

Abstract
Melanoma is characterised by its ability to metastasise at early stages of tumour development. Current clinico‐pathologic staging based on the American Joint Committee on Cancer criteria is used to guide surveillance and management in early‐stage disease, but its ability to predict clinical outcome has limitations. Herein we review the genomics of melanoma subtypes including cutaneous, acral, uveal and mucosal, with a focus on the prognostic and predictive significance of key molecular aberrations. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: melanoma, cutaneous, desmoplastic, acral, uveal, mucosal, mutations, driver genes, biomarkers, prognostic, predictive, 31‐gene expression profile
Introduction
Historically, melanoma has been classified into subtypes based on the tissue from which the primary tumour arises. The major such subtypes are cutaneous melanoma (CM), which arises in non‐glabrous skin; acral melanoma (AM), a distinct form that originates in glabrous skin of the palms, soles and nail beds; mucosal melanoma (MM), the rarest subtype, which arises from melanocytes in the mucosal lining of internal tissues; and uveal melanoma (UM) which develops from melanocytes in the uveal tract of the eye (Figure 1). These subtypes have well recognised epidemiological, clinical and histopathological characteristics, and recent studies have described the molecular alterations that underpin some of these attributes. Site of origin seems to correlate best with tumoural somatic profile, with melanomas arising from chronically sun damaged (CSD) sites having a higher mutational burden than tumours arising from non‐CSD sites 1 – a direct consequence of the UV‐induced C>T transitions at dipyrimidines that dominate the majority of CM genomes 2, 3, 4.
Figure 1.
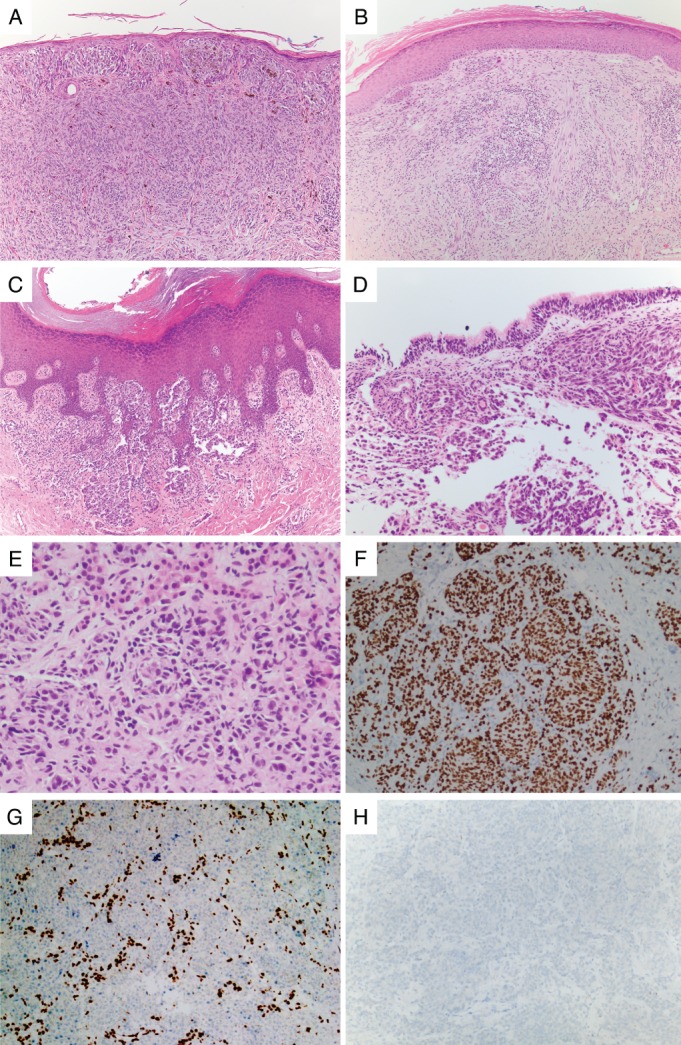
Histopathology of melanoma subtypes. (A) CM of superficial spreading type features an in situ component within the epidermis with underlying dermal invasion. (B) Desmoplastic melanoma, a type of CM, is comprised of a dermal proliferation of atypical spindled cells associated with lymphoid aggregates. (C) Acral melanoma often shows a lentiginous (linear) in situ growth pattern along the epidermal ridges with underlying invasion into the dermis. (D) Mucosal melanoma arises in non‐keratinising wet mucosa, shown here invading the subepithelial stroma of respiratory type mucosa in the nasal sinuses. (E) Uveal melanoma preferentially metastasises to the liver as pictured here with accompanying immunohistochemistry showing (F) staining for SOX10 in the melanoma cells, (G) loss of BAP1 staining in the melanoma cells with retention of normal staining in hepatocytes and lymphocytes, and (H) no staining for BRAF VE1, indicating the absence of a BRAF V600E mutation.
Based solely on the occurrence of driver mutations, melanomas have further been classified into four genomic subtypes: BRAF‐mutant, NRAS‐mutant, NF1‐loss and triple wild‐type (TWT) 3, 4. These subtypes do not have distinguishing histopathological features or sites of origin, although there are notable trends; for example, nearly all UMs and the majority of AMs and MMs fall into the TWT category 3. The BRAF, NRAS and NF1 driver alterations all activate the mitogen‐activated protein kinase (MAPK) pathway and generally occur at the earlier stages of tumour evolution 5. In CM, it has been proposed that subsequent mutations occur in the TERT promoter and in regulators of the cell cycle such as CDKN2A, which precede mutations in chromatin remodelers such as members of the SWI/SNF complex and TP53, the latter being associated with more advanced stages of primary tumour progression 5. Whether some cells are inherently able to metastasise or whether further genomic alterations are necessary to gain this ability, remains under active investigation 6, 7.
Herein, we describe how melanoma subtypes are shaped by their genomic profiles, and outline our current understanding of prognostic and predictive molecular markers.
Melanoma subtypes
CM: dominated by ultraviolet‐induced mutations
The genomic landscape of CM
CM generally affects people of European descent and is the commonest reported melanoma subtype. Consequently, the majority of genomic and transcriptomic studies have been performed on CM cases. CM has the highest burden of somatic mutations across the major cancer subtypes, with a mutational landscape that is dominated by the UV mutational signature, primarily C>T transitions as described earlier 2, 3, 4. About 45–50% of CM are BRAF‐mutant (principally through mutations at the V600 codon), ∼30% are RAS‐mutant (either NRAS, principally at codon Q61, KRAS or HRAS), 10–15% are NF1‐mutant and about 5–10% are TWT 3, 4 (Table 1). These genomic subtypes differ in their characteristics and clinical presentation. Melanomas that arise on skin with intermittent sun exposure are generally more likely to have a BRAF mutation compared with melanomas occurring on chronically sun‐exposed skin 8. Melanomas with BRAF mutations are also more common in younger patients, in the superficial spreading histopathologic subtype and on the trunk 9, 10. NRAS mutations appear more frequently in older patients, in the nodular histopathologic subtype and on skin with chronic UV‐damaged skin 11, 12. Additional recurrent mutations identified in large‐scale sequencing studies include disruptive variants in CDKN2A, TP53, ARID2 and PTEN, and 5′ UTR hotspot mutations in RPS27 and MRPS31, both ribosomal proteins 3, 4. Driver alterations and mutational burden are also related; tumours driven by BRAF V600E mutations tend to have fewer somatic mutations than tumours bearing other, possibly less potent, alterations such as loss of NF1 and activation of NRAS, KIT and BRAF non‐V600E 1. This may be due to these cancers being promoted by additional mutations spread through different biological pathways, and accordingly, tend to present in later life 1. A more recent study has used this information to propose a sequential order in which signalling pathways become disrupted as precursor lesions evolve to invasive melanoma and subsequent metastases 5, 13. More than 50% of advanced CMs have mutations in the TERT (telomerase reverse transcriptase) promoter that create binding sites for the E26 transformation‐specific (ETS) family of transcription factors 14. These promoter variants have been shown to be associated with decreased telomere length and poorer survival 15, 16, 17.
Table 1.
Overview of genomic profile of melanoma subtypes
| Biological pathways | Genes | CM | DM‐subtype | AM | UM | MM |
|---|---|---|---|---|---|---|
| MAPK genomicsubtypes | ∼Total%mut* | ∼90‐95% | ∼73% | ∼50‐60% | ∼100% | ∼50‐60% |
| BRAF | ∼ 45‐50% 3, 4 | ∼0‐5% 13, 43 | ∼ 10‐35% 3, 44, 45, 46, 47 | rarely seen 48, 49 | ∼0‐21% 3, 45, 50, 51 | |
| RAS (mainlyNRAS) | ∼ 30% 3, 4 | ∼0‐6% 13, 43 | ∼ 8‐22% 3, 44, 45, 46, 47 | rarely seen 48, 49 | ∼5‐25% 3, 45, 51 | |
| NF1 | ∼10‐15% 3, 4 | ∼52‐93% 13, 43 | ∼11‐23% 3, 44, 47 | rarely seen 48, 49 | ∼0‐18% 3, 51 | |
| TWT | ∼5‐10% 3, 4 | ∼7‐48% 13, 43 | ∼45‐58% 3, 44 | ∼100% 48, 49 | ∼65‐75% 3, 51 | |
| KIT (mut orgain) | ∼5‐10% 3, 4 | rarely seen 13, 43 | ∼3‐36% 44, 46, 47, 52 | ∼11% 53 | ∼7‐25% 3, 51, 54 | |
| GNAQ | ∼1.5‐2.1% 3, 55 | rarely seen 13, 43 | ∼0‐17% 3, 47 | ∼43‐57% 48, 49 56, 57 | ∼1‐12% 3, 51 | |
| GNA11 | rarely seen 3, 55 | rarely seen 13, 43 | rarely seen 3 | ∼41‐49% 48, 49 56 | ∼1% 51 | |
| MAP2K1 & 2 | ∼4% 3 | ∼7% 13 | ∼8% 3 | ∼9% 48 | ∼0‐11% 3, 50 | |
| Cell Cycle | ∼Total%mut* | ∼57% 3 | ∼70‐75% | ∼90% | ∼85% | ∼36‐75% |
| CDKN2A(mut) | ∼13‐40% 3, 4 | ∼20‐29% 13, 58 | ∼0‐3% 3, 44 | rarely seen, methylated in ∼50% 59 | rarely seen 3, 50, 51, 54 | |
| CDKN2A(loss) | ∼45% 3 | ∼18% 13 | ∼35% 44 | ∼12% 48 | ∼10‐38% 3, 50 | |
| CDK4 (mutor gain) | ∼5‐6% 3, 4 | ∼5% 13 | ∼9% 3, 44 | ∼3% 48 | ∼5‐25% 3, 50 | |
| RB1 | ∼4‐15% 3, 4 | ∼15% 13 | ∼9‐17% 3, 44 | ∼3% 48 | ∼0‐21% 3, 50 | |
| TP53 | ∼15‐18% 3, 4 | ∼40‐60% 13, 43, 58 | ∼6‐54% 3, 44 | ∼9% 48 | ∼7‐15% 3, 50 | |
| CCND1 | ∼5‐13% 3, 4 | ∼2% 13 | ∼6‐54% 3, 44 | ∼6% 48 | ∼25% 3 | |
| BAP1 (mut orloss) | rarely seen 3 | rarely seen 13 | rarely seen 3 | ∼70‐83% (but the great majority of metastatic UM) 48, 49 | rarely seen 3, 50, 51, 54 | |
| PI3K/AKT | PTEN (mut orloss) | ∼8.5‐40% 3, 4 | rarely seen 13 | ∼26‐28% 3, 44 | ∼6‐11%, up to 76% with LOH 48, 60 | 4‐25% 3, 50, 51, 54 |
| Number ofmutations |
|

|
|

|
|
|
| Chromosomalaberrations |

|

|

|

|

|
|
| Transcriptionfactors | NFKBIEpromoter | ∼5% 3 | ∼15‐33% 3, 13 | not seen 3 | NA | rarely seen 3 |
| MITF | ∼10‐20% 3, 18 | rarely seen 13 | ∼15% 3 | ∼63% samples are reported to include deletions or amplifications in MITF 48 | ∼5‐25% 3, 51 | |
| Telomerasepathway | TERT (mut orgain) | ∼85% 3 | ∼85% 13 | ∼9‐45% 3, 44, 46 | ∼2‐9% 48, 61 | ∼5‐13% 3, 50, 51 |
Estimates based on the literature, and on the genes listed on the table including mutations and copy number aberrations.
 Represents the mutational load.
Represents the mutational load.
 Represents the number of chromosomal aberrations.
Represents the number of chromosomal aberrations.
The number of individual symbols within each category is proportionate to the number of mutations/chromosomal aberrations.
Microphthalmia‐associated transcription factor (MITF) is a melanocyte‐specific transcription factor that binds to the promoter site of multiple target genes involved in melanocyte cell development, pigmentation and neoplasia (Figure 2). MITF amplification is present in about 10% of primary melanomas, with a higher incidence reported among metastatic melanomas 18. The role of MITF in melanoma progression and resistance to targeted therapy appears paradoxical; some studies have found that CMs expressing MITF are well differentiated and have a favourable prognosis 19 and those with low MITF expression have an invasive phenotype and are intrinsically resistant to MAPK inhibition 20, whereas others have found that activation of a robust MITF transcriptional program triggers differentiation into highly pigment‐producing drug resistant cells 21. Recent studies have found great heterogeneity in MITF expression within tumours 22. An overview of other melanoma pathways and genes is shown in Table 1 and Figure 2.
Figure 2.
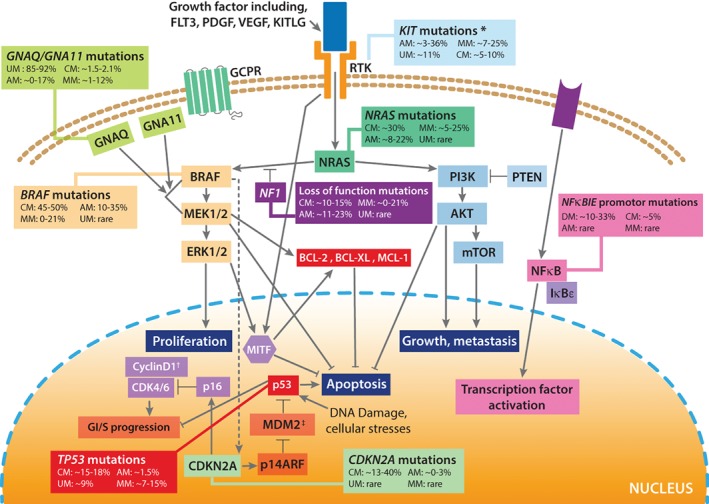
Molecular representation of the mutations associated with the RAS/RAF/MEK/ERK pathways in melanoma, including the MITF signalling cascade. GPCR, G‐protein coupled receptor; RTK, receptor tyrosine kinase. *KIT amplifications are seen in ∼10% of CMs, ∼9.5% of AMs, ∼15% MMs 64. †Cyclin D1 is also amplified in ∼18% of CMs 65. ‡MDM2 is also amplified in ∼6% of CMs 66. Adapted from 67.
The relationship between tumour driver mutation status and survival has been the subject of significant research efforts and it is now well appreciated that BRAF‐mutant tumours confer a poorer prognosis relative to BRAF wild‐type melanoma. In particular, BRAF‐mutated melanoma has been linked to a shorter overall survival in patients with stage IV disease when compared to those with BRAF WT disease 9, 23. While the majority of studies investigating the relationship between BRAF mutations and clinical outcomes are focused on patients with metastatic disease, recent studies have demonstrated that BRAF‐mutant melanomas are also associated with a shorter disease‐free and melanoma‐specific survival in patients with early‐stage disease 24, 25. Historically, NRAS‐mutant disease has been associated with thicker primary lesions and higher mitotic activity 12. However, there have been conflicting reports on its prognostic significance. In particular, no impact on survival was seen when NRAS mutations were measured in primary disease 26, 27, however when measured from metastases, NRAS mutations were associated with improved survival compared to tumours with BRAF mutations or TWT tumours 28, 29. Despite the undoubted prognostic relevance of American Joint Committee on Cancer (AJCC) classification and certain driver mutations, our ability to predict those early‐stage patients at highest metastatic risk remains conspicuously limited.
Gene expression profiles and their prognostic implication
A gene expression profile (31‐GEP) test has been proposed that evaluates the expression of 31 gene targets in the primary tumour, providing a binary classification of ‘low risk’ (Class 1) or ‘high risk’ (Class 2) of metastases within 5 years of diagnosis 30. The test assesses the expression of three control genes, four genes with proven prognostic utility for UMs 31 and 24 genes previously reported to be differentially expressed in metastatic compared to primary tumours 32, 33, 34, 35, 36, 37, 38. The performance of this test has been evaluated in several retrospective 30, 39, 40 as well as prospective validation studies 41, 42 and has been shown to enhance current prognostic accuracy in particular through identifying clinically and pathologically sentinel lymph node (SLN)‐negative patients with high‐risk of metastases. However, although there is great promise in reproducibility and clinical validity, the clinical utility for the 31‐GEP test on clinical decision‐making is still incompletely defined, and will require evidence from further large‐scale prospective multi‐institutional registry studies before it can be considered for inclusion in any national or professional association guideline recommendations.
By undertaking unsupervised hierarchical clustering of gene expression profiles, Jönsson and collaborators were able to categorise melanomas into four biologically relevant subgroups; MITF‐low/proliferative, high‐immune response, MITF‐high/pigmentation and normal‐like 19. Importantly, the MITF‐low/proliferative subtype, characterised by an absence of the expression of immune‐response genes, had only BRAF/NRAS‐mutated samples and more tumours with CDKN2A deletions, and was significantly associated with a poorer prognosis. Classification of primary melanomas by gene expression also resulted in these four classes, which could be collapsed into two classes associated with clinical outcome 62. The multi‐institutional TCGA (The Cancer Genome Atlas) initiative subsequently identified three transcriptomic subclasses, an immune group, a keratin group, and a MITF‐low group 4, 63. A subsequent analysis showed that these classifications comprised very similar biological entities (TCGA immune ∼ Lund high‐immune, TCGA keratin ∼ Lund normal‐like and MITF‐high/pigmentation, TCGA MITF‐low ∼ Lund MITF‐low/proliferative) 63.
Molecular markers of response/resistance to targeted therapy and immune‐checkpoint inhibition
The development of approved targeted therapies for patients with metastatic and early‐stage melanomas has been remarkable and driven by significant discoveries around the molecular mechanisms of melanomagenesis. Combined treatment with BRAF and MEK inhibitors achieves radiological responses in ∼70% of patients with BRAF V600 mutations 68. A proportion of patients are intrinsically resistant to BRAF inhibitors, and most patients who initially respond will eventually exhibit resistance. The need to maximise the long‐term clinical benefit of this strategy remains a key challenge and molecular profiling may play a particularly important role in deciphering the mechanisms of response and resistance to targeted therapy.
One of the most frequently reported mutations leading to intrinsic BRAF resistance is loss of the phosphatase and tensin homolog (PTEN) gene. Decreased responses to BRAF inhibition in patients with PTEN loss is thought to be attributed to constitutive activation of the PI3K/AKT pathway which leads to cell proliferation and survival 69. MITF has also been shown to be an important regulator of response, and high MITF levels allow melanoma cells to evade cell death triggered by BRAF and MEK inhibitors 70, 71. Intriguingly, it has been shown that very low levels of MITF when co‐existing with high levels of receptor tyrosine kinase AXL (MITFlow/AXLhigh phenotype) represent a de‐dedifferentiated cellular state that displays innate resistance to BRAF inhibitors and increased invasiveness 20. A number of other mechanisms of intrinsic resistance have been suggested, reviewed in 72. The most common mechanism of acquired resistance is via reactivation of the MAPK/ERK pathway 72. Recently, studies have reported non‐mutational mechanisms for the acquisition of resistance through phenotype switching 21, 73, 74.
Immune checkpoint inhibitor (ICI) therapy has revolutionised melanoma therapy and resulted in unprecedented rates of long‐term disease control and survival in patients with metastatic disease. It is hypothesised that the mutational status of a cancer influences anti‐tumour immune and ICI responses, presumably by virtue of enhanced neoantigen formation due to increased number of non‐synonymous single‐nucleotide variants 75, 76. This phenomenon probably reflects an increased likelihood of forming neoantigens that will elicit T‐cell reactivity. Consistent with this notion, tumours with microsatellite instability resulting from acquired deficiency of DNA mismatch repair are also associated with enhanced response to PD‐1 blockade 77, 78. This has formed the basis for the first site‐agnostic drug approval made by the FDA, for anti‐PD1 therapy 79, 80. Studies ex vivo strongly support the dominance of mutational neoantigens as the targets for lymphocyte recognition of a tumour, and neoantigen expression and HLA binding characteristics have been shown to be surrogates for treatment response 75, 76, 81, 82. In keeping with this, mutational and neoantigen load have also recently been linked with clinical benefit from adoptive T cell immunotherapy 83. Further evidence suggests that clonal neoantigens may be particularly relevant 84. While genomic instability may feasibly provide sufficient genomic variation to promote an effective immune response, the mechanism relating DNA damage and genomic instability to ICI response is not fully understood and mutational load does not sufficiently explain all cases 85. Significant genomic heterogeneity between tumours can contribute to heterogenous clinical responses and this may account for some of the conflicting results seen in separate cohorts 86, 87.
Immune activation gene‐expression signatures have been shown to define distinct CM subtypes 19 and the prevalence of pre‐existing tumour infiltrating T cells has been shown to correlate with clinical response to anti‐PD1 immunotherapy 88. Although previous reports have suggested that the expression of cytolytic markers might correlate with response to anti‐CTLA4 76, these are based on small retrospective analyses and there has yet to be any specific gene expression signature that has been independently validated in this context. It is increasingly appreciated that the relationship of the tumour's mutational profile to immune dynamics is moderated by additional factors that affect expression, processing and immunogenicity of putative neoantigens. Accordingly, predictive approaches are now being paired with additional filters as well as expression data to evaluate somatic mutations which are adequately expressed and processed.
Desmoplastic melanoma (DM): a CM subtype with an elevated mutational load
DM is a variant of CM, consisting of intradermal proliferations of spindled melanocytes, commonly associated with lymphoid aggregates, and typically found on chronically sun‐damaged skin of older individuals (Figure 1). The term DM initially referred to the association of invasive tumour cells with abundant stromal collagen, and therefore DM can be classified as pure and mixed, based on the degree of desmoplasia 89. Pure DMs have less frequent lymph node involvement and tend to display a less aggressive clinical course than mixed DM. DMs rank among the most heavily mutated types of cancer, with a mutation rate on average four‐fold higher than CMs, of which the great majority are attributed to UV mutagenesis 13. DMs also tend to have lower DNA copy number alterations than other melanoma subtypes; the few focal deletions that have been observed target CDKN2A and NF1, whereas amplifications affect EGFR, CDK4, MDM2, TERT, MAP3K1, MET, YAP1 and NFKBIE 13. The promoter of NFKBIE has been identified as a recurrently mutated locus in 15–33% of samples 3, 13. This gene, coding for IkBϵ, inhibits downstream nuclear factor kappa B (NFκB) signalling by sequestering NFκB transcription factors in the cytoplasm (Figure 2) 13. Although also mutated in CM, promoter mutations are enriched in DM 3. No melanoma hotspot mutations in BRAF or NRAS have been identified in studies focusing on DM 13, 90, 91; the MAPK pathway seems instead to be activated by other mutations 13 (Table 1). Indeed, possible oncogenic MAPK mutations in this subtype of melanoma include alterations detected in NF1, CBL, ERBB2, MAP2K1 and MAP3K1, as well as mutations that are hotspot in other types of cancers such as BRAF G469E, G466E and D594N and NRAS Q61H 13.
Following the recognition that somatic non‐synonymous mutational load might be associated with improved immune checkpoint responses, Eroglu and colleagues hypothesised that patients with DM may respond well to ICI therapies 92. In a retrospective analysis of pathology reports from 1058 patients with advanced melanoma treated with anti–PD‐1 or anti–PD‐L1 antibodies, Eroglu et al identified 60 patients with advanced DM, who overall had a high response rate to PD‐1 blockade. Whole‐exome sequencing data from 17 patients revealed driver NF1 mutations in 14/17 samples (82.4%) and enrichment of loss‐of‐function mutations in TP53 and ARID2. However, these mutations were not associated with response to PD‐1 blockade. These findings suggest that, despite the dense fibrous stroma that had been expected to limit immune infiltration, PD‐1/PD‐L1 blockade may be effective in patients with DM, supporting further clinical investigation of immune checkpoint blockade in these patients.
Additional rarer categories of CMs that have distinctive histopathological and molecular features include spitzoid melanoma 93, melanoma arising from giant congenital naevus 94 and melanoma in childhood 95, not reviewed herein.
AM: numerous copy number changes and low point mutation burden
AM is a rarer histological variant arising on the palms, soles and nail beds and accounts for a greater proportion of melanomas in patients of African, Asian and Latin American descent 96, 97, 98, 99. When compared to CM tumours, AMs have a much lower single nucleotide mutational burden yet display a higher number of somatic structural aberrations 3, 44. The few AM samples sequenced to date demonstrate a lower contribution of the UV signature 3, 44. A handful of cases from subungual sites, however, do demonstrate a significant proportion of UV‐associated mutations, which might suggest that skin in these locations might not be completely protected from sun‐induced UV‐radiation 100.
A large proportion of AMs fall into the TWT subtype, with only 42–55% of tumours having mutations in BRAF, NRAS or NF1 3, 44 (Table 1). KIT mutation and amplifications are also AM drivers, with between 3 and 36% of tumours bearing these alterations 44, 52. A fraction of AMs also carry activating mutations in the promoter of TERT (between 9 and 41% of patients depending on the study 44, 46) and TERT gene amplifications are currently the only recognised adverse molecular prognostic indicators 101. TERT inhibition has been shown to be cytotoxic for AM cells in vitro 44 and, following both in vivo and in vitro evidence, TERT inhibitors are currently being proposed for clinical use 102. Interestingly, although TERT deregulation in UV‐exposed melanomas is caused by point mutations, about 45% of AMs have TERT copy number gains 3. TERT copy number may predict the outcome of high‐dose (HD)‐IFNα‐2b treatment in AM 103.
Genes frequently targeted by amplifications are KIT, TERT, PAK1, CDK4 and CCND1, and genes recurrently deleted include CDKN2A, PTEN and NF1 44, 104 (Table 1). A study of 514 primary AM samples showed that the overall frequency of at least one aberration in CDK4, CCND1 or P16 INK4a was 82.7%. In this study, AM cell lines and patient‐derived xenografts containing cyclin dependent kinase 4 (CDK4) pathway aberrations were sensitive to CDK4/6 inhibitors 105 and clinical studies are anticipated (NCT03454919). There are other, infrequently altered genes identified by AM sequencing studies; for example, mutations of MAP2K2 and loss of ARID2 44. Another subset of AMs show, like CMs, MITF amplifications 3. Interestingly, very few point mutations have been described in TP53, PTEN, RAC1 or RB1, with these genes instead being targeted via amplifications or deletions.
Although AMs harbouring BRAF or KIT mutations may respond to the appropriate inhibitors, the majority of patients do not currently have any genotype‐specific treatment options. In light of the lower somatic mutation burden, it might be thought that the efficacy of ICIs may be lower in this subtype 106. However, small retrospective series have so far demonstrated that response rates are comparable to those in CM 107, 108. It remains to be seen whether the association of mutational and neoantigen load to ICI response is also observed in this subtype.
UM: a sparsely mutated melanoma subtype with poor responses to modern systemic therapies
The genomic landscape of UM
UM has one of the lowest observed mutational densities across all tumours, estimated to be about 1.1 somatic mutations per Mb 4. Indeed, the burden of coding somatic mutations is comparable to that of paediatric cancers such as medulloblastoma and neuroblastoma 48. Nonetheless, a small number of recurrent somatic mutations have been observed. Activating mutations in the guanine‐nucleotide proteins GNAQ and GNA11 occur in the great majority of tumours (a combined frequency of ∼85–92.5%), and in CYSLTR2 (4%) and PLCB4 (2.5%), all in a mutually exclusive manner 4, 56, as these may all activate the MAPK pathway 56, 109 (Figure 2, Table 1). Other significantly mutated genes in UM are BAP1, EIF1AX and SF3B1, which also form a second mutually‐exclusive subgroup 56 (Table 2). In addition to providing an insight into key molecular signalling and progression pathways, UM driver genes also associate with molecular subclasses and bear important prognostic implications (Table 2). Different studies have found a number of mutational signatures in these tumours, most notably one associated with ageing and explained by spontaneous deamination of 5‐methylcytosine, and others related to defects in nucleotide excision and in DNA mismatch repair 4, 48. There are currently no known drugs that target GNAQ/GNA11 and alternative pathway inhibitors have so far not shown clinical benefit in early phase trials, reviewed in 110. A number of trials are ongoing, targeting a range of UM signalling cascades 111. The clinical responses to ICIs have similarly been disappointing 112.
Table 2.
Uveal melanoma driver genes and their prognostic significance
| Gene | Gene function | Mutation frequency (%) | Association with metastases | Association with survival |
|---|---|---|---|---|
| GNAQ | Mediating signalling between G‐protein‐coupled receptors and downstream effectors and upregulated MAPK pathway | 43–57 | Similar frequencies reported between metastatic and non‐metastatic lesions | Mutations have not been linked to patient outcome 113 |
| GNA11 | Mediating signalling between G‐protein‐coupled receptors and downstream effectors and upregulated MAPK pathway | 41–49 | Present in 18/30 (60%) of UM metastases | Disease‐specific survival in GNA11‐mutant patients was 60 months, overall survival 50.6 months (from date of primary tumour), significantly poorer than those tumours lacking GNA11 mutations 117 |
| BAP1 | Involved in tumour suppression, DNA damage response and proliferation | 70–83 | Inactivating somatic mutations in 26/31 (84%) of metastasising tumours. Also associated with Class 2 GEP, M3 and 8q gain. | Overall survival in BAP1 positive nuclear staining by IHC was 9.97 months (95% confidence interval 8.05–11.9) versus BAP1 negative by IHC 4.74 (3.49–6.0) 49, 118, 119 |
| EIF1AX | Eukaryotic translation initiation factor | 8–21 | Mutant cases are associated with very low risk of metastases (only 2/28 cases) | EIF1AX mutant cases had a longer disease‐free survival than EIF1AX non‐mutant cases (190.1 vs. 100.2 months; p < 0.001) 113, 120, 121 |
| SF3B1 | Required for pre‐mRNA splicing | 10–24 | Intermediate risk of metastases – late‐onset (>5 years) metastases can occur | Although an association of mutated SF3B1 with favourable prognosis was observed in the first few years 122, with longer follow up time, SF3B1 mutant patients developed more metastases and tumours with D3 and SF3B1 mutation showed a significant worse prognosis compared to wild‐type tumours 121, 123 |
Chromosomal copy number gains and losses (copy number alterations [CNAs]) are more common in UM, and the largest genomic studies have focused on these aberrations. Unsupervised hierarchical clustering of UM genomes based on CNAs reveals two main chromosomal subsets 4, 48, 113. Aberrations in chromosome 3 are the main distinguishing feature between the two main subsets and TCGA refers to these subsets as Disomy 3 (D3) and Monosomy 3 (M3) 49. It has long been recognised that M3 is associated with poor prognosis and high metastatic risk, while tumours with D3 correlate with good prognosis and rarely lead to disseminated disease 114. The metastatic rate for tumours with M3 ranges from 0 to 48% 115, and the M3 genotype has been shown to be superior to clinicopathologic factors as a prognostic indicator 114. Gain of the long arm of chromosome 8q is also associated with poor prognosis 116. The M3 cluster is characterised by aberrations in BAP1, as well as 8q gain, but the extent and type of this chromosomal gain varies between the two sub‐clusters. The gain of 8q, where MYC is located, does not contain this oncogene and MYC transcript levels do not correlate with 8q status 4, 48, so perhaps this gain is targeting another genomic region. The D3 cluster further subdivides into two subsets, one characterised by little aneuploidy, gains of chromosome 6p (short‐arm) and somatic mutations in EIF1AX, and the second with gains of chromosomes 6p and 8q (long‐arm) and somatic mutations in SF3B1. Given the prevalence of observed alterations, it has been proposed that mutations in GNAQ, GNA11, CYSLTR2 or PLCB4 represent an early event, followed by loss of chromosome 3 and mutation of BAP1 in the case of M3, and by mutation of EIF1AX or SF3B1 in the case of D3 48. The TCGA study also clustered samples using transcriptional and methylation profiles, which largely aligned with the original CNA clusters 4.
Gene expression profiling in UM
UMs can also be stratified according to the GEP classification described earlier, and into the same prognostically relevant molecular classes 33 and this has become the standard of care for molecular testing in a number of oncology centres 124. Recently, Class 1 tumours have been subdivided into two subgroups, Class 1A (2% of patients 5‐year metastatic risk) and Class 1B (21% of patients 5‐year metastatic risk) 125, based on the differential expression of CDH1 and RAB31. Class 1A tumours are also associated with D3 and EIF1AX mutations. Class 2 UM tumours exhibit a dedifferentiated stem‐cell‐like and epithelioid phenotype that is associated with M3 and BAP1 mutations and confers a high metastatic risk 118, 125. They can be subclustered into Class 2A and 2B, where Class 2B cases harbour a loss of chromosome 8p that makes them even more aggressive with an earlier onset of metastases relative to Class 2A 126. Unlike CM, multiple groups have shown that the prognostic accuracy of GEP outperforms clinicopathologic features and chromosomal gains and losses in predicting metastases 127, 128, 129.
MM: a rare and aggressive subtype
MMs are rare and have a particularly aggressive clinical course 130, 131. Similar to AM, MM is characterised by a higher number of chromosomal structural aberrations and a lower mutational burden than CM 3, 54. Mutations in BRAF, NRAS or NF1 in MM are less prevalent than in CM, with loss of PTEN (4–25% of samples 3, 54) mutation or amplification of KIT (7–25% of MM samples 3, 54, 132) and CCND1 or CDK4 104 being more common (Table 1). In fact, Hayward and colleagues identified a previously unappreciated set of driver genes shared between UM and MM, with two‐thirds of TWT MM showing activating mutations in GNAQ and SF3B1. Additionally, some studies 52 suggest that losses of CDKN2A are more common in AM and MM than in CM, though estimates vary 3, 44, 50.
Targeted therapies against mutation of KIT have failed to show convincing therapeutic efficacy in MMs 133. The immune checkpoint blocking antibodies have shown variable efficacy in phase II and retrospective studies 134, 135.
Conclusion
Despite significant progress in the understanding of CM biology, our ability to assess the likelihood of recurrence and death for any individual patient remains conspicuously limited. Assessment of an individual CM patient's risk is currently based on the AJCC recommendations, which consider traditional staging factors such as Breslow thickness and ulceration 136. However, over two‐thirds of CM‐related deaths occur in patients diagnosed with stage I or II disease 137, and, as the incidence of melanoma continues to increase, the absolute number of such ‘low risk’ patients who ultimately relapse and die is rising 138. National guidelines do not currently recommend intensive surveillance and adjuvant therapy for stage I‐IIA disease 139. Additional strategies for prognostication in this early‐stage CM cohort, particularly those with biological propensity to metastasise and who might benefit from modern survival‐prolonging adjuvant therapies 140, 141, 142, would clearly be beneficial. However it seems clear that while there is tremendous enthusiasm to integrate molecular biomarkers into clinical practice, no such markers or signatures fulfil the necessary criteria for inclusion into the AJCC melanoma staging or as a component of any validated clinical tool 136. More studies are also needed to determine which type of specimen and approach yields the highest success rate. The creation of large, prospective, multi‐institution registry studies that harness the power of electronic data sharing should improve on some of the shortcomings of current prognostic tools including; relatively small study populations, short follow‐up and lack of internal and external validation. These studies will be needed to address this unmet clinical need for patient stratification in CM.
Distinct melanoma subtypes harbour somatic aberrations on the same key pathways, but the affected genes may be different (Table 1). Accordingly, it is evident that the current genomic classification of melanomas devised by TCGA may work well with CM and with DM to some extent, but a similar classification that informs therapeutic options is needed for AM and MM, where more than 50% of tumours may fall into the TWT subtype.
Why melanomas from non‐CSD regions have such different genomic landscapes to those from CSD tissues remains an open question. Apart from the fact that cells from these different regions have varying exposure to UV‐induced mutagenesis, another possible explanation may lie in the different lineages from which these melanocytes originate, and the different microenvironments they inhabit. Therefore, the question remains, if the CM driver mutations arose in melanocytes from glabrous skin, and vice versa, would melanocytes transform and form tumours? Or, are the melanocyte lineages sufficiently different that different mutations are needed to progress to malignancy? Would the microenvironment play a significant role in melanocyte transformation? In vitro experiments with cell lines from different melanocytic lineages and in vivo experiments in model organisms such as mice and zebrafish should help address this fundamental question.
Clearly, although good progress has been achieved for patients with CM, therapeutic options and response remain poor in patients with other melanoma subtypes. Recent studies exploring prognostic markers and potential therapeutic targets are helping bridge this gap, and as more genomes from rarer melanoma subtypes are sequenced, our understanding of targeted therapy and response should improve. Most of these tumoural genomes originate from patients of European descent, and a further important question is whether findings in these populations can be translated to patients from other ethnicities – particularly in AM and MM which constitute a higher proportion of melanoma cases in non‐European descent populations.
Author contributions statement
RR described the prognostic and therapeutic implications of molecular aberrations across the melanoma subtypes, and drafted sections of the introduction and conclusion. CDRE and CMA described the genomic landscape of melanoma subtypes, as well as sections of the introduction and conclusion. PF provided the H&E images and caption, and commented on the histopathologic aspects of the manuscript. DJA and CDRE provided overall supervision and leadership, as well as comments and suggestions across the entire manuscript. All authors approved the final version.
Acknowledgements
We would like to thank Nick Hayward, Patricia Possik and Sarah Welsh for very helpful discussions, as well as Phillip Ball and Richard Poulsom for their support in figure formatting. This work was supported by Cancer Research UK and The Wellcome Trust. CDR‐E is supported by a Wellcome Trust Seed Award in Science (204562/Z/16/Z), by a UNAM PAPIIT Award (IA200318), a Stimulus for Medical Research from the Miguel Aleman Trust, and by a UC‐MEXUS Collaborative Award (CN‐18‐121). CM‐A is supported by a postdoctoral salary from the Wellcome Trust (204562/Z/16/Z).
No conflicts of interest were declared.
References
- 1. Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer 2016; 16: 345–358. [DOI] [PubMed] [Google Scholar]
- 2. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayward NK, Wilmott JS, Waddell N, et al Whole‐genome landscapes of major melanoma subtypes. Nature 2017; 545: 175–180. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Network . Genomic classification of cutaneous melanoma. Cell 2015; 161: 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shain AH, Joseph NM, Yu R, et al Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell 2018; 34: 45–55.e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Celia‐Terrassa T, Kang Y. Distinctive properties of metastasis‐initiating cells. Genes Dev 2016; 30: 892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017; 168: 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim SY, Kim SN, Hahn HJ, et al Metaanalysis of BRAF mutations and clinicopathologic characteristics in primary melanoma. J Am Acad Dermatol 2015; 72: 1036–1046. e1032. [DOI] [PubMed] [Google Scholar]
- 9. Long GV, Menzies AM, Nagrial AM, et al Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 2011; 29: 1239–1246. [DOI] [PubMed] [Google Scholar]
- 10. Bauer J, Buttner P, Murali R, et al BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res 2011; 24: 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carlino MS, Haydu LE, Kakavand H, et al Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer 2014; 111: 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Devitt B, Liu W, Salemi R, et al Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 2011; 24: 666–672. [DOI] [PubMed] [Google Scholar]
- 13. Shain AH, Garrido M, Botton T, et al Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet 2015; 47: 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horn S, Figl A, Rachakonda PS, et al TERT promoter mutations in familial and sporadic melanoma. Science 2013; 339: 959–961. [DOI] [PubMed] [Google Scholar]
- 15. Griewank KG, Murali R, Puig‐Butille JA, et al TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst 2014; 106: djv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andres‐Lencina JJ, Rachakonda S, Garcia‐Casado Z, et al TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int J Cancer 2018; 144: 1027–1036. [DOI] [PubMed] [Google Scholar]
- 17. Nagore E, Heidenreich B, Rachakonda S, et al TERT promoter mutations in melanoma survival. Int J Cancer 2016; 139: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garraway LA, Widlund HR, Rubin MA, et al Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005; 436: 117–122. [DOI] [PubMed] [Google Scholar]
- 19. Jonsson G, Busch C, Knappskog S, et al Gene expression profiling‐based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res 2010; 16: 3356–3367. [DOI] [PubMed] [Google Scholar]
- 20. Muller J, Krijgsman O, Tsoi J, et al Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014; 5: 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rambow F, Rogiers A, Marin‐Bejar O, et al Toward minimal residual disease‐directed therapy in melanoma. Cell 2018; 174: 843–855.e819. [DOI] [PubMed] [Google Scholar]
- 22. Wellbrock C, Arozarena I. Microphthalmia‐associated transcription factor in melanoma development and MAP‐kinase pathway targeted therapy. Pigment Cell Melanoma Res 2015; 28: 390–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol 2011; 8: 426–433. [DOI] [PubMed] [Google Scholar]
- 24. Mar VJ, Liu W, Devitt B, et al The role of BRAF mutations in primary melanoma growth rate and survival. Br J Dermatol 2015; 173: 76–82. [DOI] [PubMed] [Google Scholar]
- 25. Nagore E, Requena C, Traves V, et al Prognostic value of BRAF mutations in localized cutaneous melanoma. J Am Acad Dermatol 2014; 70: 858–862.e851‐852. [DOI] [PubMed] [Google Scholar]
- 26. Akslen LA, Angelini S, Straume O, et al BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J Invest Dermatol 2005; 125: 312–317. [DOI] [PubMed] [Google Scholar]
- 27. Ellerhorst JA, Greene VR, Ekmekcioglu S, et al Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res 2011; 17: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Omholt K, Platz A, Kanter L, et al NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res 2003; 9: 6483–6488. [PubMed] [Google Scholar]
- 29. Platz A, Egyhazi S, Ringborg U, et al Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol 2008; 1: 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerami P, Cook RW, Russell MC, et al Gene expression profiling for molecular staging of cutaneous melanoma in patients undergoing sentinel lymph node biopsy. J Am Acad Dermatol 2015; 72: 780–785.e783. [DOI] [PubMed] [Google Scholar]
- 31. Onken MD, Worley LA, Tuscan MD, et al An accurate, clinically feasible multi‐gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn 2010; 12: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bittner M, Meltzer P, Chen Y, et al Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000; 406: 536–540. [DOI] [PubMed] [Google Scholar]
- 33. Onken MD, Worley LA, Ehlers JP, et al Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res 2004; 64: 7205–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haqq C, Nosrati M, Sudilovsky D, et al The gene expression signatures of melanoma progression. Proc Natl Acad Sci U S A 2005; 102: 6092–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith AP, Hoek K, Becker D. Whole‐genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced‐stage melanomas. Cancer Biol Ther 2005; 4: 1018–1029. [DOI] [PubMed] [Google Scholar]
- 36. Jaeger J, Koczan D, Thiesen HJ, et al Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser‐microdissected melanoma tissues. Clin Cancer Res 2007; 13: 806–815. [DOI] [PubMed] [Google Scholar]
- 37. Scatolini M, Grand MM, Grosso E, et al Altered molecular pathways in melanocytic lesions. Int J Cancer 2010; 126: 1869–1881. [DOI] [PubMed] [Google Scholar]
- 38. Mauerer A, Roesch A, Hafner C, et al Identification of new genes associated with melanoma. Exp Dermatol 2011; 20: 502–507. [DOI] [PubMed] [Google Scholar]
- 39. Zager JS, Gastman BR, Leachman S, et al Performance of a prognostic 31‐gene expression profile in an independent cohort of 523 cutaneous melanoma patients. BMC Cancer 2018; 18: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ferris LK, Farberg AS, Middlebrook B, et al Identification of high‐risk cutaneous melanoma tumors is improved when combining the online American joint committee on cancer individualized melanoma patient outcome prediction tool with a 31‐gene expression profile‐based classification. J Am Acad Dermatol 2017; 76: 818–825.e813. [DOI] [PubMed] [Google Scholar]
- 41. Hsueh EC, DeBloom JR, Lee J, et al Interim analysis of survival in a prospective, multi‐center registry cohort of cutaneous melanoma tested with a prognostic 31‐gene expression profile test. J Hematol Oncol 2017; 10: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gastman BR, Gerami P, Kurley SJ, et al Identification of patients at risk for metastasis using a prognostic 31‐gene expression profile in subpopulations of melanoma patients with favorable outcomes by standard criteria. J Am Acad Dermatol 2018; 80: 149–157. 10.1016/j.jaad.2018.07.028. [DOI] [PubMed] [Google Scholar]
- 43. Wiesner T, Kiuru M, Scott SN, et al NF1 mutations are common in desmoplastic melanoma. Am J Surg Pathol 2015; 39: 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang WS, Hendricks W, Kiefer J, et al Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res 2017; 27: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Curtin JA, Fridlyand J, Kageshita T, et al Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353: 2135–2147. [DOI] [PubMed] [Google Scholar]
- 46. Vazquez Vde L, Vicente AL, Carloni A, et al Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res 2016; 26: 93–99. [DOI] [PubMed] [Google Scholar]
- 47. Moon KR, Choi YD, Kim JM, et al Genetic alterations in primary acral melanoma and acral melanocytic nevus in Korea: common mutated genes show distinct cytomorphological features. J Invest Dermatol 2018; 138: 933–945. [DOI] [PubMed] [Google Scholar]
- 48. Royer‐Bertrand B, Torsello M, Rimoldi D, et al Comprehensive genetic landscape of uveal melanoma by whole‐genome sequencing. Am J Hum Genet 2016; 99: 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robertson AG, Shih J, Yau C, et al Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017; 32: 204–220.e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hintzsche JD, Gorden NT, Amato CM, et al Whole‐exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res 2017; 27: 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cosgarea I, Ugurel S, Sucker A, et al Targeted next generation sequencing of mucosal melanomas identifies frequent NF1 and RAS mutations. Oncotarget 2017; 8: 40683–40692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Curtin JA, Busam K, Pinkel D, et al Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006; 24: 4340–4346. [DOI] [PubMed] [Google Scholar]
- 53. Wallander ML, Layfield LJ, Emerson LL, et al KIT mutations in ocular melanoma: frequency and anatomic distribution. Mod Pathol 2011; 24: 1031–1035. [DOI] [PubMed] [Google Scholar]
- 54. Furney SJ, Turajlic S, Stamp G, et al Genome sequencing of mucosal melanomas reveals that they are driven by distinct mechanisms from cutaneous melanoma. J Pathol 2013; 230: 261–269. [DOI] [PubMed] [Google Scholar]
- 55. Yilmaz I, Gamsizkan M, Kucukodaci Z, et al BRAF, KIT, NRAS, GNAQ and GNA11 mutation analysis in cutaneous melanomas in Turkish population. Indian J Pathol Microbiol 2015; 58: 279–284. [DOI] [PubMed] [Google Scholar]
- 56. Moore AR, Ceraudo E, Sher JJ, et al Recurrent activating mutations of G‐protein‐coupled receptor CYSLTR2 in uveal melanoma. Nat Genet 2016; 48: 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Van Raamsdonk CD, Bezrookove V, Green G, et al Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009; 457: 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jahn SW, Kashofer K, Halbwedl I, et al Mutational dichotomy in desmoplastic malignant melanoma corroborated by multigene panel analysis. Mod Pathol 2015; 28: 895–903. [DOI] [PubMed] [Google Scholar]
- 59. van der Velden PA, Metzelaar‐Blok JA, Bergman W, et al Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res 2001; 61: 5303–5306. [PubMed] [Google Scholar]
- 60. Abdel‐Rahman MH, Yang Y, Zhou XP, et al High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J Clin Oncol 2006; 24: 288–295. [DOI] [PubMed] [Google Scholar]
- 61. Dono M, Angelini G, Cecconi M, et al Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br J Cancer 2014; 110: 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harbst K, Staaf J, Lauss M, et al Molecular profiling reveals low‐ and high‐grade forms of primary melanoma. Clin Cancer Res 2012; 18: 4026–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lauss M, Nsengimana J, Staaf J, et al Consensus of melanoma gene expression subtypes converges on biological entities. J Invest Dermatol 2016; 136: 2502–2505. [DOI] [PubMed] [Google Scholar]
- 64. Carvajal RD, Antonescu CR, Wolchok JD, et al KIT as a therapeutic target in metastatic melanoma. JAMA 2011; 305: 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sauter ER, Yeo UC, von Stemm A, et al Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res 2002; 62: 3200–3206. [PubMed] [Google Scholar]
- 66. Muthusamy V, Hobbs C, Nogueira C, et al Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer 2006; 45: 447–454. [DOI] [PubMed] [Google Scholar]
- 67. Carvajal RD, Schwartz GK, Tezel T, et al Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol 2017; 101: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Long GV, Weber JS, Infante JR, et al Overall survival and durable responses in patients with BRAF V600‐mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol 2016; 34: 871–878. [DOI] [PubMed] [Google Scholar]
- 69. Catalanotti F, Cheng DT, Shoushtari AN, et al PTEN loss‐of‐function alterations are associated with intrinsic resistance to BRAF inhibitors in metastatic melanoma. JCO Precis Oncol 2017; 1: 1–15; doi: 10.1200/po.16.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith MP, Ferguson J, Arozarena I, et al Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J Natl Cancer Inst 2013; 105: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Van Allen EM, Wagle N, Sucker A, et al The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014; 4: 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Griffin M, Scotto D, Josephs DH, et al BRAF inhibitors: resistance and the promise of combination treatments for melanoma. Oncotarget 2017; 8: 78174–78192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Salgia R, Kulkarni P. The genetic/non‐genetic duality of drug ‘resistance’ in cancer. Trends Cancer 2018; 4: 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kong X, Kuilman T, Shahrabi A, et al Cancer drug addiction is relayed by an ERK2‐dependent phenotype Switch. Nature 2017; 550: 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Snyder A, Makarov V, Merghoub T, et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014; 371: 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Van Allen EM, Miao D, Schilling B, et al Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 2015; 350: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Le DT, Durham JN, Smith KN, et al Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017; 357: 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rizvi NA, Hellmann MD, Snyder A, et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. [Accessed 28 September 2018]. Available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm
- 80. Brahmer JR, Tykodi SS, Chow LQ, et al Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. van Rooij N, van Buuren MM, Philips D, et al Tumor exome analysis reveals neoantigen‐specific T‐cell reactivity in an ipilimumab‐responsive melanoma. J Clin Oncol 2013; 31: e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gubin MM, Zhang X, Schuster H, et al Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature 2014; 515: 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lauss M, Donia M, Harbst K, et al Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun 2017; 8: 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McGranahan N, Furness AJ, Rosenthal R, et al Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351: 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hugo W, Zaretsky JM, Sun L, et al Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016; 165: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Riaz N, Havel JJ, Makarov V, et al Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell 2017; 171: 934–949.e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Roh W, Chen PL, Reuben A, et al Integrated molecular analysis of tumor biopsies on sequential CTLA‐4 and PD‐1 blockade reveals markers of response and resistance. Sci Transl Med 2017; 9: eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Daud AI, Loo K, Pauli ML, et al Tumor immune profiling predicts response to anti‐PD‐1 therapy in human melanoma. J Clin Invest 2016; 126: 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Busam KJ, Mujumdar U, Hummer AJ, et al Cutaneous desmoplastic melanoma: reappraisal of morphologic heterogeneity and prognostic factors. Am J Surg Pathol 2004; 28: 1518–1525. [DOI] [PubMed] [Google Scholar]
- 90. Kim J, Lazar AJ, Davies MA, et al BRAF, NRAS and KIT sequencing analysis of spindle cell melanoma. J Cutan Pathol 2012; 39: 821–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Davison JM, Rosenbaum E, Barrett TL, et al Absence of V599E BRAF mutations in desmoplastic melanomas. Cancer 2005; 103: 788–792. [DOI] [PubMed] [Google Scholar]
- 92. Eroglu Z, Zaretsky JM, Hu‐Lieskovan S, et al High response rate to PD‐1 blockade in desmoplastic melanomas. Nature 2018; 553: 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lazova R, Pornputtapong N, Halaban R, et al Spitz nevi and Spitzoid melanomas: exome sequencing and comparison with conventional melanocytic nevi and melanomas. Mod Pathol 2017; 30: 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kinsler VA, O'Hare P, Bulstrode N, et al Melanoma in congenital melanocytic naevi. Br J Dermatol 2017; 176: 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lu C, Zhang J, Nagahawatte P, et al The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol 2015; 135: 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bradford PT, Goldstein AM, McMaster ML, et al Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986–2005. Arch Dermatol 2009; 145: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lee HY, Chay WY, Tang MB, et al Melanoma: differences between Asian and Caucasian patients. Ann Acad Med Singapore 2012; 41: 17–20. [PubMed] [Google Scholar]
- 98. Pollitt RA, Clarke CA, Swetter SM, et al The expanding melanoma burden in California hispanics: importance of socioeconomic distribution, histologic subtype, and anatomic location. Cancer 2011; 117: 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lino‐Silva LS, Dominguez‐Rodriguez JA, Aguilar‐Romero JM, et al Melanoma in Mexico: Clinicopathologic features in a population with predominance of acral lentiginous subtype. Ann Surg Oncol 2016; 23: 4189–4194. [DOI] [PubMed] [Google Scholar]
- 100. Rawson RV, Johansson PA, Hayward NK, et al Unexpected UVR and non‐UVR mutation burden in some acral and cutaneous melanomas. Lab Invest 2017; 97: 130–145. [DOI] [PubMed] [Google Scholar]
- 101. Diaz A, Puig‐Butille JA, Munoz C, et al TERT gene amplification is associated with poor outcome in acral lentiginous melanoma. J Am Acad Dermatol 2014; 71: 839–841. [DOI] [PubMed] [Google Scholar]
- 102. Xu Y, Goldkorn A. Telomere and telomerase therapeutics in cancer. Genes 2016; 7: E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yu S, Xu T, Dai J, et al TERT copy gain predicts the outcome of high‐dose interferon alpha‐2b therapy in acral melanoma. Onco Targets Ther 2018; 11: 4097–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kabbarah O, Chin L. Revealing the genomic heterogeneity of melanoma. Cancer Cell 2005; 8: 439–441. [DOI] [PubMed] [Google Scholar]
- 105. Kong Y, Sheng X, Wu X, et al Frequent genetic aberrations in the CDK4 pathway in acral melanoma indicate the potential for CDK4/6 inhibitors in targeted therapy. Clin Cancer Res 2017; 23: 6946–6957. [DOI] [PubMed] [Google Scholar]
- 106. Bae SH, Seon HJ, Choi YD, et al Other primary systemic cancers in patients with melanoma: analysis of balanced acral and nonacral melanomas. J Am Acad Dermatol 2016; 74: 333–340. [DOI] [PubMed] [Google Scholar]
- 107. Johnson DB, Peng C, Abramson RG, et al Clinical activity of ipilimumab in acral melanoma: a retrospective review. Oncologist 2015; 20: 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Shoushtari AN, Munhoz RR, Kuk D, et al The efficacy of anti‐PD‐1 agents in acral and mucosal melanoma. Cancer 2016; 122: 3354–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Helgadottir H, Hoiom V. The genetics of uveal melanoma: current insights. Appl Clin Genet 2016; 9: 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Park JJ, Diefenbach RJ, Joshua AM, et al Oncogenic signaling in uveal melanoma. Pigment Cell Melanoma Res 2018; 31: 661–672. [DOI] [PubMed] [Google Scholar]
- 111. Yang J, Manson DK, Marr BP, et al Treatment of uveal melanoma: where are we now? Ther Adv Med Oncol 2018; 10: 1758834018757175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Heppt MV, Steeb T, Schlager JG, et al Immune checkpoint blockade for unresectable or metastatic uveal melanoma: a systematic review. Cancer Treat Rev 2017; 60: 44–52. [DOI] [PubMed] [Google Scholar]
- 113. Decatur CL, Ong E, Garg N, et al Driver mutations in uveal melanoma: associations with gene expression profile and patient outcomes. JAMA Ophthalmol 2016; 134: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Prescher G, Bornfeld N, Hirche H, et al Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996; 347: 1222–1225. [DOI] [PubMed] [Google Scholar]
- 115. Abdel‐Rahman MH, Christopher BN, Faramawi MF, et al Frequency, molecular pathology and potential clinical significance of partial chromosome 3 aberrations in uveal melanoma. Mod Pathol 2011; 24: 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dogrusoz M, Bagger M, van Duinen SG, et al The prognostic value of AJCC staging in uveal melanoma is enhanced by adding chromosome 3 and 8q status. Invest Ophthalmol Vis Sci 2017; 58: 833–842. [DOI] [PubMed] [Google Scholar]
- 117. Griewank KG, van de Nes J, Schilling B, et al Genetic and clinico‐pathologic analysis of metastatic uveal melanoma. Mod Pathol 2014; 27: 175–183. [DOI] [PubMed] [Google Scholar]
- 118. Harbour JW, Onken MD, Roberson ED, et al Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010; 330: 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kalirai H, Dodson A, Faqir S, et al Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br J Cancer 2014; 111: 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ewens KG, Kanetsky PA, Richards‐Yutz J, et al Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci 2014; 55: 5160–5167. [DOI] [PubMed] [Google Scholar]
- 121. Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late‐onset metastases. Ophthalmology 2016; 123: 1118–1128. [DOI] [PubMed] [Google Scholar]
- 122. Harbour JW, Roberson ED, Anbunathan H, et al Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 2013; 45: 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Furney SJ, Pedersen M, Gentien D, et al SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 2013; 3: 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Aaberg TM Jr, Cook RW, Oelschlager K, et al Current clinical practice: differential management of uveal melanoma in the era of molecular tumor analyses. Clin Ophthalmol 2014; 8: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Field MG, Harbour JW. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol 2014; 25: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Onken MD, Worley LA, Harbour JW. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res 2008; 14: 3737–3745. [DOI] [PubMed] [Google Scholar]
- 127. Singh AD, Sisley K, Xu Y, et al Reduced expression of autotaxin predicts survival in uveal melanoma. Br J Ophthalmol 2007; 91: 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Worley LA, Onken MD, Person E, et al Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res 2007; 13: 1466–1471. [DOI] [PubMed] [Google Scholar]
- 129. Petrausch U, Martus P, Tonnies H, et al Significance of gene expression analysis in uveal melanoma in comparison to standard risk factors for risk assessment of subsequent metastases. Eye (Lond) 2008; 22: 997–1007. [DOI] [PubMed] [Google Scholar]
- 130. Mihajlovic M, Vlajkovic S, Jovanovic P, et al Primary mucosal melanomas: a comprehensive review. Int J Clin Exp Pathol 2012; 5: 739–753. [PMC free article] [PubMed] [Google Scholar]
- 131. Postow MA, Hamid O, Carvajal RD. Mucosal melanoma: pathogenesis, clinical behavior, and management. Curr Oncol Rep 2012; 14: 441–448. [DOI] [PubMed] [Google Scholar]
- 132. Yun J, Lee J, Jang J, et al KIT amplification and gene mutations in acral/mucosal melanoma in Korea. APMIS 2011; 119: 330–335. [DOI] [PubMed] [Google Scholar]
- 133. Kim KB, Alrwas A. Treatment of KIT‐mutated metastatic mucosal melanoma. Chin Clin Oncol 2014; 3: 35. [DOI] [PubMed] [Google Scholar]
- 134. D'Angelo SP, Larkin J, Sosman JA, et al Efficacy and safety of nivolumab alone or in combination with ipilimumab in patients with mucosal melanoma: a pooled analysis. J Clin Oncol 2017; 35: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Studentova H, Kalabova H, Koranda P, et al Immunotherapy in mucosal melanoma: a case report and review of the literature. Oncotarget 2018; 9: 17971–17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gershenwald JE, Scolyer RA, Hess KR, et al Melanoma staging: evidence‐based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin 2017; 67: 472–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Morton DL, Thompson JF, Cochran AJ, et al Final trial report of sentinel‐node biopsy versus nodal observation in melanoma. N Engl J Med 2014; 370: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Whiteman DC, Baade PD, Olsen CM. More people die from thin melanomas (1 mm) than from thick melanomas (>4 mm) in Queensland, Australia. J Invest Dermatol 2015; 135: 1190–1193. [DOI] [PubMed] [Google Scholar]
- 139. Coit DG, Thompson JA, Algazi A, et al Melanoma, version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2016; 14: 450–473. [DOI] [PubMed] [Google Scholar]
- 140. Maio M, Lewis K, Demidov L, et al Adjuvant vemurafenib in resected, BRAF(V600) mutation‐positive melanoma (BRIM8): a randomised, double‐blind, placebo‐controlled, multicentre, phase 3 trial. Lancet Oncol 2018; 19: 510–520. [DOI] [PubMed] [Google Scholar]
- 141. Long GV, Hauschild A, Santinami M, et al Adjuvant dabrafenib plus trametinib in stage III BRAF‐mutated melanoma. N Engl J Med 2017; 377: 1813–1823. [DOI] [PubMed] [Google Scholar]
- 142. Weber J, Mandala M, Del Vecchio M, et al Adjuvant nivolumab versusipilimumab in resected stage III or IV melanoma. N Engl J Med 2017; 377: 1824–1835. [DOI] [PubMed] [Google Scholar]