

Abstract
Top-down mass spectrometry (MS)-based proteomics has become a powerful tool for comprehensive characterization of intact proteins. However, due to the high complexity of the proteome, highly effective separation of intact proteins from complex mixtures prior to MS analysis remains challenging. Monolithic columns have shown great promise for intact protein separation due to their high permeability, low backpressure, and fast mass transfer. Herein, for the first time, we developed bridged hybrid bis(triethoxysilyl)ethylene (BTSEY) monolith with C8 functional groups (C8@BTSEY) for highly effective separation and coupled it to high-resolution MS for identification of intact proteins from complex protein mixtures. We have optimized mobile phase conditions of our monolith-based reverse-phase chromatography (RPC) for online liquid chromatography (LC)-MS analysis and evaluated separation reproducibility of the C8@BTSEY column. We further assessed the chromatographic performance of this column by separating a complex protein mixture extracted from swine heart tissue. Using our monolithic column (i.d. 100 μm x 35 cm), we separated over 300 proteoforms (up to 104 kDa) from 360 ng of protein mixture in an 80-min one-dimensional (1D) LC run. The highly effective separation and recovery of intact proteins from this monolithic column allowed unambiguous identification of ~100 proteoforms including a large protein, αactinin2 (103.77 kDa), by online 1D LC-MS/MS analysis, for the first time. As demonstrated, this C8@BTSEY column is reproducible and effective in separation of intact proteins, which shows high promise for top-down proteomics.
Graphical Abstract
Top-down mass spectrometry (MS)-based proteomics, which forgoes digestions to directly analyze intact proteins, has been proven to be a promising analytical strategy for the comprehensive characterization of proteoforms1 which arise from genetic variations, alternative RNA splicing, and post-translational modifications (PTMs).2–4 Highly effective intact protein separation prior to top-down MS is necessary due to the wide dynamic range of protein expression, vast range of protein masses, and high complexity of the proteome.5 Liquid chromatography (LC) coupling to MS is a powerful tool for protein separation and identification.6–8 The recent development in the multi-dimensional (MD) LC separation of intact proteins reduces the co-elution of proteins, which enables the detection and identification of low abundance and/or high molecular weight (MW) proteins towards a deeper proteome coverage.9–11 However, MDLC requires larger amount of samples and considerably longer experiment time than one-dimensional (1D) LC. Therefore, new chromatography materials and methods to achieve high-resolution 1DLC separation of intact proteins for a deep proteome coverage including high MW proteins is still highly desirable for top-down proteomics.12–14
Reverse-phase chromatography (RPC) remains the most commonly used separation method for online LC-MS.2 Recently, Shen et al. prepared meter-scale particle-packed columns to achieve high-resolution protein separation by 1D RPC separation.15 However, such long particle-packed column still required long separation gradient (600–1,000 min). Moreover, the ultrahigh backpressure (14k psi) from meter-long particle-packed column affects the retention behavior of proteins16 and needs the state-of-the-art UHPLC instrumentation, and thus limits its application. Wirth and co-workers developed submicrometer particles using slip flow for efficient intact protein separation which still has the ultrahigh backpressure problem (9k psi) and the separated proteins are all below 30 kDa.12 Herein, we sought to develop a new bridged hybrid monolith-based RPC/LC-MS strategy for intact protein separation with a wide MW range using regular column length, low backpressure, and low sample amount. Monolithic columns have shown great promise in high-resolution intact protein separation with regular column length owing to their properties such as fast mass transfer,17 high permeability and low backpressue.18–20
To achieve high-resolution protein separation, monolithic material with homogeneous pore structure and uniform high-loaded functional groups is required.21 In addition, stable chemical and mechanical properties of the monolith are necessary to ensure high reproducibility of the separation, especially for targeted tandem MS (MS/MS) techniques for protein identification based on the retention time of proteins. Recently, periodic mesoporous organosilicas (PMOs) monoliths have attracted attention in the field of chromatographic separation with unique features of narrow pore size distribution, high loading of organic content, homogeneously distributed functional groups and stable Si-C bonds in the framework.22–24 This type of material introduces organic groups into the framework directly by sol-gel condensation of bridged organoalkoxysilane precursors ((R’O)3Si–R–Si(OR’)3).25 In our previous work, the bridged hybrid monolithic matrix with 100% vinyl groups in the framework was synthesized facilely in the capillary by one-step sol-gel condensation of bis(triethoxysilyl)ethylene (BTSEY).26 With vinyl groups highly loaded and homogeneously distributed throughout the structure, the monolith can be readily functionalized through thiol-ene click reaction. The evaluation of BTSEY matrix modified with C18 functional groups by RPC-UV system demonstrated great efficiency and reproducibility in chromatographic separation of small molecules and proteins with masses below 20 kDa.26
In this study, we have shortened the chain of functional groups to C8 in the bridged hybrid monolithic BTSEY matrix (C8@BTSEY) and combined this monolithic column with high-resolution MS to enable effective separation and identification of large proteins. For LC-MS analysis, the mobile phase was first optimized for sharp peak shape in the LC and high ionization efficiency in MS analysis. The reproducibility of separation was then evaluated using standard proteins of different size (6.5–72.3 kDa) under the optimized mobile phase condition. This LC-MS platform involving C8@BTSEY monolithic column (i.d. 100 μm x 35 cm) was further assessed using a complex protein mixture extracted from swine cardiac tissue. In total, ~300 proteoforms (up to 104 kDa) were separated and detected in an 80-min 1D LC-MS run; among them, ~100 proteoforms were identified by online 1D LC-MS/MS analysis. Notably, the highly efficient separation of proteins significantly enhanced the MS detection and identification of high MW proteins and allowed successful identification of proteins with masses up to 104 kDa.
We first prepared the C8@BTSEY monolith by one-step sol-gel condensation of BTSEY and thiol-ene click functionalization of C8 groups (Figure S1). The column was then characterized by scanning electron microscopy (SEM), which demonstrated the homogeneous distribution of the monolith (Figure S2). The homogeneously distributed pore structure, high permeability, and fast mass transfer of C8@BTSEY monolith make it an ideal material for large molecule separation.
With the success in synthesizing the C8@BTSEY monolithic column, we optimized the mobile phase for intact protein separation in LC-MS system. Trifluoroacetic acid (TFA) is frequently added in mobile phase in RPC as an ion-pairing reagent to sharpen peak shape. Nevertheless, the ion suppression from TFA reduces the protein signal intensity in MS analysis and thus affects the sensitivity of the entire analysis.27 To balance the peak shape of RPC and the sensitivity of MS experiments, the amount of TFA in mobile phase was optimized here (Figure 1A). Three different mobile phase conditions with varied amount of TFA, 0.02% TFA+0.1% formic acid (FA), 0.05% TFA+0.1% FA, and 0.1% TFA, were tested in separating standard proteins using C8@BTSEY column. Based on our MS results, although the mobile phase containing 0.1% TFA provided the most symmetrical and sharp peaks among three conditions (Figure 1A), the signal of these proteins, for example, ~2.7 × 106 for CytoC, are not as high as those separated under 0.05% and 0.02% TFA conditions (both ~3.2 × 106 for CytoC). Taking both separation resolution and ionization efficiency into consideration, mobile phase with 0.05% TFA and 0.1% FA was used in the following intact protein separation.
Figure 1.
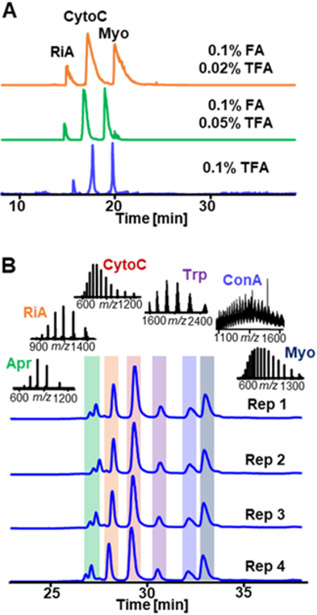
A Total ion chromatograms (TICs) of standard protein separation using mobile phases with different concentration of TFA. Sample: 60 ng ribonuclease A (RiA), 30 ng cytochrome C (CytoC) and 30 ng myoglobin (Myo). Separation gradient: 0-5-40-50-50.1-60 min, 0%-0%-80%-80%-0%-0% B. Flow rate: 500 nL/min. B TICs of six standard proteins separated by C8@BTSEY monolith under optimized mobile phase condition. Sample: aprotinin (Apr), RiA, CytoC, trypsinogen (Trp), ConA and Myo, 25 ng for each protein. Separation gradient: 0-10-10.1-50-60 min, 0%-0%-40%-80%-80% B. Flow rate: 800 nL/min.
Next, we evaluated the reproducibility of the C8@BTSEY column. As shown in Figure 1B, a protein mixture containing six standard proteins with mass range of 6.5–72.3 kDa was separated at baseline resolution with high reproducibility. Conalbumin A (ConA) with the mass of 72.3 kDa could be eluted with peak width of ~0.4 min (full width at half maximum). Additionally, the relative standard deviations (RSDs) of the protein retention time from run-to-run were less than 0.6% (n=4) (Table S1). The batch-to-batch reproducibility of C8@BTSEY columns was also evaluated (Figure S3) and the RSDs of the protein retention time from column-to-column were ~1.0% (n=5) (Table S2). These results demonstrate highly reproducible separation and are beneficial for targeted MS/MS experiments in protein identification.
The LC-MS platform involving C8@BTSEY monolithic column separation was further evaluated using a complex protein mixture extracted from swine cardiac tissue. Proteins with a wide mass range of 3–104 kDa were detected by MS after 1D RPC separation and some examples were shown in Figure 2. The mass spectra with charge state distribution showed effective separation of each protein without other co-eluted species. In the deconvoluted mass spectra, the phosphorylated forms of some proteins, cysteine and glycine-rich protein 3 (CSRP3), alpha tropomyosin (αTpm), and myosin light chain 2 ventricular form (MLC-2V), were also detected apart from their main isoforms. In total, ~300 proteoforms were effectively separated and detected in an 80-min 1D RPC separation gradient (Table S3). In addition to the highly abundant sarcomeric proteins such as myosin light chain 3 (MLC3), αTpm, and MLC-2V with masses < 40 kDa, we separated many other proteins with masses up to 104 kDa. Ten high mass proteins from 41.8 to 103.7 kDa were found in the RPC separation using C8@BTSEY column (Figure S4). Based solely on 1D RPC, high mass proteins are difficult to be separated and detected because smaller proteins usually co-elute with larger proteins resulting in signal suppression of high mass proteins. In our previous 1D RPC separation of sarcomeric proteins from swine heart with PLRP column, less than 30 proteins were separated and identified and nearly all of the detected proteins are below 40 kDa.28 Even though Shen et al. used meter-long column to enable effective separation of intact proteins in 1D RPC,15 such long particle-packed column introduces ultrahigh backpressure (14k psi) and requires long separation gradient (600–1,000 min) and larger sample amount (~2.5 μg). In other work, most strategies used for effective protein separation with wide mass range involved MDLC separation incorporating multiple different separation methods, which also demands large sample amount and long experiment time.10,11,29 In comparison, the capillary C8@BTSEY monolithic column presented in this work only needs a small amount of sample (360 ng) for separation and identification of proteins with masses of 3–104 kDa in an 80-min gradient. The low backpressure of monolithic column (~1,000 psi for 35-cm column) also allows further increase in column length to improve separation resolution.
Figure 2.
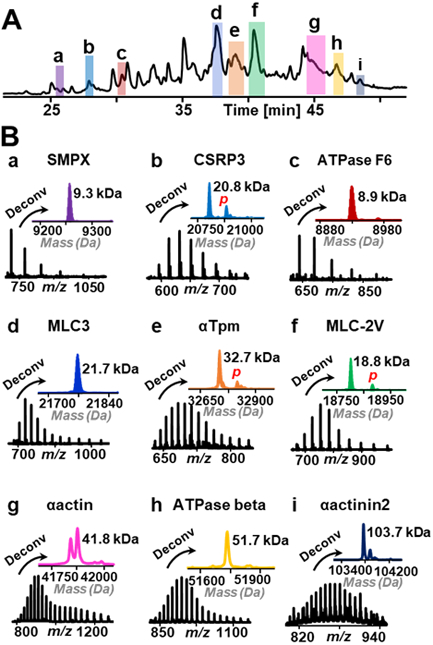
A TIC of proteins extracted from swine heart tissue separated by C8@BTSEY column. B Top-down mass spectra and deconvoluted mass spectra of selected proteins with different masses. Italic “p” in red refers to phosphorylation. In panel g, the two peaks in the deconvoluted mass spectrum of αactin are two different isoforms, alpha cardiac actin (41813.82 Da) and alpha skeletal actin (41845.77 Da).
The high recovery of proteins from this monolithic column enables subsequent protein identification by MS/MS analysis. Based on our auto MS/MS results, ~80 proteins corresponding to ~100 proteoforms were identified (Table S4). As mentioned previously, a 103.7 kDa protein was detected in our MS analysis. However, due to its high mass, this protein could not be identified by auto MS/MS analysis. Therefore, a targeted online LC-MS/MS approach was employed by fragmenting a precursor ion of this protein over the time segment where this protein was eluted. As shown in Figure 3, the precursor ion at charge state of 121 was isolated and then subjected to collisionally activated dissociation (CAD) fragmentation. Based on the b and y ions generated from CAD, this protein was identified as αactinin2 (F1RHL9_PIG, UniProtKB). The b ions also identified an acetylation at N-terminus based on mass difference of 42.01 Da from the theoretical mass. Even though this protein could not be isotopically resolved due to the limited resolution of the quadrupole time of flight (q-TOF) mass spectrometer, the most abundant mass from the experiment, 103.77 kDa, matches well with the calculated most abundant mass, 103,773.82 Da, according to the protein sequence. αactinin is a Z-disc protein in sarcomere which regulates the interaction between actin filaments and Z-lines.30 Even though it is an abundant protein in Z-disc, it was not identified in our previous 1D RPC (PLRP material) separation of sarcomeric proteins due to its large size. Here, for the first time, we separated and identified this protein using C8@BTSEY column coupled with online MS/MS analysis.
Figure 3.
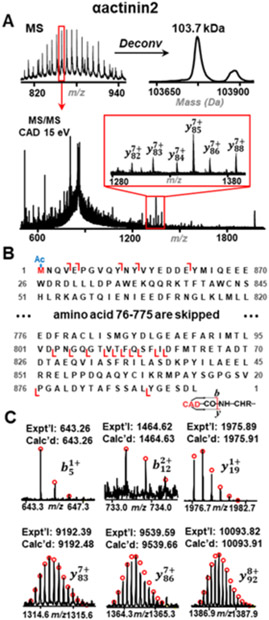
A Mass spectra, deconvoluted mass spectra and MS/MS mass spectra of αactinin2 (F1RHL9_PIG, UniProtKB). Precursor ion at 858 m/z was isolated and fragmented by CAD. Insets, zoomed-in view of the representative fragment ions. B Protein sequence map of αactinin2. N-terminal acetylation is highlighted in red. C Representative b and y ions of αactinin2 from CAD fragmentation.
Furthermore, we applied this targeted MS/MS strategy to identify a 51.7 kDa protein. This 51.7 kDa protein was identified as ATP synthase subunit beta (K7GLT8_PIG, UniProtKB) with 14 b ions and 29 y ions (Figure S5). Importantly, an 88-amino acid removal was characterized at the N-terminus based on the b ions detected, which is not present in the database. The 88-amino acid sequence is likely an N-terminal signal peptide that may play a role in the transportation of ATP synthase into mitochondria and is cleaved off after the transportation.31 The successful identification of this 51.7 kDa protein with N-terminal amino acid removal also shows the strength of this top-down proteomics platform by combining C8@BTSEY-based RPC with MS analysis.
In summary, we have developed a bridged hybrid monolithic column with C8 functional groups to achieve a highly effective 1D RPC separation of complex protein mixtures with low sample amount (360 ng), short separation gradient (80 min) and low backpressure (1,000 psi). Mobile phase was optimized and reproducibility of the column was evaluated. In the analysis of complex protein mixtures extracted from swine cardiac tissue, ~300 proteoforms with wide mass range of 3–104 kDa were separated and detected in a single 80-min LC-MS run. The effective protein separation significantly enhanced the MS/MS fragmentation efficiency, thus allowing successful identification of ~100 proteoforms including a 103.77 kDa αactinin2. Based on the high permeability and low backpressure of monolithic columns, we anticipate further development and optimization of this C8@BTSEY column will enable separation and identification of even more proteins towards a deeper proteome coverage for top-down proteomics.
Supplementary Material
Acknowledgements
We would like to thank Ziqing Lin and Yanlong Zhu for their help in instrument setup. Financial support was kindly provided by the high-end instrument grant S10OD018475 and NIH R01 grants GM117058, GM125085, HL109810, and HL096971 (to Y.G.). Y.L. also would like to acknowledge the generous support from China Scholarship Council and National Natural Science Foundation of China (21575139).
Footnotes
Supporting Information
Experimental section, relative standard deviations (RSDs) of the protein retention time from run-to-run, RSDs of the protein retention time from column-to-column, summary of all the proteoforms detected in a single LC-MS run, summary of 83 proteins (97 proteoforms) identified by LC-MS/MS analysis, workflow of C8@BTSEY column fabrication and LC-MS analysis of protein mixtures, SEM characterization of the C8@BTSEY monolith, batch-to-batch reproducibility of columns demonstrated by total ion chromatograms (TICs) of standard proteins separated by C8@BTSEY columns, TIC and deconvoluted mass spectra of high MW proteins extracted from swine heart tissue separated by C8@BTSEY column, characterization of ATP synthase subunit beta.
References
- (1).Smith LM; Kelleher NL; Consortium for Top Down, P. Nat Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chen B; Brown KA; Lin Z; Ge Y Anal Chem 2018, 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cai W; Tucholski TM; Gregorich ZR; Ge Y Expert Rev Proteomics 2016, 13, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL, et al. Nature Chemical Biology 2018, 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Doucette AA; Tran JC; Wall MJ; Fitzsimmons S Expert Review of Proteomics 2011, 8, 787–800. [DOI] [PubMed] [Google Scholar]
- (6).Gargano AFG; Roca LS; Fellers RT; Bocxe M; Dominguez-Vega E; Somsen GW Anal Chem 2018, 90, 6601–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jin Y; Wei L; Cai W; Lin Z; Wu Z; Peng Y; Kohmoto T; Moss RL; Ge Y Anal Chem 2017, 89, 4922–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Qu M; An B; Shen S; Zhang M; Shen X; Duan X; Balthasar JP; Qu J Mass Spectrom Rev 2017, 36, 734–754. [DOI] [PubMed] [Google Scholar]
- (9).Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y Anal Chem 2017, 89, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Xiu LC; Valeja SG; Alpert AJ; Jin S; Ge Y Analytical Chemistry 2014, 86, 7899–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Valeja SG; Xiu LC; Gregorich ZR; Guner H; Jin S; Ge Y Analytical Chemistry 2015, 87, 5363–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wu Z; Wei B; Zhang X; Wirth MJ Analytical Chemistry 2014, 86, 1592–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen BF; Peng Y; Valeja SG; Xiu LC; Alpert AJ; Ge Y Analytical Chemistry 2016, 88, 1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McCool EN; Lubeckyj RA; Shen XJ; Chen DY; Kou Q; Liu XW; Sun LL Analytical Chemistry 2018, 90, 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shen Y; Tolic N; Piehowski PD; Shukla AK; Kim S; Zhao R; Qu Y; Robinson E; Smith RD; Pasa-Tolic L J Chromatogr A 2017, 1498, 99–110. [DOI] [PubMed] [Google Scholar]
- (16).Gritti F; Guiochon G Analytical Chemistry 2009, 81, 2723–2736. [DOI] [PubMed] [Google Scholar]
- (17).Eeltink S; Wouters S; Dores-Sousa JL; Svec F Journal of Chromatography A 2017, 1498, 8–21. [DOI] [PubMed] [Google Scholar]
- (18).Simone P; Pierri G; Capitani D; Ciogli A; Angelini G; Ursini O; Gentile G; Cavazzini A; Villani C; Gasparrini F J Chromatogr A 2017, 1498, 46–55. [DOI] [PubMed] [Google Scholar]
- (19).Du K; Zhang Q; Dan S; Yang M; Zhang Y; Chai D J Chromatogr A 2016, 1443, 111–117. [DOI] [PubMed] [Google Scholar]
- (20).Desire CT; Arrua RD; Talebi M; Lacher NA; Hilder EF J Sep Sci 2013, 36, 2782–2792. [DOI] [PubMed] [Google Scholar]
- (21).Liu Z; Ou J; Lin H; Wang H; Liu Z; Dong J; Zou H Anal Chem 2014, 86, 12334–12340. [DOI] [PubMed] [Google Scholar]
- (22).Hoffmann F; Froba M Chem Soc Rev 2011, 40, 608–620. [DOI] [PubMed] [Google Scholar]
- (23).Wu C; Liang Y; Liang Z; Zhang LH; Zhang YK Anal Chim Acta 2018, 1019, 128–134. [DOI] [PubMed] [Google Scholar]
- (24).Wu C; Liang Y; Zhu X; Zhao Q; Fang F; Zhang X; Liang Z; Zhang L; Zhang Y Anal Chim Acta 2018, 1038, 198–205. [DOI] [PubMed] [Google Scholar]
- (25).Inagaki S; Guan S; Fukushima Y; Ohsuna T; Terasaki O J Am Chem Soc 1999, 121, 9611–9614. [Google Scholar]
- (26).Wu C; Liang Y; Yang K; Min Y; Liang Z; Zhang L; Zhang Y Anal Chem 2016, 88, 1521–1525. [DOI] [PubMed] [Google Scholar]
- (27).Shou WZ; Weng ND J Chromatogr B 2005, 825, 186–192. [DOI] [PubMed] [Google Scholar]
- (28).Peng Y; Gregorich ZR; Valeja SG; Zhang H; Cai W; Chen YC; Guner H; Chen AJ; Schwahn DJ; Hacker TA; Liu X; Ge Y Mol Cell Proteomics 2014, 13, 2752–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Anderson LC; DeHart CJ; Kaiser NK; Fellers RT; Smith DF; Greer JB; LeDuc RD; Blakney GT; Thomas PM; Kelleher NL; Hendrickson CL J Proteome Res 2017, 16, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Faulkner G; Lanfranchi G; Valle G Iubmb Life 2001, 51, 275–282. [DOI] [PubMed] [Google Scholar]
- (31).Zhang CL; Marcia M; Langer JD; Peng GH; Michel H Febs J 2013, 280, 3425–3435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.