporated in lipid microspheres) in patients with peripheral arterial occlusive disease: establishment of optimal dose in multi‐center double‐blind comparative study. Rinsho Iyaku (Journal of Clinical Therapeutics and Medicines) 1997; Vol. 13, issue 21:5595‐622. [CN‐00543608]
Feng 2009 {published data only}
- Feng K, Tan JF, Chen Y. Treatment of lower extremity diabetic atherosclerotic obliterans with shuxuetong injection. Zhongguo Zhong xi yi jie he za zhi Zhongguo Zhongxiyi jiehe zazhi = Chinese Journal of Integrated Traditional and Western Medicine / Zhongguo Zhong xi yi jie he xue hui, Zhongguo Zhong yi yan jiu yuan zhu ban 2009; Vol. 29, issue 3:255‐7. [CN‐00749881] [PubMed]
Fonseca 1991 {published and unpublished data}
- Fonseca V, Dandona P, Belcher G. Treatment of ischaemic ulceration or rest pain with intravenous Iloprost: a double‐blind placebo controlled study. Diabetes 40 1991;Suppl 1:522A. [Google Scholar]
Guan 2003 {published data only}
- Guan H, Wang Y, Zhang B, Ye W, Fu W, Liang W, et al. Comparison of beraprost and ticlopidine in Chinese patients with chronic peripheral arterial occlusion: a multicenter, single blind, randomized, controlled study. Current Therapeutic Research 2003;64(7):488‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heidrich 1991 {published data only}
- Heidrich H, Brodel C, Meuche C, Hellman M, Ranft J. Controlled long‐term blood pressure measurement with intravenous prostaglandin E1 infusion [Kontrollierte blutdrucklangzeitmessung unter intravenoser prostaglandin‐E1‐infusion]. VASA 1991;20(1):13‐6. [PubMed] [Google Scholar]
Menzoian 1995 {published and unpublished data}
- Menzoian J. Alprostadil in the treatment of patients with severe peripheral arterial occlusive disease (PAOD) not amenable to surgery: results of a randomized, placebo‐controlled multicenter study. International Angiology 1995;14(Suppl 1):104. [Google Scholar]
Mingardi 1993 {published and unpublished data}
- Mingardi R, Rumor F, Strazzabosco M, et al. Medical therapy in diabetic angiopathy with inoperable critical limb ischemia (CLI) ‐ a randomized, between‐patients study to compare the effects of iloprost (I) vs pentoxyfilline (P). Thrombosis and Haemostasis 1993;69(6):1243. [Google Scholar]
NCT00059644 {published data only}
- Efficacy/safety of ecraprost in lipid emulsion for treatment of critical leg ischemia due to peripheral arterial disease [A double‐blind, randomized, placebo‐controlled study to assess the efficacy and safety of CirculaseTM for the treatment of critical leg ischemia]. clinicaltrials.gov/ct2/show/NCT00059644. Date first received: 29 April 2003.
Nizankowski 1985 {published data only}
Ohtake 2014 {published data only}
- Ohtake T, Sato M, Nakazawa R, Kondoh M, Kobayashi S. Effect of beraprost sodium (PGI2 analogue) on peripheral arterial disease (PAD) in patients on hemodialysis: result from a multicenter randomized prospective interventional study. Nephrology Dialysis Transplantation Plus 2010;3:iii487. [Google Scholar]
- Ohtake T, Sato M, Nakazawa R, Kondoh M, Miyaji T, Moriya H, et al. Randomized pilot trial between prostaglandin I2 analog and anti‐platelet drugs on peripheral arterial disease in hemodialysis patients. Therapeutic Apheresis and Dialysis 2014; Vol. 18, issue 1:1‐8. [DOI: 10.1111/1744-9987.12051; CN‐00978202] [DOI] [PubMed]
Petronella 2004 {published data only}
- Petronella P, Freda F, Nunziata L, Antropoli M, Manganiello A, Cutolo PP, et al. Prostaglandin E1 versus lumbar sympathectomy in the treatment of peripheral arterial occlusive disease: randomised study of 86 patients. Nutrition, Metabolism, and Cardiovascular Diseases 2004;14(4):186‐92. [DOI] [PubMed] [Google Scholar]
Sakaguchi‐Shukichi 1990 {published data only}
- Sakaguchi‐Shukichi. Evaluation of the efficacy of beraprost sodium (PGI2 analogue) in chronic arterial occlusion: a double‐blind comparative study with ticlopidine hydrochloride. Rinsho to Kenkyu 1990; Vol. 67:575‐84. [CN‐00740416]
Schwarz 1995 {published data only}
- Schwarz E, Ratti R, Durante F, Buskermolen M, Brighenti G. Iloprost vs nitrates and aspirin in the treatment of geriatric patients suffering of peripheral arterial obliterative disease. International Angiology 1995;14(Suppl 1):201. [Google Scholar]
Sert 2008 {published data only}
- Sert M, Soydas B, Aikimbaev K, Tetiker T. Effects of iloprost (a prostacyclin analogue) on the endothelial dysfunction and foot ulcers in diabetic patients with peripheral arterial disease. International Journal of Diabetes and Metabolism 2008; Vol. 16, issue 1:7‐11. [CN‐00754583]
Weiss 1991 {published data only}
- Weiss T, Griesshaber J, Rogatti W, Kistner O, Hsu E, Jansen T, et al. Effect of intra‐arterial and intravenous PGE1 infusions on transcutaneous oxygen pressure in patients with critical ischemia of the extremities. VASA Supplementum 1991; Vol. 33:341‐2. [CN‐00081779] [PubMed]
Ylitalo 1990 {published data only}
- Ylitalo P, Kaukinen S, Reinikainen P, Salenius JP, Vapaatalo H. A randomized, double‐blind, crossover comparison of iloprost with dextran in patients with peripheral arterial occlusive disease. International Journal of Clinical Pharmacology, Therapy, and Toxicology 1990; Vol. 28, issue 5:197‐204. [CN‐00068745] [PubMed]
Additional references
Abu 2015
- Abu Dabrh AM, Steffen MW, Undavalli C, Asi N, Wang Z, Elamin MB, et al. The natural history of untreated severe or critical limb ischemia. Journal of Vascular Surgery 2015;62(6):1642‐51.e3. [PUBMED: 26391460] [DOI] [PubMed] [Google Scholar]
Albers 1992
- Albers M, Fratezi AC, Luccia N. Assessment of quality of life of patients with severe ischemia as a result of infrainguinal arterial occlusive disease. Journal of Vascular Surgery 1992;16(1):54‐9. [PubMed] [Google Scholar]
Anonymous 1991
- Anonymous. Second European Consensus Document on chronic critical leg ischemia. Circulation 1991;84(4 Suppl):1‐26. [PubMed] [Google Scholar]
Aquino 2001
- Aquino R, Johnnides C, Makaroun M, Whittle JC, Muluk VS, Kelley ME, et al. Natural history of claudication: long‐term serial follow‐up study of 1244 claudicants. Journal of Vascular Surgery 2001;34(6):962‐70. [PUBMED: 11743546] [DOI] [PubMed] [Google Scholar]
Becker 2011
- Becker F, Robert‐Ebadi H, Ricco JB, Setacci C, Cao P, Donato G, et al. Chapter I: Definitions, epidemiology, clinical presentation and prognosis. European Journal of Vascular and Endovascular Surgery 2011;42 (Suppl 2):S4‐12. [PUBMED: 22172472] [DOI] [PubMed] [Google Scholar]
Brodszky 2011
- Brodszky V, Farkas K, Jarai Z, Landi A, Pecsvarady Z, Baji P, et al. Effectiveness of prostanoids in patients with critical leg ischemia [Prosztanoidok hatasossaga kritikus vegtagischaemiaban.]. Orvosi Hetilap 2011;152(51):2047‐55. [PUBMED: 22130202] [DOI] [PubMed] [Google Scholar]
Cacione 2016
- Cacione DG, Baptista‐Silva JC, Macedo CR. Pharmacological treatment for Buerger's disease. Cochrane Database of Systematic Reviews 2016, Issue 2. [DOI: 10.1002/14651858.CD011033.pub3] [DOI] [PubMed] [Google Scholar]
Creutzig 2004
- Creutzig A, Lehmacher W, Elze M. Meta‐analysis of randomised controlled prostaglandin E1 studies in peripheral arterial occlusive disease stages III and IV. Vasa 2004;33(3):137‐44. [DOI] [PubMed] [Google Scholar]
Criqui 2015
- Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circulation Research 2015;116(9):1509‐26. [PUBMED: 25908725] [DOI] [PubMed] [Google Scholar]
Diehm 1987
- Diehm C, Stammler F, Hubsch‐Muller C, Eckstein HH, Simini B. Clinical effects of intravenously administered prostaglandin E1 in patients with rest pain due to peripheral obliterative arterial disease (POAD) ‐ a preliminary report on a placebo‐controlled double‐blind study. Vasa Supplementum 1987;17:52‐6. [PubMed] [Google Scholar]
Dormandy 1996
- Dormandy JA. Prostanoid drug therapy for peripheral arterial occlusive disease ‐ the European experience. Vascular Medicine 1996;1(2):155‐8. [DOI] [PubMed] [Google Scholar]
Dormandy 2000
- Dormandy JA, Rutherford RB. Management of peripheral arterial disease (PAD). TASC Working Group. TransAtlantic Inter‐Society Consensus (TASC). Journal of Vascular Surgery 2000;31(1 Pt 2):S1‐S296. [PubMed] [Google Scholar]
Dua 2016
- Dua A, Lee CJ. Epidemiology of peripheral arterial disease and critical limb ischemia. Techniques in Vascular and Interventional Radiology 2016;19(2):91‐5. [PUBMED: 27423989] [DOI] [PubMed] [Google Scholar]
Gerhard‐Herman 2016
- Gerhard‐Herman MD, Gornik HL, Barrett C, Barshes NR, Corriere MA, Drachman DE, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2016;69(11):1465‐508. [PUBMED: 27840333] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT
- GRADEpro GDT: GRADEpro Guideline Development Tool [Computer program]. Version accessed May 2017. Hamilton (ON): McMaster University, 2015 (developed by Evidence Prime, Inc.). Available from www.gradepro.org.
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Schünemann HJ. GRADE: what is "quality of evidence" and why is it important to clinicians?. BMJ (Clinical Research Ed.) 2008;336(7651):995‐8. [DOI: 10.1136/bmj.39490.551019.BE] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [DOI: 10.1016/j.jclinepi.2010.04.026] [DOI] [PubMed] [Google Scholar]
Hammer 2013
- Hammer A, Steiner S. Gene therapy for therapeutic angiogenesis in peripheral arterial disease ‐ a systematic review and meta‐analysis of randomized, controlled trials. VASA Zeitschrift fur Gefasskrankheiten. Journal for Vascular Diseases 2013;42(5):331‐9. [PUBMED: 23989068] [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hirsch 2006
- Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Greager MA, Halperin JL, et al. ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease. Circulation 2006;113(11):463‐654. [DOI] [PubMed] [Google Scholar]
Howard 2015
- Howard DP, Banerjee A, Fairhead JF, Hands L, Silver LE, Rothwell PM. Population‐based study of incidence, risk factors, outcome, and prognosis of ischemic peripheral arterial events: implications for prevention. Circulation 2015;132(19):1805‐15. [PUBMED: 26350058] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hylton 2014
- Hylton JR, Smith CA, Li CS, Pevec WC. Octogenarians develop infrapopliteal arterial occlusive disease in the absence of traditional risk factors. Annals of Vascular Surgery 2014;28(7):1712‐8. [PUBMED: 24858583] [DOI] [PubMed] [Google Scholar]
ICAI Group 1999
- The ICAI Group. Prostanoids for chronic critical leg ischemia. A randomized, controlled, open‐label trial with prostaglandin E1. Annals of Internal Medicine 1999;130(5):412‐21. [PubMed] [Google Scholar]
Jelnes 1986
- Jelnes R, Gaardsting O, Hougaard Jensen K, Backgaard N, Tonnesen KH, Schroeder T, et al. Fate in intermittent claudication: outcome and risk factors. British Medical Journal (Clinical Research Ed.) 1986;293(6555):1137‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jude 2001
- Jude EB, Oyibo SO, Chalmers N, Boulton AJ. Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care 2001;24(8):1433‐7. [PUBMED: 11473082] [DOI] [PubMed] [Google Scholar]
Loosemore 1994
- Loosemore TM, Chalmers TC, Dormandy JA. A meta‐analysis of randomized placebo control trials in Fontaine stages III and IV peripheral occlusive arterial disease. International Angiology 1994;13(2):133‐42. [PubMed] [Google Scholar]
Malyar 2016
- Malyar NM, Freisinger E, Meyborg M, Luders F, Furstenberg T, Kroger K, et al. Low rates of revascularization and high in‐hospital mortality in patients with ischemic lower limb amputation: morbidity and mortality of ischemic amputation. Angiology 2016;67(9):860‐9. [PUBMED: 26764367] [DOI] [PubMed] [Google Scholar]
Melillo 2016
- Melillo E, Micheletti L, Nuti M, Dell'Omo G, Berchiolli R, Adami D, et al. Long‐term clinical outcomes in critical limb ischemia ‐ a retrospective study of 181 patients. European Review for Medical and Pharmacological Sciences 2016;20(3):502‐8. [PUBMED: 26914126] [PubMed] [Google Scholar]
Miao 2014
- Miao YL, Wu W, Li BW, Fang WW, Liu Y, Li L, et al. Clinical effectiveness of gene therapy on critical limb ischemia: a meta‐analysis of 5 randomized controlled clinical trials. Vascular and Endovascular Surgery 2014;48(5‐6):372‐7. [PUBMED: 24951292] [DOI] [PubMed] [Google Scholar]
Moazzami 2014
- Moazzami K, Moazzami B, Roohi A, Nedjat S, Dolmatova E. Local intramuscular transplantation of autologous mononuclear cells for critical lower limb ischaemia. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD008347.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moxey 2010
- Moxey PW, Hofman D, Hinchliffe RJ, Jones K, Thompson MM, Holt PJ. Epidemiological study of lower limb amputation in England between 2003 and 2008. British Journal of Surgery 2010;97(9):1348‐53. [PUBMED: 20632310] [DOI] [PubMed] [Google Scholar]
Norgren 2007
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG, et al. Inter‐Society Consensus for the Management of Peripheral Arterial Disease (TASC II). European Journal of Vascular and Endovascular Surgery 2007;33 (Suppl 1):S1‐75. [PUBMED: 17140820] [DOI] [PubMed] [Google Scholar]
Ostchega 2007
- Ostchega Y, Paulose‐Ram R, Dillon CF, Gu Q, Hughes JP. Prevalence of peripheral arterial disease and risk factors in persons aged 60 and older: data from the National Health and Nutrition Examination Survey 1999‐2004. Journal of the American Geriatrics Society 2007;55(4):583‐9. [PUBMED: 17397438] [DOI] [PubMed] [Google Scholar]
Patel 2015
- Patel MR, Conte MS, Cutlip DE, Dib N, Geraghty P, Gray W, et al. Evaluation and treatment of patients with lower extremity peripheral artery disease: consensus definitions from Peripheral Academic Research Consortium (PARC). Journal of the American College of Cardiology 2015;65(9):931‐41. [PUBMED: 25744011] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Robertson 2013
- Robertson L, Andras A. Prostanoids for intermittent claudication. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD000986.pub3] [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Sen 2011
- Sen I, Agarwal S, Tharyan P. Lumbar sympathectomy versus prostanoids for critical limb ischaemia due to non‐reconstructable peripheral arterial disease. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD009366] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sigvant 2016
- Sigvant B, Lundin F, Wahlberg E. The risk of disease progression in peripheral arterial disease is higher than expected: a meta‐analysis of mortality and disease progression in peripheral arterial disease. European Journal of Vascular and Endovascular Surgery 2016;51(3):395‐403. [PUBMED: 26777541] [DOI] [PubMed] [Google Scholar]
Sprengers 2010
- Sprengers RW, Teraa M, Moll FL, Wit GA, Graaf Y, Verhaar MC. Quality of life in patients with no‐option critical limb ischemia underlines the need for new effective treatment. Journal of Vascular Surgery 2010;52(4):843‐9, 849.e1. [PUBMED: 20598482] [DOI] [PubMed] [Google Scholar]
Suckow 2012
- Suckow BD, Goodney PP, Cambria RA, Bertges DJ, Eldrup‐Jorgensen J, Indes JE, et al. Predicting functional status following amputation after lower extremity bypass. Annals of Vascular Surgery 2012;26(1):67‐78. [PUBMED: 22176876] [DOI] [PMC free article] [PubMed] [Google Scholar]
Suckow 2015
- Suckow BD, Goodney PP, Nolan BW, Veeraswamy RK, Gallagher P, Cronenwett JL, et al. Domains that determine quality of life in vascular amputees. Annals of Vascular Surgery 2015;29(4):722‐30. [PUBMED: 25725279] [DOI] [PMC free article] [PubMed] [Google Scholar]
Teraa 2016
- Teraa M, Conte MS, Moll FL, Verhaar MC. Critical limb ischemia: current trends and future directions. Journal of the American Heart Association 2016;5(2):e002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Trubestein 1989
- Trubestein G, Bary S, Breddin K, Diehm C, Gruss JD, Heinrich H, et al. Intravenous prostaglandin E1 versus pentoxifylline therapy in chronic arterial occlusive disease ‐ a controlled randomised multicenter study. Vasa Supplementum 1989;28:44‐9. [PubMed] [Google Scholar]
Ubbink 2013
- Ubbink DT, Vermeulen H. Spinal cord stimulation for non‐reconstructable chronic critical leg ischaemia. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD004001.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Verstraete 1994
- Verstraete M. prostaglandins in critical limb ischemia. Vascular Medicine Review 1994;5(2):93‐5. [Google Scholar]
Vitale 2016
- Vitale V, Monami M, Mannucci E. Prostanoids in patients with peripheral arterial disease: a meta‐analysis of placebo‐controlled randomized clinical trials. Journal of Diabetes and Its Complications 2016;30(1):161‐6. [PUBMED: 26516035] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Ruffolo 2007
- Ruffolo AJ, Romano M, Ciapponi A. Prostanoids for critical limb ischemia. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD006544] [DOI] [PubMed] [Google Scholar]
Ruffolo 2010
- Ruffolo AJ, Romano M, Ciapponi A. Prostanoids for critical limb ischaemia. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD006544.pub2] [DOI] [PubMed] [Google Scholar]