

Abstract
Aims
Calcific aortic valve stenosis (CAVS) is characterized by a fibrocalcific process. Studies have shown an association between CAVS and the activation of platelets. It is believed that shear stress associated with CAVS promotes the activation of platelets. However, whether platelets actively participate to the mineralization of the aortic valve (AV) and the progression of CAVS is presently unknown. To identify the role of platelets into the pathobiology of CAVS.
Methods and results
Explanted control non-mineralized and mineralized AVs were examined by scanning electron microscope (SEM) for the presence of activated platelets. In-depth functional assays were carried out with isolated human valve interstitial cells (VICs) and platelets as well as in LDLR−/− apoB100/100 IGFII (IGFII) mice. Scanning electron microscope and immunogold markings for glycoprotein IIb/IIIa (GPIIb/IIIa) revealed the presence of platelet aggregates with fibrin in endothelium-denuded areas of CAVS. In isolated VICs, collagen-activated platelets induced an osteogenic programme. Platelet-derived adenosine diphosphate induced the release of autotaxin (ATX) by VICs. The binding of ATX to GPIIb/IIIa of platelets generated lysophosphatidic acid (LysoPA) with pro-osteogenic properties. In IGFII mice with CAVS, platelet aggregates were found at the surface of AVs. Administration of activated platelets to IGFII mice accelerated the development of CAVS by 2.1-fold, whereas a treatment with Ki16425, an antagonist of LysoPA receptors, prevented platelet-induced mineralization of the AV and the progression of CAVS.
Conclusions
These findings suggest a novel role for platelets in the progression of CAVS.
Keywords: Calcific aortic valve disease, Calcific aortic valve stenosis, Aortic stenosis, Platelets, Autotaxin, ENPP2, Lysophosphatidic acid, P2Y1 receptor
See page 1374 for the editorial comment on this article (doi: 10.1093/eurheartj/ehy775)
Introduction
Calcific aortic valve stenosis (CAVS) is a prevalent heart valve disorder.1 Studies have highlighted that CAVS is associated with haemostatic abnormalities. Calcific aortic valve stenosis-related high shear stress promotes the degradation of high molecular weight multimers of von Willebrand factor (vWF) and is associated with mucosal haemorrhage (acquired Type 2A von Willebrand syndrome).2 On the other hand, the elevated shear stress created by CAVS has been linked with increased release of thrombin and activation of platelets even in patients with acquired Type 2A von Willebrand syndrome.3 Also, the level of circulating platelet-derived microparticles is increased in patients with CAVS.4 These findings are in line with the increased risk of thromboembolism documented in patients with CAVS.5,6
Translational perspective
Previous studies underlined that platelets are activated during calcific aortic valve stenosis (CAVS). However, whether platelets actively participate to the progression of CAVS is presently unknown. We found that surgically explanted aortic valves (AVs) present endothelium denudation areas, which are covered by activated platelets. Functional assays showed that autotaxin (ATX)- and glycoprotein IIb/IIIa-mediated processes promote the osteogenic transition of valve interstitial cells. In patients with CAVS, platelet-associated ATX activity is increased. In mice, administration of activated platelets accelerated the progression of CAVS, which was inhibited by the blockade of the ATX pathway. These data thus underline a novel role for platelet in CAVS.
In porcine aortic valves (AVs), several key regulators of haemostasis are expressed and their levels are modulated by ageing.7 Examination of surgically explanted mineralized AVs has demonstrated the presence of tissue factor, a promoter of blood coagulation, in leaflets.8,9 Moreover, CAVS-related shear stress and turbulent blood flow patterns could activate the endothelium and promote the adhesion and activation of platelets on the surface of AVs. Platelets liberate a vast array of mediators including bioactive lipids such as lysophosphatidic acid (LysoPA), which have pro-osteogenic activity in the AV.10 However, despite mounting evidence showing an association between the activation of platelets and CAVS, there is a lack of data on whether platelets may actively promote the progression of CAVS. In this work, using both in vitro and in vivo models of CAVS, we unravel a novel mechanism whereby activated platelets promote an osteogenic programme and the progression of CAVS through activation of the purinergic receptor P2Y1 (P2RY1)-glycoprotein IIb/IIIa(GPIIb/IIIa)-LysoPA pathway.
Methods
Expanded method section is provided in the Supplementary material online.
Procurement of tissues for analyses
We examined stenotic AVs (CAVS) that were explanted from patients at the time of AV replacement. Control non-calcified AVs with normal echocardiographic analyses were obtained during heart transplant procedures. The protocol was approved by the local ethical committee and informed consent was obtained from the subjects. The study conforms with the declaration of Helsinki.
Scanning electron microscopy
Following AV replacement or heart transplant procedures, valves were immersed in saline, transported to the laboratory, where they were immediately fixed for SEM studies (Quanta 3D FEG, FEI, USA).
In vitro analyses of calcification
Valve interstitial cells (VICs) were incubated for 7 days with a mineralizing medium containing: DMEM + 5% FBS, 10−7 M insulin, 50 μg/mL ascorbic acid, and NaH2PO4 at 2 mM. Cells were then incubated 24 h in 0.6 N HCl, supernatants was harvested and sent for quantification and cells were harvested in 1%SDS, 0.1 N NaOH for protein quantification.
Treatment with platelets
Platelets were isolated from plasma of healthy donors. Platelets were activated by incubation with collagen (2.5 µg for 3.2 × 106 platelets) for 2 min at 37°C with constant agitation. Cells were treated twice with 3.2 × 106 activated platelets during the 7 days mineralization protocol.
Animals
All animal protocols were conducted according to guidelines set out by the Laval University Animal Care and Handling Committee and are conform with the NIH guidelines for the care and use of laboratory animals. LDLR−/−/ApoB100/100/IGFII (on C57Bl/6J background) were generated from an established colony at the Heart and Lung Institute of Laval University from original founders kindly provided by Dr SeppoYlä-Herttuala (University of Eastern Finland, Finland).
Treatment with platelets in mice
Platelets were isolated from C57BL6 mice, activated with collagen for 1 min at 37°C. The activation was performed just before the injection in IGFII mice. Platelets were kept on ice and rapidly injected into treated mice (1.5×107 platelets/10 g/week).
Statistical analyses
Continuous data were expressed as mean ± standard deviation. The Shapiro–Wilk tests were performed on data. When distribution was normal, Student’s t-test, or ANOVA (post hoc Tukey) statistical analyses were performed. When distribution was not normal, Mann–Whitney, or Kurskal–Wallis (post hoc Steel-Dwass) statistical analyses were performed. Categorical data were expressed as absolute values and percentages and were compared with Fisher’s exact test. For in vitro experiments with VICs, n represents the number of experiment performed with different donors. A P-value <0.05 was considered significant. Statistical analysis was performed with a commercially available software package JMP 12.0 or Prism 6.0.
Results
Activated platelets and mineralized aortic valves
Explanted control non-mineralized (n = 9) and mineralized AVs (n = 8) were obtained from heart transplant and AV replacement surgeries, respectively (Supplementary material online, Table S1). We examined by scanning electron microscope (SEM) the aorta-facing and the ventricle-facing surfaces of mineralized AVs, which were compared with control non-mineralized AVs. Compared with control non-mineralized AVs (Figure 1A), we found in mineralized valves that the endothelium was activated in areas located on the aorta-facing side of the AVs (Figure 1B). In CAVS, the endothelium was irregular and showed the presence cuboidal cells protruding towards the lumen, suggesting activation of the endothelial cells (Figure 1B). On closer inspection, the cell membrane of endothelial cells was corrugated with many microvilli, a pattern associated with the activation of the endothelium (Figure 1C). The activation of the endothelium in mineralized AVs was confirmed by measuring the level of vascular cell adhesion-molecule 1 (VCAM1), a marker of cytokine-activated endothelium. In western blotting, we found that VCAM1 was not detected in control non-mineralized AVs, whereas it was expressed in mineralized AVs (Supplementary material online, Figure S1). Scattered areas of endothelium denudation were also observed on the aorta-facing side of mineralized AVs where microaggregates were present (Figure 1D). These microaggregates were composed of fibrin-like material and platelets showing numerous cell extensions such as filipodia, which is a hallmark of activated platelets (Figure 1E). Quantitative morphometric analyses showed that platelets were not detected in control non-mineralized AVs, whereas the area ratio covered by platelets (for the examined surface under SEM) averaged 12% per mineralized AVs (Figure 1F). Fibrin-like material was often calcified on electron dispersive X-ray (EDX) analysis (Figure 1G and H). To confirm the presence of platelet microaggregates on the surface of AVs, we performed markings for GPIIb/IIIa (αIIb β3) by using immunogold and SEM. Immunogold labelling showed in SEM the presence of GPIIb/IIIa in microaggregates, thereby confirming the presence of platelets (Figure 1G). These data suggested that micro-aggregates of platelets at the surface of AVs may interact with VICs during CAVS.
Figure 1.

Activated platelets in mineralized aortic valves. (A–E) Scanning electron microscope analyses of control (n = 9) (A) and mineralized (n = 8) (B–E) valves showing activation of endothelium (B and C) platelets aggregates and red blood cells (D and E). (F) Quantification of platelets in control (n = 9) vs. calcific aortic valve stenosis (n = 8). (G) Immunogold labelling for glycoprotein IIb/IIIa confirming the presence of platelet aggregates on calcific aortic valve stenosis. (H) Energy-dispersive X-ray spectroscopy analysis of platelet aggregates, demonstrating the presence of calcification. Scale bars: (A and B) 20 µM, (C) 2 µM, (D) 10 µM, (E) 5 µM, (G) 4 µM. Values are presented as mean ± standard deviation. CAVS, calcific aortic valve stenosis; CTL, control; GPIIb/IIIa, glycoprotein IIb/IIIa.
Activated platelets promote mineralization of valve interstitial cell cultures
Human primary VICs were isolated from non-mineralized AVs and cultured in a mineralizing medium for 7 days with or without Type I collagen-activated platelets obtained from healthy donors. Collagen Type I was selected as it is an important component of human AVs extracellular matrix where platelets were found on extracellular matrix. A high proportion of plated VICs (81%) expressed α-smooth muscle actin (α-SMA/ACTA2) and were thus activated myofibroblast-like cells, which are believed to play a key role in driving osteogenesis in the AV (Supplementary material online, Figure S2). In mineralization assays, the amount of extracellular calcium was determined biochemically with the Arsenazo III method, which is specific for calcium, and corrected with the protein content.11 The addition of Type I collagen-activated platelets to the growth medium at a concentration equivalent to 1.3 × 109/L (normal value in circulation is 150–450 × 109/L), increased by 130% the mineralization of VIC cultures (Figure 2A) (range absolute value of calcium: 1057–3419 µg Ca/mg protein). In order to establish if platelet-induced mineralization of VIC cultures relied on cell contact or the production of soluble factor(s), we carried out the mineralization experiment in a transwell co-culture system. Valve interstitial cells were plated and collagen-activated platelets were added in a transwell permeable to solutes but not to platelets (mesh size 0.2 µm). In transwell assay, collagen-activated platelets strongly increased the mineralization of VIC cultures (Figure 2B) (range absolute value of calcium: 2290–5830 µg Ca/mg protein). These data suggest that soluble factor(s) is/are released by the activated platelets and promote the mineralization of VIC cultures. We next evaluated if collagen-activated platelets could promote the osteogenic transition of VICs. Co-treatment of VICs with the osteogenic medium and activated platelets for 24 h led to higher level of RUNX2 and BGLAP transcripts (Figure 2C and D). RUNX2 is a bone-related transcription factor that positively regulates BGLAP and the mineralization process. Bone morphogenetic protein 2 (BMP2) is an important promoter of mineralization and it drives the expression of RUNX2. We documented that activated platelets increased the release of BMP2 by VICs after 24 h of treatment (Figure 2E). In addition, activated platelets increased the activity of alkaline phosphatase (ALP), an osteogenic marker, by 2.2-fold (Figure 2F). These findings indicate that platelets promote the osteogenic transition and the mineralization of VIC cultures.
Figure 2.

Activated platelets enhance valve interstitial cell mineralization in vitro. (A and B) Calcification assay showing the effect of platelets added directly (n = 6) (A) or in co-culture (n = 6) (B) with valve interstitial cells. (C and D) RUNX2 (n = 6) (C) and BGLAP (n = 6) (D) gene expression is increased in valve interstitial cells following addition of activated platelets to cells cultured in mineralizing medium (PO4). (E) Measurements of BMP2 in cell supernatants show that the protein expression rises with platelets addition (n = 6). (F) Platelets stimulates alkaline phosphatase enzymatic activity in valve interstitial cells (n = 6). Values are presented as mean ± standard deviation. ALP, alkaline phosphatase; BGLAP, bone gamma-carboxyglutamic acid-containing protein; BMP2, bone morphogenetic protein 2; CTL, control; RUNX2, Runt-related transcription factor 2.
Platelet-induced mineralization relies on valve interstitial cell-derived autotaxin and the production of lysophosphatidic acid
Platelets express phospholipase A1, which produces lysophosphatidylcholine, a substrate for autotaxin (ATX).12 The addition of palmostatin B, a blocker of phospholipase A1 and A2, abrogated platelet-induced mineralization of VIC cultures (Figure 3A) (range absolute value of calcium: 365–662 µg Ca/mg protein). Autotaxin, a lysophospholipase D that interacts with platelets,13 is highly expressed by VICs.14 We thus hypothesized that platelet-induced mineralization of VIC cultures may rely on ATX released by VICs and its downstream product LysoPA. The addition of HA130, an ATX inhibitor, abrogated platelet-induced mineralization of VIC cultures (Figure 3B) (range absolute value of calcium: 663–5257 µg Ca/mg protein). In VICs, platelet-induced release of BMP2 was prevented by the addition of HA130 to the growth medium (Figure 3C). In this experiment, HA130 treated cells released less BMP2 even compared with control VICs without platelet; this is possibly related to strong basal ATX activity in VICs, which could promote LysoPA-mediated BMP2 activation at the basal state.10,14 Also, treatment of cells with Ki16425, an inhibitor of LysoPA receptors 1-3 (LPAR1-3), negated platelet-induced mineralization of VIC cultures (Figure 3D) (range absolute value of calcium: 1911–5844 µg Ca/mg protein). These data suggest that LysoPA derived from ATX mediated the pro-mineralizing effect of platelets. We next hypothesized that ATX released by VICs could play a significant role in platelet-induced mineralization of cell cultures. In VICs, small interfering RNA (siRNA) targeting ATX, which lowered significantly ATX level measured by ELISA (Figure 3E), abrogated platelet-induced mineralization of cell cultures (Figure 3F) (range absolute value of calcium: 237–1199 µg Ca/mg protein). These data suggest that platelets induce the release of ATX by VICs, which may, in turn, interact with platelets and generate LysoPA. To further corroborate this hypothesis we measured the release of LysoPA by using a specific ELISA, which does not cross react with related lipids such as phospholipids, lysophosphatidylcholine and sphingosine-1 phosphate.15 We found that co-incubation of platelets with VICs increased the release of LysoPA in the supernatant by 4.1-fold, whereas HA130, an ATX inhibitor, abrogated this effect (Figure 3G).
Figure 3.

Effect of platelets on valve interstitial cells relies on autotaxin. (A) Effect of palmostatin B on platelet-induced mineralization of valve interstitial cell cultures (n = 6). (B and C) ATX inhibitor HA130 reduces platelet-induced mineralization (n = 6) (B) and BMP2 expression (n = 6, PO4+platelets+HA130 n = 4) (C). (D) Ki16425 (n = 6) impedes on platelet-mediated mineralization. (E) ATX protein measurements by ELISA demonstrating small interfering RNA efficiency (n = 4). (F) ATX silencing abrogates platelet-induced mineralization (n = 5). (G) ATX activity contributes to lysophosphatidic acid increase following addition of platelets to cultured valve interstitial cells (VICs, platelets n = 4, VICs+platelets, VICs+platelets+HA130 n = 5). HA130: 100 nM (DMSO: 1 µL/mL), Ki16425: 1 µM (DMSO: 0.24µL/mL), palmostatin B: 1.6µM (DMSO: 0.16µL/mL). Values are presented as mean ± standard deviation. ATX, autotaxin; BMP2, bone morphogenetic protein 2; CTL, control; DMSO, dimethyl sulfoxyde; LysoPA, lysophosphatidic acid; VICs, valvular interstitial cells.
Platelet-derived adenosine diphosphate promotes the expression of autotaxin by valve interstitial cells through P2Y1 receptor
One major mediator released by activated platelets is adenosine diphosphate (ADP). Valve interstitial cells express ADP-responsive P2RY1.16 We thus treated cell cultures with the P2RY1 inhibitor MRS2279 and we measured ATX activity in the growth medium as a readout for the release of this enzyme. Activated platelets induced the release of active ATX, which was abrogated by the addition of MRS2279 (Figure 4A). Also, the addition of apyrase, a nucleotidase that depletes ADP, to the growth medium prevented platelet-induced release of ATX by VICs (Figure 4B). In the same line, treatment of VIC cultures with the P2RY1 agonist 2-methylthioadenosine diphosphate trisodium salt (2MeS-ADP) was found to increase ATX mRNA level by 3.7-fold (Figure 4C). This effect was reduced significantly by siRNA-mediated knockdown of P2RY1 in VICs (Figure 4C–E). In addition, knockdown of P2RY1 in VICs reduced significantly platelet-induced mineralization of cell cultures (Figure 4F) (range absolute value of calcium: 237–1199 µg Ca/mg protein). These data are consistent with the hypothesis that platelet-derived ADP up-regulates the expression and release of ATX by VICs through P2RY1 and promotes the mineralization of VIC cultures.
Figure 4.

Platelets-derived adenosine diphosphate stimulates ATX expression and activity through P2Y1R signalling. (A) P2Y1R inhibitor, MRS2279, impedes on platelet-induced ATX activity in valve interstitial cells (n = 4). (B) Apyrase reduces platelet-mediated release of ATX (ATX activity) (n = 5). (C) Adenosine diphosphate analogue 2MeS-ADP signals through P2Y1R (showed by small interfering RNA) to promote ATX expression (n = 4). (D and E) qPCR (n = 4) (D) and western blot (n = 4) (E) analyses showing siP2Y1R efficiency. (F) P2Y1R is required for platelet-induced mineralization of valve interstitial cell cultures (n = 5, PO4+platelets n = 4). MRS2279: 100 nM (H2O), 2MeS-ADP: 20 µM (H2O). Values are presented as mean ± standard deviation. ATX, autotaxin; CTL, control; 2MeS-ADP, 2-methylthioadenosine diphosphate trisodium salt; P2RY1, purinergic receptor P2Y1.
Autotaxin interacts with glycoprotein IIb/IIIa and promotes mineralization of valve interstitial cell cultures
Valve interstitial cell-derived ATX may interact with platelets in driving the production of osteogenic LysoPA. In binding assay, we found that recombinant ATX interacted with activated platelets (Figure 5A). Arg-Gly-Asp-Ser peptide (RGDS), which interacts with integrins β1 and β3, dose-dependently reduced the interaction between ATX and activated platelets (Figure 5A). Also, GR144053, an inhibitor of GPIIb/IIIa (αIIb β3), abrogated the interaction between ATX and activated platelets (Figure 5B). We thus hypothesized that the binding of ATX to platelet GPIIb/IIIa was crucial in promoting the mineralization of VIC cultures. In this regard, a treatment of VIC cultures with GR144053 negated platelet-induced mineralization of cell culture (Figure 5C) (range absolute value of calcium: 300–5948 µg Ca/mg protein). These data suggest that ATX is captured by platelets, which, in turn, promotes the mineralization of the AV. Next, we hypothesized that ATX activity in circulating platelets is elevated during CAVS since blood plasma level of ATX is increased in patients with CAVS.17 We measured ATX activity in freshly isolated platelets of control subjects (n = 9) and in patients with CAVS (mild to severe; n = 18) (Supplementary material online, Table S2). Control and cases had similar risk factors except for a higher proportion of hypertension in CAVS patients (P = 0.001) (Supplementary material online, Table S2). Compared with control individuals with no valvulopathy, platelet-associated ATX activity was increased by four-fold in patients with CAVS (Figure 5D and Supplementary material online, Table S2). Platelet-associated ATX activity was positively and inversely associated with the peak transaortic velocity and the aortic valve area, respectively (Figure 5E and F). There is one outlier in graphs of Figure 5E and F; when removing this patient correlations remained highly significant (R2 = 0.52, P < 0.0001 and R2 = 0.58, P < 0.0001 respectively for Figure 5E and F).
Figure 5.

ATX interacts with glycoprotein IIb/IIIa. (A) Binding assay demonstrating an interaction between ATX and platelets that is inhibited by the addition of RGDS peptide (n = 4). (B) Binding of autotaxin to platelets is negated by GR144053 (n = 4). (C) Treatment with glycoprotein IIb/IIIa inhibitor (GR144053) abrogates platelet-induced valve interstitial cell mineralization (n = 6). (D) Platelet-associated ATX activity in control and calcific aortic valve stenosis patients (n = 27). (E and F) Spearman correlations between platelet-associated autotaxin activity and peak transaortic velocity (E) and aortic valve area (F) (n = 27). RGDS: 0.1–1µM (DMSO: 0.02µL/mL), GR144053: 60 nM (H2O). Values are mean ± standard deviation. ATX, autotaxin; AVA, aortic valve area; CAVS, calcific aortic valve stenosis; CTL, control; DMSO, dimethyl sulfoxyde; RFU, relative fluorescence unit; RGDS, arginyl-glycyl-α-aspartyl-serinyl.
Activated platelets accelerate the progression of calcific aortic valve stenosis in mice
We next assessed the presence of platelet micro-aggregates in LDLR−/− apoB100/100 IGFII transgenic mice (IGFII) which develop CAVS when fed a high-fat, high-sucrose diet (HFHS).18 Compared with control C57BL/6 mice (same background of IGFII mice), which showed normal endothelium covering AVs (Figure 6A), we found by using SEM that activated platelets were located on the aorta-facing side of AVs in IGFII mice after 6 months of HFHS diet (Figure 6A). Platelets were not detected in C57BL/6 mice, whereas the area ratio covered by platelet in examined surfaces averaged 11% in IGFII mice (Figure 6A). In order to determine whether circulating activated platelets may accelerate CAVS, we administered by intravenous injections collagen-activated platelets to IGFII mice. After 16 weeks of HFHS diet, echocardiographic analyses were performed and IGFII mice were injected collagen-activated mouse platelets isolated from C57BL/6 mice (1.5 × 107 platelets/10 g weight, which corresponds to 5% of total mouse platelets in circulation)19 once a week during 5 weeks (Figure 6B). This time point (16 weeks) was selected as it represents early CAVS process where altered haemodynamics could play a role in promoting the activation of endothelium-platelets and the progression of the disease process. Control mice were injected saline (vehicle). Echocardiographic analyses were performed before and after platelets administration. Baseline transaortic peak velocities (4 months) were similar in control (saline) and mice receiving collagen-activated platelets (Supplementary material online, Table S3). The injection of platelets during 5 weeks was found to increase the progression rate of CAVS by 2.1-fold as shown by the rise of transaortic velocities between the baseline (16 weeks) and after 5 weeks of treatment (21 weeks) (Figure 6C and Supplementary material online, Table S3). In order to document whether LysoPA was involved in this response, a group of mice was co-administered the Lpar1-3 blocker Ki16425 (5 mg/kg/d i.p.) along with activated platelets during 5 weeks (Figure 6B). Of interest, Ki16425 abrogated platelet-induced rise of peak transaortic velocities (Figure 6C and Supplementary material online, Table S3). Platelet-associated ATX activity was increased in IGFII mice and as expected neither the injection of activated platelets nor the administration of Lpar1-3 blocker affected platelet-ATX activity (Supplementary material online, Figure S3). However, platelet-associated ATX activity was positively associated with the peak transaortic velocity (Figure 6D). Post-mortem analyses performed at 21 weeks showed that platelets microaggregates and fibrin were present in mice injected with activated platelets (Figure 6E). Compared to control IGFII mice injected with vehicle (saline), the area ratio of platelets covering the AVs examined surfaces was significantly increased in mice injected with activated platelets (Figure 6E). As expected, the administration of Ki16425 (Lpar1-3 blocker) did not prevent the recruitment of platelets on AVs (Figure 6E). The expression of BMP2 determined by immunofluorescence was increased in AVs of IGFII mice receiving activated platelets (Figure 6F). The administration of Ki16425 (Lpar1-3 blocker) prevented the rise of BMP2 in leaflets induced by the injection of activated platelets (Figure 6F). The osteogenic activity, measured with a near infrared fluorescent marker (OsteoSense 680EX) recognizing hydroxyapatite of calcium, was increased significantly in AVs of mice receiving the injection of platelets (Figure 6G). However, osteogenic activity in AVs was significantly reduced in IGFII mice receiving activated platelets and Ki16425 (Lpar1-3 blocker) (Figure 6G). The osteogenic activity measured in the aorta (aortic root) was not significantly modified by the injection of activated platelets (Supplementary material online, Figure S4). The assessment of fibrosis with trichrome Masson staining revealed that administration of Ki16425 decreased the level of fibrotic tissues in AVs of mice injected with activated platelets (Figure 7A). Neither the injection of activated platelets nor the administration of Ki16425 modified the blood cholesterol levels (16.9 ± 2.5 mmol/L, 15.6 ± 3.9 mmol/L and 15.8 ± 2.9 mmol/L for control, activated platelets and activated platelets + Ki16425 mice, respectively, P = 0.55).
Figure 6.

Platelets accelerate calcific aortic valve stenosis progression in mice. (A) Scanning electron microscope studies of aortic valves (aorta facing side) of C57BL6 and IGFII mice under high-fat, high-sucrose diet and quantification of area ratio occupied with platelets (n = 10). (B) Mouse protocol. (C) Platelet-induced rise of ΔV2 between weeks 16 and 21 was abrogated by treatment with Ki16425 (n = 40). (D) Spearman correlation between platelets-associated autotaxin activity and peak transaortic gradient (n = 34). (E) Scanning electron microscope studies of mice aortic valves (aorta-facing side) showing aggregates of platelets (arrows) and quantification of area ratio occupied with platelets (n = 12). (F) Immunofluorescence analyses show that BMP2 level was elevated in leaflets of mice receiving platelets, but not in mice receiving Ki16425 (n = 27). (G) Leaflets mineralization was quantified by using Osteosense 680 and showed that platelet-induced mineral deposits on aortic valve leaflets was inhibited by Ki16425 (n = 24). Scale bars: (A) 10 µM, inset 1 µM (D) 20 µM (E and F) 200 µM. Ki16425: 5 mg/kg/day (DMSO: 10 µL/mL). Values are presented as mean ± standard deviation. ATX, autotaxin; BMP2, bone morphogenetic protein 2; DAPI, 4′,6-diamidino-2-phénylindole; HFHS, high fat high sucrose; IGFII, LDLR−/− apoB100/100 IGFII transgenic mice; RFU, relative fluorescence unit; ΔV2, delta peak transaortic velocity.
Figure 7.

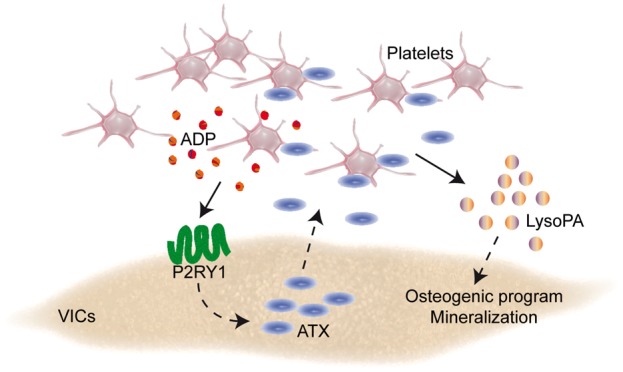
Blockade of Lpar1 decreased platelet-mediated fibrosis. (A) Trichrome Masson analysis of mouse leaflets demonstrating that Ki16425 decreases the level of fibrotic tissues in aortic valves of mice injected with platelets (n = 22). Scale bar: 100 µM. (B) Proposed working model. ADP, adenosine diphosphate; ALP, alkaline phosphatase; ATX, autotaxin; BGLAP, bone gamma-carboxyglutamic acid-containing protein; BMP2, bone morphogenetic protein 2; GPIIb/IIIa, glycoprotein IIb/IIIa; LPAR1, lysophosphatidic acid receptor 1; lysoPC, lysophosphatidylcholine; LysoPA, lysophosphatidic acid; PLA1, phospholipase A1; P2RY12, purinergic receptor P2Y1; RGDS: arginyl-glycyl-α-aspartyl-serinyl; RUNX2, Runt-related transcription factor 2; VICs: valvular interstitial cells.
Take home figure.
Discussion
In this work, we provide compelling data for a novel role of platelets in promoting the progression of CAVS (Extended Discussion available in Supplementary material online). In mineralized valves, we found that altered endothelial cell morphology and denudation on the aorta-facing side of AVs was a prevalent feature associated with the presence of platelet microaggregates. Through a series of functional studies, we showed that activated platelets promoted the osteogenic transition of VICs through a P2RY1-GPIIb/IIIa-LysoPA pathway, which triggered a faster progression of CAVS in a preclinical model. Specifically, we highlighted in vitro that platelet-derived ADP stimulated the release of ATX by VICs, which, in turn, interacted with GPIIb/IIIa in promoting the mineralization of VICs through the production of LysoPA (Figure 7B).
Coagulation defect and platelet activation in calcific aortic valve stenosis
A significant proportion of patients (∼20%) with CAVS develop haemorrhagic diathesis, which is related to shear stress-induced proteolysis of vWF.2 On the other hand, a growing number of reports suggest that platelets are activated during CAVS.3,4 Patients with CAVS have lower platelet response to nitric oxide (NO) and have increased circulating level of platelet-derived microparticles.20 Also, the anti-aggregating effect of the AV is decreased during mineralization.21 In mice, ascending aorta constriction led to shear stress-induced activation of latent TGFβ1 complex released by platelets.22 These data are in line with the present findings where we documented, in both human and mice, by SEM the presence of activated platelets on the aorta-facing side of mineralized AVs, where turbulent flow and high shear stress is present. Of note, we found evidence of endothelium activation typified by the presence of cuboidal endothelial cells with membrane microvilli.23 Also, scattered areas of endothelial denudation were observed where microthrombi and microcalcifications were detected. Taken together, these data thus strongly suggest that activated platelets could play a significant role in the development/progression of CAVS.
Activated platelets promote the osteogenic transition of valve interstitial cells
Mineralization of the AV is an organized process that involves the expression of key osteogenic genes. We documented that activated platelets exacerbated the mineralization of VIC cultures, which was associated with the expression of osteogenic genes such as RUNX2 and BGLAP. Valve interstitial cells reprograming was associated with the activation of BMP2, a key driver of osteogenic transition in the AV.24 The release of BMP2 by VICs was accompanied by a higher activity of ALP, an osteogenic marker. We have identified that platelet-induced osteogenic transition of VICs was largely dependent on LysoPA, a lipid metabolite with a strong osteogenic activity that accumulates in mineralized AVs.14 We previously showed that ATX is transported in the AV by lipoproteins and that VICs express a high level of ATX, which is released during inflammation.14 Autotaxin converts LPC, which is liberated by oxidized lipoproteins and activated platelets, into LysoPA with strong osteogenic activity. In apoE−/− mice, Huo et al.19 showed that intravenous administration of activated platelets (corresponding to 5% of circulating platelets) every 5 days for 12 weeks increased atherosclerotic lesions by 39%. In this work, once a week intravenous injection of activated platelets, which corresponded to 5% of total mouse platelets in circulation, promoted the mineralization of the AV and increased the progression rate of CAVS by 2.1-fold in only 5 weeks in IGFII mice. Hence, these findings unravel a novel process whereby a crosstalk between VICs and platelets promotes the mineralization of the AV and accelerates significantly the progression of CAVS.
Crosstalk between platelets and valve interstitial cells: role of purinergic receptor P2Y1, glycoprotein IIb/IIIa, and autotaxin
Purinergic signalling plays an important role in platelets and VICs biology. Metabotropic receptors such as P2RY1 and purinergic receptor P2Y1 (P2RY12) are involved in platelet aggregation and activation. On the other hand, VICs express P2RY1 and P2RY2.16 The latter is involved in VICs survival pathway and the control of inflammation,25 whereas the role of P2RY1 in VICs biology has not been explored previously. In this work, we highlighted that platelet-induced mineralization of cell cultures relied on the expression of P2RY1 by VICs. To this effect, the knockdown of P2RY1 in VICs reduced significantly platelets-induced mineralization of cell cultures. Conversely, stimulation of P2RY1 with 2MeS-ADP in VICs induced the expression mRNAs encoding for ATX. Activated platelets produce and liberate ADP with pro-aggregating properties. Hence, it is possible that the shear stress induced by CAVS activates platelets and promotes the release of ADP, which, in turn, promotes the synthesis of ATX. Autotaxin is secreted by different cells including VICs. The activity of ATX in the blood plasma is increased in patients with CAVS.17 Previous studies have underlined that ATX binds to GPIIb/IIIa of platelets, which is transported into the circulation.26 Also, Leblanc et al.27 showed that platelets and megakaryocytes do not express mRNA encoding for ATX. However, platelets can capture and internalize ATX, which is released upon activation.27 In this regard, we found that platelet-associated GPIIb/IIIa was necessary to bind ATX and to promote the mineralization of VIC cultures. Moreover, platelet-associated ATX activity was increased in patients with CAVS and it correlated with haemodynamic indices of disease severity. Similarly, platelet-associated ATX activity was increased in IGFII mice with CAVS and it correlated with the peak transaortic velocity.
Clinical implications
There is currently no medical treatment for CAVS.28 This findings may thus have important translational impact. We identified that P2RY1, ATX and LysoPA were involved in platelet-induced mineralization of the AV. It is likely that activated platelets are a significant contributor to CAVS during the disease process. In this regard, it is possible that during CAVS elevated shear stress activate circulating platelets, which form microaggregates in endothelium denuded areas of AVs and contribute to amplify the osteogenic process. Thus blocking this pathway may provide a protection against the progression of CAVS. Whether treatment with antiplatelet agents would translate in protective effect during CAVS remains to be investigated. However, our findings suggest that blockade of Lpar1-3 in IGFII mice, an established preclinical model of CAVS pathogenesis, can prevent the progression of CAVS. These data are of interest considering that novel inhibitors to block LysoPA and ATX are under development and clinical evaluation.29 Also, further work is needed to explore the role of P2RY1 as it is involved in platelet aggregation, inflammation,30 and the synthesis of ATX by VICs.
Limitations
In this work, we analysed surgically explanted mineralized AVs with an advanced disease process as human tissues representing an earlier stage of the disease were not available for SEM studies. Inherently, control groups used for this study differ from CAVS with regard to some risk factors. The process was examined in one mouse model of CAVS. Experiments in other in vivo models should be carried out in future studies to examine the mechanistic process whereby platelets are recruited to the AV.
Conclusion
Our work shows that platelets are activated in CAVS and promote the progression of the disease. The process relies on the activation of P2RY1-GPIIb/IIIa-LysoPA pathway, which drives an osteogenic programme in VICs. This work opens novel research perspective towards the role of platelets in CAVS and may foster the development of new therapeutic modalities.
Funding
This work was supported by the Canadian Institutes of Health Research (FRN114893, FRN142244, FRN148778, FRN130254 to P.M.) and (FDN-143247 to A.M.), the Heart and Stroke Foundation of Canada (G-14-0005913 to P.M.) and the Quebec Heart and Lung Institute Fund. Y.B. holds a Canada Research Chair in Genomics of Heart and Lung Diseases. P.P. holds the Canada Research Chair in Heart Valve Disease. A.M. holds a CIHR/industry partnered Chair on the pathogenesis of insulin resistance and CVD. B.J.A. holds a junior sholarship from Fonds de Recherche du Québec-Santé (FRQS). P.M. holds a Fonds de Recherche du Québec-Santé (FRQS) Research Chair on the Pathobiology of Calcific Aortic Valve Disease.
Conflict of interest: none declared.
Supplementary Material
References
- 1. Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, Pibarot P.. Calcific aortic stenosis. Nat Rev Dis Primers 2016;2:16006.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vincentelli A, Susen S, Le TT, Six I, Fabre O, Juthier F, Bauters A, Decoene C, Goudemand J, Prat A, Jude B.. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–349. [DOI] [PubMed] [Google Scholar]
- 3. Natorska J, Bykowska K, Hlawaty M, Marek G, Sadowski J, Undas A.. Increased thrombin generation and platelet activation are associated with deficiency in high molecular weight multimers of von Willebrand factor in patients with moderate-to-severe aortic stenosis. Heart 2011;97:2023–2028. [DOI] [PubMed] [Google Scholar]
- 4. Diehl P, Nagy F, Sossong V, Helbing T, Beyersdorf F, Olschewski M, Bode C, Moser M.. Increased levels of circulating microparticles in patients with severe aortic valve stenosis. Thromb Haemost 2008;99:711–719. [DOI] [PubMed] [Google Scholar]
- 5. Mendez RJ, Cianciulli TF, Parisi CE, Prezioso HA, Vidal LA.. Recurrent systemic embolism caused by thrombosis in a stenotic bicuspid aortic valve. Eur J Echocardiogr 2008;9:196–198. [DOI] [PubMed] [Google Scholar]
- 6. Wan S, DeSmet JM, Vincent JL, LeClerc JL.. Thrombus formation on a calcific and severely stenotic bicuspid aortic valve. Ann Thorac Surg 1997;64:535–536. [DOI] [PubMed] [Google Scholar]
- 7. Balaoing LR, Post AD, Liu H, Minn KT, Grande-Allen KJ.. Age-related changes in aortic valve hemostatic protein regulation. Arterioscler Thromb Vasc Biol 2014;34:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natorska J, Marek G, Hlawaty M, Sobczyk D, Sadowski J, Tracz W, Undas A.. Evidence for tissue factor expression in aortic valves in patients with aortic stenosis. Pol Arch Med Wewn 2009;119:636–643. [PubMed] [Google Scholar]
- 9. Breyne J, Juthier F, Corseaux D, Marechaux S, Zawadzki C, Jeanpierre E, Ung A, Ennezat PV, Susen S, Van BE, Le MH, Vincentelli A, Le TT, Jude B.. Atherosclerotic-like process in aortic stenosis: activation of the tissue factor-thrombin pathway and potential role through osteopontin alteration. Atherosclerosis 2010;213:369–376. [DOI] [PubMed] [Google Scholar]
- 10. Nsaibia MJ, Boulanger MC, Bouchareb R, Mkannez G, Le Quang K, Hadji F, Argaud D, Dahou A, Bosse Y, Koschinsky ML, Pibarot P, Arsenault BJ, Marette A, Mathieu P.. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-kappaB pathway. Cardiovasc Res 2017;113:1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hadji F, Boulanger M-C, Guay S-P, Gaudreault N, Amellah S, Mkannez G, Bouchareb R, Marchand JT, Nsaibia MJ, Guauque-Olarte S, Pibarot P, Bouchard L, Bossé Y, Mathieu P.. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation 2016;134:1848–1862. [DOI] [PubMed] [Google Scholar]
- 12. Bolen AL, Naren AP, Yarlagadda S, Beranova-Giorgianni S, Chen L, Norman D, Baker DL, Rowland MM, Best MD, Sano T, Tsukahara T, Liliom K, Igarashi Y, Tigyi G.. The phospholipase A1 activity of lysophospholipase A-I links platelet activation to LPA production during blood coagulation. J Lipid Res 2011;52:958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS.. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J Biol Chem 2011;286:34654–34663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bouchareb R, Mahmut A, Nsaibia MJ, Boulanger MC, Dahou A, Lepine JL, Laflamme MH, Hadji F, Couture C, Trahan S, Page S, Bosse Y, Pibarot P, Scipione CA, Romagnuolo R, Koschinsky ML, Arsenault BJ, Marette A, Mathieu P.. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation 2015;132:677–690. [DOI] [PubMed] [Google Scholar]
- 15. Goldshmit Y, Matteo R, Sztal T, Ellett F, Frisca F, Moreno K, Crombie D, Lieschke GJ, Currie PD, Sabbadini RA, Pebay A.. Blockage of lysophosphatidic acid signaling improves spinal cord injury outcomes. Am J Pathol 2012;181:978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Côté N, El Husseini D, Pépin A, Guauque-Olarte S, Ducharme V, Bouchard-Cannon P, Audet A, Fournier D, Gaudreault N, Derbali H, McKee MD, Simard C, Després J-P, Pibarot P, Bossé Y, Mathieu P.. ATP acts as a survival signal and prevents the mineralization of aortic valve. J Mol Cell Cardiol 2012;52:1191–1202. [DOI] [PubMed] [Google Scholar]
- 17. Nsaibia MJ, Mahmut A, Boulanger MC, Arsenault BJ, Bouchareb R, Simard S, Witztum JL, Clavel MA, Pibarot P, Bosse Y, Tsimikas S, Mathieu P.. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J Intern Med 2016;280:509–517. [DOI] [PubMed] [Google Scholar]
- 18. Le Quang K, Bouchareb R, Lachance D, Laplante MA, El Husseini D, Boulanger MC, Fournier D, Fang XP, Avramoglu RK, Pibarot P, Deshaies Y, Sweeney G, Mathieu P, Marette A.. Early development of calcific aortic valve disease and left ventricular hypertrophy in a mouse model of combined dyslipidemia and type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol 2014;34:2283–2291. [DOI] [PubMed] [Google Scholar]
- 19. Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K.. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 2003;9:61–67. [DOI] [PubMed] [Google Scholar]
- 20. Sverdlov AL, Ngo DT, Chan WP, Chirkov YY, Gersh BJ, McNeil JJ, Horowitz JD.. Determinants of aortic sclerosis progression: implications regarding impairment of nitric oxide signalling and potential therapeutics. Eur Heart J 2012;33:2419–2425. [DOI] [PubMed] [Google Scholar]
- 21. Chirkov YY, Mishra K, Chandy S, Holmes AS, Kanna R, Horowitz JD.. Loss of anti-aggregatory effects of aortic valve tissue in patients with aortic stenosis. J Heart Valve Dis 2006;15:28–33. [PubMed] [Google Scholar]
- 22. Wang W, Vootukuri S, Meyer A, Ahamed J, Coller BS.. Association between shear stress and platelet-derived transforming growth factor-beta1 release and activation in animal models of aortic valve stenosis. Arterioscler Thromb Vasc Biol 2014;34:1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walski M, Chlopicki S, Celary-Walska R, Frontczak-Baniewicz M.. Ultrastructural alterations of endothelium covering advanced atherosclerotic plaque in human carotid artery visualised by scanning electron microscope. J Physiol Pharmacol 2002;53:713–723. [PubMed] [Google Scholar]
- 24. Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ, Yutzey KE.. Bone morphogenetic protein signaling is required for aortic valve calcification. Arterioscler Thromb Vasc Biol 2016;36:1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. El Husseini D, Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, Pibarot P, Bosse Y, Mathieu P.. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J Mol Cell Cardiol 2014;72:146–156. [DOI] [PubMed] [Google Scholar]
- 26. Pamuklar Z, Federico L, Liu S, Umezu-Goto M, Dong A, Panchatcharam M, Fulkerson Z, Fulerson Z, Berdyshev E, Natarajan V, Fang X, van Meeteren LA, Moolenaar WH, Mills GB, Morris AJ, Smyth SS.. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J Biol Chem 2009;284:7385–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, Miller D, Sahay D, Ribeiro J, Clezardin P, Tigyi GJ, Peyruchaud O.. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014;124:3141–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mathieu P, Boulanger MC, Bouchareb R.. Molecular biology of calcific aortic valve disease: towards new pharmacological therapies. Expert Rev Cardiovasc Ther 2014;12:851–862. [DOI] [PubMed] [Google Scholar]
- 29. Llona-Minguez S, Ghassemian A, Helleday T.. Lysophosphatidic acid receptor (LPAR) modulators: the current pharmacological toolbox. Prog Lipid Res 2015;58:51–75. [DOI] [PubMed] [Google Scholar]
- 30. Zerr M, Hechler B, Freund M, Magnenat S, Lanois I, Cazenave JP, Leon C, Gachet C.. Major contribution of the P2Y(1)receptor in purinergic regulation of TNFalpha-induced vascular inflammation. Circulation 2011;123:2404–2413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.