

Abstract
Background
People experiencing psychosis may become aggressive. Antipsychotics, such as aripiprazole in intramuscular form, can be used in such situations.
Objectives
To evaluate the effects of intramuscular aripiprazole in the treatment of psychosis‐induced aggression or agitation (rapid tranquillisation).
Search methods
On 11 December 2014 and 11 April 2017, we searched the Cochrane Schizophrenia Group’s Study‐based Register of Trials which is based on regular searches of CINAHL, BIOSIS, AMED, Embase, PubMed, MEDLINE, PsycINFO, and registries of clinical trials.
Selection criteria
All randomised controlled trials (RCTs) that randomised people with psychosis‐induced aggression or agitation to receive either intramuscular aripiprazole or another intramuscular intervention.
Data collection and analysis
We independently inspected citations and, where possible, abstracts, ordered papers and re‐inspected and quality assessed these. We included studies that met our selection criteria. At least two review authors independently extracted data from the included studies. We chose a fixed‐effect model. We analysed dichotomous data using risk ratio (RR) and the 95% confidence intervals (CI). We analysed continuous data using mean differences (MD) and their CIs. We assessed risk of bias for included studies and used GRADE to create 'Summary of findings' tables.
Main results
Searching found 63 records referring to 21 possible trials. We could only include three studies, all completed over the last decade, with 885 participants, of which 707 were included for quantitative analyses in this systematic review. Due to limited comparisons, small size of trials and a paucity of investigated and reported 'pragmatic' outcomes, evidence was mostly graded as low or very low quality. No trials reported useful data for one of our primary outcomes of tranquil or asleep by 30 minutes. Economic outcomes were also not reported in the trials.
When compared with placebo, fewer people in the aripiprazole group needed additional injections compared to the placebo group (2 RCTs, n = 382, RR 0.69, 95% CI 0.56 to 0.85, very low‐quality evidence). Clinically important improvement in agitation at two hours favoured the aripiprazole group (2 RCTs, n = 382, RR 1.50, 95% CI 1.17 to 1.92, very low‐quality evidence). The numbers of non‐responders after the first injection also favoured aripiprazole (1 RCT, n = 263, RR 0.49, 95% CI 0.34 to 0.71, low‐quality evidence). Although no effect was found, more people in the aripiprazole compared to the placebo group experienced adverse effects (1 RCT, n = 117, RR 1.51, 95% CI 0.93 to 2.46, very low‐quality evidence).
Aripiprazole required more injections compared to haloperidol (2 RCTs, n = 477, RR 1.28, 95% CI 1.00 to 1.63, very low‐quality evidence), with no significant difference in agitation (2 RCTs, n = 477, RR 0.94, 95% CI 0.80 to 1.11, very low‐quality evidence), and similar non‐responders after first injection (1 RCT, n = 360, RR 1.18, 95% CI 0.78 to 1.79, low‐quality evidence). Aripiprazole and haloperidol did not differ when taking into account the overall number of people that experienced at least one adverse effect (1 RCT, n = 113, RR 0.91, 95% CI 0.61 to 1.35, very low‐quality evidence).
Compared to aripiprazole, olanzapine was better at reducing agitation (1 RCT, n = 80, RR 0.77, 95% CI 0.60 to 0.99, low‐quality evidence) and had a more favourable effect on global state change scores (1 RCT, n = 80, MD 0.58, 95% CI 0.01 to 1.15, low‐quality evidence), both at two hours. No differences were found in terms of experiencing at least one adverse effect during the 24 hours after treatment (1 RCT, n = 80, RR 0.75, 95% CI 0.45 to 1.24, very low‐quality evidence). However, participants allocated to aripiprazole experienced less somnolence (1 RCT, n = 80, RR 0.25, 95% CI 0.08 to 0.82, low‐quality evidence).
Authors' conclusions
The available evidence is of poor quality but there is some evidence aripiprazole is effective compared to placebo and haloperidol, but not when compared to olanzapine. However, considering that evidence comes from only three studies, caution is required in generalising these results to real‐world practice. This review firmly highlights the need for more high‐quality trials on intramuscular aripiprazole in the management of people with acute aggression or agitation.
Plain language summary
How effective is aripiprazole for calming people who are aggressive or agitated due to psychosis?
Background People with psychosis may experience hearing voices (hallucinations) or abnormal thoughts (delusions) which can make them frightened, distressed and agitated (restless, excitable or irritable). Experiencing such emotions can sometimes result in behaviour that is aggressive or violent. This poses a challenge and dilemma for mental health professionals who have to diagnose and, often quickly, give the best available treatment to prevent those who are aggressive, harming themselves or others.
Aripiprazole is a medication that can be used to treat psychosis and also calm people who are aggressive or agitated due to psychosis. It can be taken by mouth or by injection (intramuscular). However, aripiprazole can also cause unpleasant side effects such as headaches, upset stomach, and excessive sleepiness or drowsiness.
This review looks for evidence from randomised controlled trials that assesses the effectiveness of intramuscular aripiprazole for people who are agitated and aggressive as a result of having psychosis.
Searching The Information Specialist of the Cochrane Schizophrenia group ran an electronic search in 2014 and in 2017 for studies randomising adults with aggression due to psychosis to receive either injections of aripiprazole or injections of a placebo (dummy treatment) or injections of another antipsychotic. The search found 63 relevant records, referring to 21 trials. Review authors screened these records for inclusion or exclusion.
Main results Only three studies could be included. Evidence is limited due to the small number of trials and poor quality of data reported. Fewer people receiving aripiprazole required more injections to become calm than those receiving placebo or haloperidol. Overall, aripiprazole caused a similar number of adverse effects to placebo or haloperidol. Compared to olanzapine, aripiprazole was less effective at calming people but caused less sleepiness or drowsiness
Conclusions Some evidence is available, but is of poor quality, and it is difficult to make conclusions about aripiprazole's effectiveness from such data. Health professionals and people with mental health problems are therefore left without clear guidance concerning use of aripiprazole as a rapid tranquilliser. More research is needed to help people consider which medication is better at calming people down, has fewer adverse effects, and works quickly and rapidly.
Summary of findings
Background
Description of the condition
Aggression and agitation are psychiatric emergencies that can often be associated with psychosis, substance misuse or epilepsy (Tardiff 1982). Aggression is described as hostile, injurious, or destructive behaviour, and can occur in about 3% of psychiatric outpatients (Tardiff 1985) and in up to 7% to 10% of inpatient settings. Agitation is described as a state of troubled mind or feelings (Makins 1994) and is characterised by restlessness, excitability and irritability, and for some people, this can result in verbal and physical aggressive behaviour (Mohr 2005). As is often the case, patients experiencing these episodes find new environments stressful and become aggressive (Ferracuti 1993). Aggression and agitation within the psychiatric setting imposes a significant challenge to clinicians who, while attempting to make an accurate diagnosis and formulation (Schleifer 2011), have to intervene quickly in order to manage the risk that the service user may present to themselves, other service users and staff (NICE 2015).
There have been numerous national and international guidelines that created protocols for managing people with aggression and violence (APA 2006; CPA 2005; NICE 2015). Some evidence from these guidelines may be conflicting in nature. For example, the use of zuclopenthixol acetate is not recommended by NICE, but recommended by Australian and Canadian guidelines. This is not surprising as there has only been one trial of zuclopenthixol acetate in this area and the small sample size from this trial could lead to varied interpretations. An individual requiring rapid tranquillisation is frequently unable or unwilling to consent and the treatment is being provided in his or her best interest.
In the UK, NICE guidelines recommend that a comprehensive risk assessment is the first step in management of aggression and non‐pharmacological methods recommended for use include de‐escalation, physical restraints and seclusion. Where rapid tranquillisation through oral therapy is refused, is not indicated by previous clinical response, is not a proportionate response, or is ineffective, administration of an intramuscular combination of haloperidol and promethazine or intramuscular lorazepam is recommended (NICE 2015).
In the USA, the first intervention involves staff members talking to the patient in an attempt to calm him or her. Attempts to restrain the patient should be done only by a team trained in safe restraint procedures to minimise risk of harm to patients or staff. Antipsychotics and benzodiazepines are often helpful in reducing the patient’s level of agitation. If the patient will take oral medication, rapidly dissolving forms of olanzapine and risperidone can be used for quicker effect and to reduce non adherence. If a patient refuses oral medication, most states allow for emergency administration despite the patient’s objection. Short‐acting parenteral formulations of first‐ and second‐generation antipsychotic agents (e.g. haloperidol, ziprasidone, and olanzapine), with or without a parenteral benzodiazepine (e.g. lorazepam), are available for emergency administration in acutely agitated patients (APA 2006).
There have been only a handful of surveys looking at the prevalence and practices in managing acute agitation. Cunane 1994 found that in the UK, clinicians preferred using chlorpromazine, whereas Binder 1999 found that in the USA, clinicians preferred a combination of haloperidol and benzodiazepines. In a more recent survey, Huf 2002a found that Brazilian psychiatrists preferred a combination of haloperidol and promethazine. Surveys of this nature are notoriously difficult to conduct specially in resource‐poor settings, which partly explains the paucity of such surveys from low‐income countries. There have been a few good quality pragmatic trials comparing various interventions such as haloperidol, olanzapine, promethazine, benzodiazepines including lorazepam and midazolam, and other medications for managing acute agitation (Alexander 2004; Huf 2002b; Raveendran 2007).
Description of the intervention
Aripiprazole is a quinolinone derivative, 7‐[4‐[4‐(2,3‐dichlorophenyl)‐1‐piperazinyl]butoxy]‐3,4‐dihydrocarbostyril (Kikuchi 1995; Figure 1). It is available as oral tablets, orally disintegrating tablets, oral solution and intramuscular injections. The intramuscular preparations are available in single‐dose vials as a ready‐to‐use, 9.75 mg/1.3 mL (7.5 mg/mL) clear, colourless, sterile, aqueous solution. Inactive ingredients for this solution include 150 mg/mL of sulfobutylethercyclodextrin (SBECD), tartaric acid, sodium hydroxide and water for injection (Abilify 2017). Intramuscular aripiprazole seems to have two medians for peak plasma concentrations, one at one hour and the second at three hours. It has 100% bioavailability and the pharmacokinetics is linear over a dose range of 1 mg to 45 mg. It is likely to be eliminated by the liver using the P450 system of isoenzymes (CYP2D6 and CYP3A4, Abilify 2017).
1.
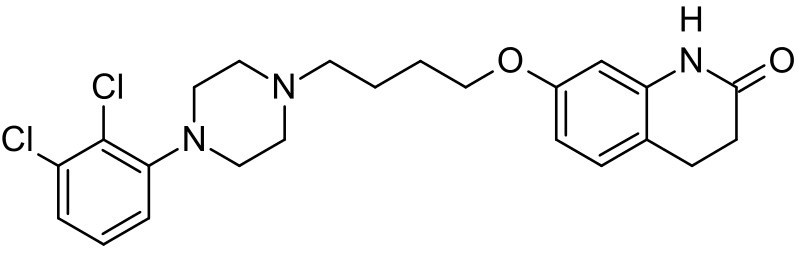
Aripiprazole structure
Aripiprazole solution, for injection, is licensed for rapid control of agitation and disturbed behaviours in adults when oral therapy is not appropriate. The dose for adults is 9.75 mg (1.3 mL) as a single intramuscular injection initially with an effective dose range of 5.25 mg to 15 mg as a single injection. Second injections can be administered two hours after the first, No more than three injections should be given in any 24‐hour period.
How the intervention might work
According to treatment guidelines, aripiprazole is classified amongst the second‐generation antipsychotics with amisulpride, clozapine, olanzapine, quetiapine, risperidone, sertindole, ziprasidone and zotepine. However, aripiprazole has been described as the prototype of a new and third generation of antipsychotics, the so called 'dopamine‐serotonin system stabilisers' (McGavin 2002; Swainston Harrison 2004).
It is reported to exert its antipsychotic effects by acting as a partial agonist at dopamine D2 and 5‐HT1A receptors and antagonist activity at 5‐HT2A receptors (Shapiro 2003). It has been postulated that through dopamine and serotonin system stabilisation, a partial D2 agonist would be able to act as an antagonist in pathways where an abundance of dopamine was leading to 'psychosis', yet it would stimulate receptors as an agonist at sites in which low‐dopaminergic tone would produce adverse effects.
Aripiprazole, however, also has an affinity to other receptors including D3, D4, 5‐HT2c, 5HT7, alpha‐1 adrenergic and histamine receptors, which could be related to some adverse effects (such as headache, gastrointestinal upset, headedness, somnolence; FDA 2004). The recommended target dose for aripiprazole is 10 mg to 15 mg per day (dose range 10 mg to 30 mg/day). Phase III trials were initially conducted in Japan in 1995 and the drug was granted Approved Status by the FDA (USA) on the 15 November 2002 for the treatment of schizophrenia. Aripiprazole has since been licensed in most countries worldwide.
Why it is important to do this review
Aripiprazole has been around since 2002 and its use has been gradually increasing in the UK. Recent data state that for aripiprazole the spending on NHS prescriptions in England was about £6m in 2008, about £10m in 2010, rising to £15m in 2013 (NHS 2014). An intramuscular formulation of aripiprazole has recently been licensed for use in treatment of acute agitation. The increase in prescribing costs of oral aripiprazole seems to be in keeping with the general increase in atypical antipsychotic prescribing in the UK. If previous trends are to go by, we could expect a general increase in cost of intramuscular aripiprazole use to the UK's NHS over the next few years. In terms of raw cost data, there are considerable differences between the older generation and newer generation of antipsychotics. For example, in the UK a vial of haloperidol 5 mg/mL costs 30 pence, a vial of promethazine 25 mg/mL 70 pence, whereas, a vial of olanzapine 5 mg/mL about £3.48, and finally a vial of aripiprazole 9.75 mg costs £3.43 in the UK (BNF 2014).
With the increasing number of the antipsychotic medications that are available for intramuscular use and treatment of acute agitation, there is an urgent need to systematically review and evaluate the effects of intramuscular aripiprazole. This is one of a series of similar reviews (Table 4).
1. Other relevant Cochrane reviews.
| Focus of review | Reference |
| Completed and maintained reviews | |
| 'As required' medication regimens for seriously mentally ill people in hospital | Douglas‐Hall 2015 |
| Benzodiazepines for psychosis‐induced aggression or agitation | Gillies 2015 |
| Chlorpromazine for psychosis‐induced aggression or agitation | Ahmed 2010 |
| Clotiapine for acute psychotic illnesses | Berk 2004 |
| Containment strategies for people with serious mental illness | Muralidharan 2006 |
| De‐escalation techniques for psychosis‐induced aggression | Du 2017 |
| Droperidol for acute psychosis | Khokhar 2016 |
| Haloperidol for long‐term aggression in psychosis | Khushu 2016 |
| Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation) | Ostinelli 2017 |
| Haloperidol plus promethazine for psychosis‐induced aggression | Huf 2016 |
| Olanzapine IM or velotab for acutely disturbed/agitated people with suspected serious mental illnesses | Belgamwar 2005 |
| Seclusion and restraint for serious mental illnesses | Sailas 2000 |
| Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses | Jayakody 2012 |
| Reviews in the process of being completed | |
| Risperidone for psychosis‐induced aggression or agitation | Ahmed 2011 |
| Loxapine inhaler for psychosis‐induced aggression | Vangala 2012 |
| Clozapine for people with psychosis‐induced aggression or agitation | Toal 2012 |
| Quetiapine for psychosis‐induced aggression or agitation | Wilkie 2012 |
Objectives
To evaluate the effects of intramuscular aripiprazole in the treatment of psychosis‐induced aggression or agitation (rapid tranquillisation).
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised control trials. If a trial was described as ’double‐blind’ but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion resulted in statistically significant differences, we did not add the data from these lower‐quality studies to the results of the better trials, but presented such data within a subcategory. We planned to exclude quasi‐randomised studies, such as those allocating by alternate days of the week or where allocation was undertaken on surname.
Types of participants
People exhibiting aggression or agitation (or both) thought to be due to psychosis, regardless of age and sex. Should studies have involved people with other diagnoses, such as drug or alcohol intoxication, organic problems including dementia, non‐psychotic mental illnesses or learning disabilities, we would have included these as long as the majority of participants (> 50%) were experiencing psychosis.
Types of interventions
1. Aripiprazole
Given alone, intramuscularly in any dose.
versus:
A. Other antipsychotic medications
Given alone, intramuscularly in any dose.
B. Placebo
Active or non‐active, intramuscular, in any dose.
Types of outcome measures
Outcomes grouped by time: by 30 minutes, up to two hours, up to four hours, up to 24 hours, and over 24 hours.
We endeavoured to report binary outcomes recording clear and clinically meaningful degrees of change (e.g. global impression of much improved, or more than 50% improvement on a rating scale ‐ as defined within the trials) before any others. Thereafter, we listed other binary outcomes and then those that were continuous.
Primary outcomes
1. Tranquil or asleep
1.1 Tranquil or asleep ‐ by up to 30 minutes *
2. Repeated need for rapid tranquillisation
Secondary outcomes
1. Tranquillisation or asleep
1.1 Tranquil or asleep ‐ over 30 minutes * 1.2 Time to tranquillisation/sleep 1.3 Time to tranquillisation 1.4 Time to sleep
2. Specific behaviours
2.1 Self‐harm, including suicide 2.2 Injury to others 2.3 Agitation 2.3.1 Another episode of agitation by 24 hours 2.3.2 Clinically important change in agitation * 2.3.3 Any change in agitation 2.3.4 Average endpoint score ‐ agitation scales 2.3.5 Average change score ‐ agitation scales 2.4 Aggression 2.4.1 Another episode of aggression by 24 hours 2.4.2 Clinically important change in aggression * 2.4.3 Any change in aggression 2.4.4 Average endpoint score ‐ aggression scales 2.4.5 Average change score ‐ aggression scales
3. Global state
3.1 Overall improvement 3.2 Use of additional medication 3.3 Use of restraints/seclusion 3.4 Relapse ‐ as defined by each study 3.5 Recurrence of violent incidents 3.6 Needing extra visits from the doctor 3.7 Refusing oral medication
4. Service use
4.1 Duration of hospital stay 4.2 Re‐admission 4.3 Clinically important engagement with services 4.4 Any engagement with services 4.5 Average endpoint engagement score 4.6 Average change in engagement scores
5. Mental state
5.1 Clinically important change in general mental state 5.2 Any change in general mental state 5.3 Average endpoint general mental state score 5.4 Average change in general mental state scores
6. Adverse effects
6.1 Death 6.2 Any general adverse effects 6.3 Any serious specific adverse effects 6.4 Average endpoint general adverse effect score 6.5 Average change in general adverse effect scores 6.6 Clinically important change in specific adverse effects 6.7 Any change in specific adverse effects 6.8 Average endpoint specific adverse effects 6.9 Average change in specific adverse effects
7. Leaving the study early
7.1 For specific reasons 7.2 For general reasons
8. Satisfaction with treatment
8.1 Recipient of treatment satisfied with treatment 8.2 Recipient of treatment average satisfaction score 8.3 Recipient of treatment average change in satisfaction scores 8.4 Informal treatment provider satisfied with treatment 8.5 Informal treatment providers' average satisfaction score 8.6 Informal treatment providers' average change in satisfaction scores 8.7 Professional providers satisfied with treatment 8.8 Professional providers' average satisfaction score 8.9 Professional providers' average change in satisfaction scores
9. Acceptance of treatment
9.1 Accepting treatment 9.2 Average endpoint acceptance score 9.3 Average change in acceptance scores
10. Quality of life
10.1 Clinically important change in quality of life * 10.2 Any change in quality of life * 10.3 Average endpoint quality of life score 10.4 Average change in quality of life score 10.5 Clinically important change in specific aspects of quality of life * 10.6 Any change in specific aspects of quality of life * 10.7 Average endpoint specific aspects of quality of life 10.8 Average change in specific aspects of quality of life
10. Economic outcomes
10.1 Direct costs 10.2 Indirect costs
Outcomes used for 'Summary of findings' tables
We used the GRADE approach to interpret findings (Schünemann 2011) and used the GRADEpro GDT web application to export data from this review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision‐making. We selected the following main outcomes for inclusion in the 'Summary of findings' tables.
1. Tranquillisation or asleep ‐ by 30 minutes. 2. Repeated need for rapid tranquillisation ‐ within 24 hours. 3. Specific behaviours ‐ agitation or aggression. 4. Global state ‐ clinically important overall improvement *. 5. Adverse effects ‐ any serious, specific adverse effects. 6. Economic outcomes
Search methods for identification of studies
No language restriction was applied within the limitations of the search tools.
Electronic searches
Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On 11 December 2014 and 11 April 2017, the Information Specialist searched the register using the following search strategy:
(*Aripiprazole*) in Intervention AND (*Aggress* or *Violent* or *Agitation* or *Tranq*) in Healthcare Condition Fields of Study
In such study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
This register is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
Where necessary, we attempted to contact the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
For the 2014 search, review authors SJ, and KS independently inspected citations from the searches and identified relevant abstracts. A random 20% sample was independently re‐inspected by SS to ensure reliability. Where disputes arose, the full report was acquired for more detailed scrutiny. SJ and KS obtained and inspected full reports of the abstracts meeting the review criteria. If it had not been possible to resolve disagreement by discussion, we would have attempted to contact the authors of the study for clarification.
For the 2017 search, review author EGO independently inspected citations and identified relevant abstracts. EGO also re‐inspected the results of the 2014 search.
Data extraction and management
1. Extraction
For the 2014 search, review author JS extracted data from all included studies. In addition, to ensure reliability, review authors KS and SS independently extracted data from a random sample of these studies, comprising 10% of the total. We would have discussed any disagreement, documented decisions and, if necessary, contacted authors of studies. We extracted data presented only in graphs and figures whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
For the 2017 search, review author EGO extracted data from the new included study and re‐inspected the data extraction from previously included studies.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument had not been written or modified by one of the trialists for that particular trial; c) the instrument should be a global assessment of an area of functioning and not sub‐scores which are not, in themselves, validated or shown to be reliable, however there are exceptions; we included sub‐scores from mental state scales measuring positive and negative symptoms of schizophrenia.
Ideally, the measuring instrument should either be i. self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, therefore, in Description of studies we noted if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis and we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Deeks 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion:
a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors;
b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996; Higgins 2011);
c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐ S min), where S is the mean score and ’S min’ is the minimum score.
Endpoint scores on scales often have a finite start and end point and these rules can be applied. Skewed data pose less of a problem when looking at means if the sample size is large (> 200) and we entered these into the syntheses. We planned to present skewed data from studies of less than 200 participants in ‘Additional tables’ rather than in analyses.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. These data were entered into analyses.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for aripiprazole is effective for psychosis‐induced aggression or agitation. Where keeping to this makes it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we reported data where the left of the line indicates an unfavourable outcome. This is noted in the relevant graphs.
Assessment of risk of bias in included studies
Review authors EGO and JS assessed risk of bias by using the criteria described in the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011a) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. We noted the level of risk of bias in both the text of the review and in the 'Summary of findings' tables.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial outcome/number needed to treat for an additional harmful outcome(NNTB/NNTH) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' tables, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (SMD). However, if scales of very considerable similarity had been used, we would have presumed there was a small difference in measurement, and calculated the effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering was not been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We would have attempted to contact the first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
If clustering had been incorporated into the analysis of primary studies, we would have reported these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
Cochrane Schizophrenia have sought statistical advice and have been advised that the binary data presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported, it is assumed to be 0.1 (Ukoumunne 1999). Where cluster studies are appropriately analysed taking into account ICCs, and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we would have only used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, where relevant, we presented the additional treatment arms in separate comparisons. If data were binary, we simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011). If the additional treatment arms had not been relevant, we would not have reproduced such data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses (except for the outcome 'leaving the study early'). When more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' tables by down‐grading quality.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis, ITT). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, we used the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when 'completer' data only were compared to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50% and 'completer'‐data were reported, we reproduced these.
3.2 Standard deviations (SDs)
If SDs were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals were available for group means, and either 'P' value or 't' values were available for differences in mean, we would have calculated them according to the rules described in the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011). When only the SE are reported, SDs can be calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae do not apply, we could calculate the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We would have examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups could be often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, preferably we used the more sophisticated approaches, e.g. we preferred MMRM or multiple‐imputation to LOCF and only presented completer analyses if some kind of ITT data were not available at all; this issue was addressed in the item 'incomplete outcome data' of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We inspected all studies for clearly outlying people or situations which we had not predicted would arise. If such situations or participant groups had arisen, we would have discussed them.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We inspected all studies for clearly outlying methods which we had not predicted would arise. If such methodological outliers had arisen, we would have discussed them.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 statistic alongside the Chi2 'P' value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a CI for I2). We interpreted an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity (Chapter 9. Cochrane Handbook for Systematic Reviews of Interventions) (Deeks 2011). If substantial levels of heterogeneity had been found in the primary outcome, we planned to explore reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Chapter 10. of the Cochrane Handbook for Systematic reviews of Interventions (Sterne 2011).
1. Protocol versus full study
We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report . If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We decided that we would not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In future, where funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model: it puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We did not anticipate any subgroup analyses.
2. Investigation of heterogeneity
If inconsistency was high, we reported it. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successfully removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present such data. If not, we would not pool data, but would discuss this issue. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state. When unanticipated clinical or methodological heterogeneity were obvious, we stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the main outcomes of interest, we we would have included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we would have used all relevant from data these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the main outcomes of interest when we used our assumption compared with 'completer' data only. Where there was a substantial difference, we reported results and discussed them but continued to employ our assumption. Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings of the main outcomes of interest when we used our assumption compared with 'completer' data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (see Assessment of reporting biases). If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analyses.
4. Imputed values
We also would have undertaken a sensitivity analysis to assess the effects of including data from trials if we had used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
All data were synthesised using a fixed‐effect model; however, we also synthesised data for the main outcomes using a random‐effects model to evaluate whether this altered the significance of the results.
Results
Description of studies
For substantive descriptions of studies please see Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification and Characteristics of ongoing studies.
Results of the search
The electronic search (December 2014) yielded 51 citations of potentially eligible studies. We obtained 51 full‐text papers, related to 17 different studies (two included studies, 13 excluded, one awaiting classification, one ongoing study, Figure 2). The last search (April 2017) yielded 25 citations of potentially eligible studies, with 13 of those already cited in the previous search; therefore, we obtained and closely inspected 12 new full‐text papers related to four studies (one study included, three studies excluded, Figure 3).
2.
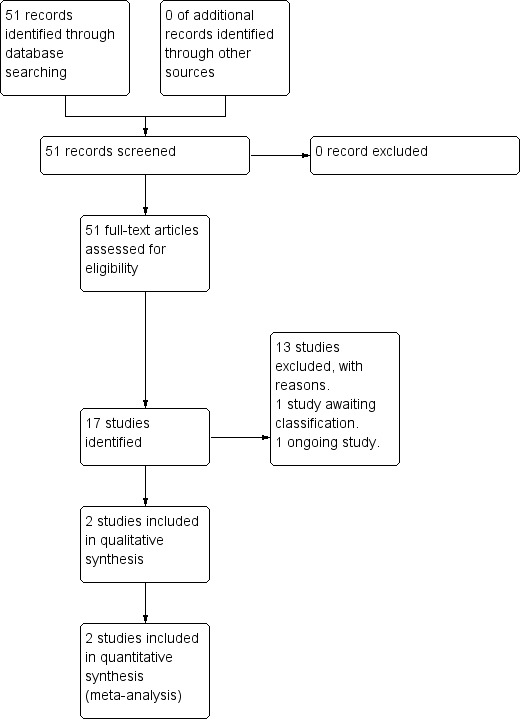
Study flow diagram (2014 search).
3.
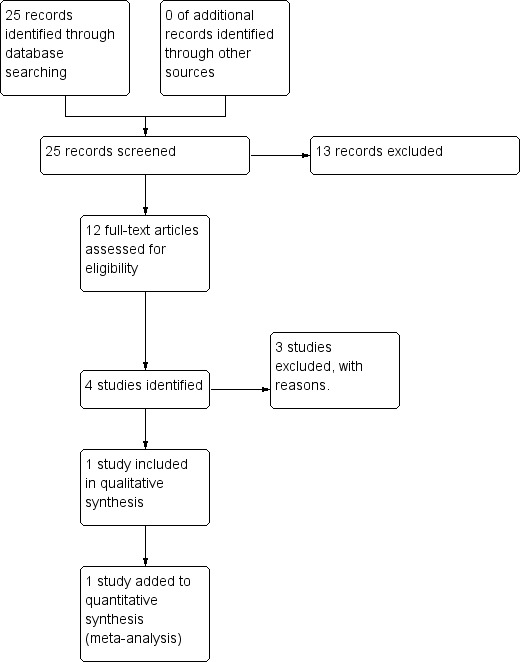
Study flow diagram (2017 search).
Included studies
Details of the three studies included in the review are provided in the Characteristics of included studies table.
1. Length of studies
The duration of the studies, limited to intramuscular (IM) intervention phase, was up to 24 hours (Andrezina 2006, Bristol‐Myers 2005; Kittipeerachon 2016).
2. Participants
A total number of 707 patients were considered for this review; please note that for Bristol‐Myers 2005, the participants reported below refer to those allocated to selected comparisons only (placebo, aripiprazole 9.75 mg, haloperidol).
2.1 Clinical state
Participants in the trials presented with acute aggression or agitation due to psychosis and therefore were deemed by the treating clinician to be appropriate candidates for IM rapid tranquillisation therapy.
2.2 Diagnosis
Overall, 526 (74.4%) of participants had a diagnosis of schizophrenia, 176 (24.9%) of schizoaffective disorder, and 5 (0.7%) of schizophreniform disorder.
2.3 Exclusion
Exclusion criteria in Andrezina 2006 and Bristol‐Myers 2005 studies were similar: psychoactive substance dependence within two months of the study start, significant risk of committing suicide, neurologic condition, psychiatric condition requiring pharmacotherapy other than those included, significant medical condition, known non‐responders to antipsychotic medication; PANSS (Excited Component (PEC) score ≥ 32; in Andrezina 2006, schizophreniform disorder was considered an exclusion criteria, whilst in Bristol‐Myers 2005 it was not.Kittipeerachon 2016, a more pragmatic randomised controlled trial (RCT), had less restrictive exclusion criteria: known allergy to aripiprazole or olanzapine, pregnancy or breastfeeding.
2.4 Age
Age range was of 18‐69 years in Andrezina 2006, 18‐66 years in Bristol‐Myers 2005, and 18‐65 years in Kittipeerachon 2016.
2.5 Sex
Participants of the studies involved in this review were 435 men (61.5%) and 272 women (38.5%).
3. Study size
The total number of participants randomised was 805 (Andrezina 2006, n = 448; Bristol‐Myers 2005, n = 357, of which 179 were considered for comparisons; Kittipeerachon 2016, n = 80).
4. Setting
All the included studies were located in emergency room departments and subsequently in hospital, involving newly admitted patients.
5. Interventions
5.1 Aripiprazole IM
Data in this systematic review relate to the 9.75 mg dose of aripiprazole for all the studies (Andrezina 2006; Bristol‐Myers 2005; Kittipeerachon 2016).
5.2 Placebo IM
Both Andrezina 2006 and Bristol‐Myers 2005 used placebo given intramuscularly.
5.3 Haloperidol IM
Dosage varied from 6.5 mg (Bristol‐Myers 2005) to 7.5 mg (Andrezina 2006).
5.4 Olanzapine IM
Dosage was 10 mg (Kittipeerachon 2016).
6. Outcomes
Binary data concerning the repeated need for tranquillisation were available for Andrezina 2006 and Bristol‐Myers 2005 only, since in Kittipeerachon 2016 repeated administration of intervention drugs was prohibited by the methodological design; with respect to adverse effects, binary data were available for all the included trials. All the trials employed continuous scales to measure other clinical outcomes; in Andrezina 2006 and Bristol‐Myers 2005 continuous measurements for adverse effects were presented as means without standard deviations (SDs), standard error (SE) or confidence intervals (CIs), and therefore were unusable for quantitative analyses. The various rating scales, from which we were able to obtain usable data, are listed below:
6.1 Specific behaviour ‐ Agitation
a. Agitated Behavior Scale (ABS/CABS)
The ABS (Corrigan 1989), is a 14‐item scale used for measuring agitation levels. The ABS was originally designed to monitor agitated behaviour in the recovery period after stroke and there are three factor‐based sub scores: I. aggression; II. disinhibition; III. lability. High scores indicated higher levels of aggression.
b. Agitation calmness Evaluation scale (ACES)
The ACES (Breier 2001), is a single‐item rating scale developed by Eli Lilly and company. On this scale, 1 = 'marked agitation', 4 = 'normal', 9 = 'unable to be aroused'.
c. Positive and Negative Syndrome Scale ‐ Excited Component (PANSS‐EC)
The PANSS‐EC (Montoya 2011) is a five‐item subscale of the PANSS scale (excitement, tension, hostility, uncooperativeness, and poor impulse control). As in the original scale, items are rated from one ('not present') to seven ('extremely severe'). Scores range from five to 35, with mean scores ≥ 20 indicating agitation. A high score indicates high levels of agitation.
6.2 Global state
a. Clinical Global Impression (CGI)
The CGl (Guy 1976) is not a diagnostic tool but rather, enables clinicians to quantify the severity of symptoms of any mental health problem at one point in time. Clinicians are then able to use this to track whether there has been any improvement or worsening of symptoms over time. A seven‐point rating scale is used with high scores indicating increased severity or less recovery.
b. Clinical Global Impression ‐ Severity (CGI‐S)
The CGI‐S (Guy 1976) requires clinicians to consider the severity of a person’s symptoms in relation to the clinicians past experience of people with the same diagnosis. Clinicians then have to give a rating from one (= 'normal') to seven (= 'extremely ill'). High scores indicate increased severity.
c. Clinical Global Impression ‐ Improvement (CGI‐I)
The CGI‐I (Guy 1976) enables clinicians to assess whether a person’s symptoms have improved or worsened following an intervention. Based on the clinicians judgement, a rating on a seven‐point scale is given from one (= 'very much improved') to seven (= 'very much worse'). Therefore, low scores indicate greater improvement.
6.3 Mental state
a. Brief Psychiatric Rating Scale (BPRS)
The Brief Psychiatric Rating Scale was originally developed by Overall and Gorham (Overall 1962) as a 14‐item scale to measure the severity of a range of psychiatric symptoms, including psychosis. This rating scale items evolved over time and now consists of 24 items which can be rated on a seven‐point scale from ‘not present’ to ‘extremely severe’. A high score would suggest poor mental health. It is not clear for the majority of the studies included in this review, which version of the BPRS was used.
b. Positive and Negative Syndrome Scale (PANSS)
The PANSS was developed and published by Kay, Flszbein and Opler (Kay 1987). The PANSS is designed as a brief interview, whereby the severity of 30 symptoms of schizophrenia can be assessed on a scale of one ('not present') to seven ('extremely severe'). A high score would indicate more severe symptoms. The PANSS can be divided into separate subscales by focusing on the statements relating to positive symptoms (e.g. hallucinations), negative symptoms (e.g. social withdrawal) or general psychopathology (e.g. anxiety and uncooperativeness).
6.4 Adverse effects
a. Udvalg for Kliniske Undersogelser (UKU) side effects rating scale
The UKU (Lingjaerde 1987) is a questionnaire that divides adverse effects into four general categories of symptoms: psychic, neurologic, autonomic, and miscellaneous; for each adverse effects, the interviewer is asked to assess the probability of causal relationship with psychotropic drugs.
b. Simpson‐Angus Scale (SAS)
The SAS (Simpson 1970) is a 10‐item scale which measures drug‐induced parkinsonism (extrapyramidal side effects). Each item is scored from zero to four. A high score would indicate increased levels of parkinsonism.
7. Missing outcomes
No studies evaluated satisfaction with care, acceptance of treatment, quality of life, or economic outcomes.
8. Funders
Andrezina 2006 and Bristol‐Myers 2005 studies received sponsorship from pharmaceutical companies.
Excluded studies
In total we excluded 16 studies. Of these, seven had to be excluded based on the characteristics of participants. These studies (Kane 2002; Cutler 2006; Fleischhacker 2009; Liang 2005; McEvoy 2007; Potkin 2003; Tandon 2006) were excluded because although participants were suspected to have psychosis, these people were not displaying an aggressive or agitated behaviour. Five studies had to be excluded because they involved oral administration of aripiprazole (Chen 2009; Kinon 2008; Li 2009; Liu 2012; Xie 2011) or the intramuscular administration of aripiprazole long acting (NCT01469039). Three studies (Bao 2007; Simpson 2010; Wang 2014) had to be excluded because the intervention medication was not aripiprazole alone.
Risk of bias in included studies
Although each ’Summary of findings’ table describes bias by outcome, an overview is reported here and a graphical impression is seen in Figure 4 and in Figure 5.
4.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
5.
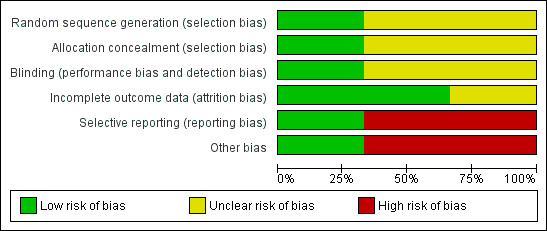
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
All included studies were described as randomised. In Andrezina 2006, the randomisation process is not stated (unclear sequence generation risk); however, allocation concealment was preserved by the utilisation of a centralised call‐in system (low allocation concealment risk). In Bristol‐Myers 2005 patients were randomly assigned in 1:1:1:1:1:1 ratio to the six arms of the study but no further information was provided regarding the process of randomisation; no details were provided regarding allocation concealment (overall unclear risk of selection bias). In Kittipeerachon 2016, the randomisation process is low risk as it is described as blindly picking the name of the assigned medication from a box with equal numbers of folded papers printed with each medication; allocation concealment management is not stated (unclear risk ).
Blinding
Andrezina 2006 is described as a double‐blinded study but no further details are provided (unclear risk of performance bias; unclear risk of detection bias). In the Bristol‐Myers 2005 study, authors stated double‐blinding but different dilution instructions for drug injections were provided, so no blinding was possible during the preparation of injections; in most cases the same person prepared and administered the drug (high risk of performance bias). To limit the magnitude of this bias, in the efficacy evaluations study investigators were blinded to treatment (low risk of detection bias), giving an overall unclear risk for blinding. In one case of medical emergency, the treating physician broke the blind design as knowledge of investigational product was considered to be critical to the patient management; however, investigators remained blinded. In Kittipeerachon 2016, patients and the physician investigator were blinded to treatment assignment.
Incomplete outcome data
Two included studies used a 'Last Observation Carried Forward' (LOCF) approach to manage missing data. In Andrezina 2006, there was no evidence of attrition bias (low risk of attrition bias). Concerning Bristol‐Myers 2005, reasons for leaving the study early were reported; however, number of participants completing different assessments at the same time point vary and reasons are not given (unclear risk of attrition bias). In Kittipeerachon 2016, procedures to manage missing data are not reported in the method section but no attritions were recorded throughout the trial (low risk of attrition bias).
Selective reporting
In both Andrezina 2006 and Bristol‐Myers 2005, adverse effects were reported only if they occurred in ≥ 5% of participants (high risk of reporting bias). In Kittipeerachon 2016, there was no evidence of reporting bias.
Other potential sources of bias
Both Andrezina 2006 and Bristol‐Myers 2005 were supported by manufacturers of one of the compared drugs (high risk of other bias). Kittipeerachon 2016 had no obvious risk of other bias.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. ARIPIPRAZOLE (IM) compared to PLACEBO/NIL for psychosis‐induced aggression or agitation (rapid tranquillisation).
| ARIPIPRAZOLE compared to PLACEBO/NIL for psychosis‐induced aggression or agitation (rapid tranquillisation) | ||||||
| Patient or population: psychosis‐induced aggression or agitation (rapid tranquillisation) Setting: hospital Intervention: ARIPIPRAZOLE (intramuscular) Comparison: PLACEBO/NIL | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with PLACEBO/NIL | Risk with ARIPIPRAZOLE | |||||
| Tranquillisation or asleep | Not reported | |||||
| Repeated need for tranquillisation ‐ needing additional injections during 24 hours | Low | RR 0.69 (0.56 to 0.85) | 382 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1, 2 | ||
| 250 per 1.000 | 173 per 1.000 (140 to 213) | |||||
| Moderate | ||||||
| 600 per 1.000 | 414 per 1.000 (336 to 510) | |||||
| High | ||||||
| 750 per 1.000 | 518 per 1.000 (420 to 638) | |||||
| Specific behaviour: Agitation ‐ clinically important change (PANSS ‐EC reduction ≥ 40% from baseline) ‐ up to 2 hours | Low | RR 1.50 (1.17 to 1.92) | 382 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1, 2 | ||
| 100 per 1.000 | 150 per 1.000 (117 to 192) | |||||
| Moderate | ||||||
| 350 per 1.000 | 525 per 1.000 (410 to 672) | |||||
| High | ||||||
| 700 per 1.000 | 1000 per 1.000 (819 to 1.000) | |||||
| Global state: non‐responders to the first injection | Low | RR 0.49 (0.34 to 0.71) | 263 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | ||
| 200 per 1.000 | 98 per 1.000 (68 to 142) | |||||
| Moderate | ||||||
| 450 per 1.000 | 221 per 1.000 (153 to 320) | |||||
| High | ||||||
| 700 per 1.000 | 343 per 1.000 (238 to 497) | |||||
| Adverse effects: one or more adverse events during 24 hours (only reported if occurred in ≥ 5% of people) | Study population | RR 1.51 (0.93 to 2.46) | 117 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1, 2 | ||
| 295 per 1.000 | 446 per 1.000 (274 to 726) | |||||
| Economic outcomes | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' (downgraded by 1) ‐ randomisation procedure is not reported for both the included studies, and allocation concealment procedure is not consistently reported in Bristol‐Myers 2005. While for the 'randomisation' bias, studies are at least reported as 'randomised', as for the latter that allocation concealment was correctly handled could not be implied.
2 Indirectness: rated 'very serious' (downgraded by 2) ‐ participants included in the studies had levels of agitation not so pronounced by inclusion criteria, potentially under‐estimating or more likely over‐estimating true effectiveness.
Summary of findings 2. ARIPIPRAZOLE (IM) compared to OTHER ANTIPSYCHOTIC: a. HALOPERIDOL for psychosis‐induced aggression or agitation (rapid tranquillisation).
| ARIPIPRAZOLE compared to OTHER ANTIPSYCHOTIC: a. HALOPERIDOL for psychosis‐induced aggression or agitation (rapid tranquillisation) | ||||||
| Patient or population: psychosis‐induced aggression or agitation (rapid tranquillisation) Setting: hospital Intervention: ARIPIPRAZOLE (intramuscular) Comparison: OTHER ANTIPSYCHOTIC: a. HALOPERIDOL | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTIC: a. HALOPERIDOL | Risk with ARIPIPRAZOLE | |||||
| Tranquillisation or asleep | Not reported | |||||
| Repeated need for tranquillisation ‐ needing additional injections during 24 hours | Low | RR 1.28 (1.00 to 1.63) | 477 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1, 2 | , | |
| 100 per 1.000 | 128 per 1.000 (100 to 163) | |||||
| Moderate | ||||||
| 300 per 1.000 | 384 per 1.000 (300 to 489) | |||||
| High | ||||||
| 500 per 1.000 | 640 per 1.000 (500 to 815) | |||||
| Specific behaviour: Agitation ‐ clinically important change (PANSS ‐EC reduction ≥ 40% from baseline) ‐ up to 2 hours | Study population | RR 0.94 (0.80 to 1.11) | 477 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 ,2 | ||
| 576 per 1.000 | 541 per 1.000 (460 to 639) | |||||
| Global state: non‐responders to the first injection | Study population | RR 1.18 (0.78 to 1.79) | 360 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | ||
| 184 per 1.000 | 217 per 1.000 (143 to 329) | |||||
| Adverse effects: one or more adverse events during 24 hours (only reported if occurred in ≥ 5% of people) | Study population | RR 0.91 (0.61 to 1.35) | 113 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1, 2 | ||
| 491 per 1.000 | 447 per 1.000 (300 to 663) | |||||
| Adverse effects: movement disorders ‐ EPS during 24 hours (only reported if occurred in ≥ 5% of people) | Low | RR 0.29 (0.12 to 0.70) | 471 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1, 2 | ||
| 50 per 1.000 | 14 per 1.000 (6 to 35) | |||||
| Moderate | ||||||
| 100 per 1.000 | 29 per 1.000 (12 to 70) | |||||
| High | ||||||
| 300 per 1.000 | 87 per 1.000 (36 to 210) | |||||
| Economic outcomes | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' (downgraded by 1) ‐ randomisation procedure is not reported for both the included studies, and allocation concealment procedure is not consistently reported in Bristol‐Myers 2005. While for the 'randomisation' bias studies are at least reported as 'randomised', as for the latter that allocation concealment was correctly handled could not be implied.
2 Indirectness: rated 'very serious' (downgraded by 2) ‐ participants included in the studies had levels of agitation not so pronounced by inclusion criteria, potentially under‐estimating or more likely over‐estimating true effectiveness.
Summary of findings 3. ARIPIPRAZOLE (IM) compared to OTHER ANTIPSYCHOTIC: b. OLANZAPINE for psychosis‐induced aggression or agitation (rapid tranquillisation).
| ARIPIPRAZOLE compared to OTHER ANTIPSYCHOTIC: b. OLANZAPINE for psychosis‐induced aggression or agitation (rapid tranquillisation) | ||||||
| Patient or population: psychosis‐induced aggression or agitation (rapid tranquillisation) Setting: hospital Intervention: ARIPIPRAZOLE (intramuscular) Comparison: OTHER ANTIPSYCHOTIC: b. OLANZAPINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTIC: b. OLANZAPINE | Risk with ARIPIPRAZOLE | |||||
| Tranquillisation or asleep | Not reported | |||||
| Repeated need for tranquillisation | Not reported | |||||
| Specific behaviour: Agitation ‐ clinically important change (PANSS ‐EC reduction ≥ 40% from baseline) ‐ up to 2 hours | Low | RR 0.77 (0.60 to 0.99) | 80 (1 RCT) | ⊕⊕⊝⊝ LOW1, 3 | ||
| 500 per 1.000 | 385 per 1.000 (300 to 495) | |||||
| Moderate | ||||||
| 800 per 1.000 | 616 per 1.000 (480 to 792) | |||||
| High | ||||||
| 900 per 1.000 | 693 per 1.000 (540 to 891) | |||||
| Global state: CGI‐S change score up to 2 hours | MD 0.58 (0.01 higher to 1.15 higher) | ‐ | 80 (1 RCT) | ⊕⊕⊝⊝ LOW 1, 3 | ||
| Adverse effects: one or more adverse effects during 24 hours | Study population | RR 0.75 (0.45 to 1.24) | 80 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2, 3 | ||
| 500 per 1.000 | 375 per 1.000 (225 to 620) | |||||
| Adverse effects: somnolence during 24 hours | Low | RR 0.25 (0.08 to 0.82) | 80 (1 RCT) | ⊕⊕⊝⊝ LOW 1, 3 | ||
| 100 per 1.000 | 25 per 1.000 (8 to 82) | |||||
| Moderate | ||||||
| 300 per 1.000 | 75 per 1.000 (24 to 246) | |||||
| High | ||||||
| 700 per 1.000 | 175 per 1.000 (56 to 574) | |||||
| Economic outcomes | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Imprecision ‐ rated 'serious' (downgraded by 1): Optimal Information Size (OIS) criterion is not met meaning that imprecision of effect estimates could not be properly handled.
2 Imprecision ‐ rated 'very serious' (downgraded by 2): Optimal Information Size (OIS) criterion is not met and CI overlaps no effect. High imprecision of effect estimates could not be properly excluded.
3 Indirectness ‐ rated 'serious' (downgraded by 1): attribution to the intervention drugs is suspected but can not be confirmed since in the study results it is showed that almost all the participants were administered with 'treatment as usual' drugs.
See Table 1; Table 2; Table 3. In the following section, the summary statistic for binary outcome results is reported as risk ratio (RR), and for continuous outcomes we used mean difference (MD). In all instances 95% confidence intervals (CIs) are reported.
1. COMPARISON 1. ARIPIPRAZOLE (intramuscular) versus PLACEBO/NIL
1.1 Repeated need for tranquillisation
Overall, significantly fewer participants allocated to aripiprazole needed additional injections during 24 hours, when compared to placebo (2 RCTs, n = 382, RR 0.69, 95% CI 0.56 to 0.85, very low‐quality evidence). Analyses for numbers needing one additional injection showed no differences between groups (1 RCT, n = 119, RR 1.38, 95% CI 0.69 to 2.80) but significantly more people receiving placebo required two additional injections (1 RCT, n = 119, RR 0.36, 95% CI 0.19 to 0.70, Analysis 1.1).
1.1. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 1 Repeated need for tranquillisation.
1.2 Specific behaviour ‐ agitation
1.2.1 Clinically important change
Aripiprazole had an effect on agitation. At two hours more participants in the aripiprazole group showed a positive response for agitation (defined as a PANSS ‐ EC reduction of ≥ 40% from baseline, 2 RCTs, n = 382, RR 1.50. 95% CI 1.17 to 1.92, very low‐quality evidenceAnalysis 1.2).
1.2. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 2 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐EC reduction ≥ 40% from baseline).
1.2.2 Average scores
Continuous measures of agitation (up to two hours) also favoured aripiprazole: ABS change score (2 RCTs, n = 380, MD ‐3.77, 95% CI ‐5.39 to ‐2.16), ACES change score (2 RCTs, n = 380, MD ‐0.71, 95% CI ‐1.15 to ‐0.28) and PANSS‐EC change score (1 RCT, n = 261, MD ‐2.49, 95% CI ‐4.28 to ‐0.70). These findings were still favourable for aripiprazole for both a schizophrenia subgroup (2 RCTs, n = 263, MD ‐2.55, 95% CI ‐3.78 to ‐1.32), and a non‐sedated participants subgroup ‐ based on ACES score (2 RCTs, n = 355, MD ‐2.68, 95% CI ‐3.75 to ‐1.62) and on AEs (2 RCTs, n = 365, MD ‐2.83, 95% CI ‐3.92 to ‐1.75, Analysis 1.3).
1.3. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours.
1.3 Global state
Various
There were fewer non‐responders at first injection in aripiprazole group (1 RCT, n = 263, RR 0.49, 95% CI 0.34 to 0.71,low‐quality evidence) and participants allocated to aripiprazole had a significantly lower need for additional intervention with benzodiazepines (2 RCTs, n = 382, RR 0.48, 95% CI 0.28 to 0.80, Analysis 1.4).
1.4. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 4 Global state: 1. Various.
1.3.2 Average scores ‐ up to two hours
Rating scales showed a significant advantage for participants allocated to aripiprazole: CGI‐I endpoint score (2 RCTs, n = 380, MD ‐0.73, 95% CI ‐0.97 to ‐0.49), CGI‐S change score (2 RCTs, n = 380, MD ‐0.56, 95% CI ‐0.86 to ‐0.26). CGI‐I endpoint score was also lower in the aripiprazole group for participants with a schizophrenia diagnosis (1 RCT, n = 75, MD ‐0.71, 95% CI ‐1.17 to ‐0.25, Analysis 1.5).
1.5. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 5 Global state: 2. Average scores ‐ i. up to 2 hours.
1.4 Mental state ‐ average scores ‐ up to two hours
Change score of PANSS‐derived BPRS total (1 RCT, n = 110, MD ‐3.39, 95% CI ‐7.03 to 0.25) and positive subscale (1 RCT, n = 110, MD ‐0.62, 95% CI ‐1.65 to 0.41) were similar between aripiprazole and placebo groups (Analysis 1.6).
1.6. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 6 Mental state: 1. Average scores ‐ i. up to 2 hours.
1.5 Adverse effects
1.5.1 General
More participants allocated to aripiprazole experienced one or more drug‐related adverse effects by two hours (1 RCT, n = 117, RR 1.51, 95% CI 0.93 to 2.46, very low‐quality evidence), and onset or worsening of adverse effects following the second injection (1 RCT, n = 262, RR 2.40, 95% CI 1.36 to 4.23, Analysis 1.7).
1.7. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 7 Adverse effects: 1a. General.
Similar findings were found for serious adverse effects, which were higher in the aripiprazole group when compared to placebo group (2 RCTs, n = 379, RR 3.77, 95% CI 0.63 to 22.60); in the aripiprazole group a tonic clonic seizure event occurred (1 RCT, n = 117, RR 3.26, 95% CI 0.14 to 78.49, Analysis 1.8). No deaths were reported.
1.8. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 8 Adverse effects: 1b. General ‐ serious.
1.5.2 Specific ‐ arousal
No clear effect was found for other effects such as 'insomnia during 24 hours' (1 RCT, n = 262, RR 0.62, 95% CI 0.25 to 1.52), 'over'‐sedated (1 RCT, n = 262, RR 1.59, 95% CI 0.60 to 4.20) somnolence (2 RCTs, n = 379, RR 1.52, 95% CI 0.57 to 4.00, Analysis 1.9).
1.9. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 9 Adverse effects: 2a. Specific ‐ arousal.
1.5.3 Specific ‐ cardiovascular
Again, similar numbers in each treatment group experienced dizziness (2 RCTs, n = 379, RR 1.77, 95% CI 0.66 to 4.70), tachycardia (1 RCT, n = 117, RR 4.36, 95% CI 0.50 to 37.82) during 24 hours and sinus tachycardia (1 RCT, n = 117, RR 0.36, 95% CI 0.02 to 8.72, Analysis 1.10).
1.10. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 10 Adverse effects: 2b. Specific ‐ cardiovascular.
1.5.4 Specific ‐ movement disorders
Drug‐related adverse effects were experienced only by participants allocated to aripiprazole during 24 hours but no effect between treatment groups was observed for akathisia (1 RCT, n = 117, RR 7.61, 95% CI 0.40 to 144.21), dystonia (1 RCT, n = 117, RR 3.26, 95% CI 0.14 to 78.49) and extrapyramidal symptoms (1 RCT, n = 262, RR 1.50 95% CI 0.06 to 36.45, Analysis 1.11).
1.11. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 11 Adverse effects: 2c. Specific ‐ movement disorders.
1.5.5 Miscellaneous
More participants allocated to the aripiprazole group experienced nausea during 24 hours (2 RCTs, n = 379, RR 3.97 95% CI 1.13 to 13.92). Concerning other miscellaneous adverse effects, no observable differences were found: agitation (2 RCTs, n = 379, RR 0.88 95% CI 0.33 to 2.37), headache (2 RCTs, n = 379, RR 1.66 95% CI 0.74 to 3.71), pain at injection site (2 RCTs, n = 379, RR 1.10 95% CI 0.30 to 3.94), and vomiting (1 RCT, n = 117, RR 2.18 95% CI 0.20 to 23.37, Analysis 1.12).
1.12. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 12 Adverse effects: 2d. Specific ‐ miscellaneous.
1.6 Leaving the study early
We found no clear difference for numbers of participants leaving the study early for 'any reason' (2 RCTs, n = 382, RR 1.29, 95% CI 0.35 to 4.74); 'lack of efficacy' (2 RCTs, n = 382, RR 0.47, 95% CI 0.07 to 3.06), 'consent withdrawal' (2 RCTs, n = 382, RR 1.68, 95% CI 0.23 to 12.46), 'adverse effects' (2 RCTs, n = 382, RR 2.25, 95% CI 0.25 to 20.54), and 'other' reasons (2 RCTs, n = 382, RR 1.05, 95% CI 0.15 to 7.44, Analysis 1.13).
1.13. Analysis.
Comparison 1 ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL, Outcome 13 Leaving the study early.
2. COMPARISON 2. ARIPIPRAZOLE (intramuscular) versus OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular)
2.1 Repeated need for tranquillisation
Compared to haloperidol, more participants allocated to aripiprazole needed additional injections during 24 hours (2 RCTs, n = 477, RR 1.28, 95% CI 1.00 to 1.63, very low‐quality evidence). A comparable pattern was found in terms of need of 1 additional injection (1 RCT, n = 117, RR 1.34, 95% CI 0.66 to 2.70) and 2 additional injections (1 RCT, n = 117, RR 2.37, 95% CI 0.77 to 7.26) during 24 hours (Analysis 2.1). Even though not being statistically significant, leading to an equal to higher number of additional injections needed must be taken into account when considering clinical significance.
2.1. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 1 Repeated need for tranquillisation.
2.2 Specific behaviour ‐ agitation
2.2.1 Clinically important change
Clinically important change in agitation by two hours (defined as a PANSS‐EC reduction of ≥ 40% from baseline) was similar between groups (2 RCTs, n = 477, RR 0.94, 95% CI 0.80 to 1.11, very low‐quality evidence;Analysis 2.2).
2.2. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 2 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐ EC reduction ≥ 40% from baseline).
2.2.2 Average scores ‐ up to two hours
Continuous measures of agitation showed similar findings, with no observable differences in ABS change score (2 RCTs, n = 473, MD 0.55, 95% CI ‐1.00 to 2.10), ACES change score (2 RCTs, n = 473, MD 0.12, 95% CI ‐0.30 to 0.55) and PANSS‐EC change score in the total sample (1 RCT, n = 357, MD 0.48, 95% CI ‐1.16 to 2.12), nor in schizophrenia subgroup (2 RCTs, n = 336, MD 0.07, 95% CI ‐0.99 to 1.13), or non‐sedated patients (based on ACES score: 2 RCTs, n = 422, MD 0.12, 95% CI ‐0.82 to 1.06). Based on AEs: 2 RCTs, n = 448, MD 0.26, 95% CI ‐0.68 to 1.19, Analysis 2.3).
2.3. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours.
2.3 Global state
Similar numbers of participants were 'non‐responders to the first injection (1 RCT, n = 360, RR 1.18, 95% CI 0.78 to 1.79, low‐quality evidence) and similar numbers needed benzodiazepine up to 24 hours (2 RCTs, n = 477, RR 0.79, 95% CI 0.46 to 1.35, Analysis 2.4).
2.4. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 4 Global state: 1. Various.
No differences between aripiprazole and haloperidol were observed for global state (as measured by CGI scale): CGI‐I endpoint score for total sample (2 RCTs, n = 473, MD 0.02, 95% CI ‐0.19 to 0.23) and in the schizophrenia subgroup (1 RCT, n = 79, MD 0.02, 95% CI ‐0.44 to 0.48), and CGI‐S change score (2 RCTs, n = 473, MD ‐0.07, 95% CI ‐0.36 to 0.22, Analysis 2.5).
2.5. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 5 Global outcome: 2. Average scores ‐ i. up to 2 hours.
2.4 Mental state
Participants in aripiprazole and haloperidol groups also had similar scores on mental state scales: PANSS‐derived BPRS change score (1 RCT, n = 102, MD 2.03, 95% CI ‐1.70 to 5.76) and in the positive subscale (1 RCT, n = 103, MD 0.23, 95% CI ‐0.82 to 1.28) by two hours (Analysis 2.6).
2.6. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 6 Mental state: 1. Average scores ‐ i. up to 2 hours.
2.5 Adverse effects
2.5.1 General
The same proportion of participants of aripiprazole and haloperidol group experienced one or more drug‐related adverse effects during 24 hours (1 RCT, n = 113, RR 0.91, 95% CI 0.61 to 1.35, very low‐quality evidence); however, more people allocated to haloperidol experienced onset or worsening of adverse effects after the second injection (1 RCT, n = 358, RR 0.74, 95% CI 0.57 to 0.96, Analysis 2.7).
2.7. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 7 Adverse effects: 1a. General.
More adverse effects rated as serious were recorded in the aripiprazole group but this was not an observable difference between the treatments (2 RCTs, n = 471, RR 1.73, 95% CI 0.54 to 5.52), as was 'event of tonic clonic seizure' (1 RCT, n = 113, RR 3.05, 95% CI 0.13 to 73.38). No death events were recorded (Analysis 2.8).
2.8. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 8 Adverse effects: 1b. General ‐ serious.
2.5.2 Specific ‐ arousal
More people allocated to haloperidol experienced insomnia during 24 hours (1 RCT, n = 358, RR 0.48, 95% CI 0.23 to 0.97) and, were 'over'‐sedated (1 RCT, n = 358, RR 0.60, 95% CI 0.34 to 1.07); no differences in terms of somnolence were found between aripiprazole and haloperidol groups (2 RCTs, n = 471, RR 0.81, 95% CI 0.41 to 1.58, Analysis 2.9).
2.9. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 9 Adverse effects: 2a. Specific ‐ arousal.
2.5.3 Specific ‐ cardiovascular
No real differences were found as regards the number of participants experiencing dizziness (2 RCTs, n = 471, RR 1.41, 95% CI 0.66 to 3.01) and tachycardia (1 RCT, n = 113, RR 4.07, 95% CI 0.47 to 35.31); three events of sinus tachycardia were recorded in the haloperidol group but again, no effect found (1 RCT, n = 113, RR 0.15, 95% CI 0.01 to 2.75, Analysis 2.10).
2.10. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 10 Adverse effects: 2b. Specific ‐ cardiovascular.
2.5.4 Specific ‐ movement disorders
Significantly more participants allocated to haloperidol experienced extrapyramidal symptoms (2 RCTs, n = 471, RR 0.29, 95% CI 0.12 to 0.70, very low‐quality evidence); but this effect was not observed for akathisia (1 RCT, n = 113, RR 0.51, 95% CI 0.13 to 1.94) and dystonia (1 RCT, n = 113, RR 0.25, 95% CI 0.03 to 2.21), although more events were recorded in the haloperidol group (Analysis 2.11).
2.11. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 11 Adverse effects: 2c. Specific ‐ movement disorders.
2.5.5 Miscellaneous
A favourable effect for haloperidol was found in terms of number of participants experiencing nausea (2 RCTs, n = 471, RR 5.52, 95% CI 1.63 to 18.70). No effect was found for 'pain at injection site' (2 RCTs, n = 471, RR 3.12, 95% CI 0.75 to 12.91), agitation (2 RCTs, n = 471, RR 1.04 95% CI 0.42 to 2.58), headache (2 RCTs, n = 471, RR 1.16 95% CI 0.62 to 2.18), or vomiting during 24 hours (1 RCT, n = 113, RR 2.04 95% CI 0.19 to 21.82, Analysis 2.12).
2.12. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 12 Adverse effects: 2d. Specific ‐ miscellaneous.
2.6 Leaving the study early
No differences in numbers leaving the study early were observed between aripiprazole and haloperidol groups for either any reason (2 RCTs, n = 477, RR 0.49 95% CI 0.20 to 1.18), consent withdrawal (2 RCTs, n = 477, RR 0.35 95% CI 0.10 to 1.28), lack of efficacy (2 RCTs, n = 477, RR 1.05, 95% CI 0.15 to 7.43), adverse effects: 2 RCTs, n = 477, RR 1.06, 95% CI 0.18 to 6.04 or other reason (2 RCTs, n = 477, RR 0.75, 95% CI 0.15 to 3.78, Analysis 2.13).
2.13. Analysis.
Comparison 2 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular), Outcome 13 Leaving the study early.
3. COMPARISON 3. ARIPIPRAZOLE (intramuscular) versus OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular)
3.1 Specific behaviour ‐ agitation
More participants allocated to olanzapine had an improvement in agitation at two hours (1 RCT, n = 80, RR 0.77, 95% CI 0.60 to 0.99, low‐quality evidence;Analysis 3.1). Continuous measurements showed similar findings, results showing a favourable effect for olanzapine at two hours using ACES change scores (1 RCT, n = 80, MD 0.58, 95% CI 0.10 to 1.06) and PANSS‐EC change scores (1 RCT, n = 80, MD 3.30, 95% CI 1.25 to 5.35, Analysis 3.2).
3.1. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 1 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐ EC reduction ≥ 40% from baseline).
3.2. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours.
3.2 Global state
Olanzapine had a favourable effect compared to aripiprazole on global state when measured using CGI‐S change scores (1 RCT, n = 80, MD 0.58, 95% CI 0.01 to 1.15, low‐quality evidence;Analysis 3.3).
3.3. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 3 Global state: 1. Average scores ‐ i. up to 2 hours.
3.3 Adverse effects
3.3.1 General
A comparable amount of participants in both groups experienced at least one adverse effect (1 RCT, n = 80, RR 0.75, 95% CI 0.45 to 1.24, very low‐quality evidence;Analysis 3.4). No serious adverse events occurred (Analysis 3.5).
3.4. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 4 Adverse effects: 1a. General.
3.5. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 5 Adverse effects: 1b. General ‐ serious.
3.3.2 Specific ‐ arousal
Significantly more participants allocated to olanzapine experienced somnolence during 24 hours (1 RCT, n = 80, RR 0.25, 95% CI 0.08 to 0.82, low‐quality evidence; Analysis 3.6).
3.6. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 6 Adverse effects: 2a. Specific ‐ arousal.
3.3.3 Specific ‐ cardiovascular
No significant differences were observed for systolic and diastolic pressure change from baseline, and for pulse rate change from baseline, both at two and 24 hours (Analysis 3.7).
3.7. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 7 Adverse effects: 2b. Specific ‐ cardiovascular.
3.3.4 Specific ‐ movement disorders
Aripiprazole and olanzapine results were also similar for rigidity (1 RCT, n = 80, RR 1.33, 95% CI 0.32 to 5.58) and tremor (1 RCT, n = 80, RR 1.50, 95% CI 0.26 to 8.50) during 24 hours. None of the participants experienced akathisia or dystonia (Analysis 3.8), with similar continuous ratings for the former both at two hours (1 RCT, n = 80, MD ‐0.05, 95% CI ‐0.26 to 0.16) and at 24 hours (1 RCT, n = 80, MD ‐0.03, 95% CI ‐0.29 to 0.23, Analysis 3.9).
3.8. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 8 Adverse effects: 2c. Specific ‐ movement disorders ‐ i. dichotomous.
3.9. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 9 Adverse effects: 2d. Specific ‐ movement disorders ‐ ii. average scores.
3.3.5 Specific ‐ miscellaneous
Again, no effect was observed when aripiprazole and olanzapine were compared for headache (1 RCT, n = 80, RR 2.00, 95% CI 0.19 to 21.18), nausea (1 RCT, n = 80, RR 3.00, 95% CI 0.13 to 71.51), and pain at injection site (1 RCT, n = 80, RR 2.67, 95% CI 0.76 to 9.33), even though as regards the latter more events occurred in the aripiprazole group (Analysis 3.10).
3.10. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 10 Adverse effects: 2e. Specific ‐ miscellaneous.
3.4 Leaving the study early
None of the involved participants left this study early (Analysis 3.11).
3.11. Analysis.
Comparison 3 ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular), Outcome 11 Leaving the study early.
Discussion
Summary of main results
1. COMPARISON 1. ARIPIPRAZOLE (intramuscular) versus PLACEBO/NIL
Two studies were included (Andrezina 2006; Bristol‐Myers 2005), with a total number for this comparison of 382 participants (aripiprazole, n = 232; placebo, n = 150); available outcomes were rated of very low‐low quality evidence (Table 1). No data concerning 'tranquillisation or asleep' and economic outcomes were available.
1.1 Repeated need for rapid tranquillisation, agitation
When compared to placebo, fewer people in the aripiprazole group required additional injections (Analysis 1.1). Significantly more participants in the aripiprazole group showed an at least 40% reduction in PANSS‐EC agitation score (Analysis 1.2) and Analysis 1.3 showed higher improvement in agitation over continuous measurements, supporting potential benefits of this drug. However, outcomes for agitation were at two hours, a time point that meets only partially the issue of rapid tranquillisation.
1.2 Global state
More people allocated to placebo failed to respond to the first injection and required additional benzodiazepines over 24 hours (Analysis 1.4); whilst the former statement is based on data from Andrezina 2006 only, the latter comes from the both included studies (Andrezina 2006; Bristol‐Myers 2005). Continuous measurement scales (CGI‐I, CGI‐S) emphasise this outcome, with people in placebo group having worse rating scores (Analysis 1.5).
1.3 Adverse effects ‐ any serious, specific adverse effects
Even though not formerly meeting statistical significance, a higher proportion of participants allocated to aripiprazole experienced one or more drug‐related adverse effects, and after the second injection more problems with adverse effects were recorded compared to people in the placebo group (Analysis 1.7). Adverse effects rated as serious were more common for participants allocated to aripiprazole, despite not meeting statistical significance (Analysis 1.8).
When specific adverse effects were taken into account, a significant difference was observed only for the 'nausea during the 24 hours' outcome, more common in the aripiprazole group (Analysis 1.12); concerning the other adverse effects (insomnia, 'over'‐sedation, somnolence, dizziness, tachycardia, akathisia, dystonia, extrapyramidal symptoms , agitation, headache, pain at injection site, vomiting), recorded events were higher in the aripiprazole group, but statistical significance was not met (Analysis 1.9; Analysis 1.10; Analysis 1.11). However, it is important to consider that in both included studies (Andrezina 2006; Bristol‐Myers 2005), data refer only to adverse effects that occurred in ≥ 5% of people.
2. COMPARISON 2. ARIPIPRAZOLE (intramuscular) versus OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular)
Data from a total number of 477 participants were extracted from included studies (Andrezina 2006; Bristol‐Myers 2005, aripiprazole, n = 232; haloperidol, n = 245); the overall quality of the evidence was very low (Table 2), suggesting caution in the interpretation of quantitative synthesis data.
2.1 Repeated need for rapid tranquillisation, agitation
Despite having comparable response rates and agitation rating scales at two hours (Analysis 2.2; Analysis 2.3), more participants allocated to aripiprazole required at least one additional injection during 24 hours (Analysis 2.1) when compared to haloperidol. Considering how difficult the management of repeated injections could be in an agitation setting and the potential distress of this for the user in need of tranquillisation, that use of haloperidol necessitates less injections is important. Direct data on tranquillisation were not available.
Despite in both Andrezina 2006 and Bristol‐Myers 2005 it is stated that primary outcomes were recorded at multiple time points before two hours (0.5, 0.75, 1, 1.5 hours for Andrezina 2006, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75 hours for Bristol‐Myers 2005), no numeric data were available on related publications; two hours is a considerable amount of time after an acute aggressive or agitated event.
2.2 Global state ‐ overall improvement
Proportions of non‐responders to the first injection between aripiprazole and haloperidol groups were comparable (Analysis 2.4), and so were the global outcome measurement scales (Analysis 2.6).
2.3 Adverse effects ‐ any serious, specific adverse effects
Compared groups did not differ in terms of number of people that experienced at least one drug‐related adverse effect (Analysis 2.7); one person in the aripiprazole group had a tonic‐clonic seizure and one in the haloperidol group developed severe dystonia (Analysis 2.8). More participants allocated to haloperidol developed insomnia or, by contrast, were 'over'‐sedated during the 24 hours (Analysis 2.9), whilst nausea was significantly more common in the aripiprazole group (Analysis 2.12); with the exception of drug‐related movement disorders, that tended to be more common with haloperidol (Analysis 2.11), other adverse effects showed no differences between the compared groups (Analysis 2.10).
2.4 Economic outcomes
No data concerning economic outcomes were available in the two included studies.
3. COMPARISON 3: ARIPIPRAZOLE (intramuscular) versus OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular)
One study was included (Kittipeerachon 2016) with a total number of 80 participants (aripiprazole, n = 40; olanzapine, n = 40); outcomes were rated being of very low to low quality (Table 3). No data concerning 'tranquillisation or asleep' and economic outcomes were available.
3.1 Agitation
As for participants allocated to aripiprazole, a significantly minor PANSS (Excited Component (PEC) response rate by two hours was observed (Analysis 3.1). Nevertheless, there are no outcomes available before two hours.
3.2 Global state ‐ overall improvement
A significantly minor CGI‐S change score was observed for aripiprazole when compared to olanzapine, at two hours (Analysis 3.3).
3.3 Adverse effects ‐ any serious, specific adverse effects
Compared groups did not differ in terms of number of people who experienced one or more drug‐related adverse effects (Analysis 3.4); no adverse events were rated as serious in both groups (Analysis 3.5). Aripiprazole and olanzapine results were comparable for all the investigated adverse events (Analysis 3.7; Analysis 3.8; Analysis 3.9; Analysis 3.10), with the exception of somnolence, which was more common in participants allocated to olanzapine (Analysis 3.6). Somnolence data at two hours could help to clarify if this issue could be a major PEC response rate of olanzapine.
2.4 Economic outcomes
No data concerning economic outcomes were available.
Overall completeness and applicability of evidence
1. Completeness
Data on several important questions are missing: satisfaction with care, acceptance of treatment, quality of life and economic outcomes. Current evidence‐based knowledge on aripiprazole in agitation is limited by the paucity of randomised controlled trials (RCTs) and low quality of available data, thus suggesting strong caution in the interpretation of results.
2. Applicability
Two out of three of the currently available studies were undertaken in circumstances quite different from real clinical practice: involved participants constituted a specific population of voluntarily hospitalised patients that, while meeting specific agitation criteria for enrolment, "their level of agitation was not so pronounced that they could not give informed consent or participate in the study" (Andrezina 2006): this appears to be in line with the inclusion criteria applied in the two trials (Andrezina 2006; Bristol‐Myers 2005), in which a PEC score ≥ 15 but ≤ 32 was requested. Participants in the included studies had their intervention delayed for screening and until deemed competent to give informed consent, a situation that is not common in the heat of an emergency and makes applicability problematic even if it is not impossible; it just necessitates a level of conjecture that could have been lessened had more pragmatic trials been available. While in Kittipeerachon 2016, a more pragmatic methodological design can be observed, generalisability of the outcomes is partly limited by the contrast between the lack of outcomes prior to two hours and the administration of a 'treatment as usual' during the 24 hours.
Quality of the evidence
This review includes three trials completed over the last decade with a total sample size of 885 participants with schizophrenia, schizoaffective or schizophreniform disorder, of which 707 were included for quantitative analyses in this systematic review. Disappointingly, a proportion of interesting data were unusable, which could have been avoided had the trials been better reported. Overall, the majority of the evidence had to be graded as very low, usually because of considerable ‐ and often avoidable ‐ limits in research design, selective reporting and imprecision.
Potential biases in the review process
There are a number of ways in which bias could have been introduced into this review.
1. Trial selection and data extraction
For Cochrane reviews, at least two review authors independently inspect all citations obtained from the search and extract the data to minimise bias with a third completing a random sample check. This was achieved in the 2014 search but for the 2017 search only one review author inspected the search results and extracted data. However, the number of citations found in this search was low and in addition, to limit the possibility of this bias, screening obtained from the search was performed on two different occasions and compared.
2. Review author conflict of interest
No conflict of interest is declared by the review authors.
Agreements and disagreements with other studies or reviews
The authors are not aware of any other studies or reviews that have focused solely on use of aripiprazole IM alone for people who are aggressive secondary to serious mental illnesses.
Authors' conclusions
Implications for practice.
1. For people with aggression or aggression induced by psychosis
For people experiencing psychosis‐induced aggression or agitation, or side effects related to acute tranquillisation, a justification for their treatment choice may be helpful when they have recovered. It would be reassuring and informative to be presented with evidence‐based treatment options, should they suffer from similar symptoms in the future. Evidence on the intramuscular use of aripiprazole for agitation or aggression in psychosis in the acute setting is limited. According to the evidence reported in this review, aripiprazole was found to be more effective and with less side effects compared to placebo. In comparison to haloperidol, aripiprazole was found to be overall comparable in tranquillisation, even though data are poor and evidence is weak. Aripiprazole was found to be more prone to cause nausea, with less movement disorders side effects compared to haloperidol. In comparison to olanzapine, aripiprazole was found to be less effective in tranquillisation but less prone to cause somnolence. Patients preference and best interest should be taken into consideration in treatment decision‐making and evidence provided by this review could support a more ethical approach to this matter.
2. For clinicians
Aggression or agitation due to psychosis is a difficult situation for clinicians, and, at present, limited evidence of very low to low quality exists for intramuscular treatment with atypical antipsychotics. In this review, the superiority of aripiprazole in comparison to placebo but not haloperidol is reported. Concerning the last comparison, people allocated to aripiprazole required more injections, a potentially complex issue to manage in an acute real‐world setting; whilst experiencing nausea was more common to participants in the aripiprazole group, haloperidol tended to cause more extrapyramidal symptoms side effects. Olanzapine was found to be more effective than aripiprazole, and tended to cause more somnolence side effects. Evidence is limited by the small number of trials available. Further good‐quality research is needed to confirm these results and to provide missing but clinically relevant outcomes, as well as to compare aripiprazole with other atypical antipsychotics (see Table 5).
2. Suggested design for a trial.
| Methods | Allocation: randomised, clearly described, concealed. Blindness: double, described and tested. Duration: 2 weeks. |
| Participants | Diagnosis: thought to be psychoses.
N = 300.*
Age: any.
Sex: both. History: acutely ill, aggressive and/or agitated. |
| Interventions | 1. Aripiprazole IM: dose flexible within recommended limits. N = 150. 2. Haloperidol IM: dose flexible within recommended limits. N = 150. |
| Outcomes | All outcomes are grouped by time: by 30 minutes, up to two hours, up to four hours, up to 24 hours, and over 24 hours. Tranquillisation or asleep ‐ tranquil; asleep. Repeated need for tranquillisation ‐ needing additional injections. Specific behaviours ‐ self‐harm, including suicide, injury to others. Global outcomes ‐ patient satisfaction; use of restraint or seclusion. Service outcomes ‐ length of hospitalisation; readmission rate. Mental state ‐ effect of medication on mental state. Adverse effects ‐ medication significant side effects. Leaving the study early ‐ detailed reasons provided. Quality of life outcomes. Economic outcomes. |
| Notes | * Powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty |
IM: intramuscular
3. For policy makers
Although atypical antipsychotics such as aripiprazole are theoretically safer and better tolerated compared to haloperidol, this is not clear for people with aggression or agitation due to psychosis. There is evidence that aripiprazole administered intramuscularly is effective for this group of people. However its superiority to haloperidol and olanzapine is not shown, particularly in terms of need for repeated injections for the former and PANSS (Excited Component (PEC) response rate for the latter. Further research with more independent trials is required to form clearer guidelines for people suffering from psychosis‐induced aggression or agitation.
Implications for research.
1. General
The Consolidated Standards of Reporting Trials (CONSORT, Moher 2001) should now be met by all the studies. The protocol, the method, the analysis and the interpretation as well as the assessment of the results validity should be enabled by clearer and more transparent reporting of randomised controlled trials. Specific figures and their measure of variance should be clearly reported in the main text when data are presented in graphs.
2. Specific
2.1 Reviews
We have included neither studies of oral use of aripiprazole for management of people with acute aggression or agitation, nor studies that combined aripiprazole with other drugs. It might be possible to update this review with these data. Other reviews relevant to people who are aggressive in combination with psychosis are suggested by the comparisons in the excluded studies. Although some of the excluded studies do find a ready 'home' in already existing reviews or protocols, several do not and potentially neglected and important titles are suggested (Table 6).
3. Additional reviews relevant to aggression and suggested from excluded studies.
| Aripiprazole | Combination | Comparison #1 | Comparison #2 | Excluded study | Suggested title of review | Cochrane review | |
| IM | Short acting | + quetiapine | aripiprazole + olanzapine | quetiapine + olanzapine | Wang 2014 | Antipsychotic combinations for psychosis‐induced aggression or agitation (rapid tranquillisation) | None |
| Long acting (441mg) | Long acting (882 mg) | placebo | NCT0146903* | Aripiprazole for long‐term aggression in psychosis; Placebo for long‐term aggression in psychosis | None | ||
| Oral | aripiprazole + magnesium valproate | Li 2009, Liu 2012, Xie 2011 | Magnesium valproate (+/‐aripiprazole) for psychosis‐induced aggression | None | |||
| oral olanzapine | Chen 2009, Kinon 2008 | Aripiprazole (oral) for psychosis‐induced aggression or agitation (rapid tranquillisation); Olanzapine (oral) for psychosis‐induced aggression or agitation (rapid tranquillisation); | ? expansion of this review | ||||
| + clonazepam | oral clozapine | Bao 2007 | Aripiprazole plus benzodiazepines for psychosis‐induced aggression or agitation (rapid tranquillisation); Clozapine for psychosis‐induced aggression or agitation (rapid tranquillisation); | ? expansion of this review; Toal 2012 | |||
| Non‐aripiprazole | |||||||
| IM haloperidol + lorazepam + benztropine | oral quetiapine | Simpson 2010 | Benzodiazepines for psychosis‐induced aggression or agitation (rapid tranquillisation); Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation); Quetiapine for psychosis‐induced aggression or agitation (rapid tranquillisation) | Gillies 2015;Ostinelli 2017;Wilkie 2012 | |||
* One sub‐study with people with aggression/agitation.
2.2 Trials
This review is the result of research over the last decade on the use of aripiprazole IM for aggression or agitation induced by psychosis. More large independent randomised trials are required to measure 'pragmatic' outcomes, such as 'tranquillisation or asleep' outcomes within multiple time points during the first two hours, or economic outcomes. This type of randomised controlled trial should be provided with high‐quality methodological design, measuring simple outcomes such as those indicated in Table 5. These studies just need modest financial support and firm help from ethics committees who understand the need to produce good evidence for practice in this difficult edge of health care.
Acknowledgements
We are grateful for the support we received from the Cochrane Schizophrenia Group editorial base, Nottingham, UK. We have used their methods template for the Data collection and analysis section. We would also like to thank Lopamudra Mitra and Bhuvaneshwar Pagadala for their contribution to writing the protocol.
We would like to thank Tarek Nassif and Ayesha Kotecha for peer reviewing the review version.
Appendices
Appendix 1. Previous searches
Search methods for identification of studies
No language restriction was applied within the limitations of the search tools.
Electronic searches
We searched the Cochrane Schizophrenia Group's Specialised Register using the phrase:
[aripiprazole* or *aripiprazole* or abilitat* or * abilitat* or abilify* or *abilify* in abstract, index terms of REFERENCE) or (aripiprazole* or abilitat* or abilify* in interventions of STUDY)) and (*aggress* or *violen* or *agitation* or *tranq* in title, abstract, index terms of REFERENCE)]
This Specialised Register is compiled by systematic searches of major databases including BIOSIS Inside; CENTRAL; CINAHL; EMBASE; MEDLINE and PsycINFO; handsearching of relevant journals and conference proceedings and searches of several key grey literature sources. A full description is given in the Group Module.
Searching other resources
1. Reference searching We inspected the references of all included studies for more trials.
2. Personal contact We contacted the first author of each included study for missing information.
3. Drug companies We contacted the manufacturers of aripiprazole for additional relevant published and unpublished data.
Data and analyses
Comparison 1. ARIPIPRAZOLE (intramuscular) vs PLACEBO/NIL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for tranquillisation | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 needing additional injections during 24 hours | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.56, 0.85] |
| 1.2 needing 1 additional injection during 24 hours | 1 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.69, 2.80] |
| 1.3 needing 2 additional injections during 24 hours | 1 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.19, 0.70] |
| 2 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐EC reduction ≥ 40% from baseline) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 up to 2 hours | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.17, 1.92] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 change score (ABS, high=worse) | 2 | 380 | Mean Difference (IV, Fixed, 95% CI) | ‐3.77 [‐5.39, ‐2.16] |
| 3.2 change score (ACES, low=agitated, high=sedated) | 2 | 380 | Mean Difference (IV, Fixed, 95% CI) | ‐0.71 [‐1.15, ‐0.28] |
| 3.3 change score (PANSS‐EC, high=worse) | 1 | 261 | Mean Difference (IV, Fixed, 95% CI) | ‐2.49 [‐4.28, ‐0.70] |
| 3.4 change score (PANSS‐EC, high=worse, schizophrenia subgroup) | 2 | 263 | Mean Difference (IV, Fixed, 95% CI) | ‐2.55 [‐3.78, ‐1.32] |
| 3.5 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on ACES) | 2 | 355 | Mean Difference (IV, Fixed, 95% CI) | ‐2.68 [‐3.75, ‐1.62] |
| 3.6 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on AEs) | 2 | 365 | Mean Difference (IV, Fixed, 95% CI) | ‐2.83 [‐3.92, ‐1.75] |
| 4 Global state: 1. Various | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 non‐responders to the first injection | 1 | 263 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.34, 0.71] |
| 4.2 need for benzodiazepine up to 24 hours | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.28, 0.80] |
| 5 Global state: 2. Average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint score (CGI‐I, high=worse) | 2 | 380 | Mean Difference (IV, Fixed, 95% CI) | ‐0.73 [‐0.97, ‐0.49] |
| 5.2 endpoint score (CGI‐I, high=worse, schizophrenia subgroup | 1 | 75 | Mean Difference (IV, Fixed, 95% CI) | ‐0.71 [‐1.17, ‐0.25] |
| 5.3 change score (CGI‐S, high=worse) | 2 | 380 | Mean Difference (IV, Fixed, 95% CI) | ‐0.56 [‐0.86, ‐0.26] |
| 6 Mental state: 1. Average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 change score up to 2 hours (BPRS total, high=worse) | 1 | 110 | Mean Difference (IV, Fixed, 95% CI) | ‐3.39 [‐7.03, 0.25] |
| 6.2 change score up to 2 hours (BPRS positive subscale, high=worse) | 1 | 110 | Mean Difference (IV, Fixed, 95% CI) | ‐0.62 [‐1.65, 0.41] |
| 7 Adverse effects: 1a. General | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 one or more drug related adverse events during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.51 [0.93, 2.46] |
| 7.2 onset or increased severity of adverse effects after 2nd injection | 1 | 262 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.40 [1.36, 4.23] |
| 8 Adverse effects: 1b. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 death | 1 | 262 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 rated as serious | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [0.63, 22.60] |
| 8.3 tonic clonic seizure | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [0.14, 78.49] |
| 9 Adverse effects: 2a. Specific ‐ arousal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 insomnia during 24 hours | 1 | 262 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.25, 1.52] |
| 9.2 "over" sedated during 24 hours | 1 | 262 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.60, 4.20] |
| 9.3 somnolence during 24 hours | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 [0.57, 4.00] |
| 10 Adverse effects: 2b. Specific ‐ cardiovascular | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 dizziness during 24 hours | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.66, 4.70] |
| 10.2 sinus tachycardia during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.72] |
| 10.3 tachycardia during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.36 [0.50, 37.82] |
| 11 Adverse effects: 2c. Specific ‐ movement disorders | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 akathisia during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.61 [0.40, 144.21] |
| 11.2 dystonia during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [0.14, 78.49] |
| 11.3 EPS during 24 hours | 1 | 262 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.06, 36.45] |
| 12 Adverse effects: 2d. Specific ‐ miscellaneous | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 agitation during 24 hours | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.33, 2.37] |
| 12.2 headache during 24 hours | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [0.74, 3.71] |
| 12.3 pain at injection site | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.30, 3.94] |
| 12.4 nausea during 24 hours | 2 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.97 [1.13, 13.92] |
| 12.5 vomiting during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.18 [0.20, 23.37] |
| 13 Leaving the study early | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 any reason | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.35, 4.74] |
| 13.2 lack of efficacy | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.07, 3.06] |
| 13.3 withdrew consent | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.68 [0.23, 12.46] |
| 13.4 adverse effects | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [0.25, 20.54] |
| 13.5 other | 2 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.15, 7.44] |
Comparison 2. ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: a. HALOPERIDOL (intramuscular).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for tranquillisation | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 needing additional injections during 24 hours | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [1.00, 1.63] |
| 1.2 needing 1 additional injection during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.66, 2.70] |
| 1.3 needing 2 additional injections during 24 hours | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [0.77, 7.26] |
| 2 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐ EC reduction ≥ 40% from baseline) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 up to 2 hours | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.80, 1.11] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 change score (ABS, high=worse) | 2 | 473 | Mean Difference (IV, Fixed, 95% CI) | 0.55 [‐1.00, 2.10] |
| 3.2 change score (ACES, low=agitated, high=sedated) | 2 | 473 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.30, 0.55] |
| 3.3 change score (PANSS‐EC, high=worse) | 1 | 357 | Mean Difference (IV, Fixed, 95% CI) | 0.48 [‐1.16, 2.12] |
| 3.4 change score (PANSS‐EC, high=worse, schizophrenia subgroup) | 2 | 336 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐0.99, 1.13] |
| 3.5 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on ACES) | 2 | 422 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.82, 1.06] |
| 3.6 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on AEs) | 2 | 448 | Mean Difference (IV, Fixed, 95% CI) | 0.26 [‐0.68, 1.19] |
| 4 Global state: 1. Various | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 non‐responders to the first injection | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.78, 1.79] |
| 4.2 need for benzodiazepine up to 24 hours | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.46, 1.35] |
| 5 Global outcome: 2. Average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint score (CGI‐I, high=worse) | 2 | 473 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.19, 0.23] |
| 5.2 endpoint score (CGI‐I, high=worse, schizophrenia subgroup | 1 | 79 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.44, 0.48] |
| 5.3 change score (CGI‐S, high=worse) | 2 | 473 | Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.36, 0.22] |
| 6 Mental state: 1. Average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 change score up to 2 hours (BPRS total, high=worse) | 1 | 102 | Mean Difference (IV, Fixed, 95% CI) | 2.03 [‐1.70, 5.76] |
| 6.2 change score up to 2 hours (BPRS positive subscale, high=worse) | 1 | 103 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐0.82, 1.28] |
| 7 Adverse effects: 1a. General | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 one or more drug related adverse events during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.61, 1.35] |
| 7.2 onset or increased severity of adverse effects after 2nd injection | 1 | 358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.57, 0.96] |
| 8 Adverse effects: 1b. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 death | 1 | 358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 rated as serious | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.54, 5.52] |
| 8.3 tonic clonic seizure | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.05 [0.13, 73.38] |
| 9 Adverse effects: 2a. Specific ‐ arousal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 insomnia during 24 hours | 1 | 358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.23, 0.97] |
| 9.2 "over" sedated during 24 hours | 1 | 358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.34, 1.07] |
| 9.3 somnolence during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.41, 1.58] |
| 10 Adverse effects: 2b. Specific ‐ cardiovascular | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 dizziness during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [0.66, 3.01] |
| 10.2 sinus tachycardia during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 2.75] |
| 10.3 tachycardia during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.07 [0.47, 35.31] |
| 11 Adverse effects: 2c. Specific ‐ movement disorders | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 akathisia during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.13, 1.94] |
| 11.2 dystonia during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.03, 2.21] |
| 11.3 EPS during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.12, 0.70] |
| 12 Adverse effects: 2d. Specific ‐ miscellaneous | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 agitation during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.42, 2.58] |
| 12.2 headache during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.62, 2.18] |
| 12.3 pain at injection site | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.75, 12.91] |
| 12.4 nausea during 24 hours | 2 | 471 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.52 [1.63, 18.70] |
| 12.5 vomiting during 24 hours | 1 | 113 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.04 [0.19, 21.82] |
| 13 Leaving the study early | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 any reason | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.20, 1.18] |
| 13.2 lack of efficacy | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.15, 7.43] |
| 13.3 withdrew consent | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.10, 1.28] |
| 13.4 adverse effects | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.18, 6.04] |
| 13.5 other | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.15, 3.78] |
Comparison 3. ARIPIPRAZOLE (intramuscular) vs OTHER ANTIPSYCHOTIC: b. OLANZAPINE (intramuscular).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1a. Agitation ‐ clinically important change (PANSS‐ EC reduction ≥ 40% from baseline) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 up to 2 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.60, 0.99] |
| 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 change score (ACES, low=agitated, high=sedated) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [0.10, 1.06] |
| 2.2 change score (PANSS‐EC, high=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 3.30 [1.25, 5.35] |
| 3 Global state: 1. Average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 change score (CGI‐S, high=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [0.01, 1.15] |
| 4 Adverse effects: 1a. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 one or more drug related adverse events during 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.45, 1.24] |
| 5 Adverse effects: 1b. General ‐ serious | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 rated as serious | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Adverse effects: 2a. Specific ‐ arousal | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 somnolence during 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.08, 0.82] |
| 7 Adverse effects: 2b. Specific ‐ cardiovascular | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 systolic blood pressure change up to 2 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 2.85 [‐6.77, 12.47] |
| 7.2 diastolic blood pressure change up to 2 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐1.05 [‐8.09, 5.99] |
| 7.3 pulse rate change up to 2 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐0.67 [‐6.80, 5.46] |
| 7.4 systolic blood pressure change up to 24 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 3.37 [‐5.43, 12.17] |
| 7.5 diastolic blood pressure change up to 24 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐1.30 [‐8.55, 5.95] |
| 7.6 pulse rate change up to 24 hours | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐6.05 [‐13.43, 1.33] |
| 8 Adverse effects: 2c. Specific ‐ movement disorders ‐ i. dichotomous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 akathisia up to 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 dystonia up to 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.3 rigidity up to 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.32, 5.58] |
| 8.4 tremor up to 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.26, 8.50] |
| 9 Adverse effects: 2d. Specific ‐ movement disorders ‐ ii. average scores | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 SAS change score up to 2 hours (high=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 [‐0.26, 0.16] |
| 9.2 SAS change score up to 24 hours (high=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.29, 0.23] |
| 10 Adverse effects: 2e. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 headache during 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.19, 21.18] |
| 10.2 pain at injection site during 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.76, 9.33] |
| 10.3 nausea during 24 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.51] |
| 11 Leaving the study early | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Andrezina 2006.
| Methods | Allocation: randomised, 2:2:1 ratio. Blindness: double. Duration: 2 to 12 hours screening period, 24 hours length of study. |
|
| Participants | Diagnosis: schizophrenia, schizoaffective disorder (DSM‐IV criteria, confirmed by MINI). N = 448. Age: range 18‐69 years, mean = 41.5 years. Sex: 274 males, 174 females. History: voluntarily hospitalised patients, experiencing acute agitation, having a PEC score between 15 and 32 and with at least 2 PEC items scored ≥ 4; mean age at diagnosis = 26.3 years. Exclusion criteria: psychiatric disorders other than schizophrenia and schizoaffective disorders, significant risk of suicide, any significant medical and neurological conditions, head trauma, substance misuse or alcohol dependence, history of neuroleptic malignant syndrome, use of benzodiazepines and anticholinergics within 4 hours before the first injection, lack of response to previous antipsychotic medication. Setting: hospital, multicentre (USA, Czech Republic, France, Estonia, Latvia, Poland, Croatia, Italy, Puerto Rico, South Africa, Spain). |
|
| Interventions | 1. Aripiprazole: IM dose 9.75 mg, maximum 3 doses during the first 24 hours (mean number of IMs = 1.54); N = 175. 2. Haloperidol: IM dose 6.5 mg, maximum 3 doses during the first 24 hours (mean number of IMs = 1.43); N = 185. 3. Placebo: IM, maximum 2 doses during the first 24 hours (mean number of IMs = 1.92), if 3rd dose needed aripiprazole IM dose 9.75 mg was administered; N = 88. |
|
| Outcomes | Repeated need for tranquillisation: needing additional injections. Agitation: improvement, ABS, ACES, PANSS‐EC. Global: CGI‐I, CGI‐S, need for benzodiazepine, non‐responders to the 1st injection. Adverse effects: general, serious, specific effects (arousal, cardiovascular, movement, miscellaneous disorders). Leaving the study early. Unable to use: Mental state: PANSS‐derived BPRS (SE, SD, CI not reported). Adverse effects: SAS (SE, SD, CI not reported), BAS (SE, SD, CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised with permuted block, randomisation procedure not reported. |
| Allocation concealment (selection bias) | Low risk | "Randomization was conducted via a centralized call‐in system and was organized by study center using permuted block randomization". |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double‐blind, no further details given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of attrition bias, reasons for discontinuation are reported. |
| Selective reporting (reporting bias) | High risk | The authors only report adverse effects that occurred in ≥ 5% of people. Despite it is stated that primary outcomes were recorded at multiple time points before two hours (0.5, 0.75, 1, 1.5 hours), no numeric data were available on related publications. |
| Other bias | High risk | Sponsored by manufacturers of aripiprazole. |
Bristol‐Myers 2005.
| Methods | Allocation: randomised. Blinding: double. Duration: 24 hours (preceded by 2‐hour screening period). |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 237), schizoaffective disorder (N = 113) and schizophreniform disorder (N = 7). N = 357*. Age: range 18‐66 years. History: acute agitation, having a PEC score between 15 and 32 and with at least 2 PEC items scored ≥ 4. Exclusion criteria: substance dependence within 2 months of the study start, requiring involuntary restraint, suicidal, neurologic or psychiatric disorder other than schizophrenia, schizoaffective disorder and schizophreniform disorders, significant medical condition, known non‐responders to antipsychotic medication. Setting: hospital, multicentre (USA, Canada, Czech Republic, France, Estonia, Latvia, Lithuania, Spain). |
|
| Interventions | 1. Aripiprazole: IM dose 1 mg, up to 3 injections (mean number of IMs not reported); N = 57. 2. Aripiprazole: IM dose 5.25 mg, up to 3 injections (mean number of IMs not reported); N = 63. 3. Aripiprazole: IM dose 9.75 mg, up to 3 injections (mean number of IMs not reported); N = 57*. 4. Aripiprazole: IM dose 15 mg, up to 3 injections (mean number of IMs not reported); N = 58. 5. Haloperidol: IM dose 7.5 mg, up to 3 injections (mean number of IMs = 1.33); N = 60. 6. Placebo: IM, up to 3 injections (mean number of IMs not reported); N = 62. |
|
| Outcomes | Repeated need for tranquillisation: needing additional injections. Agitation: improvement, ABS, ACES, PANSS‐EC. Global: CGI‐I, CGI‐S, need for benzodiazepine. Mental state: PANSS‐derived BPRS. Adverse effects: general, serious, specific effects (arousal, cardiovascular, movement, miscellaneous disorders). Leaving the study early. Unable to use: QTc abnormalities not reported. SAS (SE/SD/CI not reported), BAS (SE/SD/CI not reported). |
|
| Notes | * Comparisons are shown with aripiprazole 10 mg/IM as this is the dose referred to as usual (BNF 2017). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, randomisation procedure not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No blinding was used during the preparation of injections; in most cases the same person prepared and administered the drug. Study investigators conducting the assessment were blinded to treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for leaving the study early were reported for participants who did not complete the study. However, number of participants completing different assessments at the same time point vary and reasons are not given. |
| Selective reporting (reporting bias) | High risk | The authors only report adverse effects that occurred in ≥ 5% of people.
Despite it is stated that primary outcomes were recorded at multiple time points before two hours (0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75 hours), no numeric data were available on related publications. QTc abnormalities are not reported for aripiprazole 9.75mg. |
| Other bias | High risk | Sponsored by manufacturers of aripiprazole. |
Kittipeerachon 2016.
| Methods | Allocation: randomised. Blinding: double. Duration: 24 hours. |
|
| Participants | Diagnosis: DSM 5 diagnosis of schizophrenia (N = 80). N = 80. Age: range 18‐65 years. History: acute agitation, having a PEC score ≥ 15 and 32 with at least 1 PEC item scored ≥ 5. Exclusion criteria: known allergy to aripiprazole or olanzapine, pregnant or breastfeeding. Setting: hospital (Nakhon Sawan Rajanagarindra Psychiatric Hospital, Thailand). |
|
| Interventions | 1. Aripiprazole: IM single dose 9.75 mg. N = 40. 2. Olanzapine: IM single dose 10 mg. N = 40. |
|
| Outcomes | Agitation: improvement, ACES, PANSS‐EC. Global: CGI‐S. Adverse effects: general, serious, specific effects (arousal, cardiovascular, movement disorders, miscellaneous). Leaving the study early. |
|
| Notes | * Authors state that repeated dose of intervention drugs or any kinds of parenteral medication were prohibited during the 24 hours, and that a scheduled oral regimen of antipsychotics was prescribed; table 4 (p.235) clearly shows that almost all the patients were administered with 'treatment as usual' drugs. Hence, outcomes up to 24 hours were maintained only for adverse effects. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised, "a nurse who blindly picked the name of the assigned medication from a box with equal numbers of folded papers printed with each medication" (p.232). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "The patients and the physician investigator were blinded to treatment assignment as long as the 24‐hour assessment was not completed. The assessment was performed by the investigator and the nurse who administered the injection was not involved in the assessment process of the study." (p.232). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence for attrition bias. |
| Selective reporting (reporting bias) | Low risk | No evidence for selective reporting. |
| Other bias | Low risk | None obvious. |
ABS/CABS: Corrigan Agitated Behavior Scale ACES: Agitation‐Calmness Evaluation Scale BAS: Barnes Akathisia Scale BPRS: Brief Psychiatric Rating Scale CI: Confidence Interval CGI‐I: Clinical Global Impression ‐ Improvement CGI‐S: Clinical Global Impression ‐ Severity DSM‐IV: Diagnostic and Statistical Manual, IV version IM: Intra‐Muscular MINI: Mini International Neuropsychiatric Interview PANSS: Positive And Negative Syndrome Scale PANSS‐EC: Positive And Negative Syndrome Scale ‐ Excited Component PEC: PANSS (Positive and Negative Syndrome Scale) ‐ Excited Component SAS: Simpson‐Angus Scale SD: Standard Deviation SE: Standard Error
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bao 2007 | Allocation: randomised. Participants: people with schizophrenia and agitation. Intervention: oral aripiprazole + clonazepam versus oral clozapine. |
| Chen 2009 | Allocation: randomised. Participants: schizophrenia, displaying acute agitation. Intervention: oral aripiprazole versus oral olanzapine. |
| Cutler 2006 | Allocation: randomised. Participants: schizophrenia acute relapse, not displaying aggression or agitation. |
| Fleischhacker 2009 | Allocation: randomised. Participants: schizophrenia acute relapse, not displaying acute aggression or agitation. |
| Kane 2002 | Allocation: double‐blind randomised. Participants: schizophrenia and schizoaffective disorders, not displaying acute aggression or agitation. |
| Kinon 2008 | Allocation: randomised. Participants: people with schizophrenia experiencing acute agitation. Intervention: oral aripiprazole 15 mg versus oral olanzapine 20 mg. |
| Li 2009 | Allocation: randomised. Participants: people with schizophrenia, displaying acute aggression or agitation. Intervention: oral aripiprazole versus oral aripiprazole + valproate slow‐release tablet. |
| Liang 2005 | Allocation: randomised. Participants: schizophrenia acute disorder, not displaying acute aggression or agitation. |
| Liu 2012 | Allocation: randomised. Participants: schizophrenia, displaying acute aggression or agitation. Intervention: oral aripiprazole vs oral aripiprazole + magnesium valproate. |
| McEvoy 2007 | Allocation: randomised. Participants: schizophrenia spectrum acute exacerbation, not displaying acute aggression or agitation. |
| NCT01469039 | Allocation: randomised. Participants: schizophrenia, not displaying acute aggression or agitation (except for one sub‐study). Intervention: IM aripiprazole lauroxil long acting 441 mg vs IM aripiprazole lauroxil long acting 882 mg vs IM placebo. |
| Potkin 2003 | Allocation: randomised. Participants: schizophrenia and schizoaffective disorder, not displaying acute aggression or agitation. |
| Simpson 2010 | Allocation: randomised. Participants: adults patients acutely agitated attending psychiatric clinic. Intervention: oral quetiapine 300 mg vs IM haloperidol 5 mg + lorazepam 2 mg + benztropine mesylate 2 mg. |
| Tandon 2006 | Allocation: randomised. Participants: schizophrenia and schizoaffective disorder, not displaying acute aggression or agitation. |
| Wang 2014 | Allocation: randomised. Participants: schizophrenia, displaying acute aggression and agitation. Intervention: aripiprazole + quetiapine vs aripiprazole + olanzapine vs quetiapine + olanzapine. |
| Xie 2011 | Allocation: randomised. Participants: schizophrenia, displaying acute aggression and agitation. Intervention: oral aripiprazole versus oral aripiprazole + magnesium valproate. |
IM: intramuscular.
Characteristics of studies awaiting assessment [ordered by study ID]
Daniel 2006.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 24 hours IM phase. |
| Participants | Diagnosis: schizophrenia. N = 360. Age: range 18‐60 years. Sex: not stated. History: displaying agitation. Exclusion: not stated. Setting: not stated. |
| Interventions | 1. Aripiprazole: IM dose 10 mg, maximum 3 injections during 24 hours. 2. Haloperidol: IM dose 6.5 mg, maximum 3 injections during 24 hours. |
| Outcomes | Agitation: ABS, ACES, PANSS‐EC. Global outcome: CGI‐I, CGI‐S. |
| Notes | Conference proceeding, several data are not reported. Unclear if this study refers to Andrezina 2006. |
ABS/CABS: Corrigan Agitated Behavior Scale ACES: Agitation‐Calmness Evaluation Scale AIMS: Abnormal Involuntary Movement Scale CGI‐I: Clinical Global Impression ‐ Improvement CGI‐S: Clinical Global Impression ‐ Severity IM: intramuscular PANSS‐EC: Positive And Negative Syndrome Scale ‐ Excited Component
Characteristics of ongoing studies [ordered by study ID]
Istikoglou 2010.
| Trial name or title | |
| Methods | Allocation: randomised. Blindness: rater‐blind. Duration: not stated. |
| Participants | Diagnosis: psychosis. N = 120. Age: not stated. Sex: not stated. History: displaying agitation. Exclusion: not stated. Setting: patients with psychotic agitation presenting at emergency room of two Greek psychiatric departments ("Konstantopouleion" and "Asklepeion" General Hospitals, Athens). |
| Interventions | 1. Aripiprazole: IM unknown dose. 2. Haloperidol: IM unknown dose. |
| Outcomes | Global outcome: CGI‐I, QoL. Mental state: YMRS, PANSS. Adverse effects: AIMS, SAS. |
| Starting date | |
| Contact information | |
| Notes | Conference proceeding, several data are not reported. |
AIMS: Abnormal Involuntary Movement Scale CGI‐I: Clinical Global Impression ‐ Improvement IM: intramuscular PANSS: Positive And Negative Syndrome Scale QoL: Quality of Life SAS: Simpson Angus Scale YMRS: Young Mania Rating Scale
Differences between protocol and review
Title change: addition of 'intramuscular' and 'rapid tranquillisation' to title.
Objectives: slight change in wording to clarify intervention
Protocol objectives: To evaluate the effects of intramuscular aripiprazole in the treatment of psychosis‐induced agitation and aggression.
Review objectives: To evaluate the effects of intramuscular (IM) aripiprazole in the treatment of psychosis‐induced aggression and agitation (rapid tranquillisation).
Use of new methods template. Since publication of the protocol, Cochrane methodology has evolved (for example, the inclusion of 'Summary of findings' tables and GRADE). The Cochrane Schizophrenia group maintain a methods template for use in their reviews. We have updated our methods section with this template.
Outcomes: We have clarified the timings of the secondary outcome ‐ not tranquil or asleep to include > 30 minutes (IM) or > 60 minutes (orally) to distinguish it from our pre‐stated primary outcome of not tranquil or asleep by 30 minutes (IM) or 60 minutes (orally). We have renamed global outcomes to global state, and some outcomes are now measured as 'improvement' rather than 'no improvement'. This is in line with the methods template of the Cochrane Schizophrenia Group where avoidance of double negatives is encouraged. We have clarified the 'Summary of findings' outcomes list.
Contributions of authors
EGO: screened and retrieved papers against eligibility criteria, appraised quality of papers, extracted data from papers, entered data into RevMan and analysed data, interpreted data and took the lead in writing and finalising the review (2014 search, 2017 search).
Salwan Jajawi: study selection, data extraction, entered data into RevMan and analysed data, and writing up first draft of review (2014 search).
Styliani Spyridi: help with study selection, help with extraction (2014 search).
Kamlaj Sayal: help with study selection (2014 search).
Mahesh Jayaram ‐ developing and writing up (protocol only).
Sources of support
Internal sources
-
Università degli Studi di Milano, Milan, Italy.
Employs lead author Edoardo G Ostinelli.
-
Rotherham, Doncaster and South Humber NHS Foundation Trust, Rotherham, UK.
Employs review author Salwan Jajawi.
-
Cyprus University of Technology, Lemesos, Cyprus.
Employs review author Styliani Spyridi.
-
UK LLP, Dewsbury, UK.
Employs review author Styliani Spyridi.
-
Cygnet Hospital Derby, UK.
Employs review author Kamlaj Sayal.
-
Melbourne Neuropsychiatry Centre, Melbourne, Australia.
Employs review author Mahesh B Jayaram.
External sources
No sources of support supplied
Declarations of interest
Edoardo G Ostinelli: none known. Salwan Jajawi: none known. Styliani Spyridi: has received financial support from pharmaceutical companies to attend scientific conferences, no direct conflict of interest for this review. Kamlaj Sayal: none known. Mahesh Jayaram: none known.
New
References
References to studies included in this review
Andrezina 2006 {published data only}
- Andrezina R, Josiassen RC, Marcus RN, Oren DA, Manos G, Stock E, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double‐blind, placebo‐controlled comparison with intramuscular haloperidol. Psychopharmacology 2006;188(3):281‐92. [EMBASE: 2006450840; MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Andrezina R, Marcus RN, Oren DA, Manos G, Stock E, Carson WH, et al. Intramuscular aripiprazole or haloperidol and transition to oral therapy in patients with agitation associated with schizophrenia: sub‐analysis of a double‐blind study. Current Medical Research and Opinion 2006;22(11):2209‐19. [EMBASE: 2006574726; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Bristol‐Myers S. A randomized, double‐blind comparison of the efficacy and safety of aripiprazole intramuscular formula, haloperidol, or placebo in the treatment of acutely agitated patients with a diagnosis of schizophrenia or schizoaffective disorder. http://www.Clinicalstudyresults.org/ 2004.
- Citrome LL, Vester‐Blokland E, Archibald D, McQuade R, Kostic D, Pikalov A, et al. Benefits of a second dose of intramuscular (IM) aripiprazole to control agitation in patients with schizophrenia or bipolar I disorder. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
- Currier GW, Crandall D, Archibald D, Nyilas M, Kostic D, Pikalov A, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar disorder without excessive sedation. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
- Daniel DG, Currier GW, Zimbroff DL, Allen MH, Oren D, Manos G, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. Journal of Psychiatric Practice 2007;13(3):170‐7. [CINAHL: 2009612027; EMBASE: 2007257551; MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
- Dillenschneider A, Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, et al. Comparison of intramuscular aripiprazole and haloperidol with placebo in acutely agitated schizophrenia patients. European Neuropsychopharmacology 2005;15(Suppl.3):S509. [Google Scholar]
- Dillenschneider A, Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a pivotal phase III study. 18th European College of Neuropsychopharmacology Congress; 2005 October 22‐26, Amsterdam, Netherlands 2005.
- McQuade R, Auby P, Oren D, Marcus R, Kostic D, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a double‐blind, placebo‐controlled comparison with IM haloperidol. Proceedings of the 13th Biennial Winter Workshop on Schizophrenia Research; 2006 Feb 4‐10; Davos, Switzerland. Davos, Switzerland: Elsevier Science Bv, 2006:57‐8.
- Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, Archibald D. Efficacy and safety of intramuscular aripiprazole, haloperidol or placebo in acutely agitated patients with schizophrenia or schizoaffective disorder. 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26, Atlanta, Georgia, USA 2005.
- Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, Archibald D. Efficacy and safety of intramuscular aripiprazole, haloperidol or placebo in acutely agitated patients with schizophrenia or schizoaffective disorder. Schizophrenia Bulletin 2005;31:499. [116266] [Google Scholar]
- Oren DA, Marcus RN, Manos G, Carson WH, McQuade D. Non‐inferiority of intramuscular aripiprazole versus intramuscular haloperidol for the treatment of agitation associated with schizophrenia/schizoaffective disorder. Schizophrenia Bulletin 2007;33(2):451. [Google Scholar]
- Yocca F, Marcus R, Oren D, Manos G, Carson W, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a pivotal phase III study. 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26, Atlanta, Georgia, USA 2005.
Bristol‐Myers 2005 {published data only}
- Bristol‐Myers Squibb. Randomized, double‐blind, dose‐ranging study of intramuscular aripiprazole in the treatment of acute agitation in patients with a diagnosis of schizophrenia, schizoaffective, or schizophreniform disorder. http://www.clinicalstudyresults.org/ 2005.
- Daniel DG, Stock EG, Wilber CH, Marcus RN, Carson WH Jr, Manos G, et al. Intramuscular aripiprazole in acutely agitated psychotic patients. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, New York, USA. 2004.
- Gismondi R, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Intramuscular aripiprazole treatment for acute agitation in patients with psychosis. European Neuropsychopharmacology 2004;14(Suppl.3):S261. [Google Scholar]
- Kungel M, Czaniera R, Ebrecht M, Werner C, Oren D, McQuade R, et al. Intramuscular aripiprazole for the treatment of acute agitation accompanied with schizophrenia: Subanalysis of a double‐blind controlled study to find the dosage. Nervenarzt 2007;78:77. [Google Scholar]
- Kungel M, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Efficacy and safety of intramuscular aripiprazole in acutely agitated patients with psychosis. Thematic conference of the World Psychiatric Association on "Treatments in Psychiatry: an update". Florence, Italy, November 10‐13 2004.
- Kungel M, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Efficacy and safety of intramuscular aripiprazole in acutely agitated patients with psychosis. World Psychiatry 2004;3(S1):218. [Google Scholar]
- Modell S, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Intramuscular aripiprazole treatment for acute agitation in patients with psychosis. XXIVth Collegium Internationale Neuro‐Psychopharmacologicum Congress; 2004 June 20–24, Paris, France 2004.
- Oren DA, Manos G, Markovic O, McQuade RD. Intramuscular aripiprazole for the treatment of acute agitation associated with schizophrenia: Sub‐analysis of a double‐blind, controlled, dose‐ranging study. European Psychiatry 2007;22:S124‐S. [Google Scholar]
- Tran‐Johnson TK, Sack DA, Marcus RN, Auby P, McQuade RD, Oren DA. Efficacy and safety of intramuscular aripiprazole in patients with acute agitation: a randomized, double‐blind, placebo‐controlled trial. Journal of Clinical Psychiatry 2007;68(1):111‐9. [EMBASE: 2007077930; MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
- Yocca F, Daniel D, Stock E, Oren D, Marcus R, Carson W, et al. Efficacy and safety of intramuscular aripiprazole treatment for acute agitation in patients with psychosis. 43rd Annual Meeting of the American College of Neuropsychopharmacology; 2004 Dec 12‐16; San Juan, Puerto Rico. 2004.
Kittipeerachon 2016 {published data only}
- Kittipeerachon M, Chaichan W. Intramuscular olanzapine versus intramuscular aripiprazole for the treatment of agitation in patients with schizophrenia: A pragmatic double‐blind randomized trial. Schizophrenia Research 2016;176(2‐3):231‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bao 2007 {published data only}
- Bao L‐Y, Wang K‐M. The effectiveness of aripirazole combined with clonazepam treated with agitation in schizophrenia. Medical Journal of Chinese People's Health 2007;19(3):198‐200. [CHINESE: Academic Journals] [Google Scholar]
Chen 2009 {published data only}
- 陈远岭, 黄文武, 叶鑫武. Aripiprazole treatment of agitation associated with schizophrenia] Google Translate [阿立哌唑治疗伴激越的精神分裂症对照研究 []. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2009;19(4):261‐2. [Google Scholar]
Cutler 2006 {published data only}
- Cutler AJ, Marcus RN, Hardy SA, O'Donnell A, Carson WH, McQuade RD. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectrums 2006;11(9):691‐702. [EMBASE: 2006471567; MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fleischhacker 2009 {published data only}
- Fleischhacker WW, McQuade RD, Marcus RN, Archibald D, Swanink R, Carson WH. A double‐blind, randomized comparative study of aripiprazole and olanzapine in patients with schizophrenia. Biological Psychiatry 2009;65(6):510‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kane 2002 {published data only}
- Anutosh S, Ali MW, Ingenito G, Carson WH. Controlled study of aripiprazole and haloperidol in schizophrenia. European Psychiatry 2002;17(Suppl 1):103s. [Google Scholar]
- Carson WH, Ali M, Saha AR, Dunbar GC, Ingenito G. A double‐blind, placebo‐controlled trial of aripiprazole and haloperidol in patients with schizophrenia or schizoaffective disorder. 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 Dec 10‐14; San Juan, Puerto Rico. 2000. [XXIIND: C.I.N.P. [CD‐ROM]: Conifer, Excerpta Medica Medical Communications BV, 2000]
- Kane JM, Carson WH, Saha AR, McQuade RD, Ingenito GG, Zimbroff DL, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. Journal of Clinical Psychiatry 2002;63(9):763‐71. [2002343924] [DOI] [PubMed] [Google Scholar]
- Kane JM, Ingenito G, Ali M. Efficacy of aripiprazole in psychotic disorders: comparison with haloperidol and placebo. 153rd Annual Meeting of the American Psychiatric Association; 2000 May 13‐18; Chicago, Illinois, USA. 2000. [153RD: Annual Meeting of the American Psychiatric Association [CD‐ROM]: MARATHON Multimedia, 2000]
- Kane JM, Ingenito G, Ali M. Efficacy of aripiprazole in psychotic disorders: comparison with haloperidol and placebo. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, Pennsylvania, USA. 2002.
Kinon 2008 {published data only}
- Eli Lilly, Company. Olanzapine versus aripiprazole in the treatment of acutely ill patients with schizophrenia. Eli Lilly and Company Clinical Trial Registry 2004.
- Kinon BJ, Stauffer VL, Kollack‐Walker S, Chen L, Sniadecki J. Olanzapine versus aripiprazole for the treatment of agitation in acutely ill patients with schizophrenia. Journal of Clinical Psychopharmacology 2008;28(6):601‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- NCT00103571. Olanzapine versus aripiprazole in the treatment of acutely ill patients with schizophrenia. http://www.clinicaltrials.gov 2005. [DOI] [PubMed]
- Stauffer V, Kinon B, Kollack‐Walker S, Chen L, Sniadecki J. Olanzapine versus aripiprazole for the treatment of agitation in acutely ill patients with schizophrenia. 161st Annual Meeting of the American Psychiatric Association; 2008 May 3‐8, Washington DC, USA. 2008. [NR4‐109] [DOI] [PubMed]
Li 2009 {published data only}
- 李树生. Aripiprazole for schizophrenia combined magnesium valproate efficacy of impulse control disorders] Google Translate [阿立哌唑联合丙戊酸镁对精神分裂症冲动控制障碍的疗效观察]. Chongqing Medical Journal 2009;38(7):788‐9. [Google Scholar]
Liang 2005 {published data only}
- Liang S. A multi‐center, randomized, double‐blind study, comparing with risperidone, to evaluate the efficacy and safety of aripiprazole in the treatment of patients with schizophrenia. http:,www.clinicaltrials.gov 2005.
Liu 2012 {published data only}
- 刘杰. 丙戊酸镁对精神分裂症兴奋激越行为的辅助疗效观察. 社区医学杂志 2012;10(14):31‐2. [Google Scholar]
McEvoy 2007 {published data only}
- McEvoy JP, Daniel DG, Carson WH Jr, McQuade RD, Marcus RN. A randomized, double‐blind, placebo‐controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. Journal of Psychiatric Research 2007;41(11):895‐905. [EMBASE: 2007402756; MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
NCT01469039 {published data only}
- Citrome L, Du Y, Risinger R, Stankovic S, Claxton A, Zummo J, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. International Clinical Psychopharmacology 2016;31(2):69‐75. [DOI] [PubMed] [Google Scholar]
- Citrome L, Du Y, Risinger R, Stankovic S, Claxton A, Zummo J, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia: Erratum. International Clinical Psychopharmacology 2017;32(1):56. [DOI] [PubMed] [Google Scholar]
- Hard M, Stankovic S, Risinger R, Bose A, Du Y, Zummo J, et al. Efficacy of aripiprazole lauroxil, a new long‐acting injectable atypical antipsychotic, across three geographic regions. Neuropsychopharmacology 2014;39:S359‐60. [Google Scholar]
- Meltzer HY, Nasrallah HA, Risinger R, Du Y, Zummo J, Corey L, et al. Aripiprazole lauroxil: an innovative long‐acting injectable in development for treatment of schizophrenia. 168th Annual Meeting of American Psychiatric Association; Toronto, Canada; May 16‐20, 2015. 2015.
- Meltzer HY, Risinger R, Nasrallah HA, Du Y, Zummo J, Corey L, et al. A randomized, double‐blind, placebo‐controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. Journal of Clinical Psychiatry 2015;76(8):1085‐90. [DOI] [PubMed] [Google Scholar]
- NCT01469039. A study to evaluate the efficacy and safety of alks 9072 (also known as alks 9070) in subjects with schizophrenia. http://ClinicalTrials.gov/show/NCT01469039 2011.
- Nasrallah HA, Newcomer JW, Risinger R, Du Y, Zummo J, Bose A, et al. Effect of aripiprazole lauroxil on metabolic and endocrine profiles and related safety considerations among patients with acute schizophrenia. Journal of Clinical Psychiatry 2016;77(11):1519‐25. [DOI] [PubMed] [Google Scholar]
- Targum SD, Risinger R, Du Y, Pendergrass JC, Jamal HH, Silverman BL. Effect of patient age on treatment response in a study of the acute exacerbation of psychosis in schizophrenia [2017 Jan;179:64‐69.]. Schizophrenia Research 2017;179:64‐9. [DOI] [PubMed] [Google Scholar]
Potkin 2003 {published data only}
- Carson WH, Saha AR, Ali M, Dunbar GC, Ingenito G. Aripiprazole and risperidone versus placebo in schizophrenia and schizoaffective disorder. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans, Louisiana, USA. Marathon Multimedia, 2001. [11TH: Congress of the European College of Neuropsychopharmacology [Congress Information System]: Conifer, Excerpta Medica Medical Communications BV, 2001]
- Carson WHJ, Saha AR, Ali M, Dunbar GC, Ingenito G. Aripiprazole and risperidone versus placebo in schizophrenia and schizoaffective disorder. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, Pennsylvania, USA. 2002.
- Potkin SG, Kujawa M, Carson WH, Saha AR, Ali M, Ingenito G. Aripiprazole and risperidone versus placebo in schizophrenia and schizoaffective disorder. Schizophrenia Research 2003;60:300. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Saha AR, Kujawa MJ, Carson WH, Ali M, Stock E, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Archives of General Psychiatry 2003;60(7):681‐90. [EMBASE: 2003275542; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Saha A, Carson W, Ali M, Dunbar G, Ingenito G. Efficacy and safety of aripiprazole and risperidone vs. placebo in patients with schizophrenia and schizoaffective disorder. 7th World Congress of Biological Psychiatry; 2001 Jul 1‐6; Berlin, Germany. 2001; Vol. 2, issue Suppl 1. [7TH: World Congress of Biological Psychiatry [CD‐ROM]: Conifer, Excerpta Medica Medical Communications BV, 2001]
- Yeung P, Kujawa M, Carson WH, Saha A, Alid M, Ingenito G. Aripiprazole and risperidone versus placebo in schizophrenia and schizoaffective disorder. Schizophrenia Research 2002;53(3 Suppl 1):185‐6. [11TH: Congress of the European College of Neuropsychopharmacology [Congress Information System]: Conifer, Excerpta Medica Medical Communications BV, 2001] [Google Scholar]
- Yeung P, McQuade RD, Carson WH, Saha A, Ali MW, Ingenito G. Aripiprazole and risperidone versus placebo in schizophrenia. European Psychiatry 2002;17(Suppl 1):102s. [Google Scholar]
Simpson 2010 {published data only}
- Simpson G, Mohammad AR, Fauzia S, Campaneau M, Laska E, Ninah R, et al. A comparison of rapid titration quetiapine and haloperidol in agitated adults in an emergency setting. Proceedings of the 163rd Annual Meeting of the American Psychiatric Association; 2010 May 22‐26; New Orleans, LA. 2010.
Tandon 2006 {published data only}
- Tandon R, Marcus RN, Stock EG, Riera LC, Kostic D, Pans M, et al. A prospective, multicenter, randomized, parallel‐group, open‐label study of aripiprazole in the management of patients with schizophrenia or schizoaffective disorder in general psychiatric practice: Broad Effectiveness Trial With Aripiprazole (BETA). Schizophrenia Research 2006;84(1):77‐89. [EMBASE: 2006399870; MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wang 2014 {published data only}
- 王琦, 周丹, 李萍, 谢红涛. 非典型抗精神病药对精神分裂症患者激越行为的随机对照研究. 中国健康心理学杂志 2014;22(3):325‐7. [Google Scholar]
Xie 2011 {published data only}
- 颉瑞, 王刚平, 裴根祥. Clinical observation of magnesium valproate sustained release tablets adjuvant treatment of schizophrenia attacks [丙戊酸镁缓释片辅助治疗精神分裂症攻击行为的疗效观察]. 临床精神医学杂志 2011;21(03):191‐2. [Google Scholar]
References to studies awaiting assessment
Daniel 2006 {published data only}
- Daniel DG, Crandall D, Manos G, McQuade RD, Gutierrez‐Esteinou R, Pikalov AA, et al. Transitioning from intramuscular (IM) to oral aripiprazole in patients with schizophrenia. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
References to ongoing studies
Istikoglou 2010 {published data only}
- Istikoglou C, Vlavianou A, Foutsitzis D, Theodorakopoulos G, Polonifis C, Kanellos P, et al. Intramuscular aripiprazole versus injectable haloperidol in treatment of acute psychotic agitation. European Neuropsychopharmacology 2010;20(3):S469‐70. [Google Scholar]
Additional references
Abilify 2017
- Abilify (Aripiprazole). US full prescribing information. http://otsuka‐us.com/products/Documents/Abilify.PI.pdf?_ga=1.242215874.1724823722.1491413596 (accessed 5 April 2017).
Ahmed 2010
- Ahmed U, Jones H, Adams CE. Chlorpromazine for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD007445.pub2; PUBMED: 20393959] [DOI] [PubMed] [Google Scholar]
Ahmed 2011
- Ahmed U, Rehman F, Jones H, Adams CE. Risperidone for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD009412] [DOI] [PMC free article] [PubMed] [Google Scholar]
Alexander 2004
- Alexander J, Tharyan P, Adams CE, John T, Mol C, Philip J. Rapid tranquillisation of violent or agitated patients in a psychiatric emergency setting: Pragmatic randomised trial of intramuscular lorazepam v. haloperidol plus promethazine. British Journal of Psychiatry 2004;185:63‐9. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7068):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2006
- American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, 2nd Edition. Practice guideline for the treatment of patients with schizophrenia 2006.
Belgamwar 2005
- Belgamwar RB, Fenton M. Olanzapine IM or velotab for acutely disturbed/agitated people with suspected serious mental illnesses. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD003729.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berk 2004
- Berk M, Rathbone J, Mandriota‐Carpenter SL. Clotiapine for acute psychotic illnesses. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD002304.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Binder 1999
- Binder RL, McNiel DE. Contemporary practices in managing acutely violent patients in 20 psychiatric emergency rooms. Psychiatric Services 1999;50(12):1553‐4. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Trials randomised in clusters. BMJ 1997;315(7108):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
BNF 2014
- BNF. 4.2.1 Antipsychotic drugs. BNF 67 March‐September 2014. London: BMJ Group AND Pharmaceutical Press, 2014:235. [Google Scholar]
BNF 2017
- British National Formulary. British National Formulary. 73th. NHS, London: Pharmaceutical Press, 2017. [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Breier 2001
- Breier A, Meehan K, Birkett M, David S, Ferchland I, Sutton V, et al. A double‐blind, placebo‐controlled dose‐response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Archives of General Psychiatry 2002;59(5):441‐8. [DOI] [PubMed] [Google Scholar]
Corrigan 1989
- Corrigan JD. Development of a scale for assessment of agitation following traumatic brain injury. Journal of Clinical and Experimental Neuropsychology 1989;11(2):261‐77. [DOI] [PubMed] [Google Scholar]
CPA 2005
- Addington D, Bouchard RH, Goldberg J, Honer B, Malla A, Norman R, et al. Clinical practice guidelines: Treatment of schizophrenia. Canadian Journal of Psychiatry 2005;50(Suppl 1):3S‐56S. [Google Scholar]
Cunane 1994
- Cunnane JG. Drug management of disturbed behaviour by psychiatrists. Psychiatric Bulletin 1994;18(3):138‐9. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21(19):2971‐80. [DOI] [PubMed] [Google Scholar]
Douglas‐Hall 2015
- Douglas‐Hall P, Whicher EV. 'As required' medication regimens for seriously mentally ill people in hospital. Cochrane Database of Systematic Reviews 2015, Issue 12. [DOI: 10.1002/14651858.CD003441.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Du 2017
- Du M, Wang X, Yin S, Shu W, Hao R, Zhao S, et al. De‐escalation techniques for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2017, Issue 4. [DOI: 10.1002/14651858.CD009922.pub2; CD009922] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
FDA 2004
- US Federal Drug Administration CDER. Application number: NDA 21‐436/S‐002 (accessed 5 April 2017). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021436Orig1s002.pdf 2004.
Ferracuti 1993
- Ferracuti S, Palermo GB, Manfredi M. Homicide between inpatients in a mental institution ward. International Journal of Offender Therapy and Comparative Criminology 1993;37:331‐7. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gillies 2015
- Gillies D, Sampson S, Beck A, Rathbone J. Benzodiazepines for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD003079.pub3] [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumonne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England. American Journal of Epidemiology 1999;149(9):876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Clinical Global Impression. The ECDEU Assessment Manual for Psychopharmacology‐Revised, National Institute of Mental Health 1976:217–21.
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Huf 2002a
- Huf G, Coutinho ESF, Fagundes HM Jr, Oliveira ES, Lopez JRRA, Gewandszajder M, et al. Current practices in managing acutely disturbed patients at three hospitals in Rio de Janeiro‐Brazil: a prevalence study. BMC Psychiatry 2002;2(4):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huf 2002b
- Huf G, Coutinho ESF, Adams CE. TREC‐Rio trial: a randomised controlled trial for rapid tranquillisation for agitated patients in emergency psychiatric rooms. BMC Psychiatry 2002;2(11):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huf 2016
- Huf G, Alexander J, Gandhi P, et al. Haloperidol plus promethazine for psychosis‐induced aggression. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD005146.pub3; CD005146] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Jayakody 2012
- Jayakody K, Gibson RC, Kumar A, Gunadasa S. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD000525.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opfer LA. The Positive and Negative Syndrome Scale (PANSS) for Schizophrenia. Schizophrenia Bulletin 1987;13(2):261–76. [DOI] [PubMed] [Google Scholar]
Khokhar 2016
- Khokhar MA, Rathbone J. Droperidol for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD002830.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khushu 2016
- Khushu A, Powney MJ. Haloperidol for long‐term aggression in psychosis. Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd, 2016, issue 11. [DOI: 10.1002/14651858.CD009830.pub2; CD009830] [DOI] [PMC free article] [PubMed]
Kikuchi 1995
- Kikuchi T, Tottori K, Uwahodo Y, Hirose T, Miwa T, Oshiro Y, et al. 7‐(4‐[4‐(2,3‐Dichlorophenyl)‐1‐piperazinyl]butyloxy)‐3,4‐dihydro‐2(1H)‐quinolinone (OPC‐14597), a new putative antipsychotic drug with both presynaptic dopamine autoreceptor agonistic activity and postsynaptic D2 receptor antagonistic activity. Journal of Pharmacology and Experimental Therapeutics 1995;274(1):329‐36. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Lingjaerde 1987
- Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychiatrica Scandinavica. Supplementum 1987;334:1‐100. [DOI] [PubMed] [Google Scholar]
Makins 1994
- Makins M. Collins Compact English Dictionary. 3rd Edition. Glasgow: Harpercollins Publishers, 1994:15. [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
McGavin 2002
- McGavin JK, Goa KL. Aripiprazole. CNS Drugs 2002;16(11):779‐86. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomised trials. Lancet 2001;357(9263):1191‐4. [PubMed] [Google Scholar]
Mohr 2005
- Mohr P, Pecenak J, Svestka J, Swingler D, Treuer T. Treatment of acute agitation in psychotic disorders. Neuro Endocrinology Letters 2005;26(4):327‐35. [PubMed] [Google Scholar]
Montoya 2011
- Montoya A, Valladares A, Lizán L, San L, Escobar R, Paz S. Validation of the Excited Component of the Positive and Negative Syndrome Scale (PANSS‐EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Quality Life Outcomes 2011;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muralidharan 2006
- Muralidharan S, Fenton M. Containment strategies for people with serious mental illness. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD002084.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
NHS 2014
- NHS prescription services. http://www.nhsbsa.nhs.uk/prescriptions (accessed 12 December 2014).
NICE 2015
- National Institute for Clinical Excellence. Violence and aggression: short‐term management in mental health, health and community settings. nice.org.uk/guidance/ng10 2015.
Ostinelli 2017
- Ostinelli EG, Brooke‐Powney MJ, Li X, Adams CE. Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation). Cochrane Database of Systematic Reviews 2017, Issue 7. [DOI: 10.1002/14651858.CD009377.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Raveendran 2007
- Raveendran NS, Tharyan P, Alexander J, Adams CE and the TREC‐India II Collaborative Group. Rapid tranquilization of violent or agitated people in psychiatric emergency settings: A pragmatic randomised controlled trial of intramuscular olanzapine versus intramuscular haloperidol plus promethazine. BMJ OCT 2007;335(7265):865‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sailas 2000
- Sailas E, Fenton M. Seclusion and restraint for people with serious mental illnesses. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001163] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schleifer 2011
- Schleifer JJ. Management of acute agitation in psychosis: an evidence‐based approach in the USA. Advances in Psychiatric Treatment 2011;17:91‐100. [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Shapiro 2003
- Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28(8):1400‐11. [DOI] [PubMed] [Google Scholar]
Simpson 1970
- Simpson GM, Angus JSW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica. Supplementum 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Swainston Harrison 2004
- Swainston Harrison T, Perry CM. Aripiprazole: a review of its use in schizophrenia and schizoaffective disorder. Drugs 2004;64(15):1715‐36. [DOI] [PubMed] [Google Scholar]
Tardiff 1982
- Tardiff K, Sweillam A. Assaultive behavior among chronic inpatients. American Journal of Psychiatry 1982;139(2):212‐5. [DOI] [PubMed] [Google Scholar]
Tardiff 1985
- Tardiff K, Koenigsberg HW. Assaultive behavior among psychiatric outpatients. American Journal of Psychiatry 1985;142:960‐3. [DOI] [PubMed] [Google Scholar]
Toal 2012
- Toal F, Roberts K. Clozapine for people with psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012.
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: A systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Vangala 2012
- Vangala R, Ahmed U, Ahmed R. Loxapine inhaler for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD010190] [DOI] [Google Scholar]
Wilkie 2012
- Wilkie F, Fenton M. Quetiapine for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD009801] [DOI] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Pagadala 2009
- Pagadala B, Jayaram MB, Mitra L. Aripiprazole for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD008074] [DOI] [Google Scholar]