

Abstract
Background
Crizotinib has potent anti-tumor activity in patients with advanced MET-amplified non-small cell lung cancer (NSCLC). However, the therapeutic effect is still not satisfying. Thus, developing approaches that improve the efficacy of crizotinib remains a significant challenge.
Methods
MET-amplified NSCLC cell lines were treated with crizotinib and cyclosporine A (CsA). Cell viability was determined by MTS assay. The changes of apoptosis, cell cycle and calcineurin-Erk pathways were assessed by western blot. Xenograft mouse model, primary human NSCLC cells and hollow fiber assays were utilized to confirm the effects of CsA.
Findings
We demonstrated that CsA significantly increased the anti-tumor effect of crizotinib on multiple MET-amplified NSCLC cells in vitro and in vivo. Mechanistically, crizotinib treatment led to the activation of Ca2+-calcineurin (CaN)-Kinase suppressor of Ras 2 (KSR2) signaling, resulting in Erk1/2 activation and enhanced survival of cancer cells. CsA effectively blocked CaN-KSR2-Erk1/2 signaling, promoting crizotinib-induced apoptosis and G2/M arrest. Similarly, pharmacologic or genetic inhibition of Erk1/2 also enhanced crizotinib-induced growth inhibition in vitro. Xenograft studies further confirmed that CsA or Erk1/2 inhibitor PD98059 enhanced the anti-cancer activity of crizotinib through inhibition of CaN-Erk1/2 axis. The results were also validated by primary human NSCLC cells in vitro and hollow fiber assays in vivo.
Interpretation
This study provides preclinical evidences that combination therapy of CsA and crizotinib is a promising approach for targeted treatment of MET-amplified lung cancer patients.
Fund
This work was supported by the National Natural Science Foundation of China, the Key Projects of Natural Foundation of Zhejiang Province, the Ten thousand plan youth talent support program of Zhejiang Province, the Zhejiang Natural Sciences Foundation Grant, and the Zhejiang medical innovative discipline construction project-2016.
Keywords: Cyclosporine A, Erk1/2, Crizotinib, Lung cancer, Calcium
List of Abbreviations
| NSCLC | non-small cell lung cancer |
| CsA | cyclosporine A |
| CI | combination index |
| Fa | fraction affected |
| Erk | extracellular signal-regulated kinase |
| CaN | Ca2+-calcineurin |
| HGF | hepatocyte growth factor |
| STAT3 | signal transducer and activator of transcription 3 |
| ALK | anaplastic lymphoma kinase |
| EML4 | Echinoderm microtubule-associated protein-like 4 |
| ROS1 | ROS proto-oncogene 1 |
| FDA | US Food and Drug Administration |
| NFAT | nuclear factor of activated T cells |
| MDRs | multidrug resistance proteins |
| TKIs | tyrosine kinase inhibitors |
| KSR2 | Kinase suppressor of Ras 2 |
| PI | propidium iodide |
| FITC | fluorescein isothiocyanate |
| AV | annexin V |
| TG | thapsigargin |
| PARP | poly-(ADP-ribose) polymerase |
| RTK | receptor tyrosine kinase |
| EGFR | epidermal growth factor receptor |
| 5-FU | 5-fluorouracil |
| EMT | epithelial-mesenchymal transition |
| IP3R | inositol trisphosphate receptor |
| ER | endoplasmic reticulum |
| PKC | protein kinase C |
| HRP | horse radish peroxidase |
| DMSO | dimethyl sulfoxide |
| PBS | phosphate buffer saline; |
| PVDF | polyvinylidene fluoride |
Research in context.
Evidence before this study
Cyclosporine A (CsA) is an immunosuppressive drug widely used to prevent graft rejection after organ transplantation. Several studies have demonstrated that CsA could enhance the cytotoxic effects of chemotherapy drugs due to inhibiting multidrug resistance proteins. We previously reported that CsA increased the activity of gefitinib in EGFR mutant NESCL cell lines by inhibiting STAT3 pathway.
Added value of this study
We found that crizotinib activated Erk1/2 by inducing Ca2+ influx into the cytoplasm. CsA inhibited crizotinib induced Erk1/2 activation through calcineurin-KSR2-Erk pathway. CsA enhanced the anti-NSCLC effects of crizotinib in vitro and in vivo.
Implications of all the available evidence
Our results suggest that combination treatment of CsA and crizotinib is a promising approach for MET-amplified lung cancer patients.
Alt-text: Unlabelled Box
1. Introduction
Lung cancer is the leading course of cancer death [1]. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer. It has been identified that some aberrant expressed genes such as EGFR, BRAF, ALK, etc. play an important part in NSCLC proliferation, drug resistance and metastasis [[2], [3], [4]]. Though the prognosis of NSCLC patients has been greatly improved due to the targeted therapy of EGFR, the treatment will eventually fail due to the emergency of drug resistance [5]. More therapeutic targets are urgently needed. In recent years, MET, which is a transmembrane receptor tyrosine kinase, has become a promising target [6]. MET signaling dysregulation is involved in NSCLC growth, survival, migration and invasion, angiogenesis and activation of several pathways [7]. MET amplification is closely related to the poor prognosis, and targeting MET represents an effective method for the targeted NSCLC patients [8].
Crizotinib is a small-molecule which was originally developed as a MET inhibitor [9]. It has been approved by the US Food and Drug Administration (FDA) as a front-line treatment for locally advanced or metastatic NSCLC harboring the EML4-ALK fusion protein [10]. Recently, a phase I clinical trial (NCT00585195) demonstrated that crizotinib displays potent anti-tumor activity in patients with advanced MET-amplified NSCLC [11]. A phase II clinical trial (NCT02499614) to evaluate the therapeutic effects of crizotinib in NSCLC patients with MET amplification is currently recruiting patients. However, like other targeted agents, single-agent treatment with crizotinib generally fails to completely eradicate cancer cells [12]. Therefore, identifying resistance pathways and overcoming through rational combination strategies to improve the efficacy of crizotinib is of great significance.
Cyclosporine A (CsA) is an immunosuppressive drug that is widely used to prevent graft rejection after organ transplantation. CsA specifically binds to cyclophilins, forming CsA/cyclophilin complexes that inhibit the activity of calcineurin by binding to its CnB domain [13]. Calcineurin is a unique protein serine/threonine phosphatase that is activated by Ca2+/calmodulin signaling [14]. Upon activation, calcineurin dephosphorylates multiple phospho-residues of nuclear factor of activated T cells (NFAT), leading to NFAT cytoplasmic–nuclear trafficking, which initiates a cascade of transcriptional events [15]. Several studies reported that CsA was capable of enhancing the anti-tumor effects of chemotherapy drugs, such as carboplatin, doxorubicin, docetaxel and paclitaxel, owing to its ability to inhibit multidrug resistance proteins (MDRs) [[16], [17], [18], [19], [20]]. Blocking the calcineurin/NFAT pathway with CsA could enhance the anti-tumor effects of several tyrosine kinase inhibitors (TKIs), such as dasatinib, imatinib, vemurafenib and selumetinib [[21], [22], [23], [24], [25]]. Currently, several corresponding ongoing clinical trials seek to evaluate the sensitizing effect of CsA to chemo- or targeted therapeutics, including selumetinib combined with CsA in colorectal cancer (NCT02188264) and verapamil combined with CsA in Hodgkin lymphoma (NCT03013933).
In a previous study, we have demonstrated that CsA significantly enhanced the anti-cancer effect of gefitinib on several NSCLC cell lines in vitro and in vivo by inhibiting gefitinib-induced feedback activation of STAT3 [26]. In the present study, we found that crizotinib treatment led to the upregulation of intracellular Ca2+, which subsequently activated calcineurin/Kinase suppressor of Ras 2 (KSR2)/Erk signaling in MET-amplified NSCLC cells. Feedback activation of Erk1/2 promoted the survival of lung cancer cells in response to MET inhibition. CsA significantly sensitized MET-amplified NSCLC cells to crizotinib by blocking Ca2+/calcineurin/Erk signaling. Moreover, rational combination with PD98059, an indirect inhibitor of Erk1/2, also enhanced the anti-cancer effect of crizotinib in vitro and in vivo. Our study presents a novel, promising strategy to increase the clinical efficacy of crizotinib by inhibiting Ca2+/calcineurin/Erk signaling in targeted therapeutic regimens.
2. Materials and methods
2.1. Reagents and antibodies
The chemicals used in this study were crizotinib (Selleck Chemicals LLC, Houston, TX, USA), Cyclosporine A (CsA) (J&K chemical Ltd., Beijing, China), PD98059 (Selleck Chemicals LLC, Houston, TX, USA), MK-2206 (Selleck Chemicals LLC, Houston, TX, USA). The primary antibodies against phospho-Erk1/2, Erk1/2, phospho-AKT, AKT, phospho-STAT3, STAT3, phospho-Cdc25c, phospho-CDK1, CDK1, Cyclin B1, Bcl-XL, caspase-3 and PARP were purchased from Cell Signaling Technology (Boston, MA, USA). KSR2 and Cdc25c were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The secondary antibodies were horse radish peroxidase (HRP)-conjugated anti-rabbit IgG, anti-mouse IgG (Cell Signaling Technology).
2.2. Cell lines and animals
Cells were bought from the cell bank of the Chinese Academy of Science and were kept in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) at 37 °C under a 5% CO2 and 90% humidified atmosphere. Crizotinib was dissolved in dimethyl-sulfoxide (DMSO) and was further diluted with medium before use so that the final concentration of DMSO was under 0.1%.
Female athymic BALB/c nude mice (Shanghai Institute of Material Medicine, Chinese Academy of Science, China) were maintained in a specific pathogen-free facility and were treated with humane care after approval from the Animal Care and Use Committee of Zhejiang University.
2.3. Combination index (CI) calculation
The specific interaction between crizotinib and CsA on SPC-A1 and PC-9 cancer cell lines was evaluated by the CI analysis. Drug combination synergy was performed using CompuSyn (Compusyn Inc., Paramus, NJ, USA), a software based on Chou and Talalay method [27]. CI values and CI-Fa plot (plot representing CI versus Fa, the fraction affected by a particular dose) were calculated, a synergistic effect as CI < 1, an additive effect as CI = 1 and an antagonistic effect as CI > 1.
2.4. RNA interference, plasmids and transfections
Cells were transfected with scrambled or siRNA against Erk1/2 using Hiperfect (Qiagen) according to the manufacturer's protocol. siRNA oligonucleotides that target Erk1/2 and calcineurin were purchased from RIBOBIO (Guangzhou, China). A nonspecific oligo that is not complementary to any human genes was used as a negative control. siRNAs against Erk1/2 were as follows: 5′-CAAGAAGACCTGAATTGTA-3′; 5′-GCAAGCACTACCTGG -ATCA-3′. siRNAs against calcineurin were as follows: 5’-CCACAACATAAGATCAC -TA-3′; 5′-GTATTCAGAACGCGTATAT-3′; Primers for calcineurin were as follows: 5′-GATGCTGGTAAATGTC-3′; 5′-CACACTCTCACTCTCTTCTCTG-3′.
Two separate KSR2 siRNAs (Catalog#1299003) were bought from ThermoFisher Scientific (United States). Plasmid that contain constitutively activated Erk1/2 (pcDNA3.1/ Erk1/2) and the control empty plasmid were purchased from Biovector (Beijing, China). Cells were transfected with plasmids by Attractene Transfection Reagent (Qiagen) according to the manufacturer's protocol. 48 h after transfection, the cells were treated with crizotinib and then subjected to analysis.
2.5. Measurements of intracellular Ca2+ concentration
The iCa2+ concentration was monitored by flow cytometric analysis with a Ca2+-sensitive fluorescence indicator, fluo-3/AM (Invitrogen). Cells were collected by trypsinization, washed twice and resuspended in PBS which contains 5 μM/l of fluo-3/AM at a concentration of 5 × 105 cells/ml, then incubated in dark and 37 °C for 30–60 min. The samples were then sent for analysis by flow cytometry.
2.6. Human lung cancer xenograft model
Human lung cancer xenograft models were established according to our previous study [28]. 6 to 8-week-old female athymic BALB/c nude mouse were utilized. Briefly, 5 × 106 SPC-A1 cells were injected subcutaneously in the flank region of the mouse. Mouse was sacrificed and tumor was harvested as it grew into a size of 400–500 mm3. Then tumor was cut into 1–2 mm3 pieces and each piece was transplanted into the same region of the mouse. When the diameter of the tumors reached approximately 0.4 cm, mice were randomly assigned to four groups: the vehicle, CsA alone, crizotinib alone and crizotinib plus CsA group. Crizotinib and CsA were suspended in 30% polyethylene glycol, 0.5% Tween 80 and 5% propylene glycol, and administered by oral gavage at the dose of 30 mg/kg/day and 20 mg/kg/day respectively. The tumor volume was determined using the formula V = LW2/2, where L is the largest diameter and W is the smallest diameter.
The protocol of animal study was approved by the institutional animal ethical committee of Zhejiang University with approval No. zju-2015-4-01-001.
3. Primary cell culture
The protocols concerning human tissues were reviewed and approved by the Human Research Ethics Committee of Sir Run Run Shaw Hospital, Zhejiang University and all participants completed informed consents prior to their inclusion.
Fresh solid tumor specimens were obtained immediately after surgery. After rinsing twice with RPMI 1640 (Gibco, Carlsbad, CA, USA) supplemented with penicillin G (1,000 U/ml) and streptomycin (1 mg/ml), the tumors were minced into small pieces (about 2–5 mm diameter) with scalpels under aseptic conditions, and then immersed in 0.1% type IV collagenase solution (Gibco) for 1 h at 37 °C. After passing through a 38 μm mesh sieve, the resulting cell suspension was washed twice and centrifugated at a speed of 300 g × 5 min. The cells were then suspension in ACL4 medium [29] and cultured in a collagen-coated disk. To eliminate the contamination of fibroblasts and other normal cells, tumor cells were purified by harvesting selected colonies by scraping with a Pasteur pipet following protocols of Oie et al. [30].
Only patients who were confirmed as MET-amplified adenocarcinoma by preoperative punctures were enrolled for primary cell assay and the following hollow fiber assay. The MET amplification status was checked by the Pathology Department of Sir Run Run Shaw Hospital following a standard protocol [31].
4. Hollow fiber assay (HFA)
The in vivo HFA was carried out according to Hollingshead et al. [32] Briefly, 1-mm internal diameter and a molecular weight cutoff point of 500 kDa Polyvinylidene fluoride (PVDF) (Spectrum Laboratories) hollow fibers were used. Viable cells from lung cancer tissues were suspended to 1 × 107 cell/ml and injected into the fiber. The fibers were cut into 1.5–2 cm each and implanted into 6 to 8 weeks female Balb/C (nu/nu) mice. Drug treatment started one day after implantation of the hollow fiber. Crizotinib and CsA were suspended in 30% polyethylene glycol, 0.5% Tween 80 and 5% propylene glycol, and administered by oral gavage at the dose of 30 mg/kg/day and 20 mg/kg/day respectively once daily for one week. The mice were sacrificed one day after the last drug treatment. The ATP content of each tube was measured by the addition of luciferin-luciferase in a Spectra Max M3 plate reader (Molecular Devices, CA, U.S.A.).
4.1. Statistical analysis
Unless otherwise stated, the data are expressed as the mean ± SD and were analyzed by Student's t-test. P < 0.05 was considered statistically significant.
Other methods: Please see “Supplementary Materials and Methods”.
5. Results
5.1. CsA sensitizes MET-amplified NSCLC cells to crizotinib
Five NSCLC cell lines that express MET were treated with various concentrations of crizotinib in the presence or absence of CsA, and cell viability was determined by MTS array. Crizotinib showed a considerable growth-inhibitory effect against all five MET-amplified NSCLC cell lines. 2 μΜ CsA significantly enhanced the growth arresting effect of crizotinib on NSCLC cell lines SPC-A1 and, PC-8 cells (Fig. 1a), HCC827, H1975 and A549 (Supplementary Fig. S1), without obvious cytotoxicity when treated with CsA alone. To further confirm the sensitizing effect of CsA, colony formation assays were performed on SPC-A1 and PC-9 cells. As shown in Fig. 1b and c, CsA alone had no inhibitory effect on colony formation of either cell line, whereas approximately 55% and 65% of the SPC-A1 and PC-9 cells failed to grow into colonies after treatment with 500 nΜ crizotinib for 10 days, respectively. When cells were treated with crizotinib and CsA, the failure rate of colony formation in SPC-A1 and PC-9 cells increased to 75% and 80%, respectively. CI values of SPC-A1 and PC-9 cells were calculated upon crizotinib (concentrations rang from 0.5 to 4 μΜ) and 2 μΜ CsA treatment. We found that most of CI levels were <1.0, showing a clearly synergistic anti-tumor activity of CsA (Fig. 1d). Specifically, the CI value was as low as 0.48038 when combined with 1 μM crizotinib and 2 μM CsA for SPC-A1, and 0.60588 with 2 μM crizotinib and 2 μM CsA for PC-9 (Supplementary table 1). These two concentrations were selected for the following experiments.
Fig. 1.

CsA sensitizes NSCLC cell lines to crizotinib (criz) treatment. (a) SPC-A1 and PC-9 cells were treated with crizotinib in the presence or absence of CsA for 48 h. Cell viability was measured by MTS assay. (b and c) SPC-A1 and PC-9 cells were plated at a density of 3000 cells/well in 6-well plates, quantification of cell colonies was counted after cells were incubated with crizotinib in the presence or absence of CsA for 2 weeks. (d) SPC-A1 and PC-9 cells were plated at a density of 1.5*104 cells/well in 96-well plates, and then incubated with crizotinib (rang from 0.5 to 4 μΜ) in the presence or absence of CsA (rang from 0.5 to 4 μΜ) for 48 h, after which cell viability was determined by MTS assay. CI-Fa plot (plot representing CI versus Fa, the fraction affected by a particular dose) were calculated by CompuSyn software. Values of (a) were presented as mean ± SD and compared using one-way ANOVA. Values of (c) were presented as mean ± SD and compared using student's t-test. *P < 0.05, ** P < 0.01, and *** P < 0.001 compared with crizotinib alone.
Taken together, these results demonstrate that CsA enhances the growth arresting effect of crizotinib in MET-amplified NSCLC cells.
5.2. CsA augments crizotinib-induced growth inhibition by promoting apoptosis and G2/M arrest
To determine whether the synergistic effect of CsA on crizotinib was dependent on apoptosis or cell cycle arrest, treated cells were stained with propidium iodide/fluorescein isothiocyanate-annexin V (PI/FITC-AV) or a cell cycle staining kit, after which the cells were quantified by flow cytometry. As shown in Fig. 2a, crizotinib treatment induced apoptosis in SPC-A1 and PC-9 cells. When combining crizotinib treatment with CsA, crizotinib-induced apoptosis was greatly enhanced. In addition, crizotinib caused G2/M arrest in SPC-A1 and PC-9 cells, which was also markedly augmented by concomitant CsA treatment (Fig. 2b). CsA alone (2 μΜ) had no observable effect on cancer cell growth arrest. Western blot analysis confirmed that crizotinib treatment results in a decrease in the anti-apoptotic regulator Bcl-xL while increasing pro-apoptotic regulators such as cleaved caspase-3. Crizotinib treatment also decreased the expression levels of the G2/M transition-phase proteins p-CDK1, CDK1 and cyclin B. Correspondingly, CsA significantly augmented crizotinib-induced apoptosis and G2/M arrest, which was reflected by lower expression levels of Bcl-xL and G2/M transition phase proteins as well as the higher cleaved caspase-3 levels (Fig. 2c).
Fig. 2.

CsA enhances crizotinib-induced apoptosis in NSCLC cells. (a) SPC-A1 and PC-9 cells were treated with crizotinib in the presence or absence of CsA for 48 h, quantification of the apoptotic rates was analyzed by flow cytometry. (b) SPC-A1 and PC-9 cells were treated with crizotinib in the presence or absence of CsA for 24 h, quantification of the relative cell population in each phase of the cell cycle was analyzed by flow cytometry. c-Caspase 3 represented for cleaved caspase 3. (c) Immunoblotting for apoptosis markers and G2/M transition-related mediators in SPC-A1 and PC-9 cells treated with crizotinib alone or in combination with CsA for 48 h. *P < 0.05 and ** P < 0.01 compared with crizotinib. Values were presented as mean ± SD of three independent experiments and compared using student's t-test.
Collectively, these results suggest that CsA augments the growth inhibitory effects of crizotinib by promoting apoptosis and G2/M arrest in human MET-amplified NSCLC cells.
5.3. CsA sensitizes crizotinib by preventing feedback activation of Erk1/2
To investigate the mechanism of CsA-mediated sensitization of crizotinib, the MET signaling pathway was detected by Western blot. As shown in Fig. 3a, the phosphorylation of STAT3 and AKT, but not Erk1/2, was observably inhibited by crizotinib after 48 h treatment. In addition, CsA produced no extra effects on the expression levels of total or phospho-MET, STAT3 or AKT in the presence or absence of crizotinib. However, p-Erk1/2 expression was significantly reduced when treated with CsA alone or CsA plus crizotinib (Fig. 3a), indicating that CsA probably enhances the anti-cancer effect of crizotinib through the inhibition of Erk1/2 phosphorylation.
Fig. 3.

CsA suppresses crizotinib-induced activation of Erk1/2. (a) SPC-A1 and PC-9 cells were treated with crizotinib in the presence or absence of CsA for 48 h, after which they were examined for the expression of phosphorylated and total MET, STAT3, AKT, Erk1/2 and K-ras by immunoblotting. (b) Cells were treated with 2 μM CsA for the indicated intervals and then were examined for the expression of phosphorylated and total Erk1/2 by immunoblotting. (c) Cells were treated with the indicated concentration of CsA for 48 h and then were examined for the expression of phospho- and total Erk1/2. (d) Cells were treated with crizotinib for the indicated intervals and then examined for the expression of phospho- and total Erk1/2. (e) SPC-A1 and PC-9 cells were treated with the indicated concentration of crizotinib for 24 h and then were examined for the expression of phospho- and total Erk1/2. (f) Cells were treated with crizotinib in the presence or absence of CsA (2 μM) for the indicated intervals and then were examined for the expression of phosphorylated and total Erk1/2 by immunoblotting.
Erk1/2 is aberrantly activated in NSCLC and promotes cancer cell survival [[33], [34], [35], [36]]. In the current study, SPC-A1 and PC-9 cells exhibited high basal levels of p-Erk1/2, which was inhibited by CsA in a time- and concentration-dependent manner (Fig. 3b and c). Surprisingly, the expression level of p-Erk1/2 was rose first and then decreased upon crizotinib treatment. As shown in Fig. 3d, p-Erk1/2 reached a peak level after 12–24 h crizotinib treatment, and decreased at 48 h. To further address the activation of Erk1/2 by crizotinib, SPC-A1 and PC-9 cells were treated with increasing concentrations of crizotinib for 12 h. The results clearly show that Erk1/2 becomes activated in a dose-dependent manner (Fig. 3e). Moreover, as expected, CsA efficiently inhibits crizotinib-dependent activation of Erk1/2 (Fig. 3f).
Some previous studies reported that Akt suppression would led to ERK1/2 activation [37]. As crizotinib attenuated Akt phosphorylation in our study, to figure out whether crizotinib induced Erk1/2 activation was cause by Akt suppression, we treated SPC-A1 and PC-9 cells with a widely used Akt inhibitor MK-2206 (1 μM) at different time (12 h and 24 h). Western blot showed that p-ERK1/2 expression was consistently at the same level, although Akt phosphorylation was totally inhibited. This result indicated that ERK activation induced by crizotinib in these two cells was Akt independent (Supplementary Fig. S2).
To further determine whether CsA exerts its sensitizing effects via inhibition of p-Erk1/2, the commercially available p-Erk1/2 indirect inhibitor PD98059 was tested. PD98059 is not an ERK-inhibitor itself. it inhibits the upstream MAPKK, which leads to reduction of ERK-phosphorylation. We found that PD98059 significantly sensitized human NSCLC cells SPC-A1, PC-9, HCC827, H1975 and A549 to crizotinib (Supplementary Fig. S2a). Mechanistically, PD98059 promoted crizotinib-induced G2/M arrest and apoptosis, which was confirmed by the reduction in the expression levels of the G2/M transition-phase proteins p-CDK1, CDK1 and cyclin B (Supplementary Fig. S3b-d). These data collectively indicates that PD98059 acts in a similar manner to CsA by enhancing the growth inhibitory effects of crizotinib in MET-amplified NSCLC cells.
To rule out the possibility of non-specific effects of CsA on Erk1/2, genetic silencing or overexpression of Erk1/2 were analyzed. As shown in Fig. 4a and b, knockdown of Erk1/2 by siRNA sensitized human NSCLC cells to crizotinib by promoting crizotinib-induced G2/M arrest and apoptotic cell death (Fig. 4c and d), which was similar to the effects observed upon treatment with CsA or PD98059. In contrast, overexpression of Erk1/2 rendered SPC-A1 and PC-9 cells more resistant to crizotinib as indicated by reduced crizotinib-dependent G2/M arrest and apoptosis (Fig. 5).
Fig. 4.

Knockdown of Erk1/2 by siRNA sensitizes NSCLC cells to crizotinib. (a) After transfection with siRNA against Erk1/2, SPC-A1 and PC-9 cells were treated with crizotinib for 72 h and examined for the expression of phosphorylated and total Erk1/2 by immunoblotting. (b) After transfection with siRNA against Erk1/2 for 48 h, SPC-A1 and PC-9 cells were treated with crizotinib for another 48 h, after which the viability was determined by MTS assay. (c) After transfection with siRNA against Erk1/2 for 48 h, SPC-A1 and PC-9 cells were treated with crizotinib for 48 h, quantification of the apoptotic rate was analyzed by flow cytometry. (d) After transfection with siRNA against Erk1/2 for 48 h, SPC-A1 and PC-9 cells were treated with crizotinib for 24 h, quantification of the relative cell populations in each cell cycle stage was analyzed by flow cytometry. *P < 0.05 and ** P < 0.01 compared with control. Values were presented as mean ± SD of three independent experiments and compared using student's t-test.
Fig. 5.

Overexpression of Erk1/2 impairs the anti-cancer effects of crizotinib in NSCLC cells. (a) Cells were transfected with plasmids expressing constitutively activated Erk1/2 for 48 h, after which the cells were examined for the expression of phosphorylated and total Erk1/2 by immunoblotting. (b and c) After transfection with plasmids expressing constitutively activated Erk1/2, cells were treated with crizotinib for 48 h. Then, the cell viability (b) and quantification of the apoptotic rates (d) were determined by MTS assay and flow cytometry, respectively. (d) After transfection with plasmids expressing constitutively activated Erk1/2, cells were treated with crizotinib for 24 h, quantification of the relative cell populations in each cell cycle stage was analyzed by flow cytometry. *P < .05 compared with vehicle. Values were presented as mean ± SD of three independent experiments and compared using student's t-test.
Taking these findings together, we conclude that temporary feedback activation of Erk1/2 upon crizotinib treatment contributes to NSCLC survival, whereas concomitant CsA treatment enhances the anti-cancer effects of crizotinib to MET-amplified NSCLC cells through Erk1/2 inhibition.
5.4. Crizotinib activates Erk1/2 by inducing Ca2+ influx into the cytoplasm
Erk1/2 is a well-known downstream molecule of Ca2+ signaling. Ca2+-activated Erk1/2 is involved in drug resistance to cisplatin [38]. Hence, we hypothesized that crizotinib activates Erk1/2 by enhancing Ca2+ influx into the cytoplasm. To test this hypothesis, we treated SPC-A1 and PC-9 cells with crizotinib for different time and then labeled the cells with the Ca2+-sensitive fluorescence indicator fluo-3/AM to monitor intracellular Ca2+ levels by flow cytometry. Our results showed that intracellular Ca2+ first increased and then slightly decreased upon crizotinib treatment (Fig. 6a and Supplementary Fig. S4a), which was similar to the expression levels of p-Erk1/2 shown in Fig. 4c. Next, we treated SPC-A1 and PC-9 cells with different concentrations of crizotinib for 12 h. As shown in Fig. 6B and Supplementary Fig. S4b, crizotinib induced the up-regulation of iCa2+ in a dose-dependent manner. We also used direct recording of Ca2+-signals from cells loaded with a Ca2+-sensitive dye fluo-3/AM with confocal microscopy. The results clearly showed that intracellular Ca2+ increases upon crizotinib treatment, which was consistent with the results of flow cytometry analysis (Supplementary Fig. S4c). To further address the role of Ca2+ in crizotinib-induced Erk1/2 activation, SPC-A1 and PC-9 cells were treated with thapsigargin (TG), a chemical that enhances Ca2+ influx into the cytoplasm by inhibiting endoplasmic reticulum Ca2+-ATPase function. Our results showed that after 0.5 to 4 h treatment of TG, Erk1/2 became activated (Fig. 6c). Finally, we treated cells with crizotinib in the presence or absence of the Ca2+ chelator EGTA. As shown in Fig. 6d, EGTA not only completely inhibited the feedback activation of Erk1/2, but it also remarkably inhibited basal p-Erk1/2 levels. These results indicate that Ca2+ plays a crucial role in regulating Erk1/2 activity.
Fig. 6.

Crizotinib activates Erk1/2 by inducing Ca2+ influx into the cytoplasm. (a) SPC-A1 and PC-9 cells were treated with crizotinib for the indicated amount of time, quantification of fluorescence intensity was detected by flow cytometry. (b) SPC-A1 and PC-9 cells were treated with the indicated concentration of crizotinib for 12 h, quantification of fluorescence intensity was analyzed by flow cytometry. (c) Cells were treated with TG for the indicated amount of time, after which the cells were examined for the expression of phosphorylated and total Erk1/2 by immunoblotting. (d) Cells were incubated with crizotinib in the presence or absence of EGTA for 12 h, after which they were examined for the expression of phosphorylated and total Erk1/2 by immunoblotting.
5.5. CsA inhibits Erk1/2 activation through inhibiting calcineurin
CsA is a well-known inhibitor of calcineurin, a protein phosphatase activated by Ca2+ infulx [13]. Michele K. Dougherty and colleagues reported that calcineurin interacts with KSR2, depletion of which impairs Ca2+-mediated ERK activation and ERK-dependent signaling responses [39]. As a result, we speculated that CsA inhibits Erk1/2 phosphorylation through the inhibition of calcineurin. FK506, another inhibitor of calcineurin, also inhibited Erk1/2 and augmented the growth inhibitory effect of crizotinib on SPC-A1 and PC-9 cells in a manner similar to CsA (Fig. 7 a and b). Furthermore, genetic inhibition of calcineurin (Fig. 7 c, d and e) or KSR2 (Fig. 7f, g and h) by siRNAs also resulted in decreased p-Erk1/2 levels and sensitization to crizotinib in SPC-A1 and PC-9 cells. These results demonstrate that CsA exert its effects partly through calcineurin-KSR2-Erk pathway.
Fig. 7.

Inhibition of calcineurin sensitizes NSCLC cells to crizotinib treatment. (a) SPC-A1 and PC-9 cells were treated with crizotinib in the presence or absence of FK506 for 48 h, after which the viability was determined by MTS assay. (b) Cells were treated with FK506 for 72 h, after which the expression of phosphorylated and total Erk1/2 was examined by immunoblotting. (c) After transfection with siRNA against calcineurin (siC1 and siC2) or scramble siRNA (NC) for 72 h, the mRNA level of calcineurin was quantified by RT-PCR. (d and e) After transfection with siRNA against calcineurin, cells were treated with crizotinib for 48 h, after which the cell viability was determined by MTS assay (d) and the expression of phosphorylated and total Erk1/2 was examined by immunoblotting (e). (f) After transfection with siRNA against KSR2 (siK1 and siK2) or scramble siRNA (NC) for 72 h, the mRNA level of calcineurin was quantified by RT-PCR. (g and h) After transfection with siRNA against KSR2, cells were treated with crizotinib for 48 h, after which the cell viability was determined by MTS assay (g) and the expression of phosphorylated and total Erk1/2 was examined by immunoblotting (h). *P < 0.05, ** P < 0.01, and *** P < 0.001 compared with control. Values were presented as mean ± SD of three independent experiments and compared using student's t-test.
5.6. CsA and PD98059 enhance the anti-NSCLC effects of crizotinib in vivo
To determine whether CsA and PD98059 can enhance the anti-NSCLC effects of crizotinib in vivo, we inoculated 5 × 106 SPC-A1 cells into the right flanks of BALB/c nude mice. When the xenografted tumors were measurable, mice were randomly assigned to receive CsA, PD98059, crizotinib or a combination of CsA or PD98059 with crizotinib. As shown in Fig. 8a-c, CsA or PD98059 alone had no notable effect on tumor growth, crizotinib yielded moderate anti-tumor activity, and a combination of crizotinib with CsA or PD98059 significantly reduced tumor growth compared with crizotinib alone. Next, we extracted protein lysates from xenografted tumors and analyzed the expression of MET signaling molecules, apoptosis markers and G2/M transition-phase mediators by western blot. Our results showed that crizotinib efficiently inhibited MET, AKT and STAT3 signaling but did not inhibit Erk1/2. Consistent with our in vitro results, crizotinib treatment decreased the expression of the anti-apoptotic protein Bcl-xL, increased the expression of the pro-apoptotic proteins cleaved caspase-3 and cleaved poly-(ADP-ribose) polymerase (PARP) and downregulated the expression of the G2/M transition-phase proteins p-Cdc25c, p-CDK1 and cyclin B (Fig. 8d). CsA and PD98059 significantly inhibited p-Erk1/2 but not p-MET, p-AKT and p-STAT3. Although CsA or PD98059 alone had no effect on the expression of apoptosis markers and G2/M transition-phase mediators, concomitant treatments with crizotinib reduced the expression of the anti-apoptotic regulator while increasing the expression of pro-apoptotic regulators and decreasing the expression of G2/M transition-phase proteins (Fig. 8d). Furthermore, no significant weight loss or hepatic or renal toxicity was observed with any of the combination treatments (Supplementary Fig. S5).
Fig. 8.

CsA and PD98059 (PD) enhance the anti-cancer effect of crizotinib in NSCLC xenograft models. (a) Tumors from nude mice in each treatment group. (b and c) Tumor weight (b) and tumor volume (c) in each group. (d) Tumor lysates from each group were subjected to immunoblotting for the indicated proteins. Each lane represents a pooled lysate from all of the mice in the indicated group. c-Caspase 3 represented for cleaved caspase 3. *P < 0.05 and ** P < 0.01 compared with crizotinib. Values of (b) were presented as mean ± SD and compared using student's t-test. Values of (c) were presented as mean ± SD and compared using one-way ANOVA.
Taken together, these findings suggest that crizotinib induces apoptosis and cell cycle arrest in MET-overexpressing lung cancer cells in vivo. Concomitant treatment with crizotinib and either CsA or PD98059 augmented crizotinib-induced growth inhibition by inhibiting Erk1/2 phosphorylation and its downstream pathways.
5.7. CsA promotes the anticancer activities of crizotinib in primary human NSCLC models
To validate the effects of CsA on crizotinib in human samples, we successfully established 3 primary cell strains from MET-amplified lung cancer patients and tested the changes of apoptosis and cell cycle pathways. Meanwhile, we determined the sensitization activity of CsA to crizotinib by the In Vivo Hollow Fiber Assay, which has been successfully used as a routine in vivo screening model to quantitatively define anticancer activity by the National Cancer Institute since 1995, to observe the cytotoxity of Cyclosporine A and crizotinib in primary lung cancer tissues in vivo [32,40]. As shown in Fig. 9a and b, CsA significantly enhanced the growth arresting effect of crizotinib on primary NSCLC cells in vitro and in vivo. Crizotinib treatment decreased the anti-apoptotic protein Bcl-xL, increased the pro-apoptotic proteins cleaved caspase-3 and downregulated G2/M transition-phase proteins cyclin B1. These effects were enhanced by the addition of CsA. MET, AKT and STAT3 signaling was inhibited by crizotinib and CsA efficiently inhibited crizotinib-dependent activation of Erk1/2, which was consisted to the results of NSCLC cell lines and xenograft mouse models (Fig. 9c).
Fig. 9.

CsA promotes the anticancer activities of crizotinib in primary human NSCLC models. (a) 1 × 105 human NSCLC cells were seeded in 6 well plate and cultured in ACL4 medium plus 5% FBS. 2 μM CsA and 2 μm crizotinib were added the next day. The number of cells were calculated every day for 6 days. (b) In vivo hollow fiber assay results. Crizotinib and CsA were administered by oral gavage at the dose of 30 mg/kg/day and 20 mg/kg/day respectively once daily for one week. The ATP content of each tube was measured by the addition of luciferin-luciferase. (c) Primary NSCLC cells were incubated with 2 μM CsA and 4 μm crizotinib for 48 h, after which they were examined for immunoblotting. All results were from 3 separated cell strains. c-Caspase 3 represented for cleaved caspase 3. Values of (a) were presented as mean ± SD and compared using one-way ANOVA. Values of (b) were presented as mean ± SD and compared using student's t-test. *** P < 0.001.
6. Discussion
MET is a classic member of the receptor tyrosine kinase (RTK) superfamily, and its amplification occurs in 2%–5% of NSCLC patients, which partially accounts for acquired resistance to epidermal growth factor receptor (EGFR) inhibition as well as ALK inhibition [[41], [42], [43]]. Aberrant MET activation is considered a driving factor for a subgroup of lung adenocarcinomas [44,45]. MET overexpression in NSCLC is associated with poor prognosis, emerging as a promising target for anti-cancer therapy [46]. Several small-molecule MET inhibitors, alone or in combination with other drugs, are already under evaluation in clinical trials to treat advanced NSCLC patients [47]. Two clinical trials are underway to assess the efficacy of crizotinib in NSCLC patients with MET amplification (NCT02499614 and NCT00585195), both of which have already shown encouraging results [48,49].
Crizotinib was first recognized as a potent inhibitor of the MET receptor tyrosine kinase and was approved by the FDA to treat patients with ALK-positive NSCLC in 2011 [10]. In a previous study, we discovered that crizotinib exerted noticeable anti-tumor activity on MET-amplified NSCLC cell lines [28]. In the current study, we found that CsA significantly augments crizotinib-induced apoptosis and G2/M arrest in NSCLC cells in vitro and in vivo. Our previous study indicated that crizotinib exerts anti-cancer effects by inhibiting p-MET and its downstream STAT3 and PI3K-AKT-MTOR signaling [28], both of which are critical pathways involved in the growth and survival of cancer cells. These results were reconfirmed by the current study (Fig. 3a). However, to our surprise, crizotinib treatment resulted in transient feedback activation of Erk1/2 signaling, and CsA not only inhibited the basal levels of activated Erk1/2 but also efficiently blocked crizotinib-induced feedback activation of Erk1/2, implicating an Erk1/2-involved mechanism in CsA-mediated sensitization (Fig. 3b-f). Pharmacological or genetic inhibition of Erk1/2 significantly enhanced the growth inhibitory effects of crizotinib, whereas overexpression of Erk1/2 impaired CsA-mediated sensitization. Interestingly, Erk1/2 inhibitor or Erk1/2-targeting siRNAs sensitized NSCLC cells to crizotinib by promoting apoptosis and G2/M arrest, which is consistent with CsA-mediated sensitization (Fig. 4 and Supplementary Fig. S5). These results were further confirmed in primary human lung cancer cell model in vitro and hollow fiber assay in vivo (Fig. 9). Thus, CsA sensitizes NSCLC cells to crizotinib at least partially via Erk1/2 inhibition.
Theoretically, MET inhibition by crizotinib should result in dephosphorylation of Erk1/2. However, our results demonstrate that Erk1/2 becomes activated by crizotinib in a dose- and time-dependent manner (Fig. 3D and E), suggesting that another mechanism must be involved in the regulation of Erk1/2. Erk1/2 is a downstream molecule of Ca2+ signaling [[50], [51], [52]]. Several studies have indicated that Ca2+/calcineurin signaling is involved in the resistance to several TKIs, including dasatinib, imatinib, vemurafenib and selumetinib [[21], [22], [23],53]. A previous study reported that calcium-related Erk1/2 signaling could be regulated by calcineurin, a classic target of CsA. To address the possibility of Ca2+ involvement in our observations, we quantified the concentration of iCa2+ in treated NSCLC cells and found that crizotinib treatment led to an increase of iCa2+ in a dose- and time-dependent manner, which showed synchronization with temporary p-Erk1/2 upregulation patterns (Fig. 6A and B). Expectedly, both pharmacological and genetic inhibition of calcineurin resulted in a decrease in p-Erk1/2 levels and sensitization to crizotinib in SPC-A1 and PC-9 cells (Fig. 7A-E). Moreover, the Ca2+ influx inducer TG enhanced phosphorylation of Erk1/2, whereas the Ca2+ chelator EGTA significantly reduced the activity of Erk1/2 (Fig. 6C and D). These results indicate that CsA augments the anti-cancer activity of crizotinib by inhibiting the Ca2+-calcineurin-Erk1/2 pathway. To the best of our knowledge, this is the first report to show that the feedback activation of Ca2+-calcineurin-Erk1/2 signaling is involved in primary crizotinib resistance in MET-amplified NSCLC cells [38].
As a ubiquitous second messenger, Ca2+ is involved in several fundamental physiological functions, including cell cycle control, survival, migration and gene expression [54,55]. Ca2+ homeostasis is tightly regulated, and the interruption of this homeostasis causes many diseases, including cancer [[56], [57], [58]]. Enhanced Ca2+ influx was reported to mediate resistance to chemotherapeutic drugs, including cis‑platinum, 5-fluorouracil (5-FU) and gemcitabine [[59], [60], [61]], as well as TKIs such as dasatinib, imatinib and selumetinib [21,23]. Intriguingly, we observed that SKF-96365, a Ca2+ influx inhibitor, had strong cytotoxicity in several NSCLC cells (data not shown), indicating that Ca2+/calcineurin/Erk signaling not only mediates drug resistance to crizotinib but also maintains the survival of NSCLC cells. Ca2+ is not only involved in the regulation of Erk1/2 signaling but is also involved in the regulation of AKT and STAT3 signaling [62,63]. Ca2+ also plays an important role in regulating autophagy [64,65] and epithelial-mesenchymal transition (EMT) [63], both of which are related to drug resistance to TKIs [28,66]. As a result, blocking Ca2+ signaling may be a promising strategy that enhances the clinical efficacy of TKIs.
Our present study mainly explored the role that the Ca2+/calcineurin/Erk pathway plays in mediating drug resistance in MET-amplified NSCLC cells to crizotinib. Ca2+/calmodulin-dependent protein kinase II and Ca2+-dependent activation of protein kinase C (PKC) have also been reported to cause Erk1/2 activation [67,68]. The exact role of these two pathways in crizotinib-induced activation of Erk1/2 signaling requires further investigation. Recent studies have reported that AKT can also be activated by Ca2+/calmodulin-dependent protein kinase II [62,69,70]. We observed that crizotinib, which significantly inhibits MET and STAT3, only moderately inhibits AKT (Fig. 3a). This effect is probably because crizotinib can activate AKT by inducing Ca2+ signaling, which attenuates its inhibitory effect on AKT through the inhibition of MET. If so, AKT is probably another important molecule involved in drug resistance to crizotinib.
CsA is a widely used immunosuppressive drug, functioned as a calcineurin inhibitor. Recently, several studies revealed that TLR9-BTK-Ca2 + −calcineurin-NFAT/NFκB-TNF-α signaling plays an import part in tumor progression. Gallotta et al. demonstrated that mice treated with SD-101, a TLR9 blocker, induced CD4+ and CD8+ T-cell homing to tumors and achieved long-term control of lung tumors when combined with anti PD-1 therapy [71]. Heim's study showed that targeting PD-1 resulted in NFATc1 induction in CD4+ and CD8+ T cells in tumor-bearing mice [72]. Gong et al. suggested that TNF-driven adaptive response mediated resistance to EGFR inhibition and blocking TNF enhanced the effectiveness of EGFR inhibition [73]. Herbst et al. revealed the collaborations between NFAT and NF-κB for TNF-α production in macrophages [74]. All these studies indicate that TLR9-BTK-Ca2+-calcineurin-NFAT/NF-κB-TNF-α signaling exerts its effects through modulation of immune cells. The role of this signaling in cancer cells is still unknown. As CsA is an inhibitor of calcineurin, to further illustrate the role of calcineurin in cancer cells, we performed western blot to check the levels of TLR9, BTK, calcineurin, NFAT, NF-κB and TNF-α. As shown in the Supplementary Fig. S6, no significant changes were found among treatment group and control group, which suggested that the TLR9-BTK-Ca2+-calcineurin signaling pathway was not the predominant pathway involved in lung cancer cells upon crizotinib or CsA treatments. However, whether this pathway works through regulating tumor surrounding immune cells needs further exploration. Interestingly, a slight decrease of NF-κB and TNF-α was noticed in crizotinib or CsA-treated PC-9 cells, but not in SPC-A1 cells, indicating that calcineurin-independent signaling might exist to regulate the activity of NF-κB in treated PC-9 cells.
In summary, as illustrated in Fig. 10, we discovered that crizotinib activates Ca2+ influx in NSCLC cells, which results in the activation of calcineurin/KSR2/Erk signaling and mediates drug resistance of NSCLC cells to crizotinib. CsA effectively blocked calcineurin, promoting crizotinib-induced apoptosis and G2/M arrest. The inhibition of calcineurin and Erk1/2 by pharmacological or genetic approaches enhances the anti-cancer effects of crizotinib in MET-overexpressing lung cancer cells. Inhibition of signaling pathways induced by Ca2+ influx represents a promising approach to overcome intrinsic or acquired resistance to crizotinib. Our findings present a possible strategy for improving the clinical efficacy of crizotinib in MET-amplified NSCLC patients.
Fig. 10.

Schematic depicting the sensitization mechanism of CsA to crizotinib.
The following are the supplementary data related to this article.
Fig. S1.

CsA sensitizes multiple NSCLC cell lines to crizotinib (criz) treatment. HCC827, H1975 and A549 cells were treated with crizotinib in the presence or absence of CsA for 48 h. Cell viability was measured by MTS assay. Values were presented as mean ± SD of three independent experiments and compared using one-way ANOVA. *P < .05, ** P < .01, and ***P < 0.001 compared with crizotinib alone.
Fig. S2.
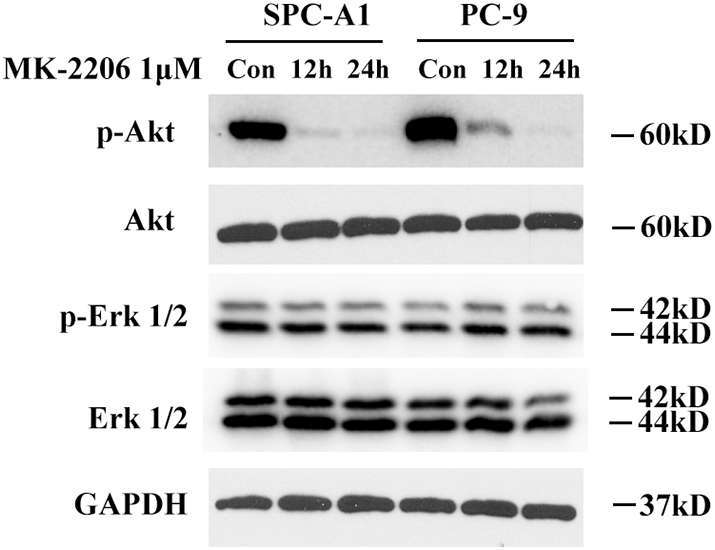
SPC-A1 and PC-9 cells were treated with MK-2206 (1 μM) for 12 h or 24 h and then cell lysates were collected for immunoblotting. Con represents for control.
Fig. S3.

PD98059 (PD) augments crizotinib-induced growth inhibition in multiple NSCLC cell lines by promoting apoptosis and G2/M arrest. (a) Cells were treated with crizotinib in the presence or absence of PD98059 for 48 h. Cell viability was measured by MTS assay. (b) Cells were treated with crizotinib in the presence or absence of PD98059 for 48 h, quantification of the apoptotic rates was analyzed by flow cytometry. (c) Cells were treated with crizotinib in the presence or absence of PD98059 for 24 h, quantification of the relative cell populations in each cell cycle stage was analyzed by flow cytometry. (d) Immunoblotting for the corresponding proteins in cells treated with crizotinib alone or in combination with PD98059 for 48 h. Values of (a) were presented as mean ± SD and compared using one-way ANOVA. Values of (b) and (c) were presented as mean ± SD and compared using student's t-test. *P < 0.05, **P < 0.01, and *** P < 0.001.
Fig. S4.

Crizotinib activates Erk1/2 by inducing Ca2+ influx into the cytoplasm. (a) SPC-A1 and PC-9 cells were treated with crizotinib for the indicated amount of time before staining with fluo-3/AM. The fluorescence intensity was detected by flow cytometry. (b) SPC-A1 and PC-9 cells were treated with the indicated concentration of crizotinib for 12 h before staining with fluo-3/AM and analyzing by flow cytometry. (c) SPC-A1 and PC-9 cells were treated with the indicated concentration of crizotinib for 12 h before staining with fluo-3/AM and analyzing by confocal microscope.
Fig. S5.

Combined treatment with crizotinib and either CsA or PD98059 does not increase systemic toxicity in xenograft models. (a) Body weight of nude mice in each group. (b) Serum glutamic-pyruvic transaminase (ALT), glutamic-oxaloacetic transaminase (AST) and creatinine (SCr) levels were determined after the nude mice had been sacrificed.
Fig. S6.
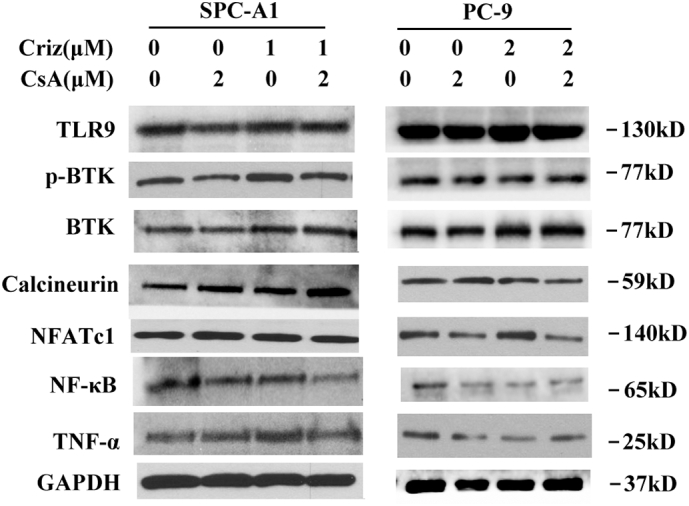
SPC-A1 and PC-9 cells were treated with crizotinib and CsA for 48 h and then examined for the expression of dedicated proteins by immunoblotting.
Supplementary material 1
Supplementary material 2
Author contribution
Conceptualization: WD.H. and HM.P.; investigation: Z.L, LM.J, YR.L, BB.X, JS.X; analysis: ZG.W, XY.Z, HL.J, Y·F; writing original draft: LM.J, YR.L, Z.L; writing, review, editing, and final approval: Z.L, WD.H; supervision: WD.H, HM.P.
Funding sources
This work was supported by the National Natural Science Foundation of China (81572592, 81602635, 81772543), the Key Projects of Natural Foundation of Zhejiang Province (LZ15H160001), the Ten thousand plan youth talent support program of Zhejiang Province, the Zhejiang Natural Sciences Foundation Grant (LQ16H160003, LY15H160025, LY18H160007), and the Zhejiang medical innovative discipline construction project-2016.
Declaration of interests
The authors declare no competing interests.
Acknowledgements
Not applicable.
Contributor Information
Hongming Pan, Email: panhongming@zju.edu.cn.
Weidong Han, Email: hanwd@zju.edu.cn.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Pao W., Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R., Karachaliou N. BRAF(V600E) and BRAF-inactivating mutations in NSCLC. Lancet Oncol. 2017;18(10):1286–1287. doi: 10.1016/S1470-2045(17)30678-2. [DOI] [PubMed] [Google Scholar]
- 4.Akerley W. Biomarker-based treatment selection in non-small cell Lung Cancer. J Natl Compr Canc Netw. 2017;15(5S):689–691. doi: 10.6004/jnccn.2017.0074. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi S., Boggon T.J., Dayaram T., Janne P.A., Kocher O., Meyerson M. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 6.Paik P.K., Drilon A., Fan P.D., Yu H., Rekhtman N., Ginsberg M.S. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cascone T., Xu L., Lin H.Y., Liu W., Tran H.T., Liu Y. The HGF/c-MET pathway is a driver and biomarker of VEGFR-inhibitor resistance and vascular Remodeling in non-small cell Lung Cancer. Clin Cancer Res. 2017;23(18):5489–5501. doi: 10.1158/1078-0432.CCR-16-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong J.H., Yeung S.F., Chan A.W., Chung L.Y., Chau S.L., Lung R.W. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell Lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048–3056. doi: 10.1158/1078-0432.CCR-15-2061. [DOI] [PubMed] [Google Scholar]
- 9.Tanizaki J., Okamoto I., Okamoto K., Takezawa K., Kuwata K., Yamaguchi H. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J Thorac Oncol. 2011;6(10):1624–1631. doi: 10.1097/JTO.0b013e31822591e9. [DOI] [PubMed] [Google Scholar]
- 10.Malik S.M., Maher V.E., Bijwaard K.E., Becker R.L., Zhang L., Tang S.W. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20(8):2029–2034. doi: 10.1158/1078-0432.CCR-13-3077. [DOI] [PubMed] [Google Scholar]
- 11.Cao Y., Xiao G., Qiu X., Ye S., Lin T. Efficacy and safety of crizotinib among Chinese EML4-ALK-positive, advanced-stage non-small cell lung cancer patients. PloS One. 2014;9(12) doi: 10.1371/journal.pone.0114008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters S., Camidge D.R., Shaw A.T., Gadgeel S., Ahn J.S., Kim D.W. Alectinib versus Crizotinib in untreated ALK-positive non-small-cell Lung Cancer. N Engl J Med. 2017;377(9):829–838. doi: 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- 13.Shou J., Jing J., Xie J., You L., Jing Z., Yao J. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015;361(2):174–184. doi: 10.1016/j.canlet.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Z.D., Yu T., Liu H.J., Jin J., He J. SOCE induced calcium overload regulates autophagy in acute pancreatitis via calcineurin activation. Cell Death Dis. 2018;9(2):50. doi: 10.1038/s41419-017-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Rao A., Hogan P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21(2):91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qadir M., O'Loughlin K.L., Fricke S.M., Williamson N.A., Greco W.R., Minderman H. Cyclosporin A is a broad-spectrum multidrug resistance modulator. Clin Cancer Res. 2005;11(6):2320–2326. doi: 10.1158/1078-0432.CCR-04-1725. [DOI] [PubMed] [Google Scholar]
- 17.Chambers S.K., Davis C.A., Schwartz P.E., Kohorn E.I., Chambers J.T. Modulation of platinum sensitivity and resistance by cyclosporin a in refractory ovarian and fallopian tube cancer patients: a phase II study. Clin Cancer Res. 1996;2(10):1693–1697. [PubMed] [Google Scholar]
- 18.Kruijtzer C.M., Schellens J.H., Mezger J., Scheulen M.E., Keilholz U., Beijnen J.H. Phase II and pharmacologic study of weekly oral paclitaxel plus cyclosporine in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(23):4508–4516. doi: 10.1200/JCO.2002.04.058. [DOI] [PubMed] [Google Scholar]
- 19.Nakahara C., Nakamura K., Yamanaka N., Baba E., Wada M., Matsunaga H. Cyclosporin-A enhances docetaxel-induced apoptosis through inhibition of nuclear factor-kappaB activation in human gastric carcinoma cells. Clin Cancer Res. 2003;9(14):5409–5416. [PubMed] [Google Scholar]
- 20.Han W., Shi L., Ren L., Zhou L., Li T., Qiao Y. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Transduct Target Ther. 2018;3:16. doi: 10.1038/s41392-018-0019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory M.A., Phang T.L., Neviani P., Alvarez-Calderon F., Eide C.A., O'Hare T. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18(1):74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perotti V., Baldassari P., Bersani I., Molla A., Vegetti C., Tassi E. NFATc2 is a potential therapeutic target in human melanoma. J Invest Dermatol. 2012;132(11):2652–2660. doi: 10.1038/jid.2012.179. [DOI] [PubMed] [Google Scholar]
- 23.Spreafico A., Tentler J.J., Pitts T.M., Tan A.C., Gregory M.A., Arcaroli J.J. Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin Cancer Res. 2013;19(15):4149–4162. doi: 10.1158/1078-0432.CCR-12-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner L.A., Klawitter J., Gregory M.A., Zaberezhnyy V., Baturin D., Pollyea D.A. Inhibition of calcineurin combined with dasatinib has direct and indirect anti-leukemia effects against BCR-ABL1(+) leukemia. Am J Hematol. 2014;89(9):896–903. doi: 10.1002/ajh.23776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding S., Patel N., Zhang H. Ciclosporin a as a reversal agent against concurrent multidrug resistance in Tumors with Nanobubbles. J Biomed Nanotechnol. 2018;14(1):190–197. doi: 10.1166/jbn.2018.2494. [DOI] [PubMed] [Google Scholar]
- 26.Shou J., You L., Yao J., Xie J., Jing J., Jing Z. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2016;379(1):124–133. doi: 10.1016/j.canlet.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 28.You L., Shou J., Deng D., Jiang L., Jing Z., Yao J. Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. Oncotarget. 2015;6(37):40268–40282. doi: 10.18632/oncotarget.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuda N., Fukuoka M., Takada M., Kudoh S., Kusunoki Y. Establishment and characterization of 20 human non-small cell lung cancer cell lines in a serum-free defined medium (ACL-4) Chest. 1991;100(2):429–438. doi: 10.1378/chest.100.2.429. [DOI] [PubMed] [Google Scholar]
- 30.Oie H.K., Russell E.K., Carney D.N., Gazdar A.F. Cell culture methods for the establishment of the NCI series of lung cancer cell lines. J Cell Biochem Suppl. 1996;24:24–31. doi: 10.1002/jcb.240630504. [DOI] [PubMed] [Google Scholar]
- 31.Bean J., Brennan C., Shih J.Y., Riely G., Viale A., Wang L. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollingshead M.G., Alley M.C., Camalier R.F., Abbott B.J., Mayo J.G., Malspeis L. In vivo cultivation of tumor cells in hollow fibers. Life Sci. 1995;57(2):131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- 33.Bai L.H., Lin G., Sun L., Liu Y., Huang X., Cao C. Upregulation of SIRT6 predicts poor prognosis and promotes metastasis of non-small cell lung cancer via the ERK1/2/MMP9 pathway. Oncotarget. 2016;7(26):40377–40386. doi: 10.18632/oncotarget.9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chi F., Wu R., Jin X., Jiang M., Zhu X. HER2 induces cell proliferation and invasion of non-small-cell lung cancer by upregulating COX-2 expression via MEK/ERK signaling pathway. OncoTargets Ther. 2016;9:2709–2716. doi: 10.2147/OTT.S96197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang P., Guo X., Li J., Yu S., Wang L., Jiang G. Immunoglobulin-like transcript 4 promotes tumor progression and metastasis and up-regulates VEGF-C expression via ERK signaling pathway in non-small cell lung cancer. Oncotarget. 2015;6(15):13550–13563. doi: 10.18632/oncotarget.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao S., Qiu Z.X., Zhang L., Li W.M. Prognostic values of ERK1/2 and p-ERK1/2 expressions for poor survival in non-small cell lung cancer. Tumour Biol. 2015;36(6):4143–4150. doi: 10.1007/s13277-015-3048-4. [DOI] [PubMed] [Google Scholar]
- 37.Mendoza M.C., Er E.E., Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayakawa J., Ohmichi M., Kurachi H., Ikegami H., Kimura A., Matsuoka T. Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem. 1999;274(44):31648–31654. doi: 10.1074/jbc.274.44.31648. [DOI] [PubMed] [Google Scholar]
- 39.Dougherty M.K., Ritt D.A., Zhou M., Specht S.I., Monson D.M., Veenstra T.D. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell. 2009;34(6):652–662. doi: 10.1016/j.molcel.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Decker S., Hollingshead M., Bonomi C.A., Carter J.P., Sausville E.A. The hollow fibre model in cancer drug screening: the NCI experience. Eur J Cancer. 2004;40(6):821–826. doi: 10.1016/j.ejca.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 41.Pao W., Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yano S., Takeuchi S., Nakagawa T., Yamada T. Ligand-triggered resistance to molecular targeted drugs in lung cancer: roles of hepatocyte growth factor and epidermal growth factor receptor ligands. Cancer Sci. 2012;103(7):1189–1194. doi: 10.1111/j.1349-7006.2012.02279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kogita A., Togashi Y., Hayashi H., Banno E., Terashima M., De Velasco M.A. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK-positive non-small cell lung cancer. Int J Oncol. 2015;46(3):1025–1030. doi: 10.3892/ijo.2014.2797. [DOI] [PubMed] [Google Scholar]
- 44.Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 45.Verschuur A., Aschero A., Petit P., Roffe-Vidal S., Chastagner P., Leblond P. Sfce Metro-01 four-drug metronomic regimen phase II trial for Pediatric Extracranial solid tumours. Pediatr Blood Cancer. 2014;61:S209–S210. doi: 10.1002/pbc.27693. [DOI] [PubMed] [Google Scholar]
- 46.Siegfried J.M., Weissfeld L.A., Singh-Kaw P., Weyant R.J., Testa J.R., Landreneau R.J. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997;57(3):433–439. [PubMed] [Google Scholar]
- 47.Gelsomino F., Facchinetti F., Haspinger E.R., Garassino M.C., Trusolino L., De Braud F. Targeting the MET gene for the treatment of non-small-cell lung cancer. Crit Rev Oncol Hematol. 2014;89(2):284–299. doi: 10.1016/j.critrevonc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Landi L., Chiari R., Dazzi C., Tiseo M., Chella A., Delmonte A. Crizotinib in ROS1 rearranged or MET deregulated non-small-cell lung cancer (NSCLC): final results of the METROS trial. Ann Oncol. 2017;28:54. [Google Scholar]
- 49.Shaw A., Riley G.J., Bang Y.J., Kim D.W., Camidge D.R., Varella-Garcia M. Crizotinib in advanced ROS1-rearranged non-small cell lung cancer (NSCLC): updated results from PROFILE 1001. Ann Oncol. 2016;27 doi: 10.1093/annonc/mdz131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agell N., Bachs O., Rocamora N., Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal. 2002;14(8):649–654. doi: 10.1016/s0898-6568(02)00007-4. [DOI] [PubMed] [Google Scholar]
- 51.Andrikopoulos P., Baba A., Matsuda T., Djamgoz M.B., Yaqoob M.M., Eccles S.A. Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J Biol Chem. 2011;286(44):37919–37931. doi: 10.1074/jbc.M111.251777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu P.K., Park J.I. MEK1/2 inhibitors: molecular activity and resistance mechanisms. Semin Oncol. 2015;42(6):849–862. doi: 10.1053/j.seminoncol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tentler J.J., Nallapareddy S., Tan A.C., Spreafico A., Pitts T.M., Morelli M.P. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther. 2010;9(12):3351–3362. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monteith G.R., McAndrew D., Faddy H.M., Roberts-Thomson S.J. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7(7):519–530. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 55.Humeau J., Bravo-San Pedro J.M., Vitale I., Nunez L., Villalobos C., Kroemer G. Calcium signaling and cell cycle: progression or death. Cell Calcium. 2018;70:3–15. doi: 10.1016/j.ceca.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 56.Carafoli E. The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol. 2003;4(4):326–332. doi: 10.1038/nrm1073. [DOI] [PubMed] [Google Scholar]
- 57.Parekh A.B. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9(5):399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- 58.Monteith G.R., Prevarskaya N., Roberts-Thomson S.J. The calcium-cancer signalling nexus. Nat Rev Cancer. 2017;17(6):367–380. doi: 10.1038/nrc.2017.18. [DOI] [PubMed] [Google Scholar]
- 59.Kondratska K., Kondratskyi A., Yassine M., Lemonnier L., Lepage G., Morabito A. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta. 2014;1843(10):2263–2269. doi: 10.1016/j.bbamcr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 60.Griesmann H., Ripka S., Pralle M., Ellenrieder V., Baumgart S., Buchholz M. WNT5A-NFAT signaling mediates resistance to apoptosis in pancreatic cancer. Neoplasia (New York, NY) 2013;15(1):11–22. doi: 10.1593/neo.121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Busselberg D., Florea A.M. Targeting intracellular calcium signaling ([Ca(2+)]i) to overcome acquired multidrug resistance of cancer cells: a mini-overview. Cancers (Basel) 2017;9(5) doi: 10.3390/cancers9050048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zou W., Meng X., Cai C., Zou M., Tang S., Chu X. Store-operated Ca2+ entry (SOCE) plays a role in the polarization of neutrophil-like HL-60 cells by regulating the activation of Akt, Src, and rho family GTPases. Cell Physiol Biochem. 2012;30(1):221–237. doi: 10.1159/000339059. [DOI] [PubMed] [Google Scholar]
- 63.Davis F.M., Azimi I., Faville R.A., Peters A.A., Jalink K., Putney J.W., Jr. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33(18):2307–2316. doi: 10.1038/onc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yao Z., Klionsky D.J. The symphony of autophagy and calcium signaling. Autophagy. 2015;11(7):973–974. doi: 10.1080/15548627.2015.1058475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vu H.T., Kobayashi M., Hegazy A.M., Tadokoro Y., Ueno M., Kasahara A. Autophagy inhibition synergizes with calcium mobilization to achieve efficient therapy of malignant gliomas. Cancer Sci. 2018;109(8):2497–2508. doi: 10.1111/cas.13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witta S.E., Gemmill R.M., Hirsch F.R., Coldren C.D., Hedman K., Ravdel L. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–950. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 67.Chang W.C., Nelson C., Parekh A.B. Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J. 2006;20(13):2381–2383. doi: 10.1096/fj.06-6016fje. [DOI] [PubMed] [Google Scholar]
- 68.Chai S., Qian Y., Tang J., Liang Z., Zhang M., Si J. Ca(2+)/calmodulin-dependent protein kinase IIgamma, a critical mediator of the NF-kappaB network, is a novel therapeutic target in non-small cell lung cancer. Cancer Lett. 2014;344(1):119–128. doi: 10.1016/j.canlet.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 69.Jing Z., Sui X., Yao J., Xie J., Jiang L., Zhou Y. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIgamma/AKT-mediated pathway. Cancer Lett. 2016;372(2):226–238. doi: 10.1016/j.canlet.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park J.H., Kim H.K., Jung H., Kim K.H., Kang M.S., Hong J.H. NecroX-5 prevents breast cancer metastasis by AKT inhibition via reducing intracellular calcium levels. Int J Oncol. 2017;50(1):185–192. doi: 10.3892/ijo.2016.3789. [DOI] [PubMed] [Google Scholar]
- 71.Gallotta M., Assi H., Degagne E., Kannan S.K., Coffman R.L., Guiducci C. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4(+) and CD8(+) T-cell interplay. Cancer Res. 2018;78(17):4943–4956. doi: 10.1158/0008-5472.CAN-18-0729. [DOI] [PubMed] [Google Scholar]
- 72.Heim L., Friedrich J., Engelhardt M., Trufa D.I., Geppert C.I., Rieker R.J. NFATc1 promotes Antitumoral effector functions and memory CD8(+) T-cell differentiation during non-small cell Lung Cancer development. Cancer Res. 2018;78(13):3619–3633. doi: 10.1158/0008-5472.CAN-17-3297. [DOI] [PubMed] [Google Scholar]
- 73.Gong K., Guo G., Gerber D.E., Gao B., Peyton M., Huang C. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J Clin Invest. 2018;128(6):2500–2518. doi: 10.1172/JCI96148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herbst S., Shah A., Mazon Moya M., Marzola V., Jensen B., Reed A. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med. 2015;7(3):240–258. doi: 10.15252/emmm.201404556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1
Supplementary material 2