

Abstract
Acute respiratory distress syndrome (ARDS) is the often fatal sequelae of a broad range of precipitating conditions. Despite decades of intensive research and clinical trials there remain no therapies in routine clinical practice that target the dysregulated and overwhelming inflammatory response that characterises ARDS. Neutrophils play a central role in the initiation, propagation and resolution of this complex inflammatory environment by migrating into the lung and executing a variety of pro‐inflammatory functions. These include degranulation with liberation of bactericidal proteins, release of cytokines and reactive oxygen species as well as production of neutrophil extracellular traps. Although these functions are advantageous in clearing bacterial infection, the consequence of associated tissue damage, the contribution to worsening acute inflammation and prolonged neutrophil lifespan at sites of inflammation are deleterious. In this review, the importance of the neutrophil will be considered, together with discussion of recent advances in understanding neutrophil function and the factors that influence them throughout the phases of inflammation in ARDS. From a better understanding of neutrophils in this context, potential therapeutic targets are identified and discussed. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: neutrophil, ARDS, apoptosis, inflammation, neutrophil extracellular trap, chemokine, interleukin, leukotriene, DAMP, PAMP, toll‐like receptor, reactive oxygen species
Introduction
Acute respiratory distress syndrome (ARDS) is the often fatal final sequalae to a broad range of direct and indirect pulmonary insults that include both infective and sterile aetiologies such as pneumonia, aspiration of gastric contents, sepsis, acute hepatic failure and acute pancreatitis. ARDS is defined by an acute onset of respiratory symptoms; profound systemic hypoxaemia; diffuse, bilateral infiltrates on chest X‐ray and the exclusion of cardiac failure or fluid overload as a precipitant 1. Despite decades of intensive research, the mortality rate for ARDS remains approximately 40%, with no effective pharmacological therapies in routine clinical practice 2. The failure to translate a large number of promising therapeutic agents from preclinical studies is well described 3. Challenges arise when attempting to develop drugs that span the diverse and heterogenous conditions that precipitate ARDS, the differences in the inflammatory phenotypes and underlying genomic variation within this patient population, as well as the difficulties in the translation of observations in animal models into human inflammatory disease 3. Distinct from interindividual variation is also the complexity of redundancy and dysregulation of the inflammatory environment that characterises ARDS. Despite these challenges, the need to develop novel therapeutics is pressing.
ARDS is characterised by an overwhelming, dysregulated and self‐perpetuating pro‐inflammatory environment; there is a significant increase in a range of pro‐inflammatory mediators accompanied by rapid recruitment of neutrophils into the alveolar space, endothelial injury and dysfunction, platelet aggregation and microthrombus formation, interstitial and alveolar oedema, alveolar epithelial cell death and macrophage activation 4. Diffuse alveolar damage is the typical histological hallmark of this exudative phase, although the histological appearances can be very variable between individuals who have died from severe ARDS 5. Following alveolar damage, there is a proliferative phase with resolution of pulmonary oedema, type II alveolar cell hyperplasia, early collagen deposition and release of pro‐resolving mediators, including lipoxins and resolvins 6, 7. Although inflammation and injury completely resolve to leave no clinical, radiological or physiological impairment in some individuals, there remains a substantial cohort who subsequently develop diffuse pulmonary fibrosis and chronic lung disease 8.
Within this inflammatory milieu there are multiple cell types with direct roles in disease pathogenesis, including macrophages, epithelial and endothelial cells. There is, however, an established body of literature that implicates the neutrophil as central to driving this inflammatory state 9, 10. Increased neutrophil numbers, the presence of neutrophil‐derived proteases and the chemotactic factors that drive neutrophil recruitment are associated with increased disease severity and higher mortality rates 9, 11. Similarly, neutrophil depletion, inhibition of key chemokines and signalling molecules, or acceleration of apoptosis to shorten neutrophil lifespan, results in improvement in oxygenation, reduction in inflammation and accelerated inflammation resolution in preclinical models 12, 13, 14, 15. To date, however, clinical trials targeting neutrophil function in ARDS have failed to show benefit in overall survival 3, 16.
Although much has been written on the detailed mechanisms of neutrophil migration and function in inflammation 17, 18, 19, this review focuses on those observations that have been demonstrated within the context of ARDS and preclinical models of acute lung injury. In doing so we hope to provide a focus on those pathological mechanisms that are of potential clinical relevance and may therefore represent therapeutic targets of the future (Table 1).
Table 1.
Neutrophil‐related mediators in ARDS, preclinical observations and associated therapeutics
| Preclinical observations | Human ARDS | Therapeutic potential | Therapeutics | Reference | ||
|---|---|---|---|---|---|---|
| PAMPs | Neutrophil recruitment and activation | Unknown | TLR1, TLR4, TLR5 antagonists | Not clinically tested | 23, 143 | |
| DAMPs | Mitochondrial formylated peptides | ↑ neutrophil migration and inflammation | ↑ in blood and BALF | FPR1 antagonists | Not clinically tested | 31 |
| Mitochondrial DNA | ↑ neutrophil migration and inflammation | ↑ in blood and BALF | TLR9 antagonists | Not clinically tested | 30 | |
| HMGB1 | ↑ neutrophils and inflammation | ↑ in blood | Metformin | No increase in survival | 132, 133, 144 | |
| Chemokines/cytokines | CXCL5 | Neutrophil chemotaxis | ↑ in BALF |
CXCL5 antibody CXCR2 antagonist |
Not clinically tested | 145 |
| CXCL8 | Neutrophil chemotaxis | ↑ in blood and BALF |
CXCL8 antibody CXCR1, CXCR2 antagonist |
Allogeneic adipose‐derived mesenchymal stem cells – no effect | 36, 38, 146 | |
| CCL2 | Neutrophil chemotaxis | ↑ in BALF |
CCL2 antibody CCR2 antagonist |
Not clinically tested | 39 | |
| CCL7 | Neutrophil chemotaxis | ↑ in BALF |
CCL7 antibody CCR1, CCR2, CCR3 antagonist |
Not clinically tested | 39 | |
| LTB4 | Neutrophil chemotaxis | ↑ in blood and BALF |
LTB4 antibody BLT2 antagonist |
Not clinically tested | 147, 148 | |
| C5a | Neutrophil chemotaxis | ↑ in BALF | Anti‐DBP | Not clinically tested | 149 | |
| TNF |
Pro‐inflammatory response Microvascular plasma protein leakage |
↑ in blood and BALF |
TNF antibody TNF‐RA |
Not clinically tested TNFR1 antibody – ↓ pulmonary neutrophilia, inflammatory cytokine release, endothelial injury | 41, 43, 50, 150 | |
| IL‐1β | Pro‐inflammatory response | ↑ in blood and BALF |
IL‐1β antibody IL‐1RA |
Not clinically tested | 50, 150 | |
| TNF receptor |
Neutralises TNF imbalance |
↑ in BALF | Administration for TNF neutralisation | Not clinically tested | 50 | |
| IL‐1RA | Neutralises IL‐1β imbalance | ↑ in BALF | Administration for IL‐1β neutralisation | Not clinically tested | 50 | |
| Selectins | L‐selectin | Aids in neutrophil migration | Soluble form ↓ in blood | L‐selectin antibody | Not clinically tested | 57, 151 |
| E‐selectin | Aids in neutrophil migration | Soluble form ↑ in blood | E‐selectin antibody | Not clinically tested | 57 | |
| P‐selectin | Aids in neutrophil migration | Soluble form ↑ in blood | P‐selectin antibody | Not clinically tested | 57, 66 | |
| Integrins | β2 integrin | Aids in neutrophil migration | ↑ sICAM‐1 in blood and BALF | β2 integrin antibody | Not clinically tested | 62, 65 |
| NETs | Lung injury | ↑ in BALF | DNAse I | Inhaled DNAse I – phase III | 68, 70, 152 | |
| Granule proteins | NE | Lung injury | ↑ in blood and BALF | Anti‐elastase therapies | No increase in survival | 72, 79, 153, 154 |
| Elafin | NE inhibitor | ↓ in blood | Administration | Not clinically tested | 154 | |
| MMP‐1 | Lung injury | ↑ in BALF | Inhibit with TIMP | Nebulised hypertonic saline – phase I | 83, 86, 88, 155 | |
| MMP‐2 | Lung injury | ↑ in BALF | Inhibit with TIMP | Nebulised hypertonic saline – phase I | 83, 86, 88, 155 | |
| MMP‐3 | Lung injury | ↑ in BALF | Inhibit with TIMP | Nebulised hypertonic saline – phase I | 83, 86, 88, 155 | |
| MMP‐8 | Lung injury | ↑ in blood and BALF | Inhibit with TIMP | Nebulised hypertonic saline – phase I | 83, 86, 88, 155, 156 | |
| MMP‐9 | Lung injury | ↑ in BALF | Inhibit with TIMP | Nebulised hypertonic saline – phase I | 83, 86, 88, 155 | |
| TIMP‐1 | Inhibits MMPs | ↑ in blood and BALF | Administration | Not clinically tested | 83, 156 | |
| HBP | Vascular leakage | ↑ in blood | Simvastatin reduced serum HBP | Simvastatin ↑ survival in hyper‐inflammatory subphenotype | 89, 90, 92, 93 | |
| α‐defensin | Lung injury | ↑ in blood and BALF | Inhibit | Not clinically tested | 95 | |
| β‐defensin | Inhibits neutrophil apoptosis | Unknown | Inhibit | Not clinically tested | 96 | |
| LL‐37 | Neutrophil activation | ↑ in BALF | Inhibit | Not clinically tested | 97 | |
| ROS | H2O2 |
Lung tissue damage Release pro‐inflammatory mediators Immune cells recruitment |
↑ in breath condensate |
Neutralising with antioxidants Pan‐PI3K inhibitor |
Inhaled carbon monoxide – phase I | 99, 112, 157, 158 |
| Glutathione | Neutralises H2O2 | ↑ oxidised glutathione in BALF | Intravenous administration | No effect | 101, 102 | |
| Apoptosis | PAI‐1 | ↓ neutrophil apoptosis | ↑ in BALF | PAI‐1 antagonist | Not clinically tested | 159 |
| Mcl‐1 | ↓ neutrophil apoptosis | ↑ in ARDS neutrophils | CDK inhibitor | Not clinically tested | 111 |
DBP, vitamin D‐binding protein; H2O2, hydrogen peroxide; BLT2, leukotriene B4 receptor 2; PAI, plasminogen activator inhibitor.
Neutrophil recruitment and function
The recruitment of neutrophils to the lung makes them a key factor in the pathogenesis of ARDS. In response to inflammatory mediators, either originating from the lung or distant organ injury, circulating neutrophils become primed and alter their cytoskeletal architecture with retention in the pulmonary capillary bed. They then migrate out of postcapillary venules across the endothelium, through the interstitium and epithelium and into alveoli, with associated local tissue dysfunction and destruction due to release of histotoxic mediators, such as neutrophil extracellular traps (NETs), reactive oxygen species (ROS) and proteases (Figure 1). This induction of epithelial and endothelial injury contributes to the development of alveolar oedema and hypoxaemia, as well as exacerbating the pro‐inflammatory state. It should be recognised that neutrophil migration into the lung without concomitant activation does not induce tissue injury 13. However, there are conflicting models with regards to the mechanisms by which initial neutrophil activation occurs. It has been proposed that activation of the intravascular immune system, through an increase in circulating pro‐inflammatory mediators, results in neutrophil priming, adhesion and/or trapping in lung capillaries. Subsequent migration along a variety of chemotactic gradients into the lung parenchyma therefore results in secondary lung injury 20. The alternative hypothesis is that the release of pro‐inflammatory mediators by alveolar macrophages plays a vital role in the initiation of inflammation 21, triggering an inflammatory cascade by activating surrounding tissues and resulting in chemotaxis of inflammatory cells, such as neutrophils, to the airways 21. It is likely that the exact mode of initial neutrophil recruitment and activation varies depending on the inciting stimulus and whether this is intrapulmonary or systemic. However, the end result in both cases is the recruitment of neutrophils to the lung, resulting in tissue injury.
Figure 1.
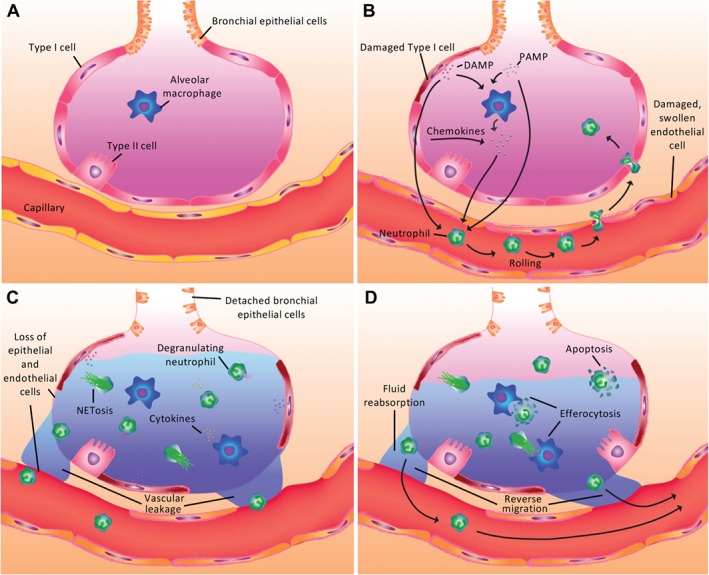
Initiation and resolution of neutrophil‐mediated inflammation in ARDS. (A) The healthy alveolar unit facilitates rapid gas transfer with the presence of resident alveolar macrophages providing rapid response to pulmonary infection and injury. (B) Following infection and/or tissue injury, release of PAMPs and/or DAMPs directly induces neutrophil recruitment into the alveolar space in addition to a range of chemokines and mediators secreted by macrophages and epithelial cells. (C) Neutrophils exert multiple pro‐inflammatory functions with the release of ROS, proteases, NETs and cytokines, as well as phagocytosis of bacteria. This is accompanied by accumulation of oedema within the alveolus, endothelial dysfunction and epithelial cell death. (D) Resolution of inflammation occurs through neutrophil apoptosis and macrophage clearance of apoptotic cells (efferocytosis) and inflammatory debris. The role of neutrophil reverse migration remains to be fully characterised in ARDS.
Pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs)
Both sterile and infective tissue injury result in neutrophil recruitment into the lung through complementary mechanisms. In the context of infection, PAMPs including lipopolysaccharide (LPS), lipoteichoic acid, DNA, RNA and proteins such as formylated peptides are released and recognised by the immune system 22. PAMPs can bind to, and are sensed by, a variety of pathogen recognition receptors, including Toll‐like receptors (TLRs) and Nod‐like receptors 23. Pathogen recognition receptors and their downstream signalling pathways drive chemotaxis, as well as priming and activating both intravascular and transmigrated neutrophils in order to fulfil their bactericidal functions 22, 24. TLRs play an important role in regulating the response to pro‐inflammatory mediators and are rapidly upregulated in mouse models of sepsis‐related acute lung injury 25. In early sepsis‐related ARDS, downregulation of TLR1, TLR4 and TLR5 transcripts in mononuclear cells correlates with increased survival 23, whereas in a pulmonary contusion mouse model of lung injury, alveolar neutrophil recruitment is TLR4/MyD88‐dependent 26. Similarly, TLR4 deficiency is associated with a reduction in sterile pulmonary inflammation and more rapid resolution of injury through alterations in downstream synthesis of cysteinyl leukotrienes and subsequent induction of SOCS3 within the lung. In this context, a reduction in TLR4‐mediated oxidative stress was observed but, surprisingly, alveolar neutrophil numbers were increased 27. Although this suggests an important role for TLRs in acute lung injury, the functional importance of neutrophils in this model is limited, thereby serving to emphasise that understanding the neutrophil‐mediated TLR function in ARDS requires further investigation.
Sterile tissue injury, either in the context of direct injury to lung parenchyma or distant organ injury, results in necrotic cell death with the release of a range of DAMPs into the extracellular environment. These DAMPs serve to induce a pro‐inflammatory response, which drives neutrophil recruitment into the lung 25, 27. A number of DAMPs have been described in ARDS, including high mobility group box 1 (HMGB1), heat shock proteins 60 and 72, hyaluronan and a range of mitochondrial‐derived factors, including DNA, formylated peptides and cardiolipin 28. Due to common evolutionary ancestry and relative structural and sequence homology with bacteria, it appears that these mitochondrial factors play an important role in driving the development of neutrophil‐mediated lung injury. Mitochondrial DNA is elevated in patients with ARDS and, through interaction with endosomal TLR9, mediates neutrophil recruitment 29. Importantly, it has also been shown to be a predictive biomarker of mortality in patients in intensive care, including those with ARDS, and therefore further studies with regards to both its role as a clinically significant predictive biomarker in ARDS and as a therapeutic target are needed.
Mitochondrial formylated peptides play a crucial role in neutrophil recruitment in ARDS, as well as altering epithelial and endothelial cell function 29, 30, 31. Elevated levels of these peptides are found in bronchoalveolar lavage fluid (BALF) and serum of ARDS patients 31. The importance of formyl peptide receptor 1 (FPR1, the cognate receptor for formylated peptides) in influencing acute inflammation is well established 32. Genetic deletion of Fpr1 in mice is associated with reduced survival in infection but an attenuated inflammatory response in the context of sterile tissue injury 29, 31, 33. FPR1, a G‐protein coupled receptor, activates a variety of intracellular signalling pathways, including PI3K, MAPKs and Akt pathways 34. This serves to directly alter neutrophil migration, ROS production, degranulation and transcriptional activity 32. In sterile lung injury in mice, neutrophil chemotaxis, together with other indices of pulmonary inflammation, were diminished in Fpr1 −/− mice or in the presence of an FPR1 antagonist delivered either prior to or following acid‐induced lung injury. This suggests that FPR1 may represent a therapeutic target in sterile ARDS but, as with many other therapies targeting neutrophil function, the challenge of concurrent infection needs to be addressed 31.
Chemokines and cytokines
Chemokines, a family of chemotactic cytokines, play a crucial role in neutrophil migration to sites of inflammation 35. CXC chemokines, in particular CXCL8 (IL‐8), play an important role in neutrophil chemotaxis in ARDS, with elevated levels associated with poor disease prognosis and increased severity and mortality 36, 37, 38. Produced by local immune cells and epithelial cells, CXCL8 is not the only chemokine responsible for the recruitment of neutrophils to the lung, as blockade results in only partial reduction in alveolar neutrophil number 13, 35. CCL2 and CCL7 are also elevated in the BALF of both LPS‐challenged volunteers and ARDS patients while neutralising either chemokine reduces neutrophil chemotactic responses in vitro 39. Interestingly, CCL2 and CCL7 also potentiate the activity of CXCL8, suggesting that a synergistic activity between these chemokines drives neutrophil recruitment in ARDS. The CXCL8 receptor, CXCR1, is more highly expressed on circulating neutrophils from ARDS patients relative to the CCL2/7 receptors CCR1, CCR2 and CCR3 39. However, a significant increase in neutrophil CCR2 expression in BALF has been observed, with the authors postulating that this confers an increased sensitivity to cognate ligands CCL2 and CCL7 in the alveolar space and therefore suggesting an important role in neutrophil chemotaxis within the lung. Other chemokines, including CXCL5, and mediators such as C5a and leukotriene B4 (LTB4), have also been shown to have a role in driving neutrophil chemotaxis in ARDS 40.
Although most cytokines are produced by other cell types, neutrophils also secrete a range of cytokines that potentiate the inflammatory response. These include TNF, which has been associated with microvascular plasma protein leakage 41, and IL‐1β, which potentiates the pro‐inflammatory cycle by inducing further cytokine and chemokine release and thereby recruiting more neutrophils to the lung 42. Furthermore, antibody‐mediated inhibition of TNFR1 reduced alveolar neutrophil recruitment, inflammatory cytokine release and biomarkers of endothelial injury in BALF and serum samples in experimental acute lung injury. As a result, inhibiting TNFR1 could be considered as a potential option in the treatment of ARDS 43.
After a non‐pulmonary acute injury, such as traumatic brain injury, burn injury or sepsis, mediators including IL‐1β, IL‐6, CXCL8, IL‐18 and TNF, as well as a variety of DAMPs, are released into the systemic circulation 23, 36, 44, 45, 46, 47. For example, intravascular neutrophil priming and activation, as part of a systemic inflammatory response syndrome that occurs following traumatic brain injury, results in neutrophil migration into the lung 47 and other organs, including the liver and kidney 48, 49, inducing tissue injury and dysfunction. Similarly, the pro‐inflammatory cytokines TNF and IL‐1β were found to be elevated in the BALF of ARDS patients alongside the natural antagonists IL‐1RA and soluble TNF receptors 50. It appears, however, that an imbalance between agonists and antagonists exists, which drives the acute phase inflammatory response 51.
At present, there have been no specific clinical trials investigating the pharmacological manipulation of the majority of chemokines, although steroids effecting their function through suppression of chemokine/cytokine axis have been proposed. Current clinical trial data on the use of steroids in ARDS are mixed and there is no definitive evidence for improved survival and in some cases has been found to worsen outcome. Although improved effects on ventilator‐free days have been described, the return to mechanical ventilation is increased among those receiving steroids, as are significant side‐effects, including neuromyopathy and hyperglycaemia 52, 53, 54, 55, 56.
Selectins and integrins
In order for neutrophils to enter the alveolar space, selectins play an important role in initiating the process of neutrophil tethering and rolling along the endothelial surface. L‐selectin, one such adhesion molecule present on neutrophils, has been found (in its soluble form) to be reduced in the plasma of ARDS patients, which directly correlated to ventilation requirements, degree of respiratory failure and mortality 57, 58. Conversely, elevated plasma levels of E‐selectin and P‐selectin, expressed by endothelial cells, correlated with increased mortality 58, 59, 60. Most recently, through genome‐wide association studies, three non‐synonymous SNPs in the selectin P ligand gene (SELPLG) have been identified to be associated with sepsis‐related ARDS 61. PSGL1 (the encoded protein) acts as an important ligand for both L‐selectin and P‐selectin. In LPS‐induced lung injury, inflammation is attenuated in Selplg −/− mice, while an inhibitory antibody to PSGL‐1 also limits inflammation in LPS‐ and ventilator‐induced lung injury models 61. The exact mechanisms through which the SELPLG SNPs exert their functional effect is not known. It was postulated that alteration in amino acid sequence may result in altered P‐selectin binding affinity and therefore alter neutrophil rolling 61.
Once tethering and rolling are initiated, integrins play a role in slowing down and immobilising neutrophils to allow transendothelial migration and activation 62. Surprisingly, neutralising antibodies to the β2 integrin CD18 in the context of sterile lung inflammation results in increased alveolar neutrophils but a reduction in neutrophil‐mediated pulmonary injury, suggesting that its predominant role is in neutrophil activation rather than chemotaxis 13. β2 integrins on the surface of activated neutrophils induce heparin‐binding protein (HBP) release through PI3K‐dependent signalling 63. Antibody‐mediated blockade of β2 integrin function resulted in lower levels of circulating HBP and a subsequent reduction in pulmonary oedema, which the authors proposed was principally through a reduction in vascular leak and endothelial dysfunction 46. The β2 integrin binds to ICAM‐1 on endothelial cells to aid in neutrophil transmigration 64. Soluble ICAM‐1 is elevated in ARDS patients and its inhibition reduces sterile lung injury in mice 65, 66.
NETosis
NETosis, the process through which neutrophils release extracellular DNA in order to trap and contain bacteria, is an important defence mechanism against invading pathogens. Increased NET production has recently been associated with increased ARDS severity 67, 68. Lefrançais et al 68 demonstrated that circulating neutrophils from ARDS patients produce significantly more NETs upon phorbol myristate acetate stimulation than those from healthy donors. As NETs contain and can release neutrophil elastases (NE), myeloperoxidase, DNA and histones, they can also potentiate the tissue damage observed in ARDS, in part through cytotoxic effects on epithelial and endothelial cells 69. Reducing NETs either by intratracheal DNase I treatment or the partial deficiency of protein arginine deiminase 4 (PAD4 +/−; a protein involved in the projection of NETs) increased survival in a mouse model of severe bacterial pneumonia/acute lung injury 68. Although partial deficiency of PAD4 reduces lung injury, complete knock out increases bacterial burden. This suggests that a NET balance is necessary and that the potential deleterious or beneficial effects of NETs in ARDS may relate to the presence of microbial infection 68. Furthermore, a phase III clinical trial is currently investigating the effectiveness of inhaled dornase alfa recombinant human DNAse 1, in reducing the incidence of moderate to severe ARDS in severe trauma patients through accelerated degradation of extracellular DNA, including NETs (Table 1) 70.
Granule proteins
The release of various granule proteins, including elastases, matrix metalloproteinases (MMP) and cationic polypeptides, has been associated with the propagation of ARDS 71. NE are implicated in lung injury, although it is unclear whether the damage is principally to endothelial or epithelial cells or as a result of degradation of the alveolar basement membrane 72, 73. Plasma levels of NE and the endogenous proteinase inhibitor elafin are predictors of ARDS mortality 74, whereas inhibition of NE reduces lung injury in various animal models 75, 76, 77, 78, 79. Mice deficient in NE are more susceptible to Gram‐negative bacteria, suggesting that NE are required for adequate host defence against invading pathogens and complete inhibition of NE can be harmful 80. Despite data from preclinical models, sivelestat, a selective NE inhibitor, did not alter 28‐day mortality in a number of clinical trials (Table 1) 81. Although alteration in oxygenation has been observed, the small sample sizes of the majority of clinical trials and heterogenous patient populations potentially mask any benefit in subgroups of patients with ARDS 81. It is difficult to separate challenges in clinical trial design from limitations of biological importance in this context. Therefore further study is required to clarify both aspects of this problem.
MMPs are zinc‐dependent endopeptidases with numerous biological functions, such as tissue remodelling, wound healing and angiogenesis 82. Fligiel and colleagues 83 investigated numerous MMPs and their natural antagonists, tissue inhibitor of metalloproteinases (TIMPs), in BALF of ARDS patients. MMP‐2, MMP‐8 and MMP‐9 are proteases secreted by neutrophils and their levels were elevated in all patients together with neutrophil number. Furthermore, elevated MMP‐1 and MMP‐3 were associated with increased mortality 83. Although neutrophils do not produce MMP‐1 and MMP‐3, they can induce MMP‐1 secretion in human vascular smooth muscle cells, which in turn acts in an autocrine feedback loop to produce CXCL8 and induce neutrophil chemotaxis 84. Consistent with this, MMP‐3‐deficient mice have reduced neutrophil migration into the lung and attenuated neutrophil‐mediated epithelial and vascular damage in the context of immune complex‐mediated pulmonary injury 85. MMP‐13 has been shown to play a role in the development of sepsis‐mediated acute lung injury 86. Hypertonic saline has been shown to reduce the production of MMPs, such as MMP‐9 and MMP‐13, in mouse models of acute lung injury, thereby reducing disease progression and inflammation; an open‐label clinical trial is currently underway to evaluate efficacy in post‐traumatic acute lung injury (Table 1) 86, 87, 88.
As mentioned previously, HBP is a cationic peptide that plays an important role in neutrophil‐mediated vascular leakage through increased endothelial permeability 89. In trauma patients admitted to intensive care, early elevation of HBP after admission was a predictor for the development of ARDS, suggesting that HBP may be a potential biomarker for the early detection of ARDS, although further work is required 90. Plasma HBP has also been shown to be an independent predictor for 30‐day mortality in ARDS 91. Administration of simvastatin to patients with acute lung injury reduced HBP plasma levels 92. Simvastatin did not improve overall survival in ARDS patients, but secondary analysis has identified improvement in 28‐ and 90‐day survival in patients with a hyper‐inflammatory subphenotype relative to placebo control (Table 1) 93.
Defensins are arginine‐rich cationic proteins that have antimicrobial properties 94. Divided into two subgroups, α‐defensins and β‐defensins exhibit different roles. Neutrophils store α‐defensins in their granules and release them in an attempt to eradicate microbes, with β‐defensins primarily expressed by mucosal surface epithelial cells. However, defensins can also result in tissue damage, as observed in ARDS 95. Elevated levels of α‐defensins were found in BALF of ARDS patients and higher levels correlate with increased severity of lung injury 95. Although plasma α‐defensin was also elevated it did not correlate with prognosis; it has been proposed that circulating α‐defensin originates from the bone marrow rather than directly from neutrophils and therefore has different functional effects in this context. Although not known to be produced by neutrophils, β‐defensins are implicated in the pathogenesis of ARDS. β‐defensin‐3 inhibits neutrophil apoptosis by downregulating Bid, a pro‐apoptotic protein, and upregulating the anti‐apoptotic protein Bcl‐xL in neutrophils 96. This delay in apoptosis is dependent upon the interaction of β‐defensin‐3 with the chemokine receptor CCR6, with the effect attenuated in the presence of a CCR6‐specific blocking antibody 96. As discussed below, delay in neutrophil apoptosis is associated with an increased severity in lung injury.
LL‐37 is another cationic protein with antimicrobial properties released from neutrophil granules 97. It also carries the ability to activate neutrophils and augment the inflammatory cascade 98 and LL‐37 is elevated in BALF samples of ARDS patients relative to healthy volunteers 97. Interestingly, although elevated LL‐37 correlated with increased lung injury, LL‐37 did not correlate with neutrophil counts, suggesting that neutrophils are not the only source of LL‐37, with macrophages and epithelial cell production also described 97.
Reactive oxygen species
ROS play an important role in eliminating pathogens within phagosomes and for the generation of NETs, but also act as chemoattractants for immune cells, resulting in tissue repair 99. However, excess ROS production results in oxidative stress and plays a major role in lung damage through the release of pro‐inflammatory cytokines, enhanced recruitment of immune cells and consequently the progression of ARDS 99. Neutrophils have been shown to produce ROS when activated and contribute to oxidative stress 99. Furthermore, increased permeability of the endothelial and epithelial barrier is observed, increasing neutrophil transmigration to the alveolar space 99. Additionally, an increase in oxidised molecules and a reduction in antioxidant proteins are observed in BALF of ARDS patients, which serves to perpetuate lung damage 100. Glutathione plays a vital role in neutralising hydrogen peroxide, a major contributor to oxidative damage, through the enzyme glutathione peroxidase, by converting glutathione to glutathione disulphide 101. Administration of the antioxidant N‐acetylcysteine restores the oxidant balance by increasing glutathione levels in erythrocytes. Several clinical trials have investigated the role of N‐acetylcysteine as a therapeutic strategy in ARDS with variable results. A recent meta‐analysis concluded that, although the duration of intensive care admission is shortened, there is no demonstrable effect on overall outcome or 30‐day survival 102.
Mechanisms of cell death
In addition to marked inflammatory cell activation and recruitment, the pathogenesis of ARDS is characterised by alterations in a variety of forms of cell death. Death and damage to the alveolar epithelial and alveolar endothelial cells are thought to play a key role in the initiation and progression of the disease process 103, 104, whereas inflammatory cell apoptosis and subsequent clearance is an important step in inflammation resolution 105, 106. Although apoptosis (described further below) is undoubtedly the most studied form of cell death, there has also been a recent ‘‐osis explosion’ with increased knowledge of alternative cell death pathways, such as pyroptosis, necroptosis, ferroptosis, entosis and NETosis 107. Although some of these non‐apoptotic pathways probably have relevance to the pathogenesis of ARDS, this is an as yet understudied area that will hopefully lead to future novel avenues for therapeutic intervention.
Targeting apoptosis
Apoptosis occurs through two distinct but converging pathways. The intrinsic pathway is activated in response to diverse stimuli, including DNA damage, ROS exposure and endoplasmic reticulum stress. The central event in intrinsic apoptosis is mitochondrial outer membrane permeabilisation that allows escape of pro‐apoptotic molecules such as cytochrome‐c, which then form a caspase‐activating complex. Active caspases act as the executioners of apoptosis, leading to cellular disassembly of the cell, DNA degradation, cell surface phosphatidylserine exposure and pannexin channel activation, all hallmarks of apoptotic cell death. Mitochondrial outer membrane permeabilisation is itself controlled by intracellular Bcl2 family proteins, which include both pro‐ and anti‐apoptotic members (such as Bid and Bcl‐xL, described above). In contrast, extrinsic apoptosis is usually activated by a cell surface death receptor upon interaction with its cognate ligand, which then leads to caspase activation. The multiple steps and checkpoints involved in apoptotic cell death allow this to be dysregulated at multiple steps in human diseases such as ARDS, but also allow the potential for therapeutic intervention at several levels 108.
Neutrophil apoptosis in ARDS has been shown to be delayed by several groups, including our own 109, 110, 111, 112, and correlates with disease severity in sepsis and sepsis‐related ARDS. Interestingly, BALF from patients with early ARDS (days 1 and 3 of disease) but not late ARDS directly inhibits apoptosis of healthy donor neutrophils 109. This effect has been attributed to soluble factors, including GM‐CSF, G‐CSF, CXCL8 and IL‐2 109, 113. Recent detailed characterisation of ARDS neutrophils has revealed multiple phenotypic alterations alongside delayed apoptosis 112. Interestingly, ARDS BALF‐induced delay of healthy neutrophil apoptosis could be overcome by PI3K inhibition, whereas the anti‐apoptosis phenotype of ARDS patient neutrophils was resistant to PI3K inhibition 112. This suggests that additional PI3K‐independent mechanisms are in play within the complex pro‐inflammatory milieu experienced during human ARDS.
Several other preclinical strategies targeting neutrophil apoptosis have also shown promise in the treatment of lung injury. Targeting of the extrinsic pathway of apoptosis has been achieved by TNF‐related apoptosis‐inducing ligand (TRAIL), part of the TNF family of ligands that can initiate apoptosis by activating cell surface receptors 114. TRAIL appears to have no role in constitutive neutrophil apoptosis nor neutrophil chemotaxis (in contrast to the TNF family ligand FasL, which is a potent neutrophil chemoattractant). However, in response to LPS‐induced lung injury, TRAIL acts to limit inflammation and enhances neutrophil apoptosis 115. Furthermore, recombinant TRAIL was able to induce an anti‐inflammatory response, suggesting that such strategies may have therapeutic potential in human ARDS. Targeting of the intrinsic pathway of neutrophil apoptosis, such as with CDK inhibitor drugs, has also been shown to have potent anti‐inflammatory effects in animal models of neutrophil dominant inflammation 14, 15. CDK inhibitor drugs principally induce neutrophil apoptosis by inhibiting CDK9‐mediated transcription of the short‐lived Bcl2 member Mcl‐1 14, 116. As neutrophils have limited expression of the main anti‐apoptotic Bcl2 homologue, Bcl2 itself, this leaves them sensitive to alterations in Mcl‐1, leading to apoptosis. CDK inhibitor drugs enhance the resolution of several lung injury models, including bleomycin‐induced, endotoxin‐induced and bacteria‐induced lung injury 14, 117. Interestingly, in an Escherichia coli‐induced model of acute lung injury, a CDK inhibitor drug administered after the onset of inflammation augmented the resolution of lung inflammation without detrimentally reducing clearance of the bacteria. Indeed, there was increased bacterial clearance, possibly resulting from lipid‐mediated enhanced bacterial phagocytosis by macrophages 14. Furthermore, and in contrast to that observed with PI3K inhibition 112, CDK inhibitor has recently been shown to override the delayed neutrophil apoptosis in sepsis‐induced human ARDS concurrent with reduced expression of Mcl‐1 111. This suggests that Mcl‐1 targeting approaches (either with CDK inhibitor or with the use of novel small molecule Mcl‐1 inhibitors) may have therapeutic potential in human ARDS.
Other strategies to modulate neutrophil apoptosis warrant further investigation in the context of ARDS. Potential strategies include the use of a p21 (Cdnk1a) peptide, which binds and sequesters proliferating cell nuclear antigen PCNA, an important endogenous neutrophil anti‐apoptotic factor 118. Although p21 peptide is able to induce apoptosis of neutrophils isolated from patients with lung inflammation 119, testing in in vivo models of ARDS is awaited. Several families of anti‐inflammatory lipid mediators that influence neutrophil lifespan and their clearance (among other pleiotropic effects) have also been delineated. These include the lipids 15‐epi‐lipoxin A4 and resolvin E1, which drive neutrophil apoptosis and attenuate experimental lung injury 120, 121.
Lung parenchymal cell death
In contrast to the potential benefits of inflammatory cell apoptosis during lung injury, there is also evidence that damage and death of the lung epithelium and endothelium can contribute to disease pathogenesis 109. Alveolar epithelial apoptosis has been observed in experimental lung injury caused by bleomycin, endotoxin and acid 31, 122, while alterations in Bcl2 members (including increases in pro‐apoptotic Bax) have been observed in lung epithelium from human lung injury cases 123. In addition, BALF from human ARDS patients contains FasL, which activates Fas receptor to induce extrinsic pathway apoptosis 124. Lung epithelium expresses Fas, with ARDS BALF able to induce epithelial apoptosis in a Fas/FasL‐dependent manner 124, but abolished Fas activity was unable to protect in experimental virus‐induced ARDS 125. Further work is needed to clarify the role and timing of Fas/FasL strategies, especially as Fas can also influence macrophage dynamics during resolution of lung injury 126.
Similarly, death of pulmonary endothelium has recently been demonstrated to be a pathogenic response in endotoxin‐induced lung injury 127. This elegant study demonstrated that endotoxin exposure led to activation of caspase‐4/5 (caspase‐11 in mice) in endothelium and a consequent pro‐inflammatory, lytic form of cell death (termed pyroptosis). Conditional deletion of caspase‐11 specifically in endothelial cells (using a Cre/lox system) led to reduced endotoxin‐induced lung oedema, neutrophil accumulation and death. Caspase‐11 inhibitors, such as wedololactone, suppress endotoxin‐induced caspase‐11 in vitro 128, but their role in lung injury in vivo remains to be tested. In summary, any potential anti‐inflammatory strategy based on modulation of inflammatory cell death has to carefully balance potentially deleterious off‐target effects should cell death be induced in lung parenchymal cells.
Clearance of apoptotic cells
Macrophages play a crucial role in limiting excessive inflammation and augmenting tissue repair, not only in the clearance of apoptotic and necrotic cells, but also in the removal of neutrophils undergoing NETosis 129, 130. In ARDS, however, macrophage phagocytic function is impaired 130. Grégoire and colleagues 130 observed enhanced NET formation and reduced neutrophil apoptosis coupled with a reduction in macrophage clearance of apoptotic cells (efferocytosis) in BALF from ARDS patients. AMP‐activated protein kinase (AMPK) has been associated with increasing macrophage phagocytosis and reduced TNF and IL‐6 production 131. The addition of metformin, an AMPK activator, to ARDS BALF samples resulted in removal of NETs and increased efferocytosis by macrophages 130. Additionally, AMPK activators administered in an LPS‐induced mouse lung injury model reduced alveolar neutrophil accumulation, pulmonary oedema and BALF TNF and IL‐6. Furthermore, retrospective analysis of diabetic patients on metformin for the 3 months prior to developing ARDS had a non‐significant reduction in 30‐day mortality from 55.32 to 42.42%. Little is known about the exact anti‐inflammatory mechanism of metformin in this context and therefore it requires further study 132.
A similar observation was made using a neutralising antibody against HMGB1 to increase macrophage efferocytosis 130. HMGB1, which is increased in ARDS 133, inhibits efferocytosis by interfering with the binding between the phosphatidylserine bridging molecule milk fat globule EGF factor 8 (MFG‐E8) and the αvβ3 integrin on the surface of macrophages 134. Importantly, MFG‐E8 knockout mice have increased apoptotic alveolar neutrophils in the alveolar space following LPS‐induced injury, an effect that can be rescued by recombinant MFG‐E8 135.
Another potential mechanism that some have speculated may be involved in neutrophil clearance is the relatively recently described concept of reverse migration. This is the process by which neutrophils migrate in the opposite direction to the chemotactic gradients that initially recruited them 136, 137. As yet, this process has only been demonstrated in zebrafish and mouse models, with no firm data demonstrating a direct role in human disease. In animal models, PGE2 is an important mediator of neutrophil reverse migration, with macrophage depletion resulting in inhibited reverse migration and therefore delayed resolution of inflammation due to reduced PGE2 production 138. Additionally, PGE2 depletion has a similar effect, further validating its importance in resolution. PGE2 signals through the EP4 receptor, increasing Alox12 production and consequently lipoxin A4, an important pro‐resolving mediator that enhances reverse migration 138. In the context of the resolution of pulmonary inflammation in mice, LTB4 released by neutrophils promotes NE release, which subsequently cleaves junctional adhesion molecule‐C (JAM‐C) from the endothelium of post‐capillary venules, facilitating reverse migration of neutrophils 139. Although the departure of neutrophils from sites of inflammation can be considered a sign of inflammatory resolution, reverse migration also has the potential to propagate inflammation. Local ischaemia–reperfusion to the ear skin or cremaster muscle in mice can progress to a systemic inflammatory response in numerous organs, including the lung, heart and liver 139. Pharmacological or genetic interference to either enhance or inhibit reverse migration led to a parallel increase or decrease in secondary inflammation in the lung distant organs 139. In humans, increased levels of soluble JAM‐C were detected in the plasma of ARDS patients, hinting that reverse migration may be occurring, with a significant direct correlation observed between soluble JAM‐C and the severity of multi‐organ failure 139. These data therefore suggest that neutrophil reverse migration may not be simply a clearance mechanism, but has the potential to cause dysregulated systemic pro‐inflammation in ARDS.
Conclusion
Neutrophils are both a hallmark and a driver of ARDS, acting in concert with other resident and recruited inflammatory cell types to induce a dysregulated, overwhelming and often fatal pro‐inflammatory state within the lung (Figure 1). To conclude that a simple ‘one size fits all’ approach in the context of both the pathogenesis and potential therapeutics is perhaps naive and out‐dated. This conclusion is supported by the identification of distinct subpopulations of ARDS patients who respond differently to fluid management, ventilation strategies and some pharmacological therapies 93, 140, 141. Recognition of these different hypo‐ and hyper‐inflammatory phenotypes within the ARDS cohort, as well as ever‐increasing numbers of predictive and prognostic biomarkers, is leading to a shift in clinical trial design to reduce clinical and biological heterogeneity 3. New combination therapies that target a variety of inflammatory components in ARDS are also being considered to address issues of redundancy, as are cell‐based therapies, including bone marrow‐derived myeloid suppressor cells in infection‐related ARDS 3.
It is therefore essential that variations in neutrophil phenotype and function, as well as understanding of neutrophil behaviour in different patient cohorts, are explored and characterised. As has been outlined, the difficulties of neutrophil‐specific therapies in the context of infection‐related ARDS warrant further exploration but, as in the context of induction of neutrophil apoptosis, this should not stop further investigation, particularly in the context of combination therapies. Further research is also required on the complex chemokine networks, NETosis, mechanisms of inflammation resolution as well as strategies that aim to protect or enhance the repair of damaged epithelial and endothelial beds. It is now over 50 years since ARDS was first described 142 and much has been learnt about its pathogenesis and beneficial supportive ventilation and fluid management strategies but the era of effective pharmacological treatments is yet to dawn. Targeting aspects of neutrophil biology will probably have a place among those therapies.
Author contributions statement
All authors contributed to manuscript writing and revision. All authors approved the final version.
Acknowledgements
Funding is acknowledged from EPSRC/MRC (PP), MRC (MR/K013386/1; AGR), The Wellcome Trust (206566/Z/17/Z; CDL) and Pathological Society (1147, TSGS 2016 10 04; DAD).
No conflicts of interest were declared.
References
- 1. ARDS Definition Task Force , Ranieri VM, Rubenfeld GD, et al Acute respiratory distress syndrome. JAMA 2012; 307: 2526–2533. [DOI] [PubMed] [Google Scholar]
- 2. Bellani G, Laffey JG, Pham T, et al Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016; 315: 788–800. [DOI] [PubMed] [Google Scholar]
- 3. Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med 2017; 5: 524–534. [DOI] [PubMed] [Google Scholar]
- 4. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med 2017; 377: 562–572. [DOI] [PubMed] [Google Scholar]
- 5. Esteban A, Fernández‐Segoviano P, Frutos‐Vivar F, et al Comparison of clinical criteria for the acute respiratory distress syndrome with autopsy findings. Ann Intern Med 2004; 141: 440–445. [DOI] [PubMed] [Google Scholar]
- 6. Zheng S, D'Souza VK, Bartis D, et al Lipoxin A4 promotes lung epithelial repair whilst inhibiting fibroblast proliferation. ERJ Open Res 2016; 2: 00079–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Y, Zhang H, Luo L, et al Resolvin D1 improves the resolution of inflammation via activating nf‐κb p50/p50‐mediated cyclooxygenase‐2 expression in acute respiratory distress syndrome. J Immunol 2017; 199: 2043–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burnham EL, Janssen WJ, Riches DWH, et al The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur Respir J 2014; 43: 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 2011; 17: 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol 2014; 306: L217–L230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steinberg KP, Milberg JA, Martin TR, et al Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med 1994; 150: 113–122. [DOI] [PubMed] [Google Scholar]
- 12. Abraham E, Carmody A, Shenkar R, et al Neutrophils as early immunologic effectors in hemorrhage‐ or endotoxemia‐induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 2000; 279: L1137–L1145. [DOI] [PubMed] [Google Scholar]
- 13. Folkesson HG, Matthay MA, Hébert CA, et al Acid aspiration‐induced lung injury in rabbits is mediated by interleukin‐8‐dependent mechanisms. J Clin Invest 1995; 96: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lucas CD, Dorward DA, Tait MA, et al Downregulation of Mcl‐1 has anti‐inflammatory pro‐resolution effects and enhances bacterial clearance from the lung. Mucosal Immunol 2014; 7: 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rossi AG, Sawatzky DA, Walker A, et al Cyclin‐dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 2006; 12: 1056–1064. [DOI] [PubMed] [Google Scholar]
- 16. Zeiher BG, Artigas A, Vincent J‐L, et al Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med 2004; 32: 1695–1702. [DOI] [PubMed] [Google Scholar]
- 17. Mantovani A, Cassatella MA, Costantini C, et al Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11: 519–531. [DOI] [PubMed] [Google Scholar]
- 18. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity 2014; 41: 694–707. [DOI] [PubMed] [Google Scholar]
- 19. Deniset JF, Kubes P. Recent advances in understanding neutrophils. F1000Research 2016; 5: 2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scott BNV, Kubes P. Death to the neutrophil! A resolution for acute respiratory distress syndrome? Eur Respir J 2018; 52: 1801274. [DOI] [PubMed] [Google Scholar]
- 21. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol 2015; 194: 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011; 30: 16–34. [DOI] [PubMed] [Google Scholar]
- 23. Ramírez Cruz NE, Maldonado Bernal C, Cuevas Urióstegui ML, et al Toll‐like receptors: dysregulation in vivo in patients with acute respiratory distress syndrome. Rev Alerg Mex 2004; 51: 210–217. [PubMed] [Google Scholar]
- 24. Prince LR, Whyte MK, Sabroe I, et al The role of TLRs in neutrophil activation. Curr Opin Pharmacol 2011; 11: 397–403. [DOI] [PubMed] [Google Scholar]
- 25. Bakopoulos A, Kapelouzou A, Tsilimigras DI, et al Expression of toll‐like receptors (TLRs) in the lungs of an experimental sepsis mouse model. PLoS One 2017; 12: e0188050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoth JJ, Wells JD, Brownlee NA, et al Toll‐like receptor 4‐dependent responses to lung injury in a murine model of pulmonary contusion. Shock 2009; 31: 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hilberath JN, Carlo T, Pfeffer MA, et al Resolution of toll‐like receptor 4‐mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J 2011; 25: 1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tolle LB, Standiford TJ. Danger‐associated molecular patterns (DAMPs) in acute lung injury. J Pathol 2013; 229: 145–156. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Q, Raoof M, Chen Y, et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakahira K, Kyung S‐Y, Rogers AJ, et al Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 2013; 10: e1001577; discussion e1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dorward DA, Lucas CD, Doherty MK, et al Novel role for endogenous mitochondrial formylated peptide‐driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax 2017; 72: 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dorward DA, Lucas CD, Chapman GB, et al The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol 2015; 185: 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N‐formylpeptide receptor. J Exp Med 1999; 189: 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Wang T, Yuan Z‐C, et al Mitochondrial peptides cause proinflammatory responses in the alveolar epithelium via FPR‐1, MAPKs, and AKT: a potential mechanism involved in acute lung injury. Am J Physiol Cell Mol Physiol 2018; 315: L775–L786. [DOI] [PubMed] [Google Scholar]
- 35. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014; 32: 659–702. [DOI] [PubMed] [Google Scholar]
- 36. Donnelly SC, Strieter RM, Kunkel SL, et al Interleukin‐8 and development of adult respiratory distress syndrome in at‐risk patient groups. Lancet 1993; 341: 643–647. [DOI] [PubMed] [Google Scholar]
- 37. Groeneveld AB, Raijmakers PG, Hack CE, et al Interleukin 8‐related neutrophil elastase and the severity of the adult respiratory distress syndrome. Cytokine 1995; 7: 746–752. [DOI] [PubMed] [Google Scholar]
- 38. Miller EJ, Cohen AB, Nagao S, et al Elevated levels of nap‐1/interleukin‐8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis 1992; 146: 427–432. [DOI] [PubMed] [Google Scholar]
- 39. Williams AE, José RJ, Mercer PF, et al Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax 2017; 72: 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2005; 288: L3–L15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finsterbusch M, Voisin M‐B, Beyrau M, et al Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J Exp Med 2014; 211: 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guma M, Ronacher L, Liu‐Bryan R, et al Caspase 1‐independent activation of interleukin‐1beta in neutrophil‐predominant inflammation. Arthritis Rheum 2009; 60: 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Proudfoot A, Bayliffe A, O'Kane CM, et al Novel anti‐tumour necrosis factor receptor‐1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 2018; 73: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vermeij J‐D, Aslami H, Fluiter K, et al Traumatic brain injury in rats induces lung injury and systemic immune suppression. J Neurotrauma 2013; 30: 2073–2079. [DOI] [PubMed] [Google Scholar]
- 45. Lechner AJ, Tredway TL, Brink DS, et al Differential systemic and intrapulmonary TNF‐alpha production in Candida sepsis during immunosuppression. Am J Physiol 1992; 263: L526–L535. [DOI] [PubMed] [Google Scholar]
- 46. Bird MD, Kovacs EJ. Organ‐specific inflammation following acute ethanol and burn injury. J Leukoc Biol 2008; 84: 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Humphries DC, O'Neill S, Scholefield E, et al Cerebral concussion primes the lungs for subsequent neutrophil‐mediated injury. Crit Care Med 2018; 46: e937–e944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nongnuch A, Panorchan K, Davenport A. Brain–kidney crosstalk. Crit Care 2014; 18: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villapol S. Consequences of hepatic damage after traumatic brain injury: current outlook and potential therapeutic targets. Neural Regen Res 2016; 11: 226–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goodman RB, Pugin J, Lee JS, et al Cytokine‐mediated inflammation in acute lung injury. Cytokine Growth Factor Rev 2003; 14: 523–535. [DOI] [PubMed] [Google Scholar]
- 51. Park WY, Goodman RB, Steinberg KP, et al Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001; 164: 1896–1903. [DOI] [PubMed] [Google Scholar]
- 52. Peter JV, John P, Graham PL, et al Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta‐analysis. BMJ 2008; 336: 1006–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weigelt JA, Norcross JF, Borman KR, et al Early steroid therapy for respiratory failure. Arch Surg 1985; 120: 536–540. [DOI] [PubMed] [Google Scholar]
- 54. Frieri M. Corticosteroid effects on cytokines and chemokines. Allergy Asthma Proc 1999; 20: 147–160. [DOI] [PubMed] [Google Scholar]
- 55. Steinberg KP, Hudson LD, Goodman RB, et al Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006; 354: 1671–1684. [DOI] [PubMed] [Google Scholar]
- 56. Meduri GU, Bridges L, Shih M‐C, et al Prolonged glucocorticoid treatment is associated with improved ARDS outcomes: analysis of individual patients' data from four randomized trials and trial‐level meta‐analysis of the updated literature. Intensive Care Med 2016; 42: 829–840. [DOI] [PubMed] [Google Scholar]
- 57. Donnelly SC, Haslett C, Dransfield I, et al Role of selectins in development of adult respiratory distress syndrome. Lancet 1994; 344: 215–219. [DOI] [PubMed] [Google Scholar]
- 58. Lawrence MB, Springer TA. Neutrophils roll on E‐selectin. J Immunol 1993; 151: 6338–6346. [PubMed] [Google Scholar]
- 59. Okajima K, Harada N, Sakurai G, et al Rapid assay for plasma soluble E‐selectin predicts the development of acute respiratory distress syndrome in patients with systemic inflammatory response syndrome. Transl Res 2006; 148: 295–300. [DOI] [PubMed] [Google Scholar]
- 60. Sakamaki F, Ishizaka A, Handa M, et al Soluble form of P‐selectin in plasma is elevated in acute lung injury. Am J Respir Crit Care Med 1995; 151: 1821–1826. [DOI] [PubMed] [Google Scholar]
- 61. Bime C, Pouladi N, Sammani S, et al Genome‐wide association study in African Americans with acute respiratory distress syndrome identifies the selectin P ligand gene as a risk factor. Am J Respir Crit Care Med 2018; 197: 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 2009; 27: 339–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu Y, Ma S, Wang X, et al The role of β2 integrin associated heparin‐binding protein release in ARDS. Life Sci 2018; 203: 92–98. [DOI] [PubMed] [Google Scholar]
- 64. Oberyszyn TM, Conti CJ, Ross MS, et al Beta2 integrin/ICAM‐1 adhesion molecule interactions in cutaneous inflammation and tumor promotion. Carcinogenesis 1998; 19: 445–455. [DOI] [PubMed] [Google Scholar]
- 65. Agouridakis P, Kyriakou D, Alexandrakis MG, et al The predictive role of serum and bronchoalveolar lavage cytokines and adhesion molecules for acute respiratory distress syndrome development and outcome. Respir Res 2002; 3: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Doerschuk CM, Quinlan WM, Doyle NA, et al The role of P‐selectin and ICAM‐1 in acute lung injury as determined using blocking antibodies and mutant mice. J Immunol 1996; 157: 4609–4614. [PubMed] [Google Scholar]
- 67. Li H, Zhou X, Tan H, et al Neutrophil extracellular traps contribute to the pathogenesis of acid‐aspiration‐induced ALI/ARDS. Oncotarget 2018; 9: 1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lefrançais E, Mallavia B, Zhuo H, et al Maladaptive role of neutrophil extracellular traps in pathogen‐induced lung injury. JCI Insight 2018; 3: 98178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Saffarzadeh M, Juenemann C, Queisser MA, et al Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 2012; 7: e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pottecher J. Inhaled dornase alpha to reduce respiratory failure after severe trauma. [Accessed 14 October 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT03368092
- 71. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration 2017; 93: 212–225. [DOI] [PubMed] [Google Scholar]
- 72. Carden D, Xiao F, Moak C, et al Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol 1998; 275: H385–H392. [DOI] [PubMed] [Google Scholar]
- 73. Ginzberg HH, Cherapanov V, Dong Q, et al Neutrophil‐mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 2001; 281: G705–G717. [DOI] [PubMed] [Google Scholar]
- 74. Wang T, Zhu Z, Liu Z, et al Plasma neutrophil elastase and elafin as prognostic biomarker for acute respiratory distress syndrome. Shock 2017; 48: 168–174. [DOI] [PubMed] [Google Scholar]
- 75. Tkalcevic J, Novelli M, Phylactides M, et al Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 2000; 12: 201–210. [DOI] [PubMed] [Google Scholar]
- 76. Kawabata K, Hagio T, Matsumoto S, et al Delayed neutrophil elastase inhibition prevents subsequent progression of acute lung injury induced by endotoxin inhalation in hamsters. Am J Respir Crit Care Med 2000; 161: 2013–2018. [DOI] [PubMed] [Google Scholar]
- 77. Hilgendorff A, Parai K, Ertsey R, et al Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol 2012; 303: L215–L227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fujino N, Kubo H, Suzuki T, et al Administration of a specific inhibitor of neutrophil elastase attenuates pulmonary fibrosis after acute lung injury in mice. Exp Lung Res 2012; 38: 28–36. [DOI] [PubMed] [Google Scholar]
- 79. Tagami T, Tosa R, Omura M, et al Effect of a selective neutrophil elastase inhibitor on mortality and ventilator‐free days in patients with increased extravascular lung water: a post hoc analysis of the PiCCO pulmonary edema study. J Intensive Care 2014; 2: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Belaaouaj A, McCarthy R, Baumann M, et al Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med 1998; 4: 615–618. [DOI] [PubMed] [Google Scholar]
- 81. Pu S, Wang D, Liu D, et al Effect of sivelestat sodium in patients with acute lung injury or acute respiratory distress syndrome: a meta‐analysis of randomized controlled trials. BMC Pulm Med 2017; 17: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Page‐McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 2007; 8: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fligiel SEG, Standiford T, Fligiel HM, et al Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol 2006; 37: 422–430. [DOI] [PubMed] [Google Scholar]
- 84. Estrada‐Gutierrez G, Cappello RE, Mishra N, et al Increased expression of matrix metalloproteinase‐1 in systemic vessels of preeclamptic women: a critical mediator of vascular dysfunction. Am J Pathol 2011; 178: 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nerusu KC, Warner RL, Bhagavathula N, et al Matrix metalloproteinase‐3 (stromelysin‐1) in acute inflammatory tissue injury. Exp Mol Pathol 2007; 83: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wohlauer M, Moore EE, Silliman CC, et al Nebulized hypertonic saline attenuates acute lung injury following trauma and hemorrhagic shock via inhibition of matrix metalloproteinase‐13. Crit Care Med 2012; 40: 2647–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Petroni RC, Biselli PJC, de Lima TM, et al Hypertonic saline (NaCl 7.5%) reduces LPS‐induced acute lung injury in rats. Inflammation 2015; 38: 2026–2035. [DOI] [PubMed] [Google Scholar]
- 88. Moore E. A clinical trial to determine if nebulized hypertonic saline attenuates acute lung injury following trauma and hemorrhagic shock. [Accessed February 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT01667666. [DOI] [PMC free article] [PubMed]
- 89. Bentzer P, Fisher J, Kong HJ, et al Heparin‐binding protein is important for vascular leak in sepsis. Intensive Care Med Exp 2016; 4: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Johansson J, Brattström O, Sjöberg F, et al Heparin‐binding protein (HBP): an early marker of respiratory failure after trauma? Acta Anaesthesiol Scand 2013; 57: 580–586. [DOI] [PubMed] [Google Scholar]
- 91. Lin Q, Shen J, Shen L, et al Increased plasma levels of heparin‐binding protein in patients with acute respiratory distress syndrome. Crit Care 2013; 17: R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McAuley DF, O'Kane CM, Craig TR, et al Simvastatin decreases the level of heparin‐binding protein in patients with acute lung injury. BMC Pulm Med 2013; 13: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Calfee CS, Delucchi KL, Sinha P, et al Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 2018; 6: 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 2003; 3: 710–720. [DOI] [PubMed] [Google Scholar]
- 95. Ashitani J, Mukae H, Arimura Y, et al High concentrations of α‐defensins in plasma and bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Life Sci 2004; 75: 1123–1134. [DOI] [PubMed] [Google Scholar]
- 96. Nagaoka I, Niyonsaba F, Tsutsumi‐Ishii Y, et al Evaluation of the effect of human ‐defensins on neutrophil apoptosis. Int Immunol 2008; 20: 543–553. [DOI] [PubMed] [Google Scholar]
- 97. Fahy RJ, Wewers MD. Pulmonary defense and the human cathelicidin hCAP‐18/LL‐37. Immunol Res 2005; 31: 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tripathi S, Wang G, White M, et al Identifying the critical domain of ll‐37 involved in mediating neutrophil activation in the presence of influenza virus: functional and structural analysis. PLoS One 2015; 10: e0133454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kellner M, Noonepalle S, Lu Q, et al ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol 2017; 967: 105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Baldwin SR, Simon RH, Grum CM, et al Oxidant activity in expired breath of patients with adult respiratory distress syndrome. Lancet 1986; 1: 11–14. [DOI] [PubMed] [Google Scholar]
- 101. Soltan‐Sharifi MS, Mojtahedzadeh M, Najafi A, et al Improvement by N‐acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti‐oxidant power: evidence for underlying toxicological mechanisms. Hum Exp Toxicol 2007; 26: 697–703. [DOI] [PubMed] [Google Scholar]
- 102. Zhang Y, Ding S, Li C, et al Effects of N‐acetylcysteine treatment in acute respiratory distress syndrome: a meta‐analysis. Exp Ther Med 2017; 14: 2863–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Matute‐Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care 2003; 7: 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tang PS, Mura M, Seth R, et al Acute lung injury and cell death: how many ways can cells die? Am J Physiol Lung Cell Mol Physiol 2008; 294: L632–L641. [DOI] [PubMed] [Google Scholar]
- 105. Poon IKH, Lucas CD, Rossi AG, et al Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 2014; 14: 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Alessandri AL, Sousa LP, Lucas CD, et al Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther 2013; 139: 189–212. [DOI] [PubMed] [Google Scholar]
- 107. Galluzzi L, Vitale I, Aaronson SA, et al Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 2018; 25: 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Duffin R, Leitch AE, Fox S, et al Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol Rev 2010; 236: 28–40. [DOI] [PubMed] [Google Scholar]
- 109. Matute‐Bello G, Liles WC, Radella F, et al Modulation of neutrophil apoptosis by granulocyte colony‐stimulating factor and granulocyte/macrophage colony‐stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med 2000; 28: 1–7. [DOI] [PubMed] [Google Scholar]
- 110. Fialkow L, Fochesatto Filho L, Bozzetti MC, et al Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis‐induced acute respiratory distress syndrome. Crit Care 2006; 10: R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dorward DA, Felton JM, Robb CT, et al The cyclin‐dependent kinase inhibitor AT7519 accelerates neutrophil apoptosis in sepsis‐related acute respiratory distress syndrome. Thorax 2017; 72: 182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Juss JK, House D , Amour A, et al Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3‐kinase inhibition. Am J Respir Crit Care Med 2016; 194: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Aggarwal A, Baker CS, Evans TW, et al G‐CSF and IL‐8 but not GM‐CSF correlate with severity of pulmonary neutrophilia in acute respiratory distress syndrome. Eur Respir J 2000; 15: 895–901. [DOI] [PubMed] [Google Scholar]
- 114. Leitch AE, Lucas CD, Rossi AG. Editorial: Neutrophil apoptosis: hot on the TRAIL of inflammatory resolution. J Leukoc Biol 2011; 90: 841–843. [DOI] [PubMed] [Google Scholar]
- 115. McGrath EE, Marriott HM, Lawrie A, et al TNF‐related apoptosis‐inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol 2011; 90: 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hoodless LJ, Lucas CD, Duffin R, et al Genetic and pharmacological inhibition of CDK9 drives neutrophil apoptosis to resolve inflammation in zebrafish in vivo . Sci Rep 2016; 6: 36980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Leitch AE, Lucas CD, Marwick JA, et al Cyclin‐dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ 2012; 19: 1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Witko‐Sarsat V, Mocek J, Bouayad D, et al Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J Exp Med 2010; 207: 2631–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Martin C, Ohayon D, Alkan M, et al Neutrophil‐expressed p21/waf1 favors inflammation resolution in Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol 2016; 54: 740–750. [DOI] [PubMed] [Google Scholar]
- 120. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis‐induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A 2012; 109: 14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. El Kebir D, József L, Pan W, et al 15‐epi‐lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med 2009; 180: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hagimoto N, Kuwano K, Nomoto Y, et al Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin‐induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol 1997; 16: 91–101. [DOI] [PubMed] [Google Scholar]
- 123. Guinee D, Brambilla E, Fleming M, et al The potential role of BAX and BCL‐2 expression in diffuse alveolar damage. Am J Pathol 1997; 151: 999–1007. [PMC free article] [PubMed] [Google Scholar]
- 124. Matute‐Bello G, Liles WC, Steinberg KP, et al Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 1999; 163: 2217–2225. [PubMed] [Google Scholar]
- 125. Lopez AD, Avasarala S, Grewal S, et al Differential role of the Fas/Fas ligand apoptotic pathway in inflammation and lung fibrosis associated with reovirus 1/L‐induced bronchiolitis obliterans organizing pneumonia and acute respiratory distress syndrome. J Immunol 2009; 183: 8244–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Janssen WJ, Barthel L, Muldrow A, et al Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 2011; 184: 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cheng KT, Xiong S, Ye Z, et al Caspase‐11‐mediated endothelial pyroptosis underlies endotoxemia‐induced lung injury. J Clin Invest 2017; 127: 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kobori M, Yang Z, Gong D, et al Wedelolactone suppresses LPS‐induced caspase‐11 expression by directly inhibiting the IKK complex. Cell Death Differ 2004; 11: 123–130. [DOI] [PubMed] [Google Scholar]
- 129. McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest 2013; 143: 1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Grégoire M, Uhel F, Lesouhaitier M, et al Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J 2018; 52: 1702590. [DOI] [PubMed] [Google Scholar]
- 131. Bae H‐B, Zmijewski JW, Deshane JS, et al AMP‐activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J 2011; 25: 4358–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jo YS, Choi SM, Lee J, et al Effect of preadmission metformin use on clinical outcome of acute respiratory distress syndrome among critically ill patients with diabetes. Tuberc Respir Dis (Seoul) 2017; 80: 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ueno H, Matsuda T, Hashimoto S, et al Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med 2004; 170: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 134. Friggeri A, Yang Y, Banerjee S, et al HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3‐integrin. Am J Physiol Cell Physiol 2010; 299: C1267–C1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Aziz M, Matsuda A, Yang W‐L, et al Milk fat globule‐epidermal growth factor‐factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol 2012; 189: 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: where, when, how, and why? Trends Immunol 2016; 37: 273–286. [DOI] [PubMed] [Google Scholar]
- 137. Lucas CD, Hoodless LJ, Rossi AG. Swimming against the tide: drugs drive neutrophil reverse migration. Sci Transl Med 2014, 6: 225fs9–225fs9. [DOI] [PubMed] [Google Scholar]
- 138. Loynes CA, Lee JA, Robertson AL, et al PGE2 production at sites of tissue injury promotes an anti‐inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo . Sci Adv 2018; 4: eaar8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Colom B, Bodkin JV, Beyrau M, et al Leukotriene B4‐neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity 2015; 42: 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Famous KR, Delucchi K, Ware LB, et al ARDS subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 2017; 195: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Calfee CS, Delucchi K, Parsons PE, et al Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014; 2: 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ashbaugh DG, Bigelow DB, Petty TL, et al Acute respiratory distress in adults. Lancet 1967; 2: 319–323. [DOI] [PubMed] [Google Scholar]
- 143. Fujishima S. Pathophysiology and biomarkers of acute respiratory distress syndrome. J Intensive Care 2014; 2: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Horiuchi T, Sakata N, Narumi Y, et al Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem 2017; 292: 8436–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Goodman RB, Strieter RM, Martin DP, et al Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med 1996; 154: 602–611. [DOI] [PubMed] [Google Scholar]
- 146. Zheng G, Huang L, Tong H, et al Treatment of acute respiratory distress syndrome with allogeneic adipose‐derived mesenchymal stem cells: a randomized, placebo‐controlled pilot study. Respir Res 2014; 15: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Auner B, Geiger EV, Henrich D, et al Circulating leukotriene B4 identifies respiratory complications after trauma. Mediators Inflamm 2012; 2012: 536156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Stephenson AH, Lonigro AJ, Hyers TM, et al Increased concentrations of leukotrienes in bronchoalveolar lavage fluid of patients with ARDS or at risk for ARDS. Am Rev Respir Dis 1988; 138: 714–719. [DOI] [PubMed] [Google Scholar]
- 149. Trujillo G, Zhang J, Habiel DM, et al Cofactor regulation of C5a chemotactic activity in physiological fluids. Requirement for the vitamin D binding protein, thrombospondin‐1 and its receptors. Mol Immunol 2011; 49: 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yang J, Azat M, Peng P, et al Serum levels of TNF‐α, IL‐1β, IL‐9, and IL‐15 in acute respiratory distress syndrome. Int J Clin Exp Pathol 2017; 10: 781–788. [Google Scholar]
- 151. Reutershan J, Ley K. Bench‐to‐bedside review: acute respiratory distress syndrome – how neutrophils migrate into the lung. Crit Care 2004; 8: 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kolaczkowska E, Jenne CN, Surewaard BGJ, et al Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 2015; 6: 6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Idell S, Kucich U, Fein A, et al Neutrophil elastase‐releasing factors in bronchoalveolar lavage from patients with adult respiratory distress syndrome. Am Rev Respir Dis 1985; 132: 1098–1105. [DOI] [PubMed] [Google Scholar]
- 154. Wang Z, Chen F, Zhai R, et al Plasma neutrophil elastase and elafin imbalance is associated with acute respiratory distress syndrome (ARDS) development. PLoS One 2009; 4: e4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Mitra S, Schiller D, Anderson C, et al Hypertonic saline attenuates the cytokine‐induced pro‐inflammatory signature in primary human lung epithelia. PLoS One 2017; 12: e0189536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Hästbacka J, Linko R, Tervahartiala T, et al Serum MMP‐8 and TIMP‐1 in critically ill patients with acute respiratory failure. Anesth Analg 2014; 118: 790–798. [DOI] [PubMed] [Google Scholar]
- 157. Kietzmann D, Kahl R, Müller M, et al Hydrogen peroxide in expired breath condensate of patients with acute respiratory failure and with ARDS. Intensive Care Med 1993; 19: 78–81. [DOI] [PubMed] [Google Scholar]
- 158. Perrella MA. A phase I trial of inhaled carbon monoxide for the treatment of sepsis‐induced acute respiratory distress syndrome (ARDS). [Accessed July 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT02425579.
- 159. El Solh AA, Bhora M, Pineda L, et al Alveolar plasminogen activator inhibitor‐1 predicts ARDS in aspiration pneumonitis. Intensive Care Med 2006; 32: 110–115. [DOI] [PubMed] [Google Scholar]