

Abstract
We have synthesized a QS-17/18 analogue (7) and evaluated its adjuvant activity in the formulation with rHagB antigen. Compound 7 and QS-21 analogues 5 and 6 are presumably the major components of GPI-0100, a widely used complex mixture of semisynthetic derivatives of Quillaja saponaria (QS) Molina saponins. The QS-17/18 analogue 7 shows an adjuvant activity profile similar to that of GPI-0100, potentiating mixed Th-1/Th-2 immune responses, which is different from those of QS-21 analogues 5 and 6 that probably only induce a Th2-like immunity. The combination of QS-17/18 and QS-21 analogues does not show a synergistic effect. These results suggest that QS-17/18 analogue 7 might be the active component of GPI-0100 responsible for its immunostimulant property. Therefore, compound 7 can not only be a structurally defined alternative to GPI-0100 but also provide a valuable clue for rational design of new QS-based vaccine adjuvants with better adjuvant properties.
Graphical Abstract
INTRODUCTION
There is increasing interest in developing new vaccines against cancers, infectious diseases, and degenerative disorders with subunit antigens. The reduced immunogenicity of the refined and homogeneous subunit antigens necessitates the use of immune adjuvants to enhance the vaccines in eliciting strong and durable immune responses to specific antigens. However, the choice of adjuvants for human use is limited. There remain imperative needs for potent new adjuvants, especially those for potentiating mixed Th1/Th2 immune responses and CTL production, because both antibody- and cell-mediated immunity are necessary for protection against intracellular pathogens that cause infectious diseases, such as AIDS, tuberculosis, leishmaniasis, leprosy, and malaria, and against cancers and degenerative disorders.1–4
We are particularly interested in studying synthetic analogues of the saponins extracted from the tree bark of Quillaja saponaria (QS) Molina. One of the QS saponin leads, QS-21, has been studied extensively and evaluated in over 100 clinical trials of vaccines against cancers and infectious diseases.5,6 It is a mixture of two isomers (1, R1 = β-D-apiofuranosyl (api) or β-D-xylopyranosyl (xyl), R2 = H, Figure 1) in a ratio of 2:1, and each isomer has a quillaic acid triterpene core connecting to a branched trisaccharide at the C3 hydroxyl group and a linear tetrasaccharide at the C28 carboxyl group through glycosidic linkages. A chiral and glycosylated fatty acyl chain is connected to the 4-O position of the fucosyl unit in the tetrasaccharide. The structures of the two QS-21 isomers differ only in the terminal sugar unit of the tetrasaccharide, but they have the same adjuvanticity and toxicity. It potentiates balanced Th1/Th2 responses and an antigen-specific CTL response.7,8 Despite its success, QS-21 has its own drawbacks such as chemical instability, limited supply, and difficult and low-yielding purification. Moreover, its dose-limiting toxicity causes local erythema, inflammation, and flu-like symptoms, which prevents it from being used at an optimal dose.
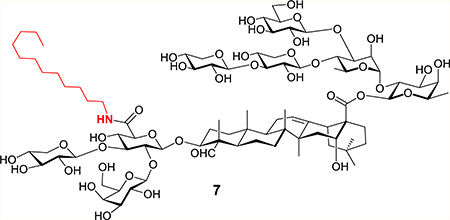
Figure 1.

Natural saponin immunostimulants (1–3) and unnatural analogues (4–7).
In pursuing QS saponin-based immunostimulants with retained favorable adjuvant activities of the parent natural products and reduced toxicity, we found that the two underexplored saponins of the QS family, that is, QS-17 (2) and QS-18 (3), can be valuable new leads. QS-18 (3) is the most abundant saponin in the extract of the QS Molina tree bark. The main structural difference between QS-21 and QS-17/18 is in the oligosaccharide domain connected to the C28 carboxyl group of quillaic acid. Instead of having a linear tetrasaccharide as in QS-21 (l), QS-17/18 has an additional β-D-glucopyranosyl (glc) unit connected to the α-L-rhamnopyranosyl (rha) unit at its 3-O position (i.e., 2 and 3, R2 = glc, Figure 1). QS-17 differs from QS-18 only in R3 of the acyl side chain, that is, QS-17 has a disaccharide unit, whereas QS-18 has a monosaccharide unit at the far end of the side chain.
Chemical instability of these natural QS saponins originates from the hydrolytically labile acyl chain at the 4-O position of the fucosyl unit (as indicated in Figure 1). Earlier SAR studies show that loss of the acyl chain of QS-21 leads to a loss in its ability to potentiate a Th1 response.9,10 Gin et al. circumvented the chemical instability issue of QS-21 by replacing the hydrolytically unstable ester groups with more stable amide groups.11
We, however, explored a different approach. Our design of chemically stable QS analogues was enlightened by the work of Marciani et al.12,13 They prepared the semisynthetic saponin mixture, GPI-0100, in two steps from a commercially available raw tree bark extract containing QS-7, QS-17/18 (2 and 3), QS-21 (1) and many other structurally unknown QS saponins. Initially, all hydrolytically unstable acyl side chains in different saponin components were removed under mild basic conditions and then a dodecylamine side chain was incorporated to the glucuronic acid of the branched trisaccharide at the C3 position of the quillaic acid unit. The resulting mixture of products, that is, GPI-0100, retained the capacity to potentiate humoral and T-cell immunity with the production of antigen-specific CTL and, more importantly, the toxicity was reduced dramatically. We have previously synthesized QS-21 derivatives 4–6.14 Among them, compounds 5 and 6, and the newly synthesized QS-17/18 derivative 7 are presumably the major components of GPI-0100 based on the composition of the starting materials, chemistry of GPI-0100 preparation, and liquid chromatography–mass spectrometry (LC–MS) analysis of the reaction products. We anticipate that evaluation of the immunostimulant activities of the pure QS-21 and QS-17/18 derivatives may identify the component that inherits the desired adjuvanticity and reduced toxicity of GPI-0100.
RESULTS AND DISCUSSION
Herein, we report our synthesis of QS-17/18 analogue 7 and compare its immune-potentiating properties with those of the QS-21 analogues. In retrosynthesis, QS17/18 analogue 7 would be prepared from dodecylamine 8 and intermediate 9, followed by global deprotection (Scheme 1). As QS-17 and QS-18 differ only in their acyl side chains, they share the same intermediate 9 without the side chain, leading to the same analogue, that is, 7. Intermediate 9 would be prepared by coupling a pentasaccharide donor 10 and known glycosyl acceptor 11.15
Scheme 1.

Retrosynthesis of QS-17/18 Analogues
To prepare the fully protected pentasaccharide donor 10, we used the two-stage allyl-glycoside-donor-activation approach for glycosidic bond construction.16–19 Thus, in a reducing end expansion fashion, an anomeric allyl group of a glycosyl donor was first isomerized to its corresponding prop-1-enyl group by using the hydrogen-activated catalyst [Ir(COD)(PMePh2)2]-PF6.20,21 Subsequent treatment of the obtained prop-1-enyl donor with NIS/TfOH in the presence of an acceptor provided the desired glycosylic bond.16–9 Repeatedly using this active-latent glycosylation protocol led to trisaccharide 12 (Scheme 2).16
Scheme 2.

Synthesis of Pentasaccharide Donor 17a
aReagents and conditions: (a) trifluoroacetic acid (TFA)/H2O (4:1), 0 °C, 95%; (b) NaOH (20% aq), nBu4NBr, BnBr, DCM, 23 °C, 83%; (c) 14, t-BuOK, DMSO, 23 °C, 94%; (d) 13, NIS/TfOH, MeCN, 23 °C, 63% (55% β + 8% α); (e) H2-activated [Ir(COD)(PMePh2)2]-PF6, THF, 23 °C, 91%; (f) 16, NIS/TfOH, MeCN, 23 °C, 51%; (g) levulinic acid, DCC, DMAP, DCM, 23 °C, 85%; (h) TFA/H2O (4:1), 0 °C; (i) Ac2O, TEA, DMAP, DCM, 0 °C; and (j) NH2NH2 H2O, allyl alcohol, 75% (over 3 steps from 19).
Next, we converted trisaccharide 12 into 13 by removing the acetonide protecting group of 12 followed by regioselective benzylation at the 2-OH group under phase-transfer reaction conditions.15 Subsequent glycosylation of acceptor 13 with donor 14 provided the desired tetrasaccharide 15 in 55% yield. The anomeric configuration of 15 was determined with HSQC 2D NMR experiment. We identified four anomeric protons at δ 5.02 ppm (d, J = 7.4 Hz), 4.81 ppm (d, J = 6.9 Hz), 4.71 ppm (singlet), and 4.59 ppm (d, J = 8.2 Hz), which is consistent with the anomeric configurations of the four sugar moieties in 15. Moreover, in a less crowded 1D 1H NMR spectrum of the tetrasaccharide prepared by removing all benzyl groups of 15 under hydrogenolysis conditions with Pd/C and H2, we confirmed the stereochemistry at the anomeric centers based on the 3J1,2 measurements. With donor 15 and allyl fucoside acceptor 16, the two-stage allyl-glycoside-donor-activation approach provided desired pentasaccharide 17 in 51% yield. Allyl fucoside acceptor 16 was synthesized in 64% overall yield from known allyl fucoside 18 with a four-step sequence through intermediate 19.14
In an alternative route to incorporate the glucosyl unit to make tetrasaccharide 15, we used trisaccharide acceptor 13 and glucosyl trichloroacetimidate donor 20 to make branched tetrasaccharide 15b in 87% yield under the same conditions as for the synthesis of QS-7 (Scheme 3).15 With tetrasaccharide donor 15b and allyl 3,4-O-isopropylidene-α-D-fucopyranoside acceptor 18, the two-stage donor-activation glycosylation protocol led to pentasaccharide 21. However, removal of the 2-O benzoyl group of the glucosyl unit under hydrolytic conditions was unsuccessful. Although reduction with DIBAL-H removed the benzoyl group,15 the route to the pentasaccharide from 13 shown in Scheme 3 is less efficient than that in Scheme 2.
Scheme 3.

Alternative Attempt to Incorporate Glucosyl Unita
aReagents and conditions: (a) TMSOTf, DCM, −45 °C, 87%; (b) H2-activated [Ir(COD)-(PMePh2)2]PF6, THF, 23 °C; and (c) 18, NIS/TfOH, MeCN, 23 °C, 31%.
In an effort to improve the yield of pentasaccharide 17, we also attempted an alternative way by using trichloroacetimidate donor 22 (Scheme 4).22 Thus, we first converted allyl tetrasaccharide 15 into its corresponding imidate 22 with a three-step sequence. Under the standard conditions, we prepared the pentasaccharide in only 35% yield, which is less efficient than the sequence employing the two-stage allyl-glycoside-donor-activation approach illustrated in Scheme 2.
Scheme 4.

Alternative Route to Synthesize Pentasaccharide 17a
aReagents and conditions: (a) t-BuOK, DMSO, 23 °C; (b) NIS, THF/H2O, 23 °C; (c) Cs2CO3, Cl3CCN, DCM, 23 °C; and (d) 16, BF3·OEt2, DCM, −45 °C, 35%.
We noticed that the two-stage allyl-glycoside-donor-activation approach was not compatible with a carboxyl acceptor under the current reaction conditions thus, we converted pentasaccharide 17 into its trichloroacetimidate donor 23 with the three-step standard procedure. We found that using fucoside 16 instead of 18 led to a pentasaccharide imidate donor more stable for silica gel column purification.14 Glycosylation of quillaic acid-trisaccharide conjugate 11 with the pentasaccharide imidate donor produced fully protected saponin skeleton 24 in 72% yield (Scheme 5). The desired β configuration of the newly formed glycosidic bond was confirmed by the characteristic doublet of the anomeric proton of the fucopyranosyl unit at δ 5.45 ppm (3J1,2 = 7.7 Hz). Removal of all benzyl protecting groups under hydrogenolysis conditions with Pd/C and H2 provided intermediate 9.14 As proven in the synthesis of QS-21 (1) and its analogues14,23–25 and also in the synthesis of QS-7,15 the alkene moiety in the quillaic acid domain is inert to the hydrogenolysis conditions. Coupling of key intermediate 9 with dodecylamine and subsequent deprotection (i.e., removal of the triethylsilyl groups under acidic conditions and deacetylation under basic conditions) generated the desired QS17/18 analogue 7. The C28 ester group is known to be stable under acidic and mild basic conditions.25
Scheme 5.

Synthesis of QS17/18 Analogue 7a
aReagents and conditions: (a) H2-activated [lr(COD)(PMePh2)2]PF6, THF, 23 °C; (b) NIS/TfOH, THF/H2O, 23 °C; (c) Cs2CO3, Cl3CCN, CH2Cl2, 23 °C, 62% (over three steps); (d) BF3·OEt2, DCM, −78 °C, 72%; (e) Pd/C, H2 (55 psi), 91%; (f) CH3(CH2)11NH2, HATU, N,N-diisopropylethylamine (DIPEA), CHCl3, 23 °C; (g) DCM, TFA/H2O (4:1), 0 °C; and (h) K2CO3, MeOH, 23 °C, 75% (over three steps).
With 7 in hand, we then assessed its adjuvant property and compared it with QS-21 analogues 5 and 6. We previously demonstrated that rHagB (a recombinant, nonfimbrial adhesion hemagglutinin B from Porphyromonas gingivalis, as an etiologic agent of periodontal disease) is an effective antigen in inducing an immune response protective against alveolar bone loss in an experimental animal model.26 We also demonstrated that 5 was effective in potentiating and maintaining a serum IgG response to rHagB following systemic immunization, but its adjuvant activity was lower than that of GPI-0100.14 In this study, we evaluated the other QS-21 analogue, that is, 6, and QS-17/18 analogue 7 in enhancing immune responses against the rHagB antigen. To investigate potential additive or synergistic effect, we also evaluated the combination of 7 with 5 and 6.
Thus, groups of female BALB/c mice (8–10 weeks of age, five per group) were immunized via a subcutaneous (s.c.) route with rHagB (20 μg) antigen alone or with GPI-0100 or different synthetic adjuvants on days 0, 14, and 28. We used GPI-0100, 6, or 7 at the dosage of 100 μg but also immunized one group of mice with 7 at 50 μg and another group with a mixture of adjuvants, that is, 5 (25 μg) + 6 (25 μg) + 7 (50 μg), to mimic GPI-0100. Before each immunization and on day 42 post the last immunization, the mice were weighed and then serum was collected from each mouse and analyzed for anti-rHagB activity using an enzyme-linked immunosorbent assay (ELISA).
A serum IgG response was detected in all immunized mice by week 2 after the initial immunization (data not shown). The magnitude of the response continued to increase following the second immunization. The IgG anti-rHagB antibody responses induced with rHagB + GPI-0100 (100 μg) were significantly higher (P < 0.001) than those induced with rHagB alone at weeks 4 and 6 (Figure 2). The QS-17/18 analogue 7 was also effective in potentiating the serum IgG anti-rHagB response. In this regard, significantly higher (P < 0.001) serum IgG anti-rHagB responses were seen in the mice receiving rHagB + 7 (100 μg), compared to those seen with the antigen alone. No significant difference was observed in the mice receiving rHagB + GPI-0100 or 7. However, no significantly increase in the serum IgG responses was observed in the mice receiving rHagB + QS-21 analogue 6 (100 μg), compared to those seen with the antigen alone. In addition, the IgG responses were significantly lower (P < 0.001) in the mice immunized with rHagB + 6 than in the mice receiving rHagB + GPI-0100 or 7 at weeks 4 and 6. These results demonstrate that QS-17/18 analogue 7 is a potential adjuvant that is as effective as GPI-0100 in potentiating serum IgG antibody activity. It is worth mentioning that 7 was injected as a clear solution (1.0 mg/mL), whereas 6 was less water-soluble than 7 and injected as a suspension (1.0 mg/mL) with the antigen.
Figure 2.

Serum IgG anti-rHagB activity at weeks 4 and 6. Data are expressed as geometric values. Horizontal bars indicate mean concentrations. Statistical significance compared to no-adjuvant control, **P < 0.01, and ***P < 0.001.
To understand the nature of the serum response (i.e., Th1 or Th2 immunity), we analyzed the IgG subclass antibody responses (Figure 3). The mice immunized with rHagB + GPI-0100 (100 μg) or rHagB + 7 (100 μg) showed similar IgG subclass profile, with significantly higher levels of IgG1 and IgG2a antibody activity than the mice receiving antigen alone at weeks 4 and 6. However, the mice immunized with rHagB + 6 (100 μg) showed significantly lower (P < 0.001) IgG1 production than those induced with rHagB + GPI-0100 (100 μg) or rHagB + 7 (100 μg) at week 4, but no significant difference was observed at week 6. In addition, significantly lower (P < 0.001) levels of IgG2a antibody activity were observed in the mice receiving antigen + 6 (100 μg) than in the mice receiving rHagB + GPI-0100 (100 μg) or rHagB + 7 (100 μg) at weeks 4 and 6.
Figure 3.

Serum IgG1 (A) and IgG2a (B) anti-rHagB activity at weeks 4 and 6. Data are expressed as geometric values. Horizontal bars indicate mean concentrations. Statistical significance compared to no-adjuvant control, **P < 0.01, and ***P < 0.001.
A group of mice immunized with rHagB + 7 at a dose of 50 μg was also included in this study to evaluate its dose-dependence. There was no significant difference (in terms of IgG, IgG1, and IgG2a responses) among the mice groups receiving rHagB + 7 at 50 μg, 100 μg, or GPI-0100 at 100 μg, respectively. We also evaluated the adjuvant activity of a mixture of 5 (25 μg) + 6 (25 μg) + 7 (50 μg) to investigate whether there would be an additive or synergistic effect. The mice immunized with rHagB and the mixture had significantly higher serum IgG anti-rHagB activity than the mice receiving rHagB alone at weeks 4 and 6 (P < 0.001) and the mice receiving rHagB + 6 (100 μg) at weeks 4 (P < 0.001) and 6 (P < 0.05). However, they showed no significant difference from the mice receiving rHagB + 7 at 50 μg, 100 μg, or GPI-0100 at 100 μg. Assessment of the IgG subclass responses revealed that the mixture potentiated similar levels of IgG1 and IgG2a anti-rHagB responses as those by adjuvant 7 at 50 μg or 100 μg (Figure 3). These results suggest that QS-21 analogues 5 and 6 at the dose combination used do not enhance the adjuvant activity of QS-17/18 analogue 7, nor do they modify the Th1/Th2 profile induced by 7 with rHagB. No sign of toxicity was observed in all of the mice based on weight monitoring (Figure S1 in the Supporting Information).
CONCLUSIONS
We synthesized a QS-17/18 analogue (7) and evaluated its adjuvant activity in the formulation with rHagB antigen in a mice immunization model. For the synthesis of the branched pentasaccharide, we used the two-stage allyl-glycoside-donor-activation approach for glycosidic bond construction, which only employed allyl glycoside building blocks. On the basis of the IgG1 and IgG2a assessments, adjuvant 7 has an adjuvant activity profile similar to that of GPI-0100 and thus might potentiate a mixed Th1- and Th2-like response to rHagB. Their profiles are different from those of QS-21 analogues 5 and 6 that might only induce a Th2-like immunity. Combination of QS-21 and QS-17/18 analogues did not enhance IgG1 or IgG2a production synergistically. These results suggest that compound 7 might be the main active component of the semisynthetic vaccine adjuvant GPI-0100, and it might be responsible for the favorable adjuvant activity and low toxicity of GPI-0100. Immunostimulant 7 can be a structurally defined alternative to GPI-0100, and it also serves as a structurally defined new lead for further SAR studies in search for a new saponin-base adjuvant with better adjuvant properties.
EXPERIMENTAL SECTION
Chemistry
General. Organic solutions were concentrated by rotary evaporation at ca. 12 Torr. Flash column chromatography was performed employing 230–400 mesh silica gel. Thin-layer chromatography was performed using glass plates precoated to a depth of 0.25 mm with 230–400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Infrared (IR) data are presented as frequency of absorption (cm−1). Proton and carbon-13 nuclear magnetic resonance (1H NMR or 13C NMR) spectra were recorded on 400 and 700 MHz NMR spectrometers; chemical shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3: δ = 7.26). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and/or multiple resonances, AB = AB quartet), coupling constant in hertz, and integration. Anhydrous solvents tetrahydrofuran (THF), acetonitrile (MeCN), and dichloromethane (DCM) were used without distillation. Solvents for workup and column chromatography, such as petroleum ether (PE), hexanes (Hex), ethyl acetate (EA), methanol (MeOH), benzene, toluene (Tol), and triethylamine (TEA), and other chemicals such as trichloroacetonitrile (Cl3CCN), cesium carbonate (Cs2CO3), TFA, and DIPEA were obtained from commercial vendors and used without further purification. The purity of all synthetic intermediates and products was measured by 1H NMR and was found to be ≥95%. The purity of all compounds evaluated in the immunological study was determined by a combination of HPLC, HRMS, and 1H NMR and found to be ≥95%.
All in vivo studies were performed in accordance with local IACUC guidelines.
Preparation of QS-17/18 Analogue 7
Compound 24 (74.0 mg, 19.6 μmol) and Pd/C (100 mg, 10% w/w) were mixed in 2.0 mL of THF/MeOH (1:1). The reaction mixture was shaken under H2 atmosphere (55 psi) for 24 h, filtered through a Celite plug, and concentrated. The debenzyled intermediate 9 (50.0 mg, 91%) was obtained as a white solid. A quarter of the intermediate (12.0 mg, 4.3 μmol) was treated with dodecylamine (2.4 mg, 12.9 μmol), HATU (5.0 mg, 12.9 μmol), and DIPEA (4.5 μL, 25.9 μmol) in 0.5 mL of chloroform at room temperature for 20 h. The reaction mixture was then concentrated and purified directly with flash column chromatography on silica gel (PE/EA gradients, 25–40% EtOAc) to provide the desired amide intermediate. The amide intermediate in 0.2 mL ofDCM cooled in an ice bath was treated with 0.5 mL of TFA/water (4:1, v/v) precooled to 0 °C. The reaction solution was stirred at 0 °C for 40 min and then concentrated to dryness at 0 °C. To the residue were added 0.5 mL of MeOH and K2CO3 (20 mg, 0.14 mmol). The reaction mixture was stirred at room temperature for 17 h. The white suspension was centrifuged, and the clear solution was acidified with AcOH, concentrated, and purified with RP-HPLC (H2O/MeCN gradients, 90–10%). The product fraction was concentrated on a rotary evaporator at room temperature to remove MeCN, and the remaining water was then removed on a lyophilizer to provide the final product 7 (5.9 mg, 82% from 9) as a white solid. 1H NMR (700 MHz, CD3OD) (characteristic protons): δ 9.50 (s, 1H), 5.33 (d, J = 1.7 Hz, 1H), 5.32 (m, 1H), 5.30 (d, J =8.1 Hz, 1H),4.83 (d, J = 7.6 Hz, 1H), 4.75 (d, J = 80 Hz, 1H), 4.60 (d, J = 7.8 Hz, 1H), 4.57 (d, J = 7.7 Hz, 1H), 4.54 (d, J = 7.5 Hz, 1H), 4.50 (s, 1H), 4.47 (d, J = 7.4 Hz, 1H), 4.29 (dd, J = 2.9, 1.9 Hz, 1H), 2.97 (dd, J = 14.7, 3.8 Hz, 1H), 2.33 (t, J = 13.4 Hz, 1H), 1.42 (s, 3h), 1.24 (d, J = 6.4 Hz, 3H), 1.20 (s, 3H), 1.04 (s, 3H), 0.98 (s, 3H), 0.95 (t, J = 6.3 Hz, 3H), 0.91 (s,3H), and 0.78 (s, 3H); 13CNMR (176 MHz, [D6]DMSO): δ 210.9, 175.5, 167.9, 166.1, 143.7, 121.8, 104.7, 103.9, 103.7, 103.1, 103.0, 102.8, 100.9, 93.9, 86.8, 84.8, 84.4, 80.9, 79.6, 77.4, 77.09, 77.0, 76.9, 76.8, 76.5, 76.1, 75.3, 74.5, 74.3, 74.2, 74.1, 73.9, 73.7, 73.65, 73.2, 72.1, 71.6, 71.1, 70.4, 69.9, 69.8, 69.6, 69.1, 68.58, 68.54, 67.9, 66.3, 66.2, 65.7, 61.5, 60.3, 54.4, 49.1, 48.3, 48.2, 47.0, 46.4, 41.4, 41.0, 38.6, 38.1, 36.0, 35.4, 35.2, 33.3, 32.3, 31.79, 31.76, 31.1, 30.6, 29.5, 29.4, 29.3, 29.2, 26.9, 26.7, 24.7, 23.3, 22.60, 22.56, 20.2, 18.6, 17.1, 16.7, 15.9, 14.5, and 10.9; IR (neat): 3381, 2925, 2855, 1676, and 1437; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C87H144NO40, 1842.9265; found, 1842.9253.
Allyl 2,3,4-Tri-O-benzyl-β-D-xylopyranosyl-(1 → 3)-2,4-di-O-beznyl-β-D-xylopyranosyl-(1 → 4)-2-O-benzyl-β-L-rhamnopyranoside (13)
The known trisaccharide 12 (959.0 mg, 1.0 mmol) was treated with 1.5 mL of TFA/H2O (4:1) and stirred at 0 °C. After 45 min, the reaction flask was connected to vacuum and the reaction solution was dried at 0 °C for 2.5 h. The intermediate, allyl 2,3,4-tri-O-benzyl-β-D-xylopyranosyl-(1 → 3)-2,4-di-O-beznyl-β-D-xylopyranosyl- (1 → 4)-β-L-rhamno-pyranoside, was obtained as a pure product (870.0 mg, 95%) and was used in the next step without further purification. To the trisaccharide intermediate (870.0 mg, 0.946 mmol) in DCM (27 mL) was added 9.0 mL of 20% NaOH (aq), nBu4NBr (61.0 mg, 0.189 mmol), and benzyl bromide (1.6 g, 9.5 mmol). The reaction solution was stirred at room temperature for 2 days. The two layers were separated, and the organic layer was concentrated and purified with flash column chromatography to provide 13 (785.0 mg, 83%) as a colorless oil. Rf = 0.33 (PE/EA 3:1); (c 2.0, CHCl3); 1H NMR (700 MHz, CDCl3): δ 7.40–7.20 (m, 30H), 5.85 (m, 1H), 5.24 (dq, J = 17.0, 1.4 Hz, 1H), 5.16 (dd, J = 10.5,1.4 Hz, 1H), 5.16 (m, 1H), 4.95 (AB1, J = 11.9 Hz, 1H), 4.91 (d, J = 7.7 Hz, 1h), 4.90 (AB2, J = 11.2 Hz, 1H), 4.87 (AB2, J = 11.2 Hz, 1H),4.81 (AB1, J = 11.9 Hz, 1H), 4.80 (d, J = 1.4 Hz, 1H), 4.78 (AB3, J = 9.8 Hz, 1H), 4.77 (AB4, J = 11.9 Hz, 1H), 4.70 (d, J = 7.7 Hz, 1H), 4.69 (AB5, J = 12.6 Hz, 1H), 4.66 (AB6, J = 12.6 Hz, 1H), 4.64 (AB5, J = 12.6 Hz, 1H), 4.63 (ab6, J = 12.6 Hz, 1H), 4.78 (AB3, J = 9.8 Hz, 1H), 4.60 (AB4, J = 11.9 Hz, 1H), 4.12 (ABMX2, J = 13.3, 4.9, 2.1 Hz, 1H), 3.96 (t, J = 8.8 Hz, 1H), 3.94–3.86 (m, 4H), 3.72 (dd, J = 3.5, 1.4 Hz, 1H), 3.65–3.58 (m, 2h), 3.56 (t, J = 9.5 Hz, 1H), 3.52–3.49 (m, 2H), 3.37 (dd, J = 9.1, 7.7 Hz, 1H), 3.32 (dd, J = 8.8, 7.7 Hz, 1H), 3.20(dd, J = 11.6, 10.2 Hz, 1H),3.04(dd, J = 11.9, 10.5 Hz, 1H), and 1.31 (d, J = 6.3 Hz, 3H); 13C NMR (176 MHz, CDCl3): δ 138.8, 138.7, 138.6, 138.3, 138.0, 137.9, 133.9, 128.7, 128.61, 128.60, 128.50, 128.47, 128.41, 128.36, 128.12, 128.10, 128.04, 128.02, 127.98, 127.78, 127.74, 127.6, 117.4, 104.2, 103.1, 96.6, 84.0, 83.2, 82.7, 81.7, 79.8, 78.30, 78.26, 75.7, 75.4, 75.3, 75.2, 73.4, 73.3, 73.2, 71.8, 67.9, 66.8, 64.1, 63.9, and 17.8; IR (neat): 3054, 2986, 2933, 1497, and 1454; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C61H68NaO13, 1031.4558; found, 1031.4534.
Allyl 2-O-Benzyl-3-O-[2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl-(1 → 3)]-4-O-[2,3,4-tri-O-benzyl-β-D-xylopyranosyl-(1 → 3)-2,4-di-O-beznyl-β-D-xylopyranosyl-(1 → 4)]-β-L-rhamnopyranoside (15)
Allyl 2,3,4,6-tetra-O-benzyl-D-glucopyranoside (3.5 g, 6.0 mmol) in 12.0 mL of anhydrous DMSO was treated with potassium tert-butoxide (1.5 g, 13.0 mmol) at room temperature. The reaction mixture was stirred for 20 h and worked up with water and ethyl acetate. The product, vinyl 2,3,4,6-tetra-O-benzyl-D-glucopyranoside (3.3 g, 94%), was used directly as the glycosyl donor in the next step of glycosylation without further purification. The glycosyl donor, vinyl 2,3,4,6-tetra-O-benzyl-D-glucopyranoside (775.0 mg, 1.335 mmol), and glycosyl acceptor 13 (898.0 mg, 0.898 mmol) were dried together by azeotropic removal of water with toluene under vacuum at room temperature. To the mixture, anhydrous acetonitrile (70 mL) was added. The clear solution was then cooled in an ice-salt water bath (−10 °C) and treated with NIS (306.0 mg, 1.335 mmol), followed immediately by a catalytic amount of TfOH (8.0 μL, 0.07 mmol). The reaction solution turned to light golden color and was quenched with TEA after 20 min. The reaction mixture was concentrated, and the residue was purified with flash column chromatography (PE/EA gradients) to provide 15 (762.0 mg, 55%) with the desired β anomeric configuration for the newly formed glycosidic bond and the corresponding α anomer (110.0 mg, 8%). Rf = 0.66 (PE/EA 3:1); (c 5.2, CHCl3); 1H NMR (700 MHz, CDCl3): δ 7.40–7.15 (m, 50H), 5.78 (m, 1H), 5.20 (dq, J = 17.5, 1.4 Hz, 1H), 5.10 (dd, J = 10.5, 1.4 Hz, 1H), 5.04 (d, J = 7.7 Hz, 1H), 5.00 (AB1, J = 11.9 Hz, 1H), 4.91 (AB2, J = 11.9 Hz, 1H), 4.89 (AB3, J = 11.2 Hz, 1H), 4.84–4.63 (m, 15H), 4.60 (d, J = 7.6 Hz, 1H), 4.57 (AB, J = 11.7 Hz, 1H), 4.54 (AB, J = 12.5 Hz, 1H), 4.52 (AB, J = 12.7 Hz, 1H), 4.46 (AB, J = 12.3 Hz, 1H), 4.43 (AB, J = 12.3 Hz, 1H), 4.11–4.07 (m, 2H), 3.95 (dd, J = 11.6, 5.3 Hz, 1H), 3.93–3.89 (m, 2H), 3.87 (AB, J = 8.9 Hz, 1H), 3.79 (dd, J = 3.3, 2.0 Hz, 1H), 3.67–3.57 (m, 3H), 3.51 (t, J = 8.8 Hz, 1H), 3.49–3.38 (m, 5H), 3.36 (dd, J = 8.8, 7.6 Hz, 1H), 3.30 (t, J = 8.4 Hz, 1H), 3.19–3.15 (m, 2H), 2.58 (t, J = 11.0 Hz, 1h), 2.55 (s, broad, 1H), and 1.30 (d, J = 6.2 Hz, 3H); 13C NMR (176 MHz, CDCl3): δ 138.8, 138.69, 138.67, 138.66, 138.59, 138.50, 138.4, 138.2, 134.0, 128.41, 128.36, 128.26, 128.23, 128.19, 128.17, 128.14, 128.08, 127.9, 127.8, 127.74, 127.72, 127.55, 127.48, 127.46, 127.44, 127.39, 127.36, 127.34, 127.32, 127.2, 127.1, 116.6, 103.1, 102.5, 101.7, 97.8, 84.4, 83.6, 83.4, 82.4, 82.3, 79.3, 78.3, 78.0, 77.8, 76.4, 75.7, 75.6, 75.4, 74.6, 74.5, 74.34, 74.30, 73.5, 73.2, 73.1, 72.5, 68.7, 67.5, 67.1, 63.7, 63.3, and 17.9; IR (neat): 3063, 3030, 2870, 1497, and 1454; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C98H102NaO18, 1553.6964; found, 1553.6963.
Allyl 3,4-Di-O-acetyl-α-D-fucopyranoside (16)
To the known allyl 3,4-O-isoproylidene-α-D-fucopyranoside (18) (2.42 g, 9.9 mmol) dissolved in 30.0 mL of DCM was added levulinic acid (2.31 g, 19.9 mmol), DCC (4.06 g, 19.7 mmol), and DMAP (121.0 mg, 0.99 mmol) at room temperature. The reaction completed after 15 h, and the solvent was evaporated under vacuum. The residue was treated with NaHCO3 (aq) and ethyl acetate. The mixture was filtered, and the filtrate was separated. The organic phase was concentrated and purified with flash column chromatography (PE/EA gradient) to provide allyl 3,4-O-isoproylidene-2-levulinoyl-α-D-fucopyranoside (19) (2.9 g, 85%). The intermediate (1.9 g, 5.6 mmol) in 5.0 mL of AcOH/H2O (4:1) was stirred at 60 °C for 1 h, and the reaction solution was concentrated. The dried residue was dissolved in 6.0 mL of DCM and kept in an ice-water bath. To the solution was added Ac2O (1.8 mL, 19.0 mmol), TEA (3.4 mL, 24.0 mmol), and DMAP (36.0 mg, 0.3 mmol). The reaction completed in 3 h, and the solvent was removed. The crude reaction mixture dissolved in 13 mL of allyl alcohol was treated with 1.3 mL of hydrazine monohydrate and stirred at room temperature for 10 min. The reaction was quenched with acetone, concentrated, and purified with flash column chromatography (PE/EA gradients) to provide 16 (1.2 g, 75%) as a colorless oil. Rf = 0.33 (PE/EA 1:1); (c 1.4, CHCl3); 1H NMR (700 MHz, CDCl3): δ 5.92 (m, 1H), 5.20 (dq, J = 17.5, 1.4 Hz, 1H), 5.26–5.22 (m, 2H), 5.15 (dd, J = 10.4, 3.3 Hz, 1H), 4.98 (d, J = 3.9 Hz, 1H), 4.24 (dd, J = 12.7, 5.3 Hz, 1H), 4.13 (q, J = 6.5 Hz, 1H), 4.06 (ddd, J = 13.4, 6.3, 1.0 Hz, 1H), 3.94 (dd, J = 10.4, 3.9 Hz, 1H), 2.16 (s, 3H), 2.06 (s, 3H), and 1.14 (d, J = 6.6 Hz, 3H); 13C NMR (176 MHz, CDCl3): δ 170.8, 170.5, 133.3, 118.2, 97.7, 71.3, 71.2, 68.8, 66.9, 64.9, 20.8, 20.6, and 15.9; IR (neat): 3464, 2985, 2939, 1740, and 1369; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C13H20NaO7, 311.1107; found, 311.1109.
Allyl 3,4-Di-O-acetyl-2-{2-O-benzyl-3-O-[2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl-(1 → 3)]-4-O-[2,3,4-tri-O-benzyl-β-D-xylopyranosyl-(1 → 3)-2,4-di-O-beznyl-β-D-xylopyranosyl-(1 → 4)]-β-L-rhamnopyranosyl-(1 → 2)}-α-D-fucopyranoside (17)
Tetrasaccharide 15 (860.0 mg, 0.561 mmol) in 12 mL of DMSO with potassium tert-butoxide (1.257 g, 11.227 mmol) was stirred at room temperature for 21 h. The reaction solution was poured into water (75 mL) and extracted with ethyl acetate (75 mL). The organic layer was washed with water (2 × 75 mL), dried over anhydrous Na2SO4, and concentrated. The crude product was purified by passing through a silica gel plug to provide the vinyl tetrasaccharide intermediate (746.0 mg, 87%). The vinyl tetrasaccharide donor (450.0 mg, 0.29 mmol) and acceptor 16 (127.0 mg, 0.44 mmol) were dried together by azeotropic removal of water with toluene under vacuum at room temperature. To the mixture, anhydrous acetonitrile (15 mL) was added, and a clear solution was obtained. To the reaction solution, NIS (80.0 mg, 0.35 mmol) was added, followed immediately by a catalytic amount of TfOH (1.3 μL, 0.015 mmol). The reaction was quenched with TEA after 20 min. The reaction mixture was concentrated, and the residue was purified with flash column chromatography (PE/EA gradients) to provide 17 (261.0 mg, 51%). Rf =0.30 (PE/EA2:1); (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.40–7.10 (m, 50H), 5.86 (m, 1H), 5.35 (dd, J = 10.5, 3.3 Hz, 1h), 5.29 (dq, J = 17.2, 1.5 Hz, 1H), 5.22 (d, J = 3.2 Hz, 1H), 5.07 (d, J = 10.3 Hz, 1H), 5.04 (d, J = 7.1 Hz, 1H), 5.00 (d, J = 3.6 Hz, 1H), 4.96 (AB1, J = 12.1 Hz, 1h), 4.90 (AB2, J = 11.6 Hz, 1H), 4.87 (AB3, J = 10.8 Hz, 1H), 4.82–4.63 (m, 13H), 4.61 (AB, J = 10.4 Hz, 1H), 4.58–4.47 (m, 6H), 4.35 (AB, J = 12.0 Hz, 1H), 4.10–4.02 (m, 3H), 4.00–3.82 (m, 5H), 3.76 (m, 1H), 3.70 (t, J = 3.0 Hz, 1H), 3.68–3.29 (m, 10H), 3.19–3.12 (m, 2H), 2.12 (s, 3H), 1.63 (s, 3H), 1.27 (d, J = 6.8 Hz, 1h), and 1.11 (d, J = 6.6 Hz, 1H); 13CNMR (176 MHz, CDCl3): δ 170.5, 169.8, 138.75, 138.66, 138.58, 138.49, 138.40, 138.1, 133.4, 128.5, 128.41, 128.38, 128.33, 128.29, 128.26, 128.22, 128.16, 128.14, 128.12, 128.09, 128.0, 127.84, 127.82, 127.76, 127.75, 127.71, 127.63, 127.59, 127.55, 127.52, 127.50, 127.4, 127.3, 127.29, 127.27, 118.1, 103.1, 101.5, 97.5, 84.7, 83.7, 82.4, 82.1, 79.3, 78.0, 75.7, 75.6, 75.5, 74.7, 74.6, 74.4, 74.3, 74.1, 73.9, 73.2, 73.1, 72.6, 71.5, 69.2, 68.7, 68.6, 67.2, 64.2, 63.7, 63.5, 60.4, 21.0, 20.7, 20.4, 15.8, and 14.2; IR (neat): 3032, 2870, 1721, and 1496; HRMS (ESI-TOf) m/z: [M + Na]+ calcd for C105H116NaO24, 1783.7754; found, 1783.7704.
Preparation of 24
A solution of [Ir(C0D)(PMePh2)2]PF6 (15.0 mg, 0.02 mmol) in 3.0 mL of THF was degassed and stirred under hydrogen atmosphere for 15 min at room temperature. The obtained light yellow clear solution of the activated catalyst was injected into 17 (173.0 mg, 0.10 mmol), and the reaction mixture was stirred at room temperature for 2 h. To the reaction solution was added drops of water and NIS (70.0 mg, 0.33 mmol). The reaction completed in 1 h. The crude product was obtained after removal of solvent under vacuum. The combined crude products (from two other runs using 32 and 70 mg of 17, respectively) were purified with flash column chromatography (Rf = 0.3, PE/EA 2:1). The hemiacetal intermediate in DCM was treated with Cs2CO3 (36.0 mg, 0.11 mmol) and trichloroacetonitrile Cl3CCN (2.0 mL, 20.0 mmol), and the reaction mixture was stirred at room temperature for 3 days. The reaction mixture was centrifuged, and the clear solution was concentrated and purified with flash column chromatography (PE/EA gradients) to provide the desired imidate 23 (182.0 mg, 62% over three steps). Imidate donor 23 (37.0 mg, 0.020 mmol) and fully protected quillaic acid-trisaccharide conjugate 11 (50.0 mg, 0.030 mmol) were dried together by azeotropic removal of water with toluene under vacuum at room temperature. The mixture dissolved in DCM (1.5 mL) was kept at −78 °C and treated with BF3·OEt2 (5.0 μL, 0.020 mmol). The reaction completed in 4 h and was then quenched with TEA at −78 °C. The reaction solution was concentrated, and the crude mixture was purified with flash column chromatography (PE/EA gradients) to provide 24 (53.7 mg, 72%) as a white amorphous solid. Rf = 0.5 (PE/EA 3:1); (c = 1.8, CHCl3); 1H NMR (400 MHz, CDCl3): δ 9.70 (s, 1H), 7.40–7.10 (m, 55H), 5.45 (d, J = 7.7 Hz, 1H), 5.33 (s, br, 1H), 5.27 (AB1, J = 12.4 Hz, 1H), 5.10 (AB1, J = 12.4 Hz, 1H), 5.08–5.05 (m, 2H), 4.92–4.86 (m, 4h), 4.82 (dd, J = 9.6, 2.9 Hz, 1h), 4.81–4.71 (m, 8H), 4.68–4.62 (m, 4h), 4.60 (d, J = 11.9 Hz, 1H), 4.56 (d, J = 7.4 Hz, 1h), 4.54–4.46 (m, 4h), 4.44 (d, J = 11.4 Hz, 1h),4.43 (d, J = 7.3 Hz, 1H), 4.39 (AB, J = 11.9 Hz, 1H), 4.19 (d, J = 7.3 Hz, 1h), 3.97 (d, J = 8.3 Hz, 1H), 3.943.78 (m, 9H), 3.77–3.72 (m, 2H), 3.67–3.64 (m, 2H), 3.62–3.55 (m, 5H), 3.52–3.46 (m, 5H), 3.44–3.32 (m, 7H), 3.25 (t, J = 8.1 Hz, 2H), 3.15–3.11 (m, 3H), 2.97 (d, J = 13.8 Hz, 1H), 2.44 (m, 2H), 2.26 (t, J = 13.3 Hz, 1H), 2.13 (s, 3h); 1.91–1.52 (m, 18H), 1.46–1.11 (m, 27H), and 1.09–0.60 (m, 137h); 13C NMR (176 MHz, CDCl3): δ 212.6, 176.1, 170.5, 169.8, 168.4, 143.1, 138.9, 138.8, 138.7, 138.6, 138.54, 138.49, 138.41, 138.2, 135.3, 128.6, 128.5, 128.4, 128.33, 128.27, 128.24, 128.21, 128.19, 128.13, 128.11, 128.0, 127.9, 127.8, 127.7, 127.59, 127.58, 127.53, 127.38, 127.35, 127.32, 129.29, 122.0, 103.6, 103.0, 102.1, 101.6, 101.4, 100.8, 99.9, 93.4, 86.2, 84.5, 83.63, 83.58, 82.6, 82.2, 79.2, 78.9, 78.8, 78.7, 78.0, 77.7, 76.7, 76.4, 76.0, 75.9, 75.8, 75.6, 75.5, 75.1, 74.9, 74.62, 74.58, 74.3, 74.2, 73.4, 73.1, 73.0, 72.60, 72.57, 72.51, 71.4, 71.1, 70.1, 69.0, 68.8, 68.3, 66.8, 65.3, 63.7, 63.4, 60.4, 53.9, 49.4, 49.0, 46.6, 46.1, 41.5, 41.2, 40.0, 38.1, 36.1, 35.0, 34.4, 32.9, 32.5, 30.7, 30.6, 29.7, 26.5, 25.4, 24.7, 23.5, 20.7, 20.5, 20.3, 18.2, 17.5, 15.9, 14.2, 12.2, 7.6, 7.5, 7.3, 7.2, 7.13, 7.06, 6.98, 6.94, 6.85, 6.78, 5.9, 5.6, 5.43, 5.36, 5.33, 5.25, 5.22, 5.12, 5.06, 5.0, 4.96, and 4.4; IR (neat): 2952, 2912, 2876, 1750, and 1455; MS (MALDI) m/z: [M + Na]+ (rel. intens) calcd for C210H314NaO43Si9, 3799.0205 (39.0%), 3800.0201 (17.8%), 3800.0239 (88.5%), 3801.0173 (11.7%), 3801.0272 (100.0%), 3801.0234 (40.4%), 3802.0207 (26.7%), 3802.0268 (45.7%); 3802.0306 (74.9%), 3803.0203 (10.8%), 3803.0241 (30.1%), 3803.0301 (34.2%), 3803.0339 (41.5%), 3804.0236 (12.2%), 3804.0274 (22.6%), 3804.0335 (19.0%), 3804.0373 (17.5%), and 3805.0308 (12.5%), found 3799.246, 3800.237, 3801.215, 3802.210, 3830.228, 3804.202, and 3805.253.
Mice and Immunization
The BALB/c mice used in this study were purchased from Frederick Cancer Research (Frederick, MD) and maintained within an environmentally controlled, pathogen-free animal facility at the University of Alabama at Birmingham (UAB). To assess the adjuvant activity of the QS saponin-based immune adjuvants, groups of female mice (8–10 weeks of age; 5 mice per group) were immunized by the subcutaneous (s.c.) route with rHagB (20 μg) along, GPI-0100 (100 μg), synthetic adjuvant 7 (100 μg or 50 μg), or with a mixture of synthetic adjuvants on days 0, 14, and 28. The mice were weighed, and blood samples were collected before and at various time points following the initial immunization. Blood samples were collected from the retro-orbital plexus by using heparinized capillary pipettes. The serum was obtained after centrifugation and stored at −20 °C until assayed. All experiments were performed according to the National Institute of Health guidelines, and protocols were approved by the UAB Institutional Animal Care and Use Committee.
Enzyme-Linked Immunosorbent Assay
The levels of specific serum IgG and IgG subclasses against rHagB in each group were determined by ELISA. Maxisorpmicrotiter plates (NUNC International, Roskilde, Denmark) were coated with rHagB (1 μg/mL) or with optimal amounts of goat antimouse IgG, IgG1, or IgG2a (Southern Biotechnology Associates, Inc., Birmingham, AL) in borate buffer saline (BBS; 100mMNaCl, 50 mM boric acid, 1.2 mMNa2B4O7,pH 8.2) at 4 °C overnight. Plates were blocked with 1% bovine serum albumin and 0.02% sodium azide in BBS for 2 h at room temperature. Serial two-fold dilutions of serum samples were made and were added in duplicate to the plates. To generate standard curves, serial dilutions of a mouse immunoglobulin reference serum (MP Biomedicals, Solon, OH) were added to two rows of wells in each plate that had been coated with the appropriate antimouse IgG or IgG subclass reagent. After incubation (overnight at 4 °C) and washing of the plates, horseradish peroxidase-conjugated goat antimouse IgG or IgG subclass antibody (Southern Biotechnology Associates, Inc.) was added to appropriate wells. After 4 h of incubation at room temperature, the plates were washed and developed by o-phenylenediamine substrate with hydrogen peroxide. Color development was recorded at 490 nm. The concentrations of antibodies were determined by interpolation on standard curves generated by using the mouse immunoglobulin reference serum and constructed by a computer program based on four-parameter logistic algorithms (Softmax/Molecular Devices Corp., Menlo Park, CA).
Statistical Analysis
Statistical significance in antibody responses and body weights between the groups was evaluated by AN0VA and the Tukey multiple-comparisons test using the InStat program (Graph Pad Software, San Diego, CA). Differences were considered significant at a P value <0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH (AI099407 and GM120159 to P.W.) for financial support. We also thank Dr. D. Devalankar for his contribution to the synthesis and Dr. M. J. Jablonsky for assistance with NMR spectroscopy.
ABBREVIATIONS
- QS
Quillaja saponaria
- IgG
immunoglobulin G
- Th
T helper cells
- CTL
cytotoxic T cell
- AIDS
acquired immunodeficiency syndrome
- SAR
structure—activity relationship
- api
apiose
- xyl
xylose
- glu
glucose
- Ir
iridium
- COD
cyclooctadiene
- P
phosphine
- Me
methyl
- Ph
phenyl
- F
fluoro
- NIS
N-iodosuccinimide
- TfOH
triflic acid
- HSQC
heteronuclear single quantum coherence
- 2D
two dimension
- NMR
nuclear magnetic resonance
- ppm
parts per million
- Pd
palladium
- Hz
hertz
- NaOH
sodium hydroxide
- nBu4NBr
tetra-n-butylammonium bromide
- BnBr
benzyl bromide
- DCM
dichloromethane
- t-BuOK
potassium tert-butoxide
- DMSO
dimethyl sulfoxide
- MeCN
acetonitrile
- THF
tetrahydrofuran
- DCC
N,N’-dicyclohexylcarbodiimide
- DMAP
4-dimethylaminopyridine
- TEA
triethylamine
- DIBAL-H
diisobutylaluminium hydride
- TMSOTf
trimethylsilyl triflate
- HATU
O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylehtylamine
- rHagB
recombinant hemagglutinin B
- sc
subcutaneous
- ELISA
enzyme-linked immunosorbent assay
- IACUC
International Animal Care and Use Committee
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01997.
Mice body weight plot, 1H and 13C NMR spectra of the new compounds 7, 13, 15–17, and 24 (PDF)
Molecular formula strings (CSV)
REFERENCES
- (1).Brito LA; O’Hagan DT Designing and building the next generation of improved vaccine adjuvants. J. Controlled Release 2014, 190, 563–579. [DOI] [PubMed] [Google Scholar]
- (2).Brunner R; Jensen-Jarolim E; Pali-Schöll I The ABC of clinical and experimental adjuvants-Abrief overview. Immunol. Lett 2010, 128, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Pasquale A; Preiss S; Silva F; Garcon N Vaccine adjuvants: from 1920 to 2015 and beyond. Vaccine 2015, 3, 320–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Didierlaurent AM; Laupeze B; Di Pasquale A; Hergli N; Collignon C; Garcon N Adjuvant system AS01: helping to overcome the challenges of modern vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [DOI] [PubMed] [Google Scholar]
- (5).Garcon N; Van Mechelen M Recent clinical experience with vaccines using MPL- and QS-21-containing adjuvant systems. Expert Rev. Vaccines 2011, 10, 471–486. [DOI] [PubMed] [Google Scholar]
- (6).Ragupathi G; Gardner JR; Livingston PO; Gin DY Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 2011, 10, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kensil CR Saponins as vaccine adjuvants. Crit. Rev. Ther. Drug. Carrier. Syst 1996, 13, 1–55. [PubMed] [Google Scholar]
- (8).Kensil CR; Liu G; Anderson C; Storey J Effects of QS-21 on Innate and Adaptive Immune Responses In Vaccine Adjuvants: Immunological and Clinical Principles; Hackett CJ, Harn DAJ, Eds.; Humana Press Inc.: Totowa, N. J., 2005; pp 221–234. [Google Scholar]
- (9).Cleland JL; Kensil CR; Lim A; Jacobsen NE; Basa L; Spellman M; Wheeler DA; Wu J-Y; Powell MF Isomerization and formulation stability of the vaccine adjuvant QS-21. J. Pharm. Sci 1996, 85, 22–28. [DOI] [PubMed] [Google Scholar]
- (10).Liu G; Anderson C; Scaltreto H; Barbon J; Kensil C R QS- 21 structure/function studies: effect of acylation on adjuvant activity. Vaccine 2002, 20, 2808–2815. [DOI] [PubMed] [Google Scholar]
- (11).Fernández-Tejada A; Tan DS; Gin DY Development of improved vaccine adjuvants based on the saponin natural product QS-21 through chemical synthesis. Acc. Chem. Res 2016, 49, 1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Marciani DJ; Press JB; Reynolds RC; Pathak AK; Pathak V; Gundy LE; Farmer JT; Koratich MS; May RD Development of semisynthetic triterpenoid saponin derivatives with immune stimulating activity. Vaccine 2000, 18, 3141–3151. [DOI] [PubMed] [Google Scholar]
- (13).Marciani D; Reynolds RC; Pathak AK; Finley-Woodman K; May RD Fractionation, structural studies, and immunological characterization of the semi-synthetic Quillaja saponins derivative GPI-0100. Vaccine 2003, 21, 3961–3971. [DOI] [PubMed] [Google Scholar]
- (14).Wang P; Dai Q; Thogaripally P; Zhang P; Michalek SM Synthesis of QS-21-based immunoadjuvants. J. Org. Chem 2013, 78, 11525–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Deng K; Adams MM; Gin DY Synthesis and structure verification of the vaccine adjuvant QS-7-Api. Synthetic access to homogeneous Quillaja saponaria immunostimulants. J. Am. Chem. Soc 2008, 130, 5860–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang P; Haldar P; Wang Y; Hu H Simple glycosylation of allyl glycosides. J. Org. Chem 2007, 72, 5870–5873. [DOI] [PubMed] [Google Scholar]
- (17).Wang Y; Liang X; Wang P Concise synthesis of Bacillus anthracis exosporium tetrasaccharide via two-stage activation of allyl glycosyl donor strategy. Tetrahedron Lett. 2011, 52, 3912–3915. [Google Scholar]
- (18).Wang Y; Zhang X; Wang P Facile glycosylation strategy with two-stage activation of allyl glycosyl donors. Application to concise synthesis of Shigella flexneri serotype Y O-antigen. Org. Biomol. Chem 2010, 8, 4322–4328. [DOI] [PubMed] [Google Scholar]
- (19).Yang H; Wang P Mechanistic study of glycosylation using a prop-1-enyl donor. J. Org. Chem. 2013, 78, 1858–1863. [DOI] [PubMed] [Google Scholar]
- (20).Baudry D; Ephritikhine M; Felkin H Isomerisation of allyl ethers catalysed by the cationic iridium complex [Ir(cyclo-octa-1,5-diene)(PMePh2)2] PF6. A highly stereoselective route to trans-propenyl ethers. Chem. Commun 1978, 694–695. [Google Scholar]
- (21).Oltvoort JJ; Van Boeckel CAA; De Koning JH; Van Boom JH Use of the cationic iridium complex 1,5-cyclooctadienebis-[methyldiphenylphosphine]iridium hexafluorophosphate in carbohydrate chemistry: smooth isomerization of allyl ethers to 1-propenyl ethers. Synthesis 1981, 305–308. [Google Scholar]
- (22).Schmidt RR; Michel J Facile synthesis of α- and β-O-glycosyl imidates; Preparation of glycosides and disaccharides. Angew. Chem., Int.Ed 1980, 19, 731–732. [Google Scholar]
- (23).Adams MM; Damani P; Perl NR; Won A; Hong F; Livingston PO; Ragupathi G; Gin DY Design and synthesis of potent Quillaja saponin vaccine adjuvants. J.Am. Chem. Soc 2010, 132, 1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Deng K; Adams MM; Damani P; Livingston PO; Ragupathi G; Gin DY Synthesis of QS-21-xylose: establishment of the immunopotentiating activity of synthetic QS-21 adjuvant with a melanoma vaccine. Angew. Chem., Int. Ed 2008, 47, 6395–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang P; Kim Y-J; Navarro-Villalobos M; Rohde BD; Gin DY Synthesis of the potent immunostimulatory adjuvant QS-21A. J. Am. Chem. Soc 2005, 127, 3256–3257. [DOI] [PubMed] [Google Scholar]
- (26).Katz J; Black KP; Michalek SM Host responses to recombinant hemagglutinin B of Porphyromonas gingivalis in an experimental rat model. Infect. Immun 1999, 67, 4352–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.