

Abstract
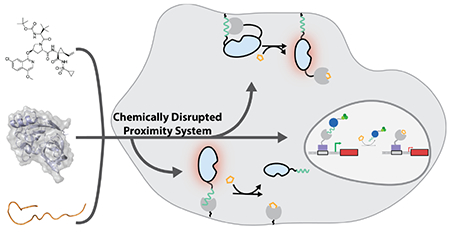
Chemical methods that allow the spatial proximity of proteins to be temporally modulated are powerful tools for studying biology and engineering synthetic cellular behaviors. Here, we describe a new chemically-controlled method for rapidly disrupting the interaction between two basally colocalized protein binding partners. Our chemically-disrupted proximity (CDP) system is based on the interaction between the hepatitis C virus protease (HCVp) NS3a and a genetically-encoded peptide inhibitor. Using clinically-approved antiviral inhibitors as chemical disrupters of the NS3a/peptide interaction, we demonstrate that our CDP system can be used to confer temporal control over diverse intracellular processes. This NS3a-based CDP system represents a new modality for engineering chemical control over intracellular protein function that is complementary to currently available techniques.
Graphical Abstract
Rationally manipulating protein localization can provide fundamental insights into cellular processes and is a powerful tool for engineering cellular behaviors.1–3 Techniques that allow temporal regulation of protein localization are particularly valuable for interrogating and programming dynamic cellular processes, with light and small molecules serving as the most widely used means of user-defined control.4 The dominant strategy for the chemical control of protein localization is the use of chemically-induced proximity (CIP), wherein two proteins are colocalized upon addition of a bridging small molecule.5
Systems that allow the interaction of two basally colocalized proteins to be rapidly disrupted with a small molecule provide a complementary method for temporally controlling protein function (Figure 1). Such chemically-disrupted proximity (CDP) systems could be used in numerous intramolecular and intermolecular engineering applications. For example, we have demonstrated that the interaction between the anti-apoptotic protein BCL-xL and a BH3 peptide can be used as a chemically-disruptable autoinhibitory switch for intramolecularly controlling the activities of various enzymes (Figure 1B).6
Figure 1.
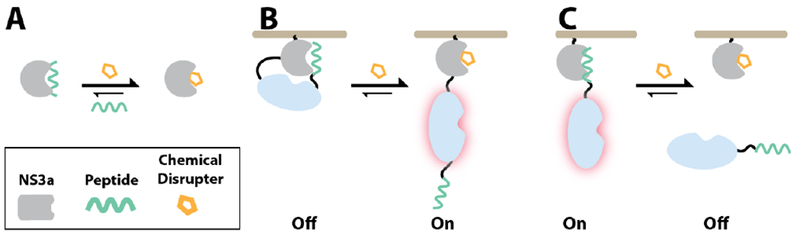
Chemically-disrupted proximity (CDP). (A) Components of a CDP system based on NS3a. CDP-mediated intramolecular (B) and intermolecular (C) regulation.
Intermolecular CDP systems that allow a basally localized activity to be chemically disrupted could be used as off-switches in numerous applications (Figure 1C). Unlike CIP systems, there is a dearth of CDP components available for engineering applications.
Here, we describe the development and use of a CDP system based on the hepatitis C virus protease (HCVp) NS3a and its interaction with a peptide inhibitor. Clinically-approved protease inhibitors that efficiently disrupt the NS3a/peptide interaction are available as bio-orthogonal inputs for this system.7 We first show that our NS3a-based CDP system can be used as a chemically-disruptable autoinhibitory switch for controlling the activity of an enzyme that activates RAS GTPase. We also demonstrate that the NS3a-based CDP system can be used to rapidly disrupt subcellular protein colocalization. Demonstrating the functional utility of chemically disrupting protein colocalization, we show that our NS3a-based CDP system can be used as a transcriptional off switch.
In order to use NS3a as a platform for a CDP system, a genetically-encoded binding partner that can be displaced with protease inhibitors is required. To provide this, we used a previously reported peptide inhibitor of NS3a’s protease activity (Figure S1).8 Consistent with previous studies, we found that this peptide, hereafter called apo NS3a reader (ANR), has low double-digit nanomolar affinity for NS3a. (Figure S2). Furthermore, we observed that the drug danoprevir potently and dose-dependently displaced ANR from NS3a (Figure S3), demonstrating that this interaction can be used as the basis for a CDP system.
We first explored using the NS3a/ANR interaction as a chemically-disruptable autoinhibitory switch for intramolecularly controlling the guanine nucleotide exchange factor (GEF) activity of the RAS GTPase activator Son of sevenless (SOS). We previously generated a chemically-inducible activator of RAS (CIAR) by computationally designing a fusion construct that contains the catalytic domain of SOS (SOScat) flanked by an N-terminal BH3 peptide and C-terminal BCL-xL.6b We observed that the intramolecular interaction between BCL-xL and the BH3 peptide autoinhibited SOScat’s catalytic activity, which small molecule disrupters rapidly released. The BCL-xL/BH3 peptide interaction was ideal for the computational design of a CIAR because it forms a compact and rigid complex that can be modeled as a single domain, a property that the NS3a/ANR interaction shares. Therefore, we explored a similar computational design strategy for generating a CIAR based on the NS3a/ANR interaction.
We used the computational modeling tool RosettaRe-model to guide the selection of flexible linker lengths with which to fuse ANR and NS3a to opposing termini of SOScat.9 Our goal was to identify linkers of sufficient length that NS3a and ANR can form an intramolecular complex but short enough that the complex is primarily centered over SOScat’s active site, with an energetic penalty for adopting non-inhibitory conformations. To do this, we computationally treated variable linker length SOScat fusions with ANR at the N-terminus and NS3a at the C-terminus as a single loop closure problem (Figure S4). An arbitrary break in one of the linkers of these fusion constructs was introduced, and subsequent chain closures were only permitted in geometrically allowed models. For each linker length combination, the percentage of successful chain closures was used to calculate the chain closure frequency (Figure S4). For models that successfully closed, torsional angles within the linkers were allowed to further vary in order to sample the most energetically favorable conformations of the ANR/NS3a complex relative to SOScat. Using this algorithm, we determined how linker lengths ranging from 5-29 and 1-13 residues for the N- and C-terminal linkers, respectively, affects the frequency of closure and the overlap of the ANR/NS3a complex with SOScat’s active site (Figure 2B, 2C). We found that output PDBs showed the NS3a/ANR complex most tightly clustered over SOScat’s active site–smallest center-of-mass distance and standard deviation–when the linkers connecting ANR to the N-terminus of SOScat was 17 amino acids and between the C-terminus of SOScat and NS3a was 7 amino acids (Figure S5). Therefore, we next determined whether a construct with these linkers can function as a CIAR in cells.
Figure 2.
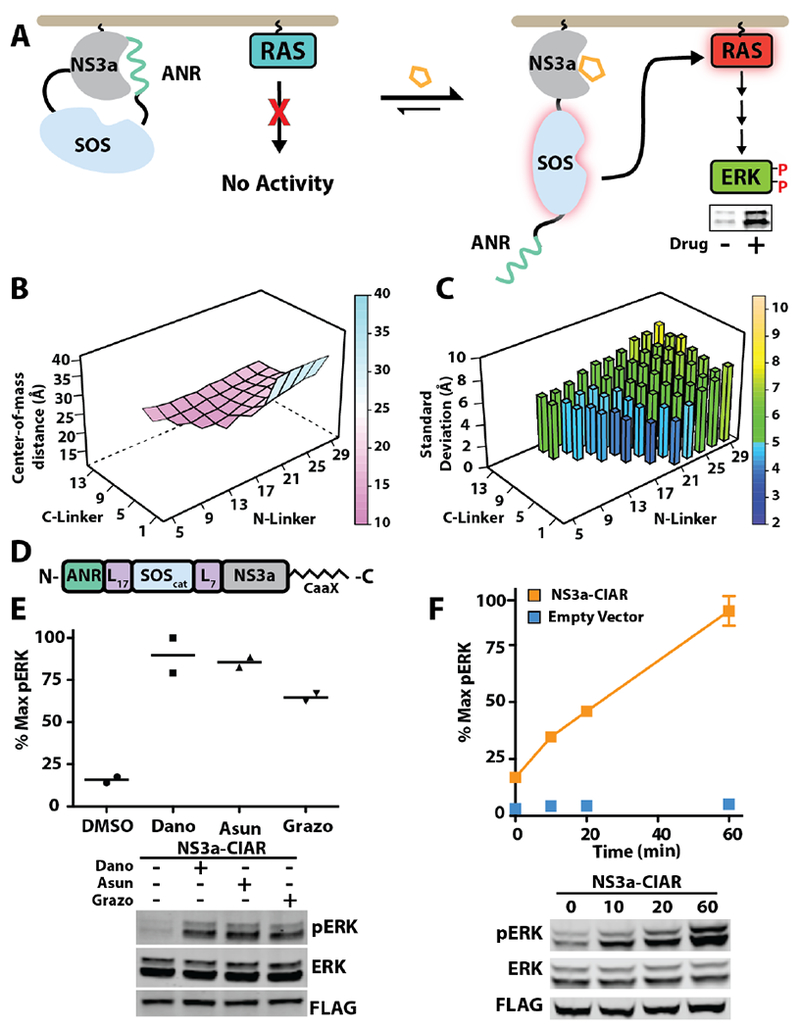
NS3a-based chemically-inducible activator of RAS. (A) Schematic of NS3a-CIAR’s activation of RAS/ERK signaling. (B) Dependence of the NS3a/ANR complex’s center-of-mass (in Å) relative to SOScat’s active site on N- and C-terminal linker length (NL and CL). (C) Standard deviation of the NS3a/ANR complex’s center-of-mass (in Å) as a function of NL and CL. (D) NS3a-CIAR construct used for cellular studies (E) Phospho-ERK blot (bottom) and quantification (top) of cells expressing NS3a-CIAR and treated with 1 μΜ danoprevir, grazoprevir, asunaprevir, or DMSO for 60 min (n=2). (F) Phospho-ERK blot (bottom) and quantification (top) of NS3a-CIAR-expressing cells treated with 10 μΜ asunaprevir for the times indicated (n=3).
To determine the utility of our NS3a-CIAR design for activating the RAS/ERK pathway, we transfected HEK293 cells with a membrane-targeted variant of our computationally-designed construct (Figure 2D) and monitored downstream activation of ERK kinase (phospho-ERK) (Figure 2E). Consistent with the NS3a/ANR interaction providing significant autoinhibition of SOScat’s GEF activity, we found that phospho-ERK levels were low in untreated cells expressing NS3a-CIAR-albeit higher than in cells not expressing this construct (Figure S6). In contrast, untreated cells expressing an NS3a-CIAR construct where ANR has been replaced with a peptide that has no affinity for NS3a demonstrated high basal phospho-ERK levels. We observed a robust increase in phospho-ERK levels when danoprevir, asunaprevir, or grazoprevir were added to cells expressing NS3a-CIAR (Figure 2E). However, these drugs did not lead to an increase in cellular phospho-ERK levels in the absence of the NS3a-CIAR construct (Figure S7). We found that NS3a-CIAR rapidly activated RAS/ERK signaling, with activation kinetics similar to what we observed in HEK293 cells with our BCL-xL-based CIAR (Figures 2F, S8). Thus, the NS3a/ANR interaction can serve as a drug-disruptable switch for rapidly activating RAS with clinically approved drugs that are orthogonal to mammalian systems.
We next investigated the utility of the NS3a/ANR interaction as an intermolecular CDP system by determining whether it could provide temporal control over protein colocalization. An N-terminal amphipathic helix-helix α0-from the NS3a variant used in our NS3a-CIAR construct has previously been demonstrated to interact with membranes (Figure S9), which we thought would be problematic for an intermolecular CDP system.10 Therefore, we determined whether a solubility-optimized NS3a variant-NS3a*-could be used with ANR as part of an intermolecular CDP system.11 Unfortunately, we observed that ANR has very low affinity for NS3a* (Figure S10). Therefore, we generated and tested a series of NS3a/NS3a* chimeras for their ability to colocalize with ANR in cells (Figures S9, S11).
To functionally test our NS3a chimeras, we used a fluorescent protein colocalization assay (Figure 3A). Each NS3a chimera was expressed as a mitochondrially-localized mCherry fusion and the amount of colocalization with an EGFP-ANR fusion protein was determined in cells treated with DMSO or asunaprevir (Figure S11). We found that all NS3a chimeras were capable of localizing EGFP-ANR to mitochondria in the absence of drug but constructs lacking hydrophobic residues at the C-terminal end of helix α0 provided the highest degree of colocalization. Furthermore, we observed that these more polar chimeras–in particular NS3a(H1)–demonstrated the largest difference in colocalization between DMSO and asunaprevir-treated cells. Binding assays with purified NS3a(H1) showed that this chimera’s affinity for ANR is similar to NS3a (Figure S13). Therefore, we used the NS3a(H1) variant for all subsequent engineering efforts. We next determined how rapidly the intracellular NS3a(H1)/ANR interaction can be disrupted. We found that the interaction between EGFP-ANR with mitochondrially-localized NS3a(H1) was completely disrupted within five minutes of asunaprevir addition (Figure 3B, 3C). Furthermore, we observed similar disruption kinetics when EGFP-NS3a(H1) was colocalized to membranes with N-terminally myristoylated ANR (Figure 3D, 3E). Robust, albeit slower, disruption of EGFP-NS3a(H1) nuclear localization was obtained when NLS-ANR-expressing cells were treated with asunaprevir. (Figure 3F, 3G). Thus, the NS3a/ANR interaction can colocalize proteins in diverse subcellular compartments, which chemical disruptors rapidly reverse.
Figure 3.
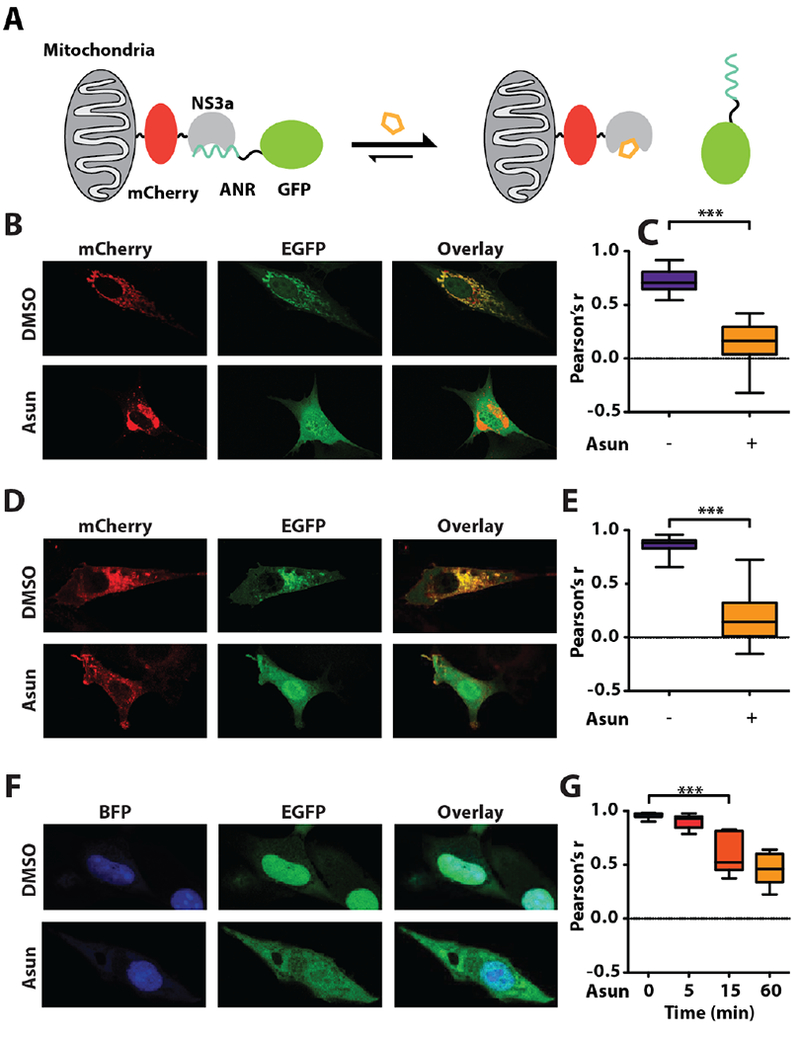
CDP control of protein localization. (A) Schematic of the mitochondrial colocalization assay. (B) Representative images of cells expressing mitochondrially-localized NS3a(H1) (Tom20-mCherry-NS3a(H1)) and EGFP-ANR2 treated with DMSO or asunaprevir (Asun) for 5 min. (C) Quantification of EGFP and mCherry colocalization in DMSO and Asun-treated cells. (D) Representative images of cells expressing membrane-localized ANR (myr-mCherry-ANR2) and EGFP-NS3a(H1) treated with DMSO or Asun for 15 min. (E) Quantification of EGFP and mCherry colocalization in DMSO and Asun-treated cells. (F) Representative images of cells expressing nuclear-localized ANR (NLS3-BFP-ANR2) and EGFP-NS3a(H1) treated with DMSO or Asun. (G) Quantification of EGFP and BFP colocalization in cells treated with Asun for the times shown. Quantification details and statistical analyses provided in Figure S12.
The localization of transcriptional activation domains upstream of genes can drive transcription and subsequent protein expression.3 We reasoned that the NS3a(H1)/ANR interaction could function as a chemically-disruptable off switch of transcription. To test this notion, we first determined whether ANR was capable of colocalizing the transcriptional activator VP64-p65-Rta (VPR) with a Gal4 DNA-binding domain-NS3a(H1) fusion bound upstream of an mCherry reporter gene (Figure 4A). Consistent with the NS3a(H1)/ANR interaction promoting transcription, we observed a significant increase in mCherry levels in ANR-VPR fusion-expressing cells (Figure 4B). We found that treatment of cells with danoprevir or grazoprevir decreased mCherry expression to background levels-defined by cells expressing a VPR fusion (DNCR2-VPR) that lacks ANR.
Figure 4.
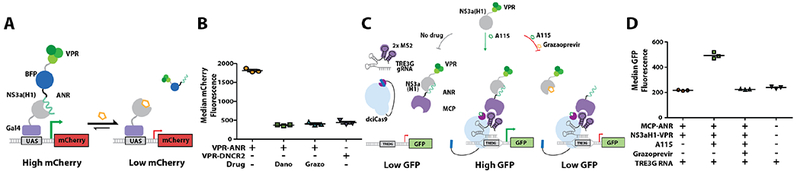
Intermodular disruption of transcriptional activation. (A) Schematic of chemically-disruptable Gal4(DBD)-NS3a(H1)/ANR-VPR transcriptional regulation. (B) Quantification of median mCherry fluorescence for the conditions shown (n=3). (C) Schematic of chemically-inducible/disruptable, dciCas9-mediated transcriptional regulation. (D) Quantification of median GFP fluorescence for the conditions shown (n=3). Expanded schematics are shown in Figure S14.
Finally, we explored whether our CDP system could be combined with chemical methods for activating transcription. To do this, we used a nuclease-null, chemically-inducible Cas9 (dciCas9) variant that is autoinhibited by the BCL-xL/BH3 interaction and can be activated with a chemical disrupter. An NS3a(H1)-VPR fusion was recruited upstream of a GFP reporter gene through its interaction with an MCP-ANR fusion bound to an MS2 stem loop of a scaffold RNA targeted to the Tet operator (Figure 4C).12,13 Activation of dciCas9 with a drug–A115–that disrupts the autoinhibitory BCL-xL/BH3 interaction led to an increase in GFP expression (Figure 4D). We observed that this increase in expression was reversed when grazoprevir was co-administered with A115. Thus, the chemically-disruptable NS3a/ANR interaction can be combined with chemical systems for transcriptional activation to provide temporally-regulated on/off switches.
In sum, we have shown that a system based on the interaction between the viral protease NS3a and a genetically-encoded peptide inhibitor can be used to engineer chemical control over a number of intracellular protein functions. The use of NS3a as a component of a CDP system further expands the utility of this protease as a synthetic biology engineering module that can be regulated with clinically-approved drugs.14 We envision that our new method will serve as a chemical genetic complement to light-mediated techniques for disrupting intracellular protein proximity.15 The availability of both light- and chemically-controlled techniques for rapidly disrupting protein proximity will allow researchers to select a system that best matches the requirements of their specific engineering application. Furthermore, it should be possible to multiplex our NS3a-based CDP system with existing CIP and light-induced proximity methods to provide even more sophisticated control over intracellular protein function.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the US National Institutes of Health (NIH) grants R01GM086858(D.J.M.) and F30CA189793 (J.C.R.), and a National Science Foundation GRFP (E.M.D.).
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website. Supplemental Figures S1-S14 and Methods (PDF)
REFERENCES
- (1).Haugh JM; Lauffenburger DA Physical modulation of intracellular signaling processes by locational regulation. Biophys. J. 1997, 72, 2014–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kholodenko BN; Hoek JB; Westerhoff HV Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 2000, 10, 173–8. [DOI] [PubMed] [Google Scholar]
- (3).Ptashne M; Gann A Transcriptional activation by recruitment. Nature 1997, 386, 569–77. [DOI] [PubMed] [Google Scholar]
- (4).(a) Fegan A; White B; Carlson JCT; Wagner CR Chemically controlled protein assembly: techniques and applications. Chem. Rev. 2010, 110, 3315–36. [DOI] [PubMed] [Google Scholar]; (b) Putyrski M; Schultz C Protein translocation as a tool: The current rapamycin story. FEBS Lett. 2012, 586, 2097–105. [DOI] [PubMed] [Google Scholar]; (c) Rakhit R; Navarro R; Wandless TJ Chemical biology strategies for posttranslational control of protein function. Chem. Biol. 2014, 21, 1238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) DeRose R; Miyamoto T; Inoue T Manipulating signaling at will: chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflugers Arch. 2013, 465, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Niu J; Ben-Johny M; Dick IE; Inoue T Following optogenetic dimerizers and quantitative prospects. Biophys. J. 2016, 111, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tischer D; Weiner OD Illuminating cell signaling with optogenetic tools. Nat. Rev. Mol. Cell Bio. 2014, 15, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stanton BZ; Chory EJ; Crabtree GR Chemically induced proximity in biology and medicine. Science 2018, 359,eaao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Goreshnik I; Maly DJ A small molecule-regulated guanine nucleotide exchange factor. J. Am. Chem. Soc. 2010, 132, 938–940. [DOI] [PubMed] [Google Scholar]; (b) Rose JC; Huang P-S; Camp ND; Ye J; Leidal AM; Goreshnik I; Trevillian BM; Dickinson MS; Cunningham-Bryant D; Debnath J; Baker D; Wolf-Yadlin A; Maly DJ A computationally engineered RAS rheostat reveals RAS-ERK signaling dynamics. Nat. Chem. Biol. 2017, 13, 119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rose JC; Dieter EM; Cunningham-Bryant D; Maly DJ Examining RAS pathway rewiring with a chemically inducible activator of RAS. Small GTPases 2018, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rose JC; Stephany JJ; Valente WJ; Trevillian BM; Dang HV; Bielas JH; Maly DJ; Fowler DM Rapidly inducible Cas9 and DSB-ddPCR to probe editing kinetics. Nat. Methods 2017, 14, 891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rose JC; Stephany JJ; Wei CT; Fowler DM; Maly DJ Rheostatic Control of Cas9-Mediated DNA Double Strand Break (DSB) Generation and Genome Editing. ACS Chem. Biol. 2018, 13, 438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McCauley JA; Rudd MT Hepatitis C virus NS¾a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. [DOI] [PubMed] [Google Scholar]
- (8).Kügler J; Schmelz S; Gentzsch J; Haid S; Pollmann E; van den Heuvel J; Franke R; Pietschmann T; Heinz DW; Collins J High affinity peptide inhibitors of the hepatitis C virus NS3–4A protease refractory to common resistant mutants. J. Biol. Chem. 2012, 287, 39224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huang P-S; Ban Y-EA; Richter F; Andre I; Vernon R; Schief WR; Baker D RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS ONE 2011, 6, e24109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Brass V; Berke JM; Montserret R; Blum HE; Penin F; Moradpour D Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad Sci. U.S.A. 2008, 105, 14545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wittekind M; Weinheirner S; Zhang Y; Goldfarb V Modified forms of hepatitis C NS3 protease for facilitating inhibitor screening and structural studies of protease:inhibitor complexes. US Patent 6333186 2004.
- (12).Mali P; Aach J; Stranges PB; Esvelt KM; Moosburner M; Kosuri S; Yang L; Church GM CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zalatan JG; Lee ME; Almeida R; Gilbert LA; Whitehead EH; La Russa M; Tsai JC; Weissman JS; Dueber JE; Qi LS; Lim WA Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Jacobs CL; Badiee RK; Lin MZ StaPLs: versatile genetically encoded modules for engineering drug-inducible proteins. Nat. Methods 2018, 15, 523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tague EP; Dotson HL; Tunney SN; Sloas DC.; Ngo JT Chemogenetic control of gene expression and cell signaling with antiviral drugs. Nat. Methods 2018, 15, 519–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao XJ; Chong LS.; Kim MS; Elowitz MB Programmable protein circuits in living cells. Science 2018, 361, 1252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chung HK; Jacobs CL; Huo Y; Yang J; Krumm SA; Plemper RK.; Tsien RY; Lin MZ Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015, 11, 713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang H; Vilela M; Winkler A; Tarnawski M; Schlichting I Yumerefendi H; Kuhlman B; Liu R; Danuser G; Hahn KM. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat. Methods 2016, 13, 755–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.