

Abstract
Background
Antipsychotic (neuroleptic) medication is used extensively to treat people with chronic mental illnesses. Its use, however, is associated with adverse effects, including movement disorders such as tardive dyskinesia (TD) – a problem often seen as repetitive involuntary movements around the mouth and face. Vitamin E has been proposed as a treatment to prevent or decrease TD.
Objectives
The primary objective was to determine the clinical effects of vitamin E in people with schizophrenia or other chronic mental illness who had developed antipsychotic‐induced TD.
The secondary objectives were: 1. to examine whether the effect of vitamin E was maintained as duration of follow‐up increased; 2. to test the hypothesis that the use of vitamin E is most effective for those with early onset TD (less than five years)
Search methods
We searched the Cochrane Schizophrenia Group Trials Register (July 2015 and April 2017), inspected references of all identified studies for further trials and contacted authors of trials for additional information.
Selection criteria
We included reports if they were controlled trials dealing with people with antipsychotic‐induced TD and schizophrenia who remained on their antipsychotic medication and had been randomly allocated to either vitamin E or to a placebo, no intervention, or any other intervention.
Data collection and analysis
We independently extracted data from these trials and we estimated risk ratios (RR) or mean differences (MD), with 95% confidence intervals (CI). We assumed that people who left early had no improvement. We assessed risk of bias and created a 'Summary of findings' table using GRADE.
Main results
The review now includes 13 poorly reported randomised trials (total 478 people), all participants were adults with chronic psychiatric disorders, mostly schizophrenia, and antipsychotic‐induced TD. There was no clear difference between vitamin E and placebo for the outcome of TD: not improved to a clinically important extent (6 RCTs, N = 264, RR 0.95, 95% CI 0.89 to 1.01, low‐quality evidence). However, people allocated to placebo may show more deterioration of their symptoms compared with those given vitamin E (5 RCTs, N = 85, RR 0.23, 95% CI 0.07 to 0.76, low‐quality evidence). There was no evidence of a difference in the incidence of any adverse effects (9 RCTs, N = 205, RR 1.21, 95% CI 0.35 to 4.15, very low‐quality evidence), extrapyramidal adverse effects (1 RCT, N = 104, MD 1.10, 95% CI ‐1.02 to 3.22, very low‐quality evidence), or acceptability of treatment (measured by participants leaving the study early) (medium term, 8 RCTs, N = 232, RR 1.07, 95% CI 0.64 to 1.80, very low‐quality evidence). No trials reported on social confidence, social inclusion, social networks, or personalised quality of life, outcomes designated important to patients. There is no trial‐based information regarding the effect of vitamin E for those with early onset of TD.
Authors' conclusions
Small trials of limited quality suggest that vitamin E may protect against deterioration of TD. There is no evidence that vitamin E improves symptoms of this problematic and disfiguring condition once established. New and better trials are indicated in this under‐researched area, and, of the many adjunctive treatments that have been given for TD, vitamin E would be a good choice for further evaluation.
Plain language summary
Vitamin E for antipsychotic‐induced tardive dyskinesia
Review question
Is vitamin E useful for treating an unpleasant side effect of taking antipsychotics ‐ tardive dyskinesia ‐ in people with schizophrenia or other similar mental illnesses?
Background
People with schizophrenia often hear voices and see things (hallucinations) and have strange beliefs (delusions). These symptoms are usually treated with antipsychotic drugs. However, these drugs can have debilitating side effects. Tardive dyskinesia is an involuntary movement that causes the face, mouth, tongue and jaw to convulse, spasm and grimace. It is caused by long‐term or high‐dose use of antipsychotic drugs, is difficult to treat and can be incurable. Vitamin E has been suggested as a treatment, but so far the benefit of using Vitamin E for this purpose seems small.
Study characteristics
We searched for trials, July 2015 and April 2017, using the Cochrane Schizophrenia Group's register of trials. The review includes 13 poorly reported randomised trials investigating the effects of vitamin E for people with schizophrenia or other chronic mental illnesses who also developed TD as a result of taking antipsychotics. The trials randomised a total of 478 participants who had been ill for a long time.
Key results
Vitamin E may protect against tardive dyskinesia. However, there is no clear evidence that vitamin E improves this problematic and disfiguring condition.
Quality of the evidence
Available evidence is weak, limited and poor and we are unable to make any conclusions regarding the use of Vitamin E for antipsychotic‐induced tardive dyskinesia. Well‐designed trials involving a large number of participants investigating the effects of vitamin E over long periods of time are needed to determine whether this vitamin provides an effective treatment option for tardive dyskinesia.
This plain language summary was adapted by the review authors from a summary originally written by Ben Gray, Senior Peer Researcher, McPin Foundation (http://mcpin.org/).
Summary of findings
Summary of findings for the main comparison. VITAMIN E compared with PLACEBO for antipsychotic‐induced tardive dyskinesia.
| VITAMIN E compared with PLACEBO for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: patients with various chronic psychiatric disorders, but mainly schizophrenia, and antipsychotic‐induced tardive dyskinesia Settings: in hospital and outpatients in China (1 study), Hong Kong (1), Israel (2), India (1), Switzerland (1), the UK (1), and the USA (6). Intervention: VITAMIN E Comparison: PLACEBO | ||||||
| Outcomes | Illustrative comparative risks* (CI) | Relative effect (CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PLACEBO | VITAMIN E | |||||
| Tardive dyskinesia: 1. Not improved to a clinically important extent Follow‐up: up to 1 year | 970 per 1000 |
921 per 1000 (863 to 979) |
RR 0.95 (0.89 to 1.01) | 264 (6 studies) | ⊕⊕⊝⊝ low1,2 | |
| Tardive dyskinesia: 3. Deterioration of symptoms Follow‐up: up to 1 year | 263 per 1000 | 61 per 1000 (18 to 200) |
RR 0.23 (0.07 to 0.76) |
85 (5 studies) | ⊕⊕⊝⊝ low1,2 | |
|
Adverse effect: any Follow‐up: up to 1 year |
21 per 1000 | 25 per 1000 (7 to 86) |
RR 1.21 (0.35 to 4.15) |
205 (9 studies) | ⊕⊝⊝⊝ very low3,4 | |
|
Adverse effect: extrapyramidal symptoms measured by Simpson‐Angus Scale (low = better) Follow‐up: up to 1 year |
The mean extrapyramidal adverse events in the intervention groups was 1.10 points higher (1.02 lower to 3.22 higher) | 104 (1 study) | ⊕⊝⊝⊝ very low1,3 | |||
|
Acceptability of treatment (measured by participants leaving the study early) Follow‐up: up to 1 year |
168 per 1000 | 180 per 1000 (108 to 303) |
RR 1.07 (0.64 to 1.80) |
232 (8 studies) | ⊕⊝⊝⊝ very low2,3,5 | This outcome was designated to be of importance at 2017 update. |
| Social confidence, social inclusion, social networks, or personalised quality of life | See comment | See comment | Not estimable | 0 (0) | See comment | This outcome was designated to be of importance, especially to patients, at 2017 update. We found no studies rating this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded one step for risk of bias: most studies did not adequately describe randomisation procedure, allocation concealment or blinding, and some studies were at high risk of attrition bias. 2 Downgraded one step for imprecision: few events (< 300) were reported. 3 Downgraded two steps for imprecision: small sample size, and effect estimate includes both appreciable benefit and appreciable harm for vitamin E. 4 Downgraded one step for reporting bias: only one study reported on this common, typically monitored adverse effect. 5 Downgraded one step for indirectness: leaving the study early can give an indication, but is not a direct measurement, of treatment acceptability.
Background
Description of the condition
Since the 1950s antipsychotic (neuroleptic) medication has been used extensively to treat people with chronic mental illnesses such as schizophrenia. These drugs can effectively control symptoms such as abnormal perceptions (hallucinations), disordered thinking and fixed false beliefs (delusions). In addition, maintenance therapy with antipsychotics is associated with a reduced risk of relapse (Schooler 1993). However, antipsychotic medication has been also associated with a wide range of adverse effects, including movement disorders. The appearance of these movement disorders can contribute to poor compliance with antipsychotic treatment (Barnes 1993).
Tardive dyskinesia (TD) is one such movement disorder and is characterised by abnormal, repetitive and involuntary movements. TD is a chronic condition of insidious onset, the severity of which spontaneously fluctuates (APA 1992). Studies on the natural history of TD have reported widely variable remission rates (1% to 62%) depending on patient age, psychiatric diagnosis, course of the psychiatric disorder, and duration of therapy (Bergen 1989; Fernandez 2001; Glazer 1990). It occurs in more than 20% of those using antipsychotic medication continually for longer than three months. Every year 4% to 5% of adults and 25% to 30% of elderly persons who continually use these drugs begin to show signs of TD (APA 1992; Correll 2004). This disorder can result in considerable social and physical disability (Barnes 1993).
The prevalence of TD is often thought to be decreasing based on the use of atypical antipsychotics in place of typical antipsychotics (Cloud 2014). A systematic review found that the incidence of TD associated with atypical drugs (2% to 4%) was significantly lower than that for typicals (5% to 8%) (Correll 2008). Despite this, the widespread use of atypical drugs in clinical settings may still result in an overall increase in the number of cases of TD (Glazer 2000).
Although the most frequent cause of TD is the use of antipsychotic medication, it is striking that dose reduction can lead to a temporary exacerbation in symptoms. Conversely, increasing the dose is often associated with a temporary remission. Antipsychotic drugs block certain chemical receptor sites in the brain ‐ one of these is specific for dopamine (Casey 1994). One hypothesis explaining the cause of antipsychotic‐induced TD is that chronic blockade of dopamine receptors in specific cells of the brain (neurones from the nigrostriatum) causes an overgrowth of these receptors (Casey 1994). However, there is some suggestion that the chronic use of antipsychotics may also cause an abnormal production of highly active atoms and chemical groups (cytotoxic free radicals), which may damage specific cells in the brain. This, in turn, could be responsible for the appearance of TD (Cadet 1989).
This work updates one in a series of reviews relevant to the management of antipsychotic‐induced tardive dyskinesia (Table 2).
1. Other reviews in the series.
| Interventions | Reference |
| Anticholinergic medication | Soares‐Weiser 1997; Soares 2000c |
| Benzodiazepines | McGrath 2000b; Umbrich 2003; Bhoopathi 2006 |
| Calcium channel blockers | Soares 2000b; Soares 2001c; Essali 2011 |
| Cholinergic medication | McGrath 2000d; Tammenmaa 2002 |
| Gamma‐aminobutyric acid agonists | Soares 2000d; Soares 2001b; Alabed 2011 |
| Miscellaneous treatments | McGrath 2000a; Soares‐Weiser 2003 |
| Neuroleptic reduction and/or cessation and neuroleptics | McGrath 2000c; Soares‐Weiser 2006 |
| Non‐neuroleptic catecholaminergic drugs | Lyra da Silva 1997; El‐Sayeh 2006 |
| Vitamin E | This review |
Description of the intervention
Vitamin E (tocopherol) is a lipid‐soluble antioxidant, illustrated in Figure 1, that acts as a free radical scavenger and has been proposed as a treatment for antipsychotic‐induced TD (Rotrosen 1996).
1.

Vitamin E
How the intervention might work
Vitamin E may assist in minimising damage caused by cytotoxic free radical overproduction, and may prevent or decrease the severity of TD, particularly for people who have had the onset of the problem in the preceding five years (Feltner 1993; Jeste 1993).
Why it is important to do this review
Several atypical antipsychotic drugs have been produced in the last decades that claim to cause less or no TD (Lieberman 1996). These claims may or may not be true, and certainly evidence does point to the fact that thoughtful use of older generation drugs is not associated with any more problems of TD than with newer treatments (Chouinard 2008). However, in a global context, it is likely that the less expensive and more familiar drugs ‐ such as chlorpromazine or haloperidol ‐ will continue to be the mainstay of treatment of people with schizophrenia (WHO Essential List 2010). Use of drugs such as these is associated with emergence of TD and, therefore, this condition will remain a problem for years to come.
Cessation or reduction of the dose of antipsychotic medication is the ideal management for TD. In clinical practice this is not always possible, not least because in many individuals such a reduction would lead to relapse. This review focuses on whether the addition of vitamin E to those already receiving antipsychotic medication is likely to help TD.
Objectives
The primary objective was to determine the clinical effects of vitamin E in people with schizophrenia or other chronic mental illness who had developed antipsychotic‐induced TD.
The secondary objectives were: 1. to examine whether the effect of vitamin E was maintained as duration of follow‐up increased; 2. to test the hypothesis that the use of vitamin E is most effective for those with early onset TD (less than five years) (see Subgroup analysis and investigation of heterogeneity).
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. Where a trial was described as 'double‐blind' but it was implied that the study was randomised and the demographic details of each group were similar, we have included it. We have excluded quasi‐randomised studies, such as those allocated by using alternate days of the week.
Types of participants
People with schizophrenia or other chronic mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who:
required the use of antipsychotics for more than three months;
developed tardive dyskinesia (TD) (diagnosed by any criteria at baseline and at least one other occasion) during antipsychotic treatment; and
for whom the dose of antipsychotic medication had been stable for one month or more (the same applies for those free of antipsychotics).
Types of interventions
1.a. Vitamin E
Any dose or means of administration
compared with
1.b. Placebo or no intervention
2.a. Vitamin E
Any dose or means of administration
compared with
2.b. Any other interventions
Types of outcome measures
We have defined clinical efficacy as an improvement in the symptoms of TD of more than 50%, on any scale. We grouped outcomes into short term (less than six weeks), medium term (between six weeks and six months) and long term (more than six months).
Primary outcomes
1. Tardive dyskinesia
No clinically important improvement in the symptoms of individuals, defined as more than 50% improvement on any TD scale ‐ any time period.
2. Adverse effects
No clinically significant extrapyramidal adverse effects ‐ any time period.
Secondary outcomes
1. Tardive dyskinesia (TD)
1.1 Any improvement in the symptoms of individuals on any TD scale, as opposed to no improvement. 1.2 Deterioration in the symptoms of individuals, defined as any deleterious change on any TD scale. 1.3 Average change in severity of TD during the trial period. 1.4 Average difference in severity of TD at the end of the trial.
2. General mental state changes
2.1 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale. 2.2 Average difference in severity of psychiatric symptoms at the end of the trial.
3. Acceptability of the treatment
3.1 Acceptability of the intervention to the participant group as measured by numbers of people dropping out during the trial.
4. Adverse effects
4.1 Use of any anti‐parkinsonism drugs. 4.2 Average score/change in extrapyramidal adverse effects. 4.3 Acute dystonia.
5. Other adverse effects, general and specific
6. Hospital and service utilisation outcomes
6.1 Hospital admission. 6.2 Average change in days in hospital. 6.3 Improvement in hospital status (for example: change from formal to informal admission status, use of seclusion, level of observation).
7. Economic outcomes
7.1 Average change in total cost of medical and mental health care. 7.2 Total indirect and direct costs.
8. Social confidence, social inclusion, social networks, or personalised quality of life measures
8.1. No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures. 8.2 Average score/change in social confidence, social inclusion, social networks, or personalised quality of life measures.
9. Behaviour
9.1 Clinically significant agitation. 9.2 Use of adjunctive medication for sedation. 9.3 Aggression to self or others.
10. Cognitive state
10.1 No clinically important change. 10.2 No change, general and specific.
'Summary of findings' table
We used the GRADE (Grading of Recommendations Assessment, Development and Evaluation) approach to interpret findings (Schünemann 2011) and used GRADEpro to export data from this review to create Table 1. This table provides outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effects of interventions examined and the sum of available data on all outcomes rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the Table 1. (Review author NM was not biased by being familiar with the data).
1. Tardive dyskinesia
1.1 Improved to a clinically important extent 1.2 Deteriorated
2. Adverse effect
2.1 Any adverse event 2.2 Adverse effects: no clinically significant extrapyramidal adverse effects
3. Acceptability of treatment
3.1 Leaving the study early
4. Social confidence, social inclusion, social networks, or personalised quality of life measures*
4.1 No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures for either recipients of care or caregivers
This summary table was used to guide our conclusions and recommendations.
* Outcome designated important to patients. We wished to add perspectives from people’s personal experience with TD to the research agenda. A consultation with service users was planned where the previously published version of this review and a lay overview of the review gave the foundation for the discussions. The session was planned to provide time to reflect on current research on TD and consider gaps in knowledge. The report is published in the Health Technology Assessment (HTA) report for the UK National Institute of Health Research (Bergman 2017). We have added one figure showing one service user's expression of frustration concerning this neglected area of research (Figure 2). Informed by the results of the consultation, for this review, we updated outcomes for the 'Summary of findings' table.
2.

Message from one of the participants of the Public and patient involvement consultation of service user perspectives on tardive dyskinesia research.
Search methods for identification of studies
Electronic searches
This review update was carried out in parallel with updating eight other TD reviews, see Table 2 for details. The searches covered all nine TD reviews. For details of previous electronic searches see Appendix 1 and Appendix 2.
1. Cochrane Schizophrenia Group’s Register
We searched Cochrane Schizophrenia Group’s Study‐Based Register of Trials on July 16, 2015 and April 26, 2017 using the following string:*Tardive Dyskinesia* in Healthcare Condition Field of Study. In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Methods used for the 2017 update are presented below. For methods used in previous versions, please see Appendix 3 and Appendix 4.
Selection of studies
Review authors NM and HB inspected all abstracts of studies identified as above and identified potentially relevant reports. We resolved disagreement by discussion, or where there was still doubt, we acquired the full article for further inspection. We acquired the full articles of relevant reports/abstracts meeting initial criteria for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). NM and HB were not blinded to the names of the authors, institutions or journal of publication. Where difficulties or disputes arose, we asked author KSW for help and where it was impossible to decide or if adequate information was not available to make a decision, we added these studies to those awaiting assessment and the authors of the papers contacted for clarification.
Data extraction and management
1. Extraction
Review authors NM and HB independently extracted data from all included studies. Again, we discussed any disagreement and documented decisions. We extracted data presented only in graphs and figures whenever possible, but included only if both review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multi‐centre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data online in Covidence.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument has not been written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, we noted in Description of studies if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant data before inclusion.
Please note, we entered skewed data from studies of at least 200 participants in the analysis, because skewed data pose less of a problem in large studies. We also entered all relevant change data as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
For endpoint data from studies < 200 participants:
(a) when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggests a skew and we excluded these data. If this ratio was higher than one but below 2, there is suggestion of skew. We entered these data and tested whether its inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2 we included these data, because skew is less likely (Altman 1996; Higgins 2011).
(b) if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
2.5 Common measure
Where relevant, to facilitate comparison between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we converted continuous outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this can be considered as a clinically significant response (Leucht 2005; Leucht 2005). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for Vitamin E. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we presented data where the left of the line indicates an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
Review authors NM and HB independently assessed risk of bias within the included studies by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011a). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies contacted in order to obtain further information. If non‐concurrence occurred, we reported this.
We noted the level of risk of bias in the text of the review and in Figure 3 and Figure 4 and incorporated these judgements in assessing limitations in study design for critical and important outcomes in the Table 1.
3.

4.

Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios as odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If any of the included trials had randomised participants by clusters, and where clustering was not accounted for in primary studies, we would have presented such data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study, but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Had a study involved more than two treatment arms, if relevant, we would have presented the additional treatment arms in comparisons. If data were binary we simply would have added and combined within the two‐by‐two table. If data were continuous we would have combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We would not use data where the additional treatment arms were not relevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we would address this within the 'Summary of Findings' table by down‐rating quality. We also planned to downgrade quality within the 'Summary of Findings' table should loss be 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome is between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). We assumed all those leaving the study early had no improvement in TD symptoms. We undertook a sensitivity analysis to test how prone the primary outcomes were to change by comparing data only from people who completed the study to that point to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
We reported and used data where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either P value or t value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic reviews of Interventions (Deeks 2011): When only the SE is reported, standard deviations (SDs) are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic reviews of Interventions (Deeks 2011) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently, methods such as multiple imputation or mixed effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, we preferred to use the more sophisticated approaches. (e.g. MMRM or multiple‐imputation) and only presented completer analyses if some kind of ITT data were not available at all. Moreover, we addressed this issue in the item "incomplete outcome data" of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise and discussed in the text if they arose.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise and discussed in the text if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, can be interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 Cochrane Handbook for Systematic Reviews of InterventionsDeeks 2011). We explored and discussed in the text potential reasons for substantial levels of heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there are 10 or fewer studies, or where all studies were of similar sizes. In future versions of this review, if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed0effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
We anticipated one subgroup analysis to test the hypothesis that the use of vitamin E is most effective for those with early onset TD (less than five years). We had hoped to present data for this subgroup for the primary outcomes.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of vitamin E for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems.
2. Investigation of heterogeneity
We reported when inconsistency was high. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present data. If not, we would not pool such data but would discuss issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply discussed. We did not undertake sensitivity analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
If trials were described in some way as to imply randomisation, we planned to undertake a sensitivity analyses for the primary outcomes. We would have included these studies in the analyses and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we would have used relevant data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
Where assumptions have to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with completer data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported and discussed these results but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that we judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, we included data from these trials in the analysis.
4. Imputed values
Had we included cluster‐randomised trials, we planned to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we found substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised data using a fixed‐effect model, however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether this altered the significance of the results.
Results
Description of studies
Please see Characteristics of included studies, Characteristics of excluded studies, and Characteristics of studies awaiting classification.
Results of the search
The searches up to 2017 retrieved 704 references for 344 studies, see Figure 5 for study flow diagram. The 2015 and 2017 update searches were part of an update of nine Cochrane reviews, see Table 2.
5.

Study flow diagram for study selection from searching up to and including 2017
From the 2015 search we identified five new potentially relevant studies to add to the review and screened the full texts. Agreement about which reports may have been randomised was 100%. Two of these studies could be included. One is a new study for this review (Zhang 2004). Another study was previously excluded because all data were unusable (Dorevitch 1997a), however we could include in the update as we found some data from the first phase of this cross‐over trial to extract. One of the references was a new companion paper in German to an already included study (Adler 1999) with no additional data, we also added one ongoing study (ISRCTN14688109 2015) and one excluded study (Dorfman‐Etrog 1999).
The 2017 search found 8 records (5 studies). Editorial base of Cochrane Schizophrenia screened these records and no new studies were relevant to this review. They could be relevant to another reviews in this series of TD reviews (see Table 2), and have been put into awaiting assessment of Soares‐Weiser 2003.
Thirteen studies are now included in this review (Adler 1993; Adler 1999; Akhtar 1993; Dabiri 1994; Dorevitch 1997a; Dorevitch 1997b; Egan 1992; Elkashef 1990; Lam 1994; Lohr 1996; Sajjad 1998; Schmidt 1991; and Zhang 2004).
Included studies
Overall the review now includes 13 studies with 478 participants published between 1990 and 2004.
1. Methods
All studies were stated to be randomised and double‐blind. For further details please see sections below on allocation and blinding.
2. Design
All included studies presented a parallel longitudinal design. Six of the 11 studies used a cross‐over design with two periods (Dorevitch 1997a; Dorevitch 1997b; Egan 1992; Elkashef 1990; Lam 1994; Schmidt 1991). We had considered this as likely when embarking on the review and have used only the data from before the first cross‐over period for the reasons outlined above (Unit of analysis issues).
3. Duration
Most studies were of short duration (less than 13 weeks) but four had a follow‐up period longer than five months (Adler 1993; Adler 1999; Dorevitch 1997b; Sajjad 1998). All the other included studies were randomised, double‐blind, controlled trials of short duration. Four trials employed washout periods of two weeks.
4. Participants
Participants, now totaling 478 people, were mostly men in their 50s, with diagnoses of various chronic psychiatric disorders, but mainly schizophrenia. All had antipsychotic‐induced tardive dyskinesia (TD) diagnosed using Schooler and Kanes research diagnostic criteria, except Schmidt 1991, who did not report any criteria for the diagnosis of TD. The number of participants ranged from 10 to 158 (median 23).
5. Setting
Most trials were conducted in hospital. The studies themselves were from around the world, with six conducted in the USA , two in Israel (Dorevitch 1997a; Dorevitch 1997b), and one each in China (Zhang 2004), Hong Kong (Lam 1994), India (Akhtar 1993), Switzerland (Schmidt 1991) and the UK (Sajjad 1998).
6. Interventions
6.1 Vitamin E
The vitamin E dose ranged from 1200 IU/day to 1600 IU/day, with the exception of Sajjad 1998, in which the dose was 600 IU/day.
6.2 Comparison group
In most of the studies a placebo was used as a comparison, with no further details given. In one study, the comparison group was given nothing (Sajjad 1998), and in another trial the placebo was a sesame oil placebo gelcap (Lohr 1996). None of the included studies compared vitamin E with another active intervention.
Participants remained on schizophrenia treatment antipsychotic medication during the trials.
7. Outcomes
7.1 General
Some outcomes were presented in graphs, inexact P values of differences, or a statement of significant or non‐significant difference. This made it impossible to acquire raw data for synthesis. Some continuous outcomes could not be extracted due to missing number of participants or missing means, standard deviations, or standard errors. All included studies used the last observation carried forward (LOCF) strategy for the intention‐to‐treat (ITT) analysis of dichotomous data.
7.2 Scales used to measure the TD symptoms
We have shown details of the scales that provided usable data below. We have provided reasons for exclusions of data under 'Outcomes' in the Characteristics of included studies table.
7.2.1 Abnormal Involuntary Movement Scale
The AIMS (Guy 1976) is a 12‐item scale consisting of a standardised examination followed by questions rating the orofacial, extremity and trunk movements, as well as three global measurements. Each of these 10 items can be scored from zero (none) to four (severe). Two additional items assess the dental status. The AIMS ranges from zero to 40, with higher scores indicating greater severity.
7.2.2 Tardive Dyskinesia Rating Scale (TDRS)
The TDRS (Simpson 1970) is a 34‐item scale consisting of measurement of the movements around the orofacial region, neck, trunk and extremities. Each of these items can be scored from zero (absent) to five (severe). This scale ranges from 10 to 102, with higher scores indicating greater severity.
7.3 Scales used to measure adverse events related to antipsychotic medication
7.3.1 Simpson‐Angus Scale (SAS)
The SAS (Simpson 1970) is a 10‐item scale, with a scoring system of zero to four for each item, measures drug‐induced parkinsonism, a short‐term drug‐induced movement disorder. A low score indicates low levels of parkinsonism.
7.3.2 Barnes Akathisia Scale (BAS)
The BAS (Barnes 1989) is a 12‐item scale consisting of a standardised examination followed by questions rating the orofacial, extremity and trunk movements, as well as three global measurements. Each of these 10 items can be scored from zero (none) to four (severe). Two additional items assess the dental status. The BAS ranges from zero to 40, with higher scores indicating greater severity.
Excluded studies
There are eight excluded studies. Peet 1993 and Spivak 1992 were not randomised and we have therefore excluded them. We excluded Dorfman‐Etrog 1999 and Salmasi 2009 because participants had schizophrenia but not TD. Although participants in Ricketts 1995 had TD, we excluded this study because participants did not have schizophrenia but had mental retardation due to brain damage. After two years of unfruitful attempts to contact authors for further details, we have also had to exclude a further three randomised studies which reported no usable data (Junker 1992; Lohr 1988; Shriqui 1992).
Awaiting assessment
We have not, as yet, obtained the report of Kar‐Ahmadi 2002; it may also be in Farsi. We hope to include it in the next update of this review.
Ongoing studies
We identified one ongoing study conducted in Nigeria (ISRCTN14688109 2015) investigating vitamin E compared with cannabidiol extract in participants with TD. Results of the trial are expected to be published in early 2018.
Risk of bias in included studies
Please refer to Figure 3 and Figure 4 for graphical overviews of the risk of bias in the included studies.
Allocation
Only two studies were rated at low risk of selection bias. Adler 1999 was randomised centrally and Sajjad 1998 used a computer‐generated sequence although did not describe allocation concealment. Most other studies were not explicit about how allocation was achieved other than using the word "randomized". Adler 1993 allocated people into vitamin E and placebo groups in a ratio of 3:2.
Blinding
Although all studies were conducted on a double‐blind basis, none explicitly described how this was undertaken and tested the blindness of raters, clinicians and trial participants. We have, however, rated Adler 1993, Akhtar 1993, Dabiri 1994, Dorevitch 1997b, Lam 1994 and Zhang 2004 as being of higher quality because they stated that the trial was double‐blinded and specifically that either the raters or the raters and the participants were blinded. All the other studies gave no further details other than stating that they were double‐blinded. In Sajjad 1998 the blind was broken after one month.
Incomplete outcome data
The trialists excluded three people from Adler 1993 and four from Lam 1994 because of lack of post‐baseline data. Three studies had a greater than 30% loss to follow‐up (Adler 1993; Lohr 1996; Sajjad 1998). In all cases, however, we tried to ensure that every person randomised was analysed.
Selective reporting
The majority of data in this review originates from published reports. Expected outcomes (impact on TD symptoms) were reported for most of the trials. Six studies reported results of all outcomes listed in the methods section fully (Adler 1993; Adler 1999; Akhtar 1993; Dabiri 1994; Sajjad 1998; Schmidt 1991). We rated risk of reporting bias for these studies as unclear because we have had no opportunity to see protocols of these trials to compare the outcomes reported in the full publications with what was measured during the conduct of the trial. As a result, we feel that there may be an element of selective reporting that we could perpetuate in this review and that this bias would favour vitamin E. Seven studies did not fully report outcomes that were measured during the study and were rated at high risk of reporting bias. Attempts to contact authors of trials for additional data were unsuccessful.
Other potential sources of bias
All studies had small or very small sample sizes. Six of the studies used a cross‐over design (Dorevitch 1997a; Dorevitch 1997b; Egan 1992; Elkashef 1990; Lam 1994; Schmidt 1991). Five of the studies had the drugs used in the trials provided by pharmaceutical companies (Dorevitch 1997a; Dorevitch 1997b; Elkashef 1990; Lohr 1996; Schmidt 1991), and in five studies no details of funding were given (Akhtar 1993; Dabiri 1994; Egan 1992; Lam 1994; Sajjad 1998).
Effects of interventions
See: Table 1
1. Comparison 1. Vitamin E versus placebo
1.1 TD symptoms
We had chosen 'any improvement in TD symptoms of more than 50% on any TD scale ‐ any time period' as a primary outcome. Although data we found in trials did not fit this exactly, we feel that the outcome 'not improved to a clinically important extent' fits best with what we had hoped to find.
1.1.1 Not improved to a clinically important extent
The overall results for 'clinically relevant improvement' found no benefit of vitamin E against placebo (low‐quality evidence, 6 trials, 264 people, risk ratio (RR) 0.95. 95% CI 0.89 to 1.01, Analysis 1.1).
1.1. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 1 Tardive dyskinesia: 1. Not improved to a clinically important extent.
1.1.2 Not any improvement
For the outcome of 'any improvement in TD symptoms', again added across all time periods, we found no difference between vitamin E and placebo (low‐quality evidence, 7 trials, 319 people, RR 0.87, 95% CI 0.76 to 1.00, Analysis 1.2).
1.2. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 2 Tardive dyskinesia: 2. Not any improvement.
1.1.3 Average endpoint scores
TD symptoms were also measured on the continuous Abnormal Involuntary Movement Scale (AIMS) and Tardive Dyskinesia Rating Scale (TDRS) scales. A beneficial effect of vitamin E was found when measured on the AIMS (medium term, 6 studies, 157 people, mean difference (MD) ‐1.77, 95% CI ‐2.59 to ‐0.95, Analysis 1.3), but one study found no difference as measured on the TDRS (short term, 32 people, MD ‐2.10 95% CI ‐6.35 to 2.15, Analysis 1.4).
1.3. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 3 Tardive dyskinesia: 3a. Average endpoint score (AIMS, low score = best).
1.4. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 4 Tardive dyskinesia: 3b. Average endpoint score ‐ short term (TDRS, low score = best).
1.1.4 Deterioration of symptoms
People allocated to placebo showed more deterioration of their symptoms compared with those on vitamin E (low‐quality evidence, 5 trials, 85 people, RR 0.23, 95% CI 0.07 to 0.76, Analysis 1.5).
1.5. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 5 Tardive dyskinesia: 4. Deterioration of symptoms.
1.2 Adverse effects
1.2.1 Extrapyramidal adverse effects
One study measured extrapyramidal symptoms (Simpson‐Angus Scale) and found no difference between Vitamin E and placebo (very low‐quality evidence, 104 people, MD 1.10, 95% CI ‐1.02 to 3.22, Analysis 1.6). We had pre‐specified the dichotomous outcome 'No clinically significant extrapyramidal adverse effects ‐ any time period' as a primary outcome. No study reported this as a finding.
1.6. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 6 Adverse events: Extrapyramidal adverse events ‐ long term (Simpson‐Agnus Scale, low = better).
1.2.2 Specific adverse effects
The same study measured akathisia and found no difference between Vitamin E and placebo (Barnes Akathisia Scale (BAS), 104 people, MD 0.30, 95% CI ‐1.03 to 1.63, Analysis 1.7).
1.7. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 7 Adverse events: Specific adverse events: akathisia ‐ long term (BAS, low = better).
1.2.3 Any adverse effects
There was no difference in the incidence of ‘any adverse effect’ (very low‐quality evidence, 9 trials, 205 people, RR 1.21, 95% CI 0.35 to 4.15, Analysis 1.8).
1.8. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 8 Any adverse effect.
1.2.4 Discontinuation due to adverse events
Five trials reported on this outcome with only one person in the vitamin E arm who discontinued due to adverse events (123 people, RR 2.50, 95% CI 0.11 to 54.87, Analysis 1.9).
1.9. Analysis.
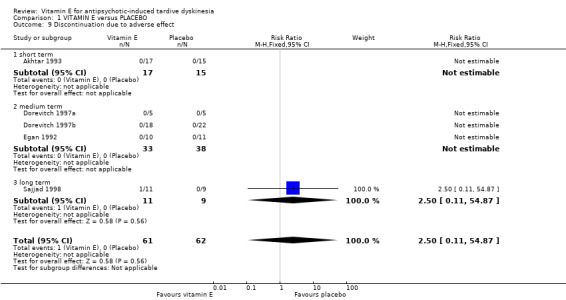
Comparison 1 VITAMIN E versus PLACEBO, Outcome 9 Discontinuation due to adverse effect.
1.3 Mental state
Two trials reported endpoint data and one trial reported change from baseline data. These were combined as described in Data extraction and management ‐ '2.3 Endpoint versus change data'. We found no difference between vitamin E and placebo for a measure of psychiatric symptoms (British Psychiatric Rating Scale (BPRS), 3 trials, 165 people, MD ‐0.20, 95% CI ‐3.21 to 2.82, Analysis 1.10).
1.10. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 10 Mental state: Average score (BPRS, low = best).
1.4 Leaving the study early
Using vitamin E did not significantly increase the chances of a person leaving the study early (very low‐quality evidence, medium term overall ˜20% loss to follow‐up, 8 trials, 232 people, RR 1.07, 95% CI 0.64 to 1.80, Analysis 1.11).
1.11. Analysis.

Comparison 1 VITAMIN E versus PLACEBO, Outcome 11 Leaving study early.
We did not identify any studies that reported on hospital and service utilisation outcomes, economic outcomes, quality of life, satisfaction with care, behaviour, or cognitive state.
1.5 Subgroup analysis
1.5.1 Clinical stage: Recent onset TD
It was not possible to evaluate whether those with recent onset TD responded differently to those with more established problems, since no trial reported data for groups with different durations of TD that could be extracted for separate analyses.
1.5.2 Duration of follow‐up
Any effects that vitamin E may have did not clearly change in relation to duration of follow‐up.
1.6 Heterogeneity
Data were homogeneous. We did not detect clinical, methodological or statistical heterogeneity as described in Assessment of heterogeneity.
1.7 Sensitivity analyses
1.7.1 Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. As all studies were stated to be randomised, we did not undertake this sensitivity analysis.
1.7.2 Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data) we compared the findings when we used our assumption compared with completer data only. Using completer only data for no improvement in TD symptoms, we found that more people improved with vitamin E than with placebo due to a minor shift in the effect estimate and the precision of the effect estimate (RR 0.77, 95% CI 0.61 to 0.96; participants = 219; studies = 7, analysis not shown) .Although there was a change in direction of effect, we continued to employ our assumption in the main results.
1.7.3 Risk of bias
When excluding three trials that we judged to be at high risk of bias across one or more of the domains, there was no substantial alteration to the direction of effect or the precision of the effect estimates (RR 0.96, 95% CI 0.90 to 1.03; participants = 218; studies = 6; analysis not shown).
1.7.4 Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from cluster‐randomised trials where we used imputed values for ICC in calculating the design effect. No cluster‐randomised trials were included.
1.7.5 Fixed and random effects
We also synthesised data for the primary outcome using a random‐effects model This did not alter the significance of the results (RR 0.96, 95% CI 0.90 to 1.02; participants = 264; studies = 6).
2. Comparison 2. Vitamin E versus any other intervention
No included studies were identified that reported on this comparison.
Discussion
Summary of main results
1. The search
This area of research does not seem to be active. this 2017 update has identified additional data, but most trials predate the year 2000, only one was carried out after that time (published 2004). This could be because of reasons such as less concern with tardive dyskinesia (TD), or less emergence of the problem in research‐active communities because of more thoughtful use of antipsychotic drugs or loss of faith in vitamin E as a potential treatment.
2. Few data
Only a little under 500 people have been involved in placebo‐controlled trials of vitamin E for TD. It is possible that real, and important, effects have not been highlighted because of the necessarily wide confidence intervals of the findings. Many outcomes were not measured at all (see Overall completeness and applicability of evidence), including one of our pre‐stated outcome measures. We may have been overambitious in hoping for some of these outcomes in TD trials but simple reporting of satisfaction with care or quality of life still does not seem too demanding and does remain of interest.
3. Comparison 1. Vitamin E versus placebo
3.1 TD symptoms
Results of this review do not suggest that vitamin E improves symptoms of TD. However, the quality of the evidence for improvement of TD symptoms is low; further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. The recent addition of Zhang 2004 did not impact on the binary outcomes for improvement in TD symptoms, but changed the continuous scale results in favour of vitamin E. The finding that people allocated to placebo may show more deterioration of their symptoms compared with those allocated to vitamin E is interesting (low‐quality evidence, 5 trials, 85 people, RR 0.23 95% CI 0.07 to 0.76) and generates some hope that the experimental treatment may have a preventative role. This is worthy of further investigation.
3.2 Adverse effects
There is no evidence that vitamin E has any significant adverse effects.
3.3 Mental state
Three trials reported on mental state. All used the BPRS scale, but this involved only asking 165 people regarding this important outcome. There was no suggestion that vitamin E had any different effect on mental state than placebo.
3.4 Leaving the study early
It is always unclear what leaving the study early means. It could be to do with the participant not accepting treatment for a series of reasons, or of participants finding the trial intolerable. It also could be a function of a trial design in which willing participants are still asked to leave because of some degree of protocol violation. In any event, between 20% and 30% of people left the study early, but this was not different for those allocated to either group.
3.5 Social confidence, social inclusion, social networks, or personalised quality of life
This group of outcomes was selected as being of importance to patients for the 2017 review update following a service user consultation. No studies were identified that reported on any of these outcomes.
4. Comparison 2. Vitamin E versus any other intervention
We found no included study that compared the efficacy of vitamin E compared to another active intervention. There is currently one ongoing study (ISRCTN14688109 2015) comparing vitamin E to cannabidiol that is due to be published in 2018.
Overall completeness and applicability of evidence
1. Completeness
No outcomes in this review involved large numbers of people. Some are general measures and more subtle findings are not recorded. For example, we identified few data on the outcome of ‘any adverse effect’ but none on the binary outcomes ‘use of any anti‐parkinsonism drugs’ or ‘no clinically significant extrapyramidal adverse effects ‐ any time period’‐ the latter being one of our pre‐stated primary outcomes. There were no data on the patient‐designated important outcomes quality of life and satisfaction with care for either recipients of care or caregivers, nor were there data on hospital and service utilisation outcomes, economic outcomes, behaviour or cognitive response.
2. Applicability
All trials were hospital‐based but were nevertheless on people who would be recognisable in everyday care. The intervention in question ‐ vitamin E ‐ is readily accessible and most outcomes understandable in terms of clinical practice. Should vitamin E have had important effects the findings may well have been applicable.
Quality of the evidence
The largest trial in this area randomised only 158 people (Adler 1999). A trial of this size is unable to detect subtle, yet important differences due to vitamin E with any confidence. In order to detect a 20% difference between groups, probably about 150 people are need in each arm of the study (alpha 0.05, beta 0.8). Overall, the quality of reporting of these trials was poor (see Figure 3). Allocation concealment was not described, generation of the sequence was not explicit, studies were not clearly blinded, we are unsure if data are incomplete or selectively reported or if other biases were operating. The small trial size, along with the poor reporting of trials, would be associated with an exaggeration of effect of the experimental treatment (Jűni 2001) if an effect had been detected. This is only evident for the outcome of ‘deterioration’ where there is indeed an effect favouring the vitamin E group. This interesting finding may be real – but could equally be a function of biases or of chance.
Potential biases in the review process
1. Missing studies
Every effort is made to identify relevant trials. However, these studies are all small and it is likely that we have failed to identify other studies of limited power. It is likely that such studies would also not be in favour of the vitamin E group. If they had been so, it is more likely that they would have been published in accessible literature. We do not, however, think it likely that we have failed to identify large relevant studies.
2. Introducing bias
This review group has now updated this review several times. We have tried to be balanced in our appraisal of the evidence but could have inadvertently introduced bias. We welcome comments or criticisms. New methods and innovations now make it possible to report data where, in the past, we could not report data at all or had to report data in a different way. We think the Table 1 to be a valuable innovation – but problematic to those not ‘blind’ to the outcome data. It is possible to ‘cherry pick’ significant findings for presentation in this table. We have tried to decrease the chance of doing this by asking a new review author (NM) to select outcomes relevant for this table before becoming familiar with the data.
Agreements and disagreements with other studies or reviews
The only other relevant quantitative review we know of is the previous version of this Cochrane review (Soares‐Weiser 2011). This update expands and improves this review but does not substantially change the findings or the conclusions.
Authors' conclusions
Implications for practice.
1. For people with TD
There is some suggestion that vitamin E supplementation may alter the course of tardive dyskinesia (TD) for those with schizophrenia and these findings are in keeping with other types of studies (Hawkins 1989). These data are very weak and should be interpreted with considerable caution. If offered vitamin E supplementation to offset symptoms of TD a person taking antipsychotic medication would be justified in asking for evidence, and encouraging generation of new better evidence by volunteering to help with a well‐designed evaluative study.
2. For clinicians
TD is such a disfiguring condition. This review generates a theory that vitamin E could be a preventative measure for TD. The data in this review do not justify routine use of vitamin E. It would be reasonable, however, to ask people with schizophrenia to complicate their treatment plan if the use of vitamin E was to generate data better than seen in this review. There is a real place for clinician‐driven research, with prescription within the context of a randomised trial and routine data collection on outcomes of relevance to people with TD and their clinicians.
3. Policy makers or managers
This is one of the largest in the series of Cochrane reviews on TD. No evidence is convincing that addition of another drug helps with the symptoms of TD. There are, however, many unanswered questions in this area. This unattractive adverse effect is caused, to a greater or lesser extent, by antipsychotic drugs. Clinicians and researchers should feel responsible enough to continue to try to help it. Those compiling guidance could encourage supportive activity and more research into this neglected area.
Implications for research.
1. General
The power of this review would have been greatly enhanced by better reporting of data. For example, only one study made explicit how randomisation was undertaken (Adler 1999). We realise that much of the work for these trials predates CONSORT, which was first published in 1996 (Begg 1996), and that it is only too easy to judge studies of the past by standards of today. Future studies, however, should report to a much higher standard.
2. Specific
Well‐designed randomised controlled trials, involving a large number of participants over protracted periods of time, are needed if we are to see if vitamin E could have a role in prevention and treatment of TD. Such studies are of importance to people with the problem (Figure 2) and have long been ignored.
2.1 Use of cross‐over design
Trialists find it difficult to identify people with both TD and schizophrenia to participate in trials (Schmidt 1991). Randomised cross‐over design is used in the hope of improving the power of the study to find outcomes of interest. This design initially asks participants to be randomised to one of the experimental interventions, and then, at a pre‐specified time, to be crossed over to the treatment that they did not at first receive. Conditions with a more stable time course than TD are better suited for cross‐over studies (Fleiss 1984). Further difficulties are the carry‐over effect. At the very least, vitamin E is dissolved in fat and probably high levels from the doses given in trials may well persist in the body for long periods after discontinuation. Unless cross‐over studies include a mid‐study washout period (where the person is free of treatment before starting the next arm of the study), any effect of vitamin E may continue into the second half placebo arm of the trial – the 'carry‐over effect'. Also, carry‐over may involve the re‐growth or retreat of neuroreceptors. This slow re‐balancing, if started, could continue long after all traces of intervention drugs are gone, so physiological half life of the experimental treatment may not be the only variable to consider when thinking though the issues of carry‐over. Tardive dyskinesia is also an unstable condition and people with TD may not remain compliant with medication. All these factors make the arguments for not using cross‐over methodology strong, despite the initial attraction (Armitage 1991; Fleiss 1984; Pocock 1983).
2.2 Sample size calculation
Only one of the studies included in this review mentioned how they calculated the sample size (Adler 1999). However, the results suggest that larger sample size should be used to provide more precise estimates of effect.
2.3 Length of study
Three studies included in this review (Adler 1993; Adler 1999; Sajjad 1998) used the intervention for more than five months. TD, however, is a chronic condition of insidious onset, the severity of which fluctuates spontaneously (APA 1992). Even if vitamin E has a swift effect, which is unlikely, it is the long‐term outcomes that must be considered of most clinical value.
2.4 Outcomes
Scale‐derived data do have their place. Trials most commonly used the Abnormal Involuntary Movement Scale (AIMS) scale. This is a very widely used tool to measure the severity of symptoms of those who have TD. The use of this scale to measure change as a result of treatment is, however, problematic (Bergen 1984). It is therefore important that a scale is validated for measuring changes secondary to treatment in those with TD. In addition, many of the outcomes we initially desired when we started this review have not been investigated. Finally, a service user consultation also informed the addition of outcomes of special importance to patients. We have reconsidered all these outcomes in case they were too ambitious and tried to tailor them to a real‐world pragmatic trial design (see Table 3).
2. Suggestions for design of future study.
| Methods | Allocation: randomised, with sequence generation and concealment of allocation clearly described. Blindness: double, tested. Duration: 12 months beyond end of intervention at least. Raters: independent. |
| Participants | People with antipsychotic‐induced tardive dyskinesia.* Age: any. Sex: both. History: any. N = 300.** |
| Interventions | 1. Vitamin E: 1600 IU/day. N = 150. 2. Placebo: N = 150. |
| Outcomes | Tardive dyskinesia: any clinically important improvement in TD, any improvement, deterioration.*** Adverse effects: no clinically significant extrapyramidal adverse effects ‐ any time period***, use of any antiparkinsonism drugs, other important adverse events. Leaving the study early. Service outcomes: admitted, number of admissions, length of hospitalisation, contacts with psychiatric services. Compliance with drugs. Economic evaluations: cost‐effectiveness, cost‐benefit. General state: relapse, frequency and intensity of minor and major exacerbations. Social confidence, social inclusion, social networks, or personalised quality of life: binary measure Distress among relatives: binary measure. Burden on family: binary measure. |
| Notes | * This could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. ** Size of study with sufficient power to highlight about a 10% difference between groups for primary outcome. *** Primary outcome. The same applies to the measure of primary outcome as for diagnosis. Not everyone may need to have operational criteria applied if clinical impression is proved to be accurate. |
What's new
| Date | Event | Description |
|---|---|---|
| 24 September 2019 | Amended | Correction to Summary title of Plain Language Summary (PLS). Previous text referred to the review question. Summary Title of PLS has been added to this section. |
History
Protocol first published: Issue 2, 1995 Review first published: Issue 1, 1999
| Date | Event | Description |
|---|---|---|
| 18 October 2017 | New citation required but conclusions have not changed | New data added do not substantially change previous conclusions. |
| 26 April 2017 | New search has been performed | Update search run 26 April, 2017. Eight records found and assessed by editorial base at Cochrane Schizophrenia, none of the records found were relevant to this review. The records were all added to Studies awaiting classification of Miscellaneous treatments for antipsychotic‐induced tardive dyskinesia (see also Results of the search). |
| 16 May 2016 | Amended | Title changed from 'Vitamin E for neuroleptic‐induced tardive dyskinesia'. Two new trials added from 2015 searching (Dorevitch 1997a; Zhang 2004), text updated, conclusions not substantially changed. |
| 15 July 2015 | Amended | Update search run July 15 2015. 704 records found and assessed by review authors. |
| 19 January 2011 | New citation required but conclusions have not changed | Substantial update, conclusions not substantially changed |
| 21 July 2010 | New search has been performed | New trials added (Adler 1999; Sajjad 1998), text rewritten. These weakened, but did not substantially change results. |
| 11 November 2009 | Amended | Contact details updated. |
| 26 April 2008 | Amended | Converted to new review format. |
| 13 August 2001 | New citation required and conclusions have changed | Substantive amendment |
| 9 November 2000 | Amended | Reformatting |
| 23 September 1999 | Amended | Reformatted |
| 12 July 1996 | Amended | First version of review |
Acknowledgements
The authors acknowledge Clive Adams, Jair Mari and Rochelle Seifas for their constant help and support, Jon Deeks for statistical assistance, and Ben Gray for writing the Plain language summary. We also wish to acknowledge John McGrath, an author of previous versions of this review, for his invaluable contribution. We thank Dr Lam and Dr Edson for their support with more information regarding their trial, and Dr Lavori for his prompt reply to our enquiries. Finally, we wish to thank Rosie Asher and Antonio Grande for screening literature and helping with data extraction for this 2017 update.
We would also like to thank and acknowledge Linda Ojo for peer reviewing this version of the review.
Appendices
Appendix 1. Seach method for the 2010 update
1. Cochrane Schizophrenia Group Trials Register (March 2010)
This register was searched using the phrase: [(*vitamins* or *vitamin E* or *tocopherol*) in interventions of STUDY field]
This register is compiled by systematic searches of major database, handsearches and conference proceedings (see Group Module).
Appendix 2. Search methods for identification of studies on the previous version of the review (January 2001)
1. Electronic searching 1.1 Biological Abstracts (January 1982 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase:
[and ((tardive near (dyskine* or diskine*) or (abnormal near movement* near disorder*) or (involuntar* near movement*))]
This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*]
1.2 The Cochrane Schizophrenia Group's Register (January 2001) was searched using the phrase:
[vitamin or tocopherol*]
1.3 Embase (January 1980 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase:
[and ((tardive dyskinesia in thesaurus ‐subheadings, prevention, drug therapy, side effect and therapy) or (neuroleptic dyskinesia in thesaurus ‐all subheadings) or (tardive or dyskines*) or (movement* or disorder*) or (abnormal or movement* or disorder*))]
This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*]
1.4 LILACS (January 1982 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase:
[and ((tardive or (dyskinesia* or diskinesia*)) or (drug induced movement disorders in thesaurus))]
This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*]
1.5 Medline (January 1966 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase:
[and ((movement‐disorders in MeSH / explode all subheadings) or (anti‐dyskinesia‐agents in MeSH / explode all subheadings) or (dyskinesia‐drug‐induced in MeSH / explode all subheadings) and (psychosis in MeSH / explode all subheadings) or (schizophrenic disorders in MeSH / explode all subheadings) or (tardive near (dyskine* or diskine*)) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))]
This downloaded set of reports was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*]
1.6 PsycLIT (January 1974 to September 1998) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase:
[and ((explode movement‐disorders in DE) or (explode tardive‐dyskinesia in DE) or (tardive near (dyskine* or diskine*) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))]
This downloaded set of reports was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*]
1.7 SCISEARCH ‐ Science Citation Index (1997) Each of the included studies was sought as a citation on the SCISEARCH database. Reports of articles that had cited these studies were inspected in order to identify further trials.
2. Reference searching The references of all identified studies were also inspected for more studies.
3. Personal contact The first author of each included study was contacted for information regarding unpublished trials.
Appendix 3. Methods of 2001 version of this review
Criteria for considering studies for this review
Types of studies All relevant randomised controlled trials. Types of participants People with schizophrenia or other chronic mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who: 1. Required the use of antipsychotics for more than three months 2. Developed tardive dyskinesia (diagnosed by any criteria at baseline and at least one other occasion) during antipsychotic treatment; and 3. For whom the dose of antipsychotic medication had been stable for one month or more (the same applies for those free of antipsychotics). Types of interventions 1. Vitamin E: any dose or means of administration. 2. Placebo or no intervention. Types of outcome measures Clinical efficacy was defined as an improvement in the symptoms of TD of more than 50%, on any scale, after at least six weeks of intervention. The outcomes of interest were as follows: 1. Symptoms of tardive dyskinesia 1.1 The number of people per treatment group that did not show an improvement in the symptoms of individuals of more than 50% on any TD scale. 1.2 The number of people per treatment group that did not show any improvement in the symptoms of individuals on any TD scale, as opposed to some improvement, 1.3 Deterioration in the symptoms, defined as any deleterious change on any TD scale. 1.4 Any adverse effect, other than deterioration of symptoms of TD, as reported in the trials. 1.5 Average change in severity of TD during the trial period. 1.6 Average severity of TD at the end of the trial. 2. General mental state changes 2.1 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale. 2.2 Average severity of psychiatric symptoms at the end of the trial. 3. Acceptability of the treatment Acceptability of the intervention to the participant group as measured by number of people dropping out during the trial. Three time periods for reporting of outcomes were pre‐stated ‐ short term (less than 6 weeks), medium term (between 6 weeks and 6 months) and long term (over 6 months). Search methods for identification of studies 1. Electronic searching 1.1 Biological Abstracts (January 1982 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((tardive near (dyskine* or diskine*) or (abnormal near movement* near disorder*) or (involuntar* near movement*))] This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*] 1.2 The Cochrane Schizophrenia Group's Register (January 2001) was searched using the phrase: [vitamin or tocopherol*] 1.3 Embase (January 1980 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((tardive dyskinesia in thesaurus ‐subheadings, prevention, drug therapy, side effect and therapy) or (neuroleptic dyskinesia in thesaurus ‐all subheadings) or (tardive or dyskines*) or (movement* or disorder*) or (abnormal or movement* or disorder*))] This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*] 1.4 LILACS (January 1982 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((tardive or (dyskinesia* or diskinesia*)) or (drug induced movement disorders in thesaurus))] This downloaded set of reports was handsearched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*] 1.5 Medline (January 1966 to January 2001) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((movement‐disorders in MeSH / explode all subheadings) or (anti‐dyskinesia‐agents in MeSH / explode all subheadings) or (dyskinesia‐drug‐induced in MeSH / explode all subheadings) and (psychosis in MeSH / explode all subheadings) or (schizophrenic disorders in MeSH / explode all subheadings) or (tardive near (dyskine* or diskine*)) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))] This downloaded set of reports was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*] 1.6 PsycLIT (January 1974 to September 1998) was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((explode movement‐disorders in DE) or (explode tardive‐dyskinesia in DE) or (tardive near (dyskine* or diskine*) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))] This downloaded set of reports was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [vitamin or tocopherol*] 1.7 SCISEARCH ‐ Science Citation Index (1997) Each of the included studies was sought as a citation on the SCISEARCH database. Reports of articles that had cited these studies were inspected in order to identify further trials. 2. Reference searching The references of all identified studies were also inspected for more studies. 3. Personal contact The first author of each included study was contacted for information regarding unpublished trials. Data collection and analysis [For definitions of terms used in this, and other sections, please refer to the Glossary.] 1. Selection of reports and studies KSW and JM inspected every report identified by the search, independently, to see if the study was likely to be relevant. Where resolving disagreement by discussion was not possible, the full article was obtained. The reviewers then inspected these articles, independently, to assess their relevance to this review. Again, where disagreements could not be resolved by discussion the article was added to those awaiting assessment and the authors of the study were contacted for clarification for ambiguous or missing descriptions of the methodology. 2. Assessment of methodological quality The reviewers also evaluated the quality of all included trials independently of one another. A rating was given for each trial based on the three quality categories as described in the Cochrane Collaboration Handbook (Clarke 2000). Only trials that stated to be randomised (category A or B of the Handbook) were included in this review. 3. Data extraction Data were independently extracted by KSW and JM. When disputes arose resolution was attempted by discussion. When this was not possible and further information was necessary to resolve the dilemma, data were not entered and this outcome of the trial was added to the list of those awaiting assessment. Outcomes are assessed using continuous (for example changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such a 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a persons behaviour). Currently RevMan does not support categorical data so they were presented only in the text of the review. 4. Data synthesis 4.1 Intention to treat analysis Data were excluded from studies where more than 50% of participants in any group were lost to follow‐up. For all events, in studies with less than 50% attrition rate, people leaving early were considered to have had the negative outcome, except for the event of death. 4.2 Binary data For binary outcomes a standard estimation of the relative risk (RR) and its 95% confidence interval (CI) was calculated. The weighted number needed to treat or harm statistic (NNT, NNH), and its 95% confidence interval (CI), was also calculated (http://www.mango3d.cwc.net/vsx.htm). If heterogeneity was found (see section 5) a random effects model was used. 4.3 Continuous data 4.3.1 Skewed data: continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, the following standards are applied to all data before inclusion: (i) standard deviations and means were reported in the paper or were obtainable from the authors; (ii) when a scale started from the finite number zero, the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); (iii) if a scale started from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above in (ii) was modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied to them. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale), there is no way of telling whether data is non‐normally distributed (skewed) or not. It is thus preferable to use scale end point data, which typically cannot have negative values. If end point data were not available, the reviewers used change data, but they were not subject to a meta‐analysis, and were reported in the 'Additional data' tables, as were non‐normally distributed end point data. 4.3.2 Summary statistic: for continuous outcomes a weighted mean difference (WMD) between groups was estimated. Again, if heterogeneity was found (see section 5) a random effects model was used. 4.3.3 Valid scales: continuous data from rating scales were included only if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000) and the instrument was either a self report or completed by an independent rater or relative (not the therapist). 4.3.4 Endpoint versus change data Where possible endpoint data were presented and if both endpoint and change data were available for the same outcomes then only the former were reported in this review. 5. Test for heterogeneity A Mantel‐Haenszel chi‐square test was used, as well as visual inspection of graphs, to investigate the possibility of heterogeneity. A significance level less than 0.10 was interpreted as evidence of heterogeneity. If heterogeneity was found the data were re‐analysed using a random effects model to see if this made a substantial difference. If it did, the studies responsible for heterogeneity were not added to the main body of homogeneous trials, but summated and presented separately and reasons for heterogeneity investigated. 6. Addressing publication bias Data from all included studies were entered into a funnel graph (trial effect against trial size) in an attempt to investigate the likelihood of overt publication bias (Egger 1997). 7. Sensitivity analyses The effect of including studies with high attrition rates was analysed in a sensitivity analysis. 8. General Where possible, reviewers entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for vitamin E.
Appendix 4. Previous methods and searches
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. Where a trial was described as 'double‐blind' but it was implied that the study was randomised and the demographic details of each group were similar, we have included it. We have excluded quasi‐randomised studies, such as those allocated by using alternate days of the week.
Types of participants
People with schizophrenia or other chronic mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who:
required the use of antipsychotics for more than three months;
developed TD (diagnosed by any criteria at baseline and at least one other occasion) during antipsychotic treatment; and
for whom the dose of antipsychotic medication had been stable for one month or more (the same applies for those free of antipsychotics).
Types of interventions
1. Vitamin E: any dose or means of administration
2. Placebo or no intervention
Types of outcome measures
We have defined clinical efficacy as an improvement in the symptoms of TD of more than 50%, on any scale. We grouped outcomes into short term (less than six weeks), medium term (between six weeks and six months) and long term (more than six months).
Primary outcomes
1. Tardive dyskinesia
Any improvement in the symptoms of individuals of more than 50% on any tardive dyskinesia scale ‐ any time period.
2. Adverse effects
No clinically significant extrapyramidal adverse effects ‐ any time period.
Secondary outcomes
1. Tardive dyskinesia (TD)
1.1 Any improvement in the symptoms of individuals on any TD scale, as opposed to no improvement. 1.2 Deterioration in the symptoms of individuals, defined as any deleterious change on any TD scale. 1.3 Average change in severity of TD during the trial period. 1.4 Average difference in severity of TD at the end of the trial.
2. General mental state changes
2.1 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale. 2.2 Average difference in severity of psychiatric symptoms at the end of the trial.
3. Acceptability of the treatment
3.1 Acceptability of the intervention to the participant group as measured by numbers of people dropping out during the trial.
4. Adverse effects
4.1 Use of any anti‐parkinsonism drugs. 4.2 Average score/change in extrapyramidal adverse effects. 4.3 Acute dystonia.
5. Other adverse effects, general and specific
6. Hospital and service utilisation outcomes
6.1 Hospital admission. 6.2 Average change in days in hospital. 6.3 Improvement in hospital status (for example: change from formal to informal admission status, use of seclusion, level of observation).
7. Economic outcomes
7.1 Average change in total cost of medical and mental health care. 7.2 Total indirect and direct costs.
8. Quality of life/satisfaction with care for either recipients of care or caregivers
8.1. No significant change in quality of life/satisfaction. 8.2 Average score/change in quality of life/satisfaction.
9. Behaviour
9.1 Clinically significant agitation. 9.2 Use of adjunctive medication for sedation. 9.3 Aggression to self or others.
10. Cognitive state
10.1 No clinically important change. 10.2 No change, general and specific.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group Trials Register (March 2010)
This register was searched using the phrase: [(*vitamins* or *vitamin E* or *tocopherol*) in interventions of STUDY field]
This register is compiled by systematic searches of major database, handsearches and conference proceedings (see Group Module).
2. Details of previous electronic search
See Appendix 2.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
Reviewer NM inspected all abstracts of studies identified as above and identified potentially relevant reports. In addition, to ensure reliability, KSW inspected a random sample of these abstracts, comprising 10% of the total. Where disagreement occurred, we resolved this by discussion, or where there was still doubt, acquired the full article for further inspection. We acquired the full articles of relevant reports for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). Once we obtained the full articles, in turn NM and KSW inspected all full reports and independently decided whether they met inclusion criteria. NM and KSW were not blinded to the names of the authors, institutions or journal of publication. Where difficulties or disputes arose, we asked author JM for help and if it was impossible to decide, added these studies to those awaiting assessment and contacted the authors of the papers for clarification.
Data extraction and management
1. Extraction
Reviewer NM extracted data from all included studies. In addition, to ensure reliability, KSW independently extracted data from a random sample of these studies, comprising 10% of the total. Again, we discussed any disagreement, documented decisions and, if necessary, contacted authors of studies for clarification. With remaining problems JM helped clarify issues and we documented those final decisions. We extracted data presented only in graphs and figures whenever possible, but included these only if two reviewers independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. Where possible, we extracted data relevant to each component centre of multi‐centre studies separately.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
a. the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument was not written or modified by one of the trialists for that particular trial.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand calculation of change needs two assessments (baseline and endpoint); normally both sets of data would be available to trialists but if change scores are presented, the SD of the change is often not provided. We decided to primarily use endpoint data and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences rather than standardised mean differences throughout (Deeks 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: a) standard deviations and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996); c) if a scale started from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2SD>(S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We have entered skewed data from studies of less than 200 participants in additional tables rather than into an analysis. Skewed data of means pose less of a problem if the sample size is large; if this condition was met, then we entered skewed data into syntheses.
2.5 Conversion to common measures
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary outcome measures
Where possible, efforts were made to convert outcome measures to dichotomous data. This could be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
We entered data for unfavourable outcomes in such a way that the area to the left of the line of no effect indicates a favourable outcome for vitamin E. Where the outcomes were favourable, the area to the right of the line of no effect indicates a favourable outcome for vitamin E. In either instance we labelled the direction of effect for the interventions at the bottom of the relevant graph.
2.8 Summary of findings table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE Profiler (GRADE 2004) to import data from Review Manager 5 (RevMan 2008) to create the 'Table 1' for the following primary and secondary outcomes considered to be critically important or important to the management of tardive dyskinesia with vitamin E (Reviewer NM was not biased by being familiar with the data).
1. Tardive dyskinesia 1.1 Improved to an important extent (ideally this be stated as in the primary outcome for the review) 1.2 Deteriorated
2. Adverse effect 2.1 Any adverse event (ideally this should be the stated primary outcome: Adverse effects: no clinically significant extrapyramidal adverse effects) 2.2 Specific adverse event
3. Quality of life 3.1 No significant change in quality of life/satisfaction
This table provides information concerning the overall quality of the evidence from the trial, the magnitude of effect of the interventions examined, and the sum of available data on all primary outcomes and the selected secondary outcomes. This summary was used to guide our conclusions and recommendations.
Assessment of risk of bias in included studies
KSW and NM independently assessed included studies for the risk of bias using the Cochrane Collaboration's 'Risk of bias' assessment tool (Deeks 2011) on the following six domains: sequence generation, allocation concealment, blinding or masking, incomplete outcome data, selective outcome reporting and other biases.
For each of these six domains, we assigned a judgement regarding the risk of bias as 'yes' for free of the risk of bias, 'no' for at high risk of bias, or 'unclear' when judgements could not be reliably made due to lack of information in the report or after contacting the trial authors. We used the criteria summarised in Table 8.5.c of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011) to make judgements, and recorded these assessments in the standard 'risk of bias' tables in RevMan 5 (RevMan 2008). For the domains of blinding or masking and for incomplete outcome reporting, we assessed the risk of bias separately for subjectively reported and for objectively ascertained outcomes. We presented these evaluations in the 'risk of bias' summary figure (Figure 3), and risk of bias summary graph (Figure 4) and discussed them further in the results section under Risk of bias in included studies. We incorporated these judgements in assessing limitations in study design for critical and important outcomes in the Table 1
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). For statistically significant results we had planned to calculate the number needed to treat to provide benefit/to induce harm statistic (NNTB/H), and its 95% confidence interval (CI) using Visual Rx (http://www.nntonline.net/) taking account of the event rate in the control group. This, however, was superseded by the Table 1 and the estimates therein.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However, had different scales with similar characteristics been used to measure the same outcome for comparisons, we would have calculated the SMD and transformed the weighted, pooled effect back to the units of one (or more) of the specific and familiar instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If any of the included trials had randomised participants by clusters, and if the results were adjusted for clustering, we combined the adjusted measures of effects of these cluster‐randomised trials with parallel group RCTs using the generic inverse variance technique. If results had not adjusted for clustering, we would have attempted to adjust the results by multiplying the standard errors of the estimates by the square root of the design effect (where the design effect is calculated as DEff = 1 + (M ‐ 1) ICC, where M is the average cluster size and ICC is the intra‐cluster coefficient) (Donner 2002). If the ICC was not reported nor available from the authors, we assumed it to be 0.1 (Ukoumunne 1999).
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only use data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we have presented the additional treatment arms in comparisons. If data were binary we have simply added these added and combined within the two‐by‐two table. If data were continuous we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook. Where the additional treatment arms were not relevant, we have not reproduced these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2007). For any particular outcome should more than 20% of data be unaccounted for, we did not reproduce these data or use them within analyses. If, however, more than 20% of those in one arm of a study were lost, but the total loss was less than 20%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary outcomes
We extracted data to allow an intention‐to‐treat analysis in which all randomised participants were analysed in the groups to which they were originally assigned. If there was a discrepancy in the number randomised and the numbers analysed in each treatment group, we calculated the percentage loss to follow‐up in each group and reported this information. We sought supplementary information from trial authors such as intention to treat and per‐protocol analyses data‐set, or a participant flow diagram in a sufficiently detailed manner as to facilitate data retrieval. If unexplained dropouts exceeded 20% in either group, we would have assigned the same proportion of those with the worst outcome to those lost to follow‐up for dichotomous outcomes (except for mortality and adverse effects) as for those who completed the study, and assessed the impact of this in sensitivity analyses with the results of completers.
3. Continuous outcomes
For continuous outcomes, if provided and where possible, we calculated missing standard deviations from other available data such as standard errors, P, T or F values as detailed in Deeks 2011. If this was not possible, we calculated the SDs according to a validated imputation method that is based on the SDs of the other included studies (Furukawa 2006). We examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.1 Last observation carried forward
We preferred not to make any assumptions about loss to follow‐up for continuous data and analysed results for those who completed the trial, since the use of methods such as the last observation carried forward (LOCF) introduce uncertainty about the reliability of the result (Leucht 2007). If LOCF data had been used in included trials, we reproduced these data and indicated that they were the result of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying situations or people which we had not predicted would arise. When such situations or participant groups arose, we have discussed these fully.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose we have discussed these fully.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected the forest plots to evaluate the possibility of heterogeneity of intervention effects across trials as evidenced by outlying trials with non‐overlapping confidence intervals. We also noted differences in the direction of effect estimates across trials.
3.2 Employing the I2 statistic
We attempted to assess if significant heterogeneity was present using the Chi2 test for homogeneity at a 10% level of significance. We used the I2 statistic to quantify inconsistency (the percentage of the variability in effect estimates that is due to heterogeneity rather than sampling error) (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). In general we interpreted I2 estimates greater than or equal to 50% accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity, although we acknowledge that values of I2 ranging from 30% to 60% may also indicate substantial heterogeneity (Section 9.5.2 ‐ Deeks 2011). When we found substantial levels of heterogeneity in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity). If severe heterogeneity was present (I2 > = 75%) and could not be explained by differences across the trials in terms of clinical or methodological features or by subgroup analyses (see below), we would not have combined the trials in a meta‐analysis, but presented the results in a forest plot.
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook (Deeks 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. Therefore, we chose the fixed‐effect model for all analyses. If the I2 statistic indicated substantial heterogeneity (values 50% or greater), we presented the results using fixed‐effect and random‐effects meta‐analysis and assessed the impact of both models on the direction and precision of the effect estimate.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We anticipated one sub‐group analysis to test the hypothesis that the use of vitamin E is most effective for those with early onset TD (less than five years). We had hoped to present data for this subgroup for the primary outcomes.
2. Investigation of heterogeneity
In the presence of substantial heterogeneity, we had hoped to undertake one pre‐stated subgroup analysis for the primary outcomes in this review to test the hypothesis that vitamin E is more effective for those with an onset of TD within five years.
We also explored the possibility that unanticipated clinical or methodological differences contributed to significant statistical heterogeneity by removing trials with these characteristics from the meta‐analysis, to assess if this reduced the I2 estimate to below 50%. If substantial heterogeneity was reduced, we reported this but presented the forest plot with the data from these trials as well. We stated hypothesis regarding these in the discussion and hoped to evaluate them more fully in future versions of the review.
When unanticipated clinical or methodological heterogeneity were obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we employed all data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we have reported results and discussed them, but continue to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with complete data only. We undertook a sensitivity analysis testing how prone results were to change when 'complete' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we have reported results and discussed them, but continue to employ our assumption.
Data and analyses
Comparison 1. VITAMIN E versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: 1. Not improved to a clinically important extent | 6 | 264 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.89, 1.01] |
| 1.1 short term | 2 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.79, 1.15] |
| 1.2 medium term | 2 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.85, 1.14] |
| 1.3 long term | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.87, 1.02] |
| 2 Tardive dyskinesia: 2. Not any improvement | 7 | 319 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.76, 1.00] |
| 2.1 short term | 2 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.14, 1.47] |
| 2.2 medium term | 3 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.61, 0.98] |
| 2.3 long term | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.82, 1.13] |
| 3 Tardive dyskinesia: 3a. Average endpoint score (AIMS, low score = best) | 10 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 short term | 2 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐2.64 [‐6.35, 1.08] |
| 3.2 medium term | 6 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐1.77 [‐2.59, ‐0.95] |
| 3.3 long term | 3 | 141 | Mean Difference (IV, Fixed, 95% CI) | ‐1.92 [‐3.11, ‐0.73] |
| 4 Tardive dyskinesia: 3b. Average endpoint score ‐ short term (TDRS, low score = best) | 1 | 32 | Mean Difference (IV, Random, 95% CI) | ‐2.10 [‐6.35, 2.15] |
| 5 Tardive dyskinesia: 4. Deterioration of symptoms | 5 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.07, 0.76] |
| 5.1 short term | 2 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.05, 9.97] |
| 5.2 medium term | 2 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.02, 1.12] |
| 5.3 long term | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.03, 1.45] |
| 6 Adverse events: Extrapyramidal adverse events ‐ long term (Simpson‐Agnus Scale, low = better) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7 Adverse events: Specific adverse events: akathisia ‐ long term (BAS, low = better) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8 Any adverse effect | 9 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.35, 4.15] |
| 8.1 short term | 3 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.07, 3.64] |
| 8.2 medium term | 5 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.25, 19.11] |
| 8.3 long term | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.11, 54.87] |
| 9 Discontinuation due to adverse effect | 5 | 123 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.11, 54.87] |
| 9.1 short term | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 medium term | 3 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.3 long term | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.11, 54.87] |
| 10 Mental state: Average score (BPRS, low = best) | 3 | 165 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐3.21, 2.82] |
| 10.1 short term (endpoint score) | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐0.77 [‐7.20, 5.66] |
| 10.2 medium term (change from baseline score) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐7.92 [‐17.20, 1.36] |
| 10.3 long term (endpoint score) | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | 1.20 [‐2.47, 4.87] |
| 11 Leaving study early | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 short term | 3 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.11, 2.00] |
| 11.2 medium term | 8 | 232 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.64, 1.80] |
| 11.3 long term | 3 | 215 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.67, 1.47] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adler 1993.
| Methods | Allocation: "random allocation", ratio of 3 vitamin E: 2 placebo. Double‐blind: no further details. Duration: 36 weeks (preceded by 2 week washout). Setting: inpatients and outpatients of the Department of Veterans Affairs Medical Center, USA. Design: parallel group. | |
| Participants | Diagnosis: schizophrenia, depression (no criteria) and antipsychotic‐induced TD (Research Diagnostic Criteria, Schooler and Kane). N = 40*. Sex: 2 female, 27 male*. Age: average vitamin E 58.0 (SD 9.5) years; placebo 61.0 (SD 9.2) years* | |
| Interventions | 1. Vitamin E: dose increasing over 3 weeks to 1600 IU/day. N = 24.** 2. Placebo. N = 16.** Stable antipsychotic medication: dose average (CPE) vitamin E = 536 mg/day (SD 642); placebo = 921 mg/day (SD 1026). Compliance assessed by pill counts. | |
| Outcomes | TD symptoms: AIMS. Leaving the study early. | |
| Notes | * initial report at 8 weeks, N = 29.
** three people left the study in the first 2 weeks and could not be considered in the analysis ‐ original group assumed from 3:2 randomisation. Source of funding: Supported in part by the Department of Veterans Affairs. Declarations of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned to treatment with Vitamin E, 400IU, or one matching placebo capsule, by mouth, b.i.d." No further details |
| Allocation concealment (selection bias) | Unclear risk | "We used a randomization of 3:2 (vitamin E to placebo) to maximize the number of patients receiving active treatment while maintaining the blind". No further details |
| Blinding of participants and personnel (performance bias) | Unclear risk | "Both rater and patient were blind to the patient's drug assignment". No further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | "Both rater and patient were blind to the patient's drug assignment". No further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “One patient dropped out after 2 weeks due to non‐compliance” “Two patients developed significant medical illnesses ... unrelated to study treatment”, “By prior design, treatment for the first 8 patients was terminated after 8 weeks.” |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | Baseline AIMS scores were somewhat higher in the vitamin E group than in the placebo group, however, this difference was not statistically significant. Small sample size |
Adler 1999.
| Methods | Allocation: randomisation co‐ordinated centrally, allocation with "biased coin" method, stratified by site, age, and baseline TD. Double‐blind: no further details. Duration: 1 year. Setting: outpatients and inpatients, Department of Veterans Affairs Medical Center, USA. Design: parallel | |
| Participants | Diagnosis: schizophrenia, schizoaffective (DSM‐IV), and antipsychotic‐induced TD (Research Diagnostic Criteria). N = 158 Sex: 5 female, 153 male. Age: average 50 years (SD 10). | |
| Interventions | 1. Vitamin E: 1600 IU/day. N = 73. 2. Placebo. N = 85. Antipsychotic medication: not stable dose, average (CPE) vitamin E 380 mg/day (SD 110); placebo 458 mg/day (SD 433). Compliance assessed by pill counts. | |
| Outcomes | TD symptoms: AIMS.
Mental state: BPRS.
Leaving the study early. Adverse Effects: Extrapyramidal symptoms (Modified Simpson‐Angus Scale); Akathisia (Barnes Akathisia Scale) |
|
| Notes | Source of funding: Cooperative Studies Program of the Department of Veterans Affairs, Veterans Affairs Headquarters, Washington, DC Declarations of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation co‐ordinated centrally |
| Allocation concealment (selection bias) | Low risk | Allocation with "biased coin" method, stratified by site, age, and baseline TD |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind: no further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind: no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “Of the 51 subjects who did not complete 1 year, most changed their minds about participating (n = 18), moved too far away from a site to continue in the study (n = 11), or were classified as “whereabouts unknown” (n = 8)”. “Per protocol, we analyzed the data according to the intention‐ to‐treat principle.” |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | No significant differences between groups' baseline characteristics. Small sample size |
Akhtar 1993.
| Methods | Allocation: "random allocation", no further details. Double‐blind: no further details. Duration: 4 weeks (preceded by 2 weeks washout). Setting: inpatients in a psychiatric hospital, India. Design: parallel group. | |
| Participants | Diagnosis: psychiatric disorder (Spitzer criteria) and antipsychotic‐induced TD (Schooler and Kane criteria). N = 32. Sex: 14 female, 18 male. Age: Vitamin E mean 53.06 years (SD 13.39); Placebo mean 56.87 years (SD11.13).. | |
| Interventions | 1. Vitamin E: initial dose 600 mg once daily, doubled in the second week 600mg twice daily (1200 mg/day). N = 17. 2. Placebo. N = 15. Stable antipsychotic medication: dose average (CPE) = 323 mg/day (SD 249); placebo = 187 mg/day (SD 189). | |
| Outcomes | TD symptoms: TDRS. Mental state: BPRS. Adverse effects. Leaving the study early. | |
| Notes | Authors contacted but did not reply. Source of funding: Not reported. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were then randomly assigned". Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | "double blind manner to receive either one capsule of 600 mg vitamin E or an identical placebo" |
| Blinding of outcome assessment (detection bias) | Low risk | Investigators and raters were blind to the intervention until the completion of analysis |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The study results seem to include all participants and no one seemed to drop out from the study. |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | There was no significant difference in the demographic profile of the two groups. Small sample size. |
Dabiri 1994.
| Methods | Allocation: "random allocation", no further details. Double‐blind: yes. Duration: 12 weeks. Setting: outpatients, Outpatients from San Mateo Country Mental Health Services, USA. Design: parallel group. | |
| Participants | Diagnosis: psychiatric disorder (no criteria) and antipsychotic‐induced TD (Research diagnosis, Schooler and Kane criteria). N = 12. Sex: 5 female, 6 male, 1 not specified. Age: average 51 years; range 35‐68. | |
| Interventions | 1. Vitamin E: 400 IU/day for the first week, 800 IU/day for the second week, and 1200 IU/day during the remaining 10 weeks. N = 7. 2. Placebo. N = 5. Stable antipsychotic medication: dose average (CPE) 444 mg/day; range 200 mg to 1000 mg/day. | |
| Outcomes | TD symptoms: AIMS. Leaving study early. Adverse effects: any. | |
| Notes | Authors contacted but did not reply. Source of funding: Not reported. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'random allocation', no further details. |
| Allocation concealment (selection bias) | Low risk | "patients were randomly divided into treatment and placebo groups by a non‐clinical staff member" |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind study", details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | "Each patient was rated blindly by one of us (L.M.D.) before and after treatment using the Abnormal Involuntary Movement Scale (AIMS)". Blinding details not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "One patient who was taking vitamin E stopped treatment after 2 weeks because of diarrhea, leaving five patients taking placebo and six vitamin E". |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | No statistically significant differences in AIMS baseline scores between groups. Very small sample size. |
Dorevitch 1997a.
| Methods | Allocation: "randomized", no further details. Double‐blind: yes. Duration: 20 weeks (4 week washout). Setting: specific setting not reported, Israel. Design: crossover. | |
| Participants | Diagnosis: DSM‐IIIR diagnosis of schizophrenia. All 10 candidates had tardive dyskinesia for a minimum of 5 years and had been exposed to antipsychotic drugs for longer than 10 years N = 10. Sex: 2 female 8 male Age: average 63.1 years; range 56‐70 | |
| Interventions | 1. Vitamin E: dose increasing over 4 weeks to 1600 IU/day. N = 5. 2. Placebo. N = 5. At the start of the study, the patients were receiving an average dose of 652 mg/day chlorpromazine equivalents, with a range of 75 to 4000 mg/day. | |
| Outcomes | Leaving study early.
Adverse effects: parkinsonism; akathisia. Unable to use ‐ Adverse effects: AIMS (data not reported) |
|
| Notes | Source of funding: Not reported. Teva Pharmaceuticals supplied the vitamin E and placebo for this study Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized". Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind". Blinding details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | "double‐blind". Blinding details not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The study results seem to include all participants and no one seemed to drop out from the study. |
| Selective reporting (reporting bias) | High risk | TD symptoms (AIMS) were assessed but not reported. |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size. |
Dorevitch 1997b.
| Methods | Allocation: "randomized", no further details. Double‐blind: yes. Duration: 20 weeks. Setting: Inpatients, Israel. Design: cross‐over. | |
| Participants | Diagnosis: DSM‐III‐R diagnosis of schizophrenia or schizoaffective disorder, research diagnostic criteria for TD (Schooler and Kane criteria). N = 40. Sex: 17 female, 23 male. Age: average 64.4 years (SD 8.5); range 32‐80. | |
| Interventions | 1. Vitamin E: 400 IU/day during the first week, titrated to 800 IU/day for the second week, 1200 IU/day for the third week, and 1600 IU/day from week 4 until the end of 8 weeks. N = 18. 2. Placebo. N = 22. Stable antipsychotic medication: dose average (CPE) 594 mg/day; range 75 mg to 5000 mg/day. | |
| Outcomes | TD symptoms: AIMS.
Leaving study early.
Adverse effects.
Unable to use ‐ Mental state: BPRS (data not reported). |
|
| Notes | Source of funding: Not reported. Teva Pharmaceuticals supplied the vitamin E and placebo for this study Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised" ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind," Blnding details were not reported |
| Blinding of outcome assessment (detection bias) | Low risk | "Two senior psychiatrists served as blinded raters" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Two patients did not complete the study. Both patients were from the placebo phase of the placebo‐vitamin E sequence group. One died while choking on food and the second as the result of a traffic accident" |
| Selective reporting (reporting bias) | High risk | “Addition of vitamin E or placebo did not adversely affect patient mental status as measured by brief psychiatric rating scale (BPRS).“ BPRS data not fully reported |
| Other bias | Unclear risk | Baseline characteristics not reported. Small sample size. |
Egan 1992.
| Methods | Allocation: 'random allocation', no further details. Double‐blind: no further details. Duration: 12 weeks (6 weeks then crossed over to another 6 weeks, no washout). Setting: inpatients and outpatients, USA. Design: cross‐over. | |
| Participants | Diagnosis: schizophrenia, schizoaffective, bipolar disorder, depression (DSM‐III‐R) and antipsychotic‐induced TD (Schooler and Kane criteria). N = 21. Sex: 8 female, 13 male. Age: average 43.9 years (SD 2.8) | |
| Interventions | 1. Vitamin E: 400 IU/day for week 1, 800 lU/day for week 2, 1200 lU/day for week 3, and 1600 IU/day for weeks 4‐6. N = 10. 2. Placebo. N = 11. Stable antipsychotic medication: dose average (CPE) 1946 mg/day (no SD, N = 15). | |
| Outcomes | TD symptoms: AIMS.
Side effects.
Leaving study early. Unable to use ‐ Mental symptoms: PSAS, NSRS (means and SDs not reported) |
|
| Notes | Three patients were not included in the data analysis: one dropped out and two had inconsistent vitamin E blood levels Source of funding: Not reported. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were assigned randomly" Details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind," Details not reported. |
| Blinding of outcome assessment (detection bias) | Low risk | "All raters were blind to treatment with either placebo or vitamin E. " |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not ITT analysis: "Eighteen patients who demonstrated high blood levels of vitamin E were included in the data analysis", 3 patients were excluded from the analysis. |
| Selective reporting (reporting bias) | High risk | Data for mental state (PSAS and NSAS) not reported. |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size |
Elkashef 1990.
| Methods | Allocation: 'random allocation', no further details. Double‐blind: no further details. Duration: 10 weeks (4 weeks then crossed over to another four weeks; randomisation was preceded by 2 weeks washout. Setting: outpatients, USA. Design: cross‐over. | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐III‐R) and antipsychotic‐induced TD (Schooler and Kane criteria). N = 10. Sex: 1 female, 7 male (among completers). Age: average 56.6 years (SD 12) (among completers). History: no description of chronicity of TD. | |
| Interventions | 1. Vitamin E: 400 IU/day for the first week. 400 IU twice daily (800 IU/day)for the second week, and 400 IU three times daily (1200 IU/day) for the final two weeks. N = 5. 2. Placebo. N = 5. Stable antipsychotic medication: dose not specified. | |
| Outcomes | TD symptoms: AIMS.
Adverse effects.
Leaving study early. Unable to use ‐ Mental state: BPRS |
|
| Notes | Source of funding: Not reported. Hollman‐La Roche, Inc., supplied the drug and placebo for this study. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The subjects were then assigned in a random, double‐blind manner...", no further details. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind: no further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | "The subjects were evaluated biweekly by a blind trained rater using the AIMS and the Brief Psychiatric Rating Scale (BPRS)" Details of blinding not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2/5 participants in the placebo group while none in the vitamin E group dropped out: "Two patients did not complete the study, one because of noncompliance and the other experienced substantial side effects (nausea) while taking placebo" |
| Selective reporting (reporting bias) | High risk | AIMS data partially reported and BPRS evaluated but not reported |
| Other bias | Unclear risk | The baseline severity of tardive dyskinesia was closely matched in the two groups. Very small sample size. |
Lam 1994.
| Methods | Allocation: 'random allocation', no further details.
Double‐blind: no further details.
Duration: 16 weeks: 2 week placebo lead‐in phase, 6 weeks intervention followed by 2 weeks wash‐out then crossed over to another 6 weeks. Setting: inpatients, Hong Kong. Design: cross‐over. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III‐R) and antipsychotic‐induced TD (Schooler and Kane criteria). N = 16. Sex: 7 female, 5 male*. Age: average 61.8 yrs (SD 12.8 yrs)*. History: no history of chronicity of TD. | |
| Interventions | 1. Vitamin E: 400 IU/day for the first week; 400 IU twice daily in the second week; 400 IU three times daily from week 3 to 6. N = 5*. 2. Placebo. N = 7*. Stable antipsychotic medication. For those taking antipsychotic medication the average daily dose was 365 mg CPZ equivalent. | |
| Outcomes | TD symptoms: AIMS
Leaving study early (assuming equal randomisation into the two groups). Unable to use ‐ Mental state: BPRS (no mean or SD reported) Adverse effects. |
|
| Notes | * Completers 4 people left study early (no information about allocation); reasons: death, deterioration of symptoms of schizophrenia, bacillary dysentery (all stated not to be related to treatment), poor compliance. Authors contacted and replied, no more information available. Source of funding: Not reported. Declarations of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Subjects were then selected randomly". No further details. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind". Details not reported. |
| Blinding of outcome assessment (detection bias) | Low risk | "Subjects were evaluated weekly with the AIMS...and Brief Psychiatric Rating Scale..., respectively, by two independent blind raters at the initial stabilization period, and the last 2 weeks of each test period" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Twelve participants completed the trial. One patient died of unrelated medical illness, one contracted bacillary dysentery and was dropped from the trial, and one had poor compliance and refused to continue medication. It was not reported to which groups these participants were allocated. |
| Selective reporting (reporting bias) | High risk | TD symptoms data not reported as mean (SD); BPRS data not reported per period. Adverse effects not reported per group. |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size. |
Lohr 1996.
| Methods | Allocation: "random assigned" ‐ no further details. Double‐blind: participants and personnel blinded Duration: 8 weeks. Setting: outpatients, USA. Design: parallel. | |
| Participants | Diagnosis: schizophrenia, bipolar disorder, unipolar depression (no specified criteria) and antipsychotic‐induced TD (Schooler and Kane criteria). N = 55. Sex: 2 female, 33 male, 20 not informed. Age: average 48.9 years (SD 13.6). |
|
| Interventions | 1. Vitamin E: 1600 IU/day. N = 17 (completers)*. 2. Placebo. N = 18 (completers)*. Stable psychotropic medication for at least 1 month prior to entry into study. Antipsychotic dose average (CPE) vitamin E = 706 mg/day (SD 680); placebo = 376 mg/day (SD 242). | |
| Outcomes | TD symptoms: mAIMS. Mental state: BPRS (reported for subgroup with schizophrenia, n = 29) Leaving the study early. |
|
| Notes | *Total numbers randomised per group were imputed from numbers analysed per group.
Authors contacted but did not reply. Source of funding: Partial funding by a VA Merit Review grant, and United States Public Health Service grants. Vitamin E and placebo supplied by Hoffmann‐La Roche Inc. Declarations of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned to receive either active vitamin E or sesame oil placebo gelcaps." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment details not reported. |
| Blinding of participants and personnel (performance bias) | Low risk | "Patients were randomly assigned to receive either active vitamin E or sesame oil placebo gel caps, which were indistinguishable from the active gel caps." |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information to make a judgement |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropout rate of 36% (20 of 55 patients) but not reported per study group "2 developed manic symptoms necessitating medical changes, and 18 were non‐compliant with either the vitamin E or the psychotropic medication. These 20 patients, who did not differ significantly from the remaining 35 patients in terms of age, gender, or diagnosis, were dropped from the study". |
| Selective reporting (reporting bias) | High risk | Adverse effects: extrapyramidal side effects (Parkinsonism) data not reported. |
| Other bias | Unclear risk | There were no significant differences in baseline characteristics between the two study groups. Small sample size. |
Sajjad 1998.
| Methods | Allocation: "random allocation" Double‐blind: probably not, there was no placebo administered to the control group. Duration: 7 months. Setting: inpatients, UK. Design: parallel. | |
| Participants | Diagnosis: antipsychotic‐induced TD (Schooler and Kane criteria). N = 20. Sex: 7 female, 13 male. Age: average 68 yrs (SD 8.7). | |
| Interventions | 1. Vitamin E: first week 400 mg/day. Dose was increased to 600 mg/day in the second week, 800 mg/day in the fourth month, 1200 mg/day in the fifth month and 1600 mg/day in the sixth month. N = 11. 2. Placebo. N = 9. Stable antipsychotic medication throughout the trial. | |
| Outcomes | TD symptoms: AIMS. Adverse effects. Leaving the study early. | |
| Notes | Source of funding: Not reported. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...the patients were randomly divided into two groups using... a computer statistic programme." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | As an active group was compared to treatment as usual, the study could not be double‐blinded. The only person blinded seems to have been the doctor "...the dose increased by another doctor not involved in the ratings and who, therefore, was blind as to whether or not the patient was receiving a‐tocopherol for the first month of the trial" |
| Blinding of outcome assessment (detection bias) | High risk | Rater initially blind. However, after one month, the rater performed statistical tests and hence blindness was not maintained. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 40% dropout (12/20 participants completed the study): 6/11 participants in the intervention and 2/9 participants in the control group did not complete the trial. By the fourth month, there were 12 patients left in the trial, five in the treatment group and seven in the control group. Patients excluded at this stage included those whose dose of antipsychotic medication was changed. |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | Mean AIMS scores and age were similar between groups at baseline. Very small sample size |
Schmidt 1991.
| Methods | Allocation: "randomized pattern", no further details. Double‐blind: no further details. Duration: 4 weeks (2 weeks then crossed over to another 2 weeks, no washout). Setting: inpatients, Switzerland. Design: cross‐over. | |
| Participants | Diagnosis: schizophrenia, depression, schizoaffective psychoses (no criteria) and antipsychotic‐induced TD (no criteria). N = 23. Sex: 12 female, 11 male. Age: average 45 years; range 21‐88 years. | |
| Interventions | 1. Vitamin E: dose 1200 IU/ day. N = 13. 2. Placebo. N = 10. Stable antipsychotic medication: dose unspecified. | |
| Outcomes | TD symptoms: AIMS. Adverse effects. Leaving study early. | |
| Notes | It was observed that 2 of the patients who benefited from the vitamin E therapy continued taking it: after stopping vitamin E medication, one of them experienced an increase in TD, while in the other the beneficial effect was still observed even 3 months later Source of funding: Not reported. Declarations of interest: Not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized pattern", no further details. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | "double‐blind". Details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Details not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13 patients initially randomised to vitamin E: two left before the end of the study (1 died and the other withdrew); 10 patients initially randomised to placebo: two left before the end of the study (1 died and the other had his treatment modified) |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported. |
| Other bias | Unclear risk | Baseline characteristics similar between study groups. Very small sample size; cross‐over design. |
Zhang 2004.
| Methods | Allocation: randomly assigned Double‐blind: yes Duration: 12 weeks. Setting: inpatients, China. Design: parallel | |
| Participants | Diagnosis: DSM‐III‐R criteria for schizophrenia, using the Structured Clinical Interview for DSM‐III‐R ; TD diagnosed by Schooler and Kane criteria. N = 41. Sex: 18 female, 23 male. Age: average vitamin E 54.5 years (SD 10.1); placebo 53.3 (SD 9.7). | |
| Interventions | 1. Vitamin E: 800 IU/day during the first week and increased up to 1200 IU/day for another 11 weeks . N = 22. 2. Placebo. N = 19. Clinically stable with duration of TD for at least 1 year; stable dose of oral antipsychotics | |
| Outcomes | TD symptoms: AIMS.
Leaving study early.
Unable to use ‐ Mental state: PANSS |
|
| Notes | Source of funding: Not reported. Declarations of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Eligible patients were randomly assigned" no further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment method not reported |
| Blinding of participants and personnel (performance bias) | Low risk | "either capsulized vitamin E (N = 22) or identically capsulized placebo (N = 19) using a double‐blind fashion." |
| Blinding of outcome assessment (detection bias) | Low risk | "TD was evaluated in all patients using the Abnormal Involuntary Movement Scale (AIMS) at baseline, 6 and 12 weeks of treatment by the same trained investigator (DCC) who was blind to the patient's medication status. The patient's psychopathology was rated at baseline and at posttreatment using the Positive and Negative Syndrome Scale (PANSS), 18 which was performed by 2 trained psychiatrists who were blinded scoring the PANSS." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Forty‐one inpatients with TD completed a double‐ blind, placebo‐controlled, parallel‐group study of vitamin E. Twenty‐ two of the patients were randomly assigned to receive a fixed dose of 1200 IU/d vitamin E, and 19 were assigned to a placebo". "Eligible patients were randomly assigned to receive either capsulized vitamin E (N = 22) or identically capsulized placebo (N = 19)". All randomised participants seem to have completed the study. |
| Selective reporting (reporting bias) | High risk | Outcome data were not reported for mental symptoms (PANSS) |
| Other bias | Low risk | "No significant differences in demographic data were observed between vitamin E and placebo groups" |
Scales AIMS ‐ Abnormal Involuntary Movement Scale NSRS ‐ Negative Symptom Rating Scale PANSS ‐ Positive and Negative Syndrome Scale PSAS ‐ Psychiatric Symptoms Assessment Scale SAS ‐ Simpsom‐Angus Scale for Extrapyramidal Side Effects TDRS ‐ Tardive Dyskinesia Rating Scale
Others CPE ‐ Chlorpromazine equivalents CPZ ‐ Chlorpromazine DSM‐III‐R‐ Diagnostic Statistical Manual of Mental Disorders TD ‐ tardive dyskinesia
BPRS: British Psychiatric Rating Scale ITT ‐ intention‐to‐treat IU ‐ international units mAIMS:Adverse and Involuntary Movement Scale SD: Standard Deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Dorfman‐Etrog 1999 | Allocation: randomised. Participants: people with schizophrenia, but not tardive dyskinesia. |
| Junker 1992 | Allocation: randomised.
Participants: people with both tardive dyskinesia and schizophrenia. N = 25.
Interventions: vitamin E supplementation vs no vitamin E supplementation.
Outcomes: movement disorders, not possible to extract any data. Authors contacted twice in 1995 and failed to reply. |
| Lohr 1988 | Allocation: randomised, cross‐over design. Participants: people with both tardive dyskinesia and schizophrenia. N = 15. Interventions: vitamin E supplementation vs no vitamin E supplementation. Outcomes: movement disorders, not possible to extract data from the first period. Authors contacted twice in 1995 and failed to reply. |
| Peet 1993 | Allocation: not randomised, cohort study. |
| Ricketts 1995 | Allocation: quasi‐randomised, cross‐over study. Participants: people with both tardive dyskinesia and mental retardation due to brain damage, not schizophrenia. |
| Salmasi 2009 | Allocation: randomised, double‐blind, placebo‐controlled study. Participants: people with schizophrenia, but not tardive dyskinesia. Interventions: vitamin E supplementation vs placebo. Outcomes: insulin resistance in patients treated with olanzapine |
| Shriqui 1992 | Allocation: randomised, cross‐over design. Participants: people with both tardive dyskinesia and schizophrenia. N = 27. Interventions: vitamin E supplementation vs no vitamin E supplementation. Outcomes: movement disorders, not possible to extract data from the first period. Authors contacted twice in 1995 and failed to reply. |
| Spivak 1992 | Allocation: not randomised, cohort study. |
Characteristics of studies awaiting assessment [ordered by study ID]
Kar‐Ahmadi 2002.
| Methods | Allocation: "randomized" no further details. Blindness: double ‐ no further details. Duration: 6 weeks. Setting: inpatients. Design: parallel. |
| Participants | Diagnosis: antipsychotic‐induced TD. N = 30. Sex: unknown. Age: unknown. |
| Interventions | 1. Vitamin E: dose 600 mg/ day. N = 15. 2. Placebo. N = 15. Stable antipsychotic medication: dose unspecified. |
| Outcomes | TD symptoms: AIMS. |
| Notes | A copy of this study was not available in the British Library. |
AIMS: Abnormal Involuntary Movement Scale TD ‐ tardive dyskinesia
Characteristics of ongoing studies [ordered by study ID]
ISRCTN14688109 2015.
| Trial name or title | Investigation of the potential beneficial effects of cannabidiol in the treatment of tardive dyskinesia |
| Methods | Randomised, double‐blind placebo‐controlled study |
| Participants | Target number of participants: 28 per group Adults 18 years and older that currently meets the ICD‐10 diagnosis of a psychotic disorder, verified with the Mini International Neuropsychiatric Interview (MINI‐PLUS) questionnaire and that currently meets the clinical diagnosis of tardive dyskinesia confirmed with the Abnormal Involuntary Movement Scale (AIMS). Patients should currently be receiving treatment for a psychotic disorder and should be on either the atypical or conventional antipsychotics |
| Interventions | 1. Group 1 has high cannabidiol extract Nabidiolex® (CBD) (300 mg) administered twice a day for six weeks as an adjunctive treatment alongside their usual antipsychotic medication. CBD will be administered orally in capsules. 2. Group 2 has vitamin E (400 IU) administered daily for six weeks as an adjunctive treatment alongside their usual antipsychotic medication |
| Outcomes | Improvement in symptoms of tardive dyskinesia measured using the Abnormal Involuntary Movement Scale (AIMS). Assessments will be conducted at baseline, 2 weeks, 4 weeks, 6 weeks (post‐treatment) and at 12 weeks follow‐up. Side effects of CBD will be periodically assessed with the Glasgow check list and reported at each assessment Improvement in psychotic symptoms |
| Starting date | 01/12/2015 |
| Contact information | jaiyeolakajero@yahoo.com |
| Notes | Source of funding: Federal Neuropsychiatric Hospital, Nigeria. Trial is part of a Stellenbosch University PhD, Intention to publish date: 01/01/2018. |
ICD ‐ International Classification of Diseases IU ‐ international units
Differences between protocol and review
The protocol as published with this review has evolved over time. The revisions of protocol are in line with the development of RevMan and in keeping with Cochrane guidance. We think the revisions have greatly improved and enhanced this review. We do not think, however, that it has materially affected our conduct of the review or interpretation of the results.
There was a substantial update to the protocol in the 2010 review update that the 2017 update also follows, but with two additions.
The comparison: 'Vitamin E: any dose or means of administration compared to any other intervention'
The outcomes 'Acceptability of treatment' and 'Social confidence, social inclusion, social networks, or personalised quality of life' were added to the 'Summary of findings' table. These outcomes were designated to be of importance to patients.
The previous methods are reproduced in Appendix 3 and Appendix 4.
Contributions of authors
Karla Soares‐Weiser ‐ protocol writing, searching, trial selection, data extraction and assimilation, report writing.
Nicola Maayan ‐ data extraction, 'Summary of findings' table.
Hanna Bergman ‐ data extraction, report writing
Sources of support
Internal sources
-
Enhance Reviews Ltd, UK.
Salary and logistics support for Karla Soares‐Weiser and Nicola Maayan for earlier published versions of this review, and for Hanna Bergman for the 2017 update.
-
Queensland Health, Australia.
Salary and logistics support for John McGrath, author of earlier published versions of this review
-
Universidade Federal de Sao Paulo, Brazil.
Salary and logistics support for Karla Soares‐Weiser for earlier published versions of this review
External sources
-
CAPES ‐ Ministry of Education, Brazil.
Salary and logistics support for Karla Soares‐Weiser for earlier published versions of this review
-
NIHR HTA Project Grant, reference number: 14/27/02, UK.
Salary support for Hanna Bergman. Support for patient involvement. Support for traceable data database.
Declarations of interest
KSW is the Deputy Editor‐in‐Chief for Cochrane and Cochrane Innovations. When the NHIR HTA programme grant was awarded that included to update this review, Karla was the Managing Director of Enhance Reviews Ltd.
NM worked for Enhance Reviews Ltd during the preparation of this review, a company that carries out systematic reviews mostly for the public sector, it currently does not provide services for the pharmaceutical industry.
HB worked for Enhance Reviews Ltd during the preparation of this review, a company that carries out systematic reviews mostly for the public sector, it currently does not provide services for the pharmaceutical industry.
Edited (no change to conclusions)
References
References to studies included in this review
Adler 1993 {published data only}
- Adler LA, Edson R, Lavori P, Peselow E, Duncan E, Rosenthal M, et al. Long‐term treatment effects of vitamin E for tardive dyskinesia. Biological Psychiatry 1998;43:868‐72. [CSzG: 4096] [DOI] [PubMed] [Google Scholar]
- Adler LA, Peselow E, Angrist B, Duncan E, Lee M, Rosenthal M, et al. Vitamin E in tardive dyskinesia: effects of longer term treatment. Proceedings of the 31st Annual Meeting of the American College of Neuropsychopharmacology; 1992 Dec 14‐18; San Juan, Puerto Rico. 1992. [CSzG: 6958]
- Adler LA, Peselow E, Duncan E, Rosenthal M, Angrist B. Vitamin E in tardive dyskinesia: time course of effect after placebo substitution. Psychopharmacology Bulletin 1993;29:371‐4. [CSzG: 174] [PubMed] [Google Scholar]
- Adler LA, Peselow E, Rotrosen J, Duncan E, Lee M, Rosenthal M, et al. Vitamin E treatment of tardive dyskinesia. American Journal of Psychiatry 1993;150(9):1405‐7. [CSzG: 1490] [DOI] [PubMed] [Google Scholar]
Adler 1999 {published and unpublished data}
- Adler LA, Rotrosen J, Edson R, Lavori P, Lohr J, Hitzemann R, et al. Vitamin E treatment for tardive dyskinesia. Archives of General Psychiatry 1999;56:836‐41. [CSzG: 4097] [DOI] [PubMed] [Google Scholar]
- Adler LA, Rotrosen J, Lavori P, Edson R. Vitamin E treatment of TD: development of a VA cooperative study. Biological Psychiatry 1994;35:730‐1. [CSzG: 8373] [Google Scholar]
- Bridler R. Vitamin E is ineffective in treatment of late dyskinesias [Vitamin E unwirksam in der Behandlung von Spatdyskinesien (TD)]. Praxis 2001;90(18):809‐10. [CSzG: 7644] [PubMed] [Google Scholar]
- Caligiuri MP, Lohr JB, Rotrosen J, Adler L, Lavori P, Edson R, et al. Reliability of an instrumental assessment of tardive dyskinesia: results from VA Cooperative Study #394. Psychopharmacology 1997;132:61‐6. [CSzG: 4903] [DOI] [PubMed] [Google Scholar]
- Edson R, Lavori P, Tracy K, Adler LA, Rotrosen J. Interrater reliability issues in multicentric trials, part II: statistical procedures used in Department of Veterans Affairs Cooperative Study #394. Psychopharmacology Bulletin 1997;33:59‐67. [CSzG: 16730] [PubMed] [Google Scholar]
- Lohr JB, Lavori P. Whither vitamin E and tardive dyskinesia?. Biological Psychiatry 1998;43:861‐2. [CSzG: 16718] [DOI] [PubMed] [Google Scholar]
- Tracy K, Adler LA, Rotrosen J, Edson R, Lavori P. Interrater reliability issues in multicentric trials, part I: theoretical concepts and operational procedures used in Department of Veterans Affairs Cooperative Study 394. Psychopharmacology Bulletin 1997;33:53‐7. [CSzG: 16731] [PubMed] [Google Scholar]
Akhtar 1993 {published data only}
- Akhtar S, Jajor TR, Kumar S. Vitamin E in the treatment of tardive dyskinesia. Journal of Postgraduate Medicine 1993;39:124‐6. [CSzG: 191] [PubMed] [Google Scholar]
Dabiri 1994 {published data only}
- Dabiri LM, Pasta D, Darby JK, Mosbacher D. Effectiveness of vitamin E for treatment of long‐term tardive dyskinesia. American Journal of Psychiatry 1994;151:925‐6. [CSzG: 657] [DOI] [PubMed] [Google Scholar]
Dorevitch 1997a {published data only}
- Dorevitch A, Lerner V, Shalfman M, Kalian M. Lack of effect of vitamin E on serum creatine phosphokinase in patients with long‐term tardive dyskinesia. International Clinical Psychopharmacology 1997;12:171‐3. [CSzG: 1909] [DOI] [PubMed] [Google Scholar]
Dorevitch 1997b {published data only}
- Dorevitch A, Kalian M, Shlafman M, Lerner V. Treatment of long‐term tardive dyskinesia with vitamin E. Biological Psychiatry 1997;41:114‐6. [CSzG: 3204] [DOI] [PubMed] [Google Scholar]
Egan 1992 {published data only}
- Egan MF, Hyde TM, Albers GW, Elkashef A, Alexander RC, Reeve A, et al. Treatment of tardive dyskinesia with vitamin E. American Journal of Psychiatry 1992;149:773‐7. [CSzG: 741] [DOI] [PubMed] [Google Scholar]
Elkashef 1990 {published data only}
- Elkashef AM, Ruskin PE, Bacher N, Barret D. Vitamin E in the treatment of tardive dyskinesia. American Journal of Psychiatry 1990;147:505‐6. [CSzG: 749] [DOI] [PubMed] [Google Scholar]
Lam 1994 {published data only}
- Lam LCW, Chiu HFK, Hung SF. Vitamin E in the treatment of tardive dyskinesia: a replication study. Journal of Nervous and Mental Diseases 1994;182:113‐4. [CSzG: 1605] [DOI] [PubMed] [Google Scholar]
Lohr 1996 {published data only}
- Lohr JB, Caliguiri M. A double‐blind placebo‐controlled study of vitamin E treatment of tardive dyskinesia. Journal of Clinical Psychiatry 1996;57:167‐73. [CSzG: 1739] [PubMed] [Google Scholar]
Sajjad 1998 {published data only}
- Sajjad S. Vitamin E in the treatment of tardive dyskinesia: a preliminary study over seven months at different doses. International Clinical Psychopharmacology 1998;13(4):147‐55. [CSzG: 5237] [DOI] [PubMed] [Google Scholar]
Schmidt 1991 {published data only}
- Schmidt M, Meister P, Baumann P. Treatment of tardive dyskinesias with vitamin E. European Psychiatry 1991;6:201‐7. [CSzG: 2517] [Google Scholar]
Zhang 2004 {published data only}
- Zhang XY, Zhou DF, Cao LY, Xu CQ, Chen da C, Wu GY. The effect of vitamin E treatment on tardive dyskinesia and blood superoxide dismutase: a double‐blind placebo‐controlled trial. Journal of Clinical Psychopharmacology 2004;24(1):83‐6. [CSzG: 10831] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Dorfman‐Etrog 1999 {published data only}
- Dorfman‐Etrog P, Hermesh H, Prilipko L, Weizman A, Munitz H. The effect of vitamin E addition to acute neuroleptic treatment on the emergence of extrapyramidal side effects in schizophrenic patients: an open label study. European Neuropsychopharmacology 1999;9(6):475‐7. [DOI] [PubMed] [Google Scholar]
Junker 1992 {published data only}
- Junker D, Steigleider P, Gattaz WF. Alpha‐tocopherol in the treatment of tardive dyskinesia. Schizophrenia Research 1992;6:122‐3. [Google Scholar]
Lohr 1988 {published data only}
- Cadet JL, Lohr JB. Possible involvement of free radicals in neuroleptic induced movement disorders. Annals of the New York Academy of Sciences 1989;570:176‐85. [DOI] [PubMed] [Google Scholar]
- Lohr JB, Cadet JL, Lohr MA, Jeste DV, Wyatt RJ. Alpha‐tocopherol in tardive dyskinesia [letter]. Lancet 1987;1(8538):913‐4. [DOI] [PubMed] [Google Scholar]
- Lohr JB, Cadet JL, Lohr MA, Larson L, Wasli E, Wade L, et al. Vitamin E in the treatment of tardive dyskinesia: the possible involvement of free radical mechanisms. Schizophrenia Bulletin 1988;14(2):291‐6. [DOI] [PubMed] [Google Scholar]
Peet 1993 {published data only}
- Peet M, Laugharne J, Rangarajan N, Reynolds G. Tardive dyskinesia. Hospital and Community Psychiatry 1993;44(8):795. [DOI] [PubMed] [Google Scholar]
- Peet M, Laugharne J, Rangarajan N, Reynolds GP. Tardive dyskinesia, lipid peroxidation and sustained amelioration with vitamin E treatment. International Clinical Psychopharmacology 1993;8:151‐3. [DOI] [PubMed] [Google Scholar]
Ricketts 1995 {published data only}
- Ricketts RW, Singh NN, Ellis CR, Chambers S, Singh YN, Carmanico SJ, et al. Calcium channel blockers and vitamin E for tardive dyskinesia in adults with mental retardation. Journal of Developmental and Physical Disabilities 1995;7(2):161‐74. [Google Scholar]
Salmasi 2009 {published data only}
- Salmasi FB, Jazayeri M, Ghaeli P, Hashemian F, Akhondzadeh S, Raisi F, et al. Comparing the effects of high‐dose vitamin E with those of placebo on insulin resistance in patients with schizophrenia treated with olanzapine. Journal of Clinical Psychopharmacology 2009;29(2):182‐3. [DOI] [PubMed] [Google Scholar]
Shriqui 1992 {published data only}
- Shriqui CL, Bradwejn J, Annable L, Jones BD. Vitamin E in the treatment of tardive dyskinesia: a double‐blind placebo‐controlled study. American Journal of Psychiatry 1992;149:391‐3. [DOI] [PubMed] [Google Scholar]
Spivak 1992 {published data only}
- Spivak B, Schwartz B, Radwan M, Weizman A. Alpha‐tocopherol treatment for tardive dyskinesia. Journal of Nervous and Mental Disease 1992;180:400‐1. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Kar‐Ahmadi 2002 {published data only}
- Kar‐Ahmadi M. Vitamin E in the management of drug induced tardive dyskinesia: a double‐blind randomized clinical trial. Journal of Research in Medical Sciences 2002;4(6):311 20. [Google Scholar]
References to ongoing studies
ISRCTN14688109 2015 {published data only}
- ISRCTN14688109. Investigation of the potential beneficial effects of cannabidiol in the treatment of tardive dyskinesia. http://www.isrctn.com 2015.
Additional references
Alabed 2011
- Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma‐aminobutyric acid agonists for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 4. [DOI: 10.1002/14651858.CD000203.pub3] [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1992
- American Psychiatric Association. Tardive Dyskinesia: A Task Force Report of the American Psychiatric Association. Washington DC: APA, 1992. [Google Scholar]
Armitage 1991
- Armitage P. Should we cross off the crossover?. Journal of Clinical Pharmacology 1991;32:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR, Charing C. Westminster Medical School HHES. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Barnes 1993
- Barnes TRE, Ewards JG. The side‐effects of antipsychotic drugs. I. CNS and neuromuscular effects. Antipsychotic Drugs and their Side‐Effects. London: Harcourt Brace & Company, 1993. [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276(8):637‐9. [PUBMED: 8773637] [DOI] [PubMed] [Google Scholar]
Bergen 1984
- Bergen JA, Griffiths DA, Rey JM, Beumont PJV. Tardive dyskinesia: fluctuating patient or fluctuating rater. British Journal of Psychiatry 1984;144:498‐502. [DOI] [PubMed] [Google Scholar]
Bergen 1989
- Bergen JA, Eyland EA, Campbell JA. The course of tardive dyskinesia in patients on long‐term neuroleptics. British Journal of Psychiatry 1989;154:523‐8. [DOI] [PubMed] [Google Scholar]
Bergman 2017
- Bergman H, Walker DM, Nikolakopoulou A, Soares‐Weiser K, Adams CE. Systematic review of interventions for treating or preventing antipsychotic‐induced tardive dyskinesia. Health Technol Assess 2017 Aug;21(43):1‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhoopathi 2006
- Bhoopathi PS, Soares‐Weiser K. Benzodiazepines for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD000205.pub2] [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Cadet 1989
- Cadet JL, Lohr JB. Possible involvement of free radical in neuroleptic‐induced movement disorders. Annals of the New York Academy of Sciences 1989;570:176‐85. [DOI] [PubMed] [Google Scholar]
Casey 1994
- Casey DE. Tardive dyskinesia: pathophysiology. In: Bloom FE, Kupfer DJ editor(s). Psychopharmacology. The Fourth Generation of Progress. New York: Raven Press, 1994. [Google Scholar]
Chouinard 2008
- Chouinard G, Chouinard VA. Atypical antipsychotics: CATIE study, drug‐induced movement disorder and resulting iatrogenic psychiatric‐like symptoms, supersensitivity rebound psychosis and withdrawal discontinuation syndromes. Psychotherapy and Psychosomatics 2008; Vol. 77, issue 2:69‐77. [PUBMED: 18230939] [DOI] [PubMed]
Cloud 2014
- Cloud LJ, Zutshi D, Factor SA. Tardive Dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics 2014;11(1):166‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Correll 2004
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second‐generation antipsychotics: a systematic review of 1‐year studies. American Journal of Psychiatry 2004;161(3):414‐25. [DOI] [PubMed] [Google Scholar]
Correll 2008
- Correll CU, Schenka EM. Tardive dyskinesia and new antipsychotics. Current Opinion in Psychiatry 2008;21:151‐6. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG, editor(s). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Sayeh 2006
- El‐Sayeh HG, Lyra da Silva JP, Rathbone J, Soares‐Weiser K. Non‐neuroleptic catecholaminergic drugs for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000458.pub2] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2011
- Essali A, Deirawan H, Soares‐Weiser K, Adams CE. Calcium channel blockers for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD000206.pub3] [DOI] [PubMed] [Google Scholar]
Feltner 1993
- Feltner DE, Hertzman M. Progress in the treatment of tardive dyskinesia: theory and practice. Hospital and Community Psychiatry 1993;44(1):25‐34. [DOI] [PubMed] [Google Scholar]
Fernandez 2001
- Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14‐year follow‐up. Neurology 2001;56:805‐7. [DOI] [PubMed] [Google Scholar]
Fleiss 1984
- Fleiss JL. The crossover study. The Design and Analysis of Clinical Experiments. Chichester: John Wiley & Sons, 1984. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Glazer 1990
- Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. British Journal of Psychiatry 1990;157:585‐92. [DOI] [PubMed] [Google Scholar]
Glazer 2000
- Glazer WM. Expected incidence of tardive dyskinesia associated with atypical antipsychotics. Journal of Clinical Psychiatry 2000;61(Suppl 4):21‐6. [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Revised Edition. Washington, DC: Department of Health, Education and Welfare, 1976. [Google Scholar]
Hawkins 1989
- Hawkins DR. Successful prevention of tardive dyskinesia. Journal of Orthomolecular Medicine 1989;4(1):35‐6. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Jeste 1993
- Jeste DV, Caligiuri MP. Tardive dyskinesia. Schizophrenia Bulletin 1993;19(2):303‐15. [DOI] [PubMed] [Google Scholar]
Jűni 2001
- Jűni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ (Clinical research ed.) 2001;323(7303):42‐6. [PUBMED: 11440947] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 1996
- Lieberman JA, Fleishhacker W. Introduction. British Journal of Psychiatry 1996;168(Supplement 29):7‐8. [Google Scholar]
Lyra da Silva 1997
- Lyra da Silva JP, Soares‐Weisser K, McGrath J. Non‐neuroleptic catecholaminergic drugs for neuroleptic induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1997, Issue 1. [DOI: 10.1002/14651858.CD000458.pub2] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams CE, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
McGrath 2000a
- McGrath J, Soares K. Miscellaneous treatments for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000208] [DOI] [PubMed] [Google Scholar]
McGrath 2000b
- McGrath J, Soares K. Benzodiazepines for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000205.pub2; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
McGrath 2000c
- McGrath J, Soares K. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000459.pub2] [DOI] [PubMed] [Google Scholar]
McGrath 2000d
- McGrath J, Soares K. Cholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000207; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pocock 1983
- Pocock SJ. Crossover trials. Clinical trials. A Practical Approach. Chichester: John Wiley & Sons, 1983. [Google Scholar]
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Rotrosen 1996
- Rotrosen J, Adler L, Lohr J, Edson R, Lavori P. Antioxidant treatment of tardive dyskinesia. Prostaglandins, Leukotrienes and Essential Fatty Acids 1996;55(1‐2):77‐81. [DOI] [PubMed] [Google Scholar]
Schooler 1993
- Schooler NR, Keith SJ. The clinical research base for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Soares 2000b
- Soares K, McGrath J. Diltiazem, nifedipine, nimodipine or verapamil for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000206; PUBMED: 10796323] [DOI] [PubMed] [Google Scholar]
Soares 2000c
- Soares K, McGrath J. Anticholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000204; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Soares 2000d
- Soares K, McGrath J, Deeks J. Gamma‐aminobutyric acid agonists for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000203.pub2; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Soares 2001b
- Soares K, McGrath J, Deeks J. Gamma‐aminobutyric acid agonists for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2001, Issue 2. [DOI: 10.1002/14651858.CD000203.pub2; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Soares 2001c
- Soares K, McGrath J. Calcium channel blockers for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2001, Issue 1. [DOI: 10.1002/14651858.CD000206.pub2; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 1997
- Soares‐Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1997, Issue 2. [DOI: 10.1002/14651858.CD000204] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2003
- Soares‐Weiser K, Joy C. Miscellaneous treatments for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000208; PUBMED: 12804390] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2006
- Soares‐Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000459.pub2] [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Tammenmaa 2002
- Tammenmaa I, McGrath J, Sailas E, Soares‐Weiser K. Cholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD000207; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Umbrich 2003
- Umbrich P, Soares K. Benzodiazepines for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000205.pub2; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Xia 2007
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, Pinfold V, et al. The Leeds Outcomes Stakeholders Survey (LOSS) Study. Proceedings of the 15th Cochrane Colloquium; 2007 Oct 23‐27, Sao Paulo. Sao Paulo, 2007.
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
McGrath 2001
- McGrath J, Soares K. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD000209] [DOI] [PubMed] [Google Scholar]
Soares 1999
- Soares KVS, McGrath JJ. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1999, Issue 2. [DOI: 10.1002/14651858.CD000209] [DOI] [Google Scholar]
Soares 2000a
- Soares K, McGrath J. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000209; MEDLINE: ; PUBMED: 10796508] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2011
- Soares‐Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD000209.pub2] [DOI] [PubMed] [Google Scholar]