

Abstract
Background:
Parkinson’s disease is characterized by the progressive loss of dopamine neurons in the substantia nigra, leading to severe motor deficits. Although the disease likely begins to develop years before observable motor symptoms, the specific morphological and functional alterations involved are poorly understood.
Objectives:
MitoPark mice lack the gene coding for mitochondrial transcription factor A specifically in dopamine neurons, which over time produces a progressive decline of neuronal function and related behavior that phenotypically mirrors human parkinsonism. Our previous work identified a progressive decrease in cell capacitance in dopamine neurons from MitoPark mice, possibly suggesting reduced membrane surface area. We therefore sought to identify and quantify somatodendritic parameters in this model across age.
Methods:
We used whole-cell patch clamp and fluorescent labeling to quantify somatodendritic morphology of single, neurobiotin-fllled dopamine neurons in acutely isolated brain slices from MitoPark mice.
Results:
We found that MitoPark mice exhibit an adultonset, age-dependent reduction of neuritic branching and soma size in dopamine neurons. This decline proceeds similarly in MitoPark mice of both sexes, but does not begin until after the age that early decrements in ion channel physiology and behavior have previously been observed.
Conclusions:
A progressive and severe decline in somatodendritic morphology occurs prior to cell death, but is not responsible for the subtle decrements observable in the earliest stages of neurodegeneration. This work could help identify the ideal time window for specific treatments to halt disease progression and avert debilitating motor deficits in Parkinson’s patients.
Keywords: branching, dendrites, in vivo, MitoPark, soma
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by severe motor symptoms including rigidity, bradykinesia, and resting tremor.1,2 These decrements are primarily caused by a progressive loss of dopamine neurons within the substantia nigra,3–5 with an estimated 50% to 70% of neurons lost prior to overt motor impairment.6,7 Current treatments for PD primarily focus on improving motor symptoms, and frustratingly no current therapeutics prevent dopamine cell loss. An understanding of the mechanisms and the time course of progressive cellular decline is critical for developing treatments that halt disease progression before debilitating and life-threatening symptoms emerge.
Although numerous gene- and toxin-based rodent models of PD have been described,8 MitoPark mice are noteworthy in that they develop an adult-onset, robust parkinsonism that mirrors the slow, progressive course of PD seen in the majority of human Parkinson’s patients.9,10 Anatomical decline of dopamine neurons in MitoPark mice begins with a decrease of striatal innervation at 12 weeks of age and ends with mass neuronal death by around 30 weeks.10–12 MitoPark mice are produced by knocking out the gene for mitochondrial transcription factor A (Tfam) specifically in dopamine neurons and are excellent tools for exploring the structural and functional degeneration of single dopamine neurons in adult animals.10,13 Decrements in dopamine neuron function in these mice can be observed prior to both motor deficits10–12 and frank cell loss.10,14 In addition, dopamine neurons in MitoPark mice display an age-related decrease of cell capacitance and increase of input resistance,14,15 which could indicate a progressive decline in somatodendritic membrane surface area.
Although axonal degeneration in PD has been studied in some depth,16–19 work investigating somatodendritic degeneration in PD is less extensive. Dendrites are shorter in nigral neurons from postmortem PD patients,20 and soma volumes are decreased in nigral neuromelanin-containing neurons in postmortem samples when compared with controls of similar age.21 However, no study to date has quantified in vivo changes in morphology in a progressive animal model of PD, which is a necessary step in determining the sequence of events that occurs throughout degeneration of dopamine neurons.
In this study, we used fluorescent labeling and confocal imaging to quantify the morphological parameters of single nigral dopamine neurons in MitoPark mice throughout the development of parkinsonism. A decline in neurite branching and soma volume emerges at 16 to 20 weeks of age in MitoPark mice, which is later than established decrements in cellular physiology and behavior. These findings provide insight into the timing of morphological degeneration and could help identify targets for therapeutic strategies to slow or halt the progression of PD.
Methods
Animals
Male and female MitoPark mice (DAT+/cre × TfamloxP/loxP) and littermate controls (DAT+/+ × TfamloxP/loxP) were bred as described previously.14 One control experiment (Supplementary Fig. 5) was performed using 4-month-old C57Bl/6 mice from the National Institute on Aging aged rodent colony. All mice were group housed in ventilated standard shoebox cages with ad libitum access to food and water. Animal rooms were maintained on a reverse 12-hour light/dark cycle (lights off at 9:00 AM) with the temperature held at 26°C. Procedures were in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at University of Texas Health, San Antonio and the Oklahoma Medical Research Foundation.
Cellular Labeling
Mice aged 12 to 31 weeks were anesthetized with isoflurane and decapitated.22 The brains were resected and horizontal sections (200-μm thick) containing the substantia nigra were collected using a vibrating microtome (Leica, Wetzlar, Germany). The sections were maintained in artificial CSF + glutamate receptor antagonists (1.3-mM kynurenic acid and 10-μM MK-801) at 34°C, continuously bubbled with 95% O2/5% CO2.
Putative dopamine neurons were identified by location and appearance from the region of highest dopamine neuron density, and electrophysiological data were acquired from each cell. Substantia nigra dopamine neurons have characteristic electrophysiological parameters that are progressively disrupted in aged MitoPark mice14; therefore, the cells were not discarded unless they showed firing characteristics of GABA neurons in this region. Only 1 cell was excluded under these criteria. Neurons were dialyzed using pipettes (2.5–6 MΩ resistance) filled with an intracellular solution containing 0.2% neurobiotin and the following (in mM): 115 K-methylsulfate, 20 NaCl, 1.5 MgCl2, 10 HEPES, 10 BAPTA, 2 adenosine triphosphate, and 0.4 guanosine triphosphate, pH 7.35 to 7.40, 267 to 275 mOsm/L. Cell location was largely limited to the medial substantia nigra and is illustrated in Supplementary Figure 1.
Fluorescent Labeling and Imaging
Anatomical analysis was performed on horizontal sections postfixed in 4% paraformaldehyde. All processing was performed at room temperature. Slices were pretreated with 1% H2O2 for 15 minutes and washed with phosphate buffered saline (PBS). The sections were incubated in a 1:750 dilution of Streptavidin 488 (Abcam, Cambridge, UK) in PBS with 0.3% triton-x for 2 hours. After final washes in PBS, the brain sections were mounted on gelatin-coated slides and cover slips were applied using Prolong Gold antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA).
Z-stacks (2 μm between planes) were obtained using a Zeiss (Oberkochen, Germany) LSM 710 confocal microscope with a 20 × objective. Neurites were traced and soma volume calculated using the z-stacks. A single 2-dimensional image was then created by flattening the 3-dimensional skeleton of each neuron onto a 2-dimensional plane. Two images, one containing the cell body and the other containing the flattened traces, were overlaid to generate final representative images for each neuron. Neurites were traced using ImageJ software (National Institutes of Health, Bethesda, Maryland) plugin Simple Neurite Tracer.23 The neurite number was counted by hand, and cumulative neurite length and arborization were quantified from the 2-dimensional image. Neurite arborization was specifically quantified via Sholl analysis24 using the ImageJ Sholl plugin.25
Graphing and Statistical Analysis
Data were analyzed using Prism 7.0 software (GraphPad, La Jolla, California). Data are reported as means with error bars representing standard error of the mean, and values from individual neurons are overlaid as circles or squares. The D’Agostino & Pearson test was used to determine normality in morphological data sets. The Mann–Whitney U test was used to compare genotypes when normality failed in 1 or more groups, otherwise Welch’s corrected t-tests were used. Two-way analyses of variance were used for Sholl analysis of arborization, and all groups in that analysis were normally distributed.
Chemicals
Kynurenic acid, MK-801, NaCl, MgCl2, HEPES sodium salt, adenosine triphosphate, guanosine triphosphate, triton-x, and normal goat serum were obtained from Sigma-Aldrich (St. Louis, MO, USA). Neurobiotin was obtained from Vector Laboratories (Peterborough, UK). Streptavidin 488 was obtained from Abcam. K- methylsulfate was obtained from MP Biomedicals (Santa Ana, CA, USA). BAPTA tetrapotassium salt, 10X PBS, and ProLong Gold antifade mountant were obtained from Thermo Fisher Scientific.
Results
We used whole-cell patch clamp to dialyze individual substantia nigra dopamine neurons with neurobiotin tracer and enable an analysis of somatodendritic morphology in the brain slices from MitoPark mice and litter-mate controls. Based on the MitoPark literature, data were collected from mice at ages that roughly corresponded to predominately asymptomatic (12 weeks), early locomotor impairment (16–20 weeks), and severe locomotor impairment (27–31 weeks). Similar to previously published findings,14,15 dopamine neurons from MitoPark mice exhibited higher input resistance, as well as a lower cell capacitance that reached statistical significance by 16 to 20 weeks (Supplementary Fig. 2). Cellular morphology appeared unaffected in 12-week-old MitoPark mice (Fig. 1A). However, cell branching appeared to be reduced by 16 to 20 weeks of age (Fig. 1B), and somatic and dendritic morphology was dramatically altered in 27- to 31-week-old MitoPark mice (Fig. 1C) when compared with age-matched littermate controls.
FIG. 1.
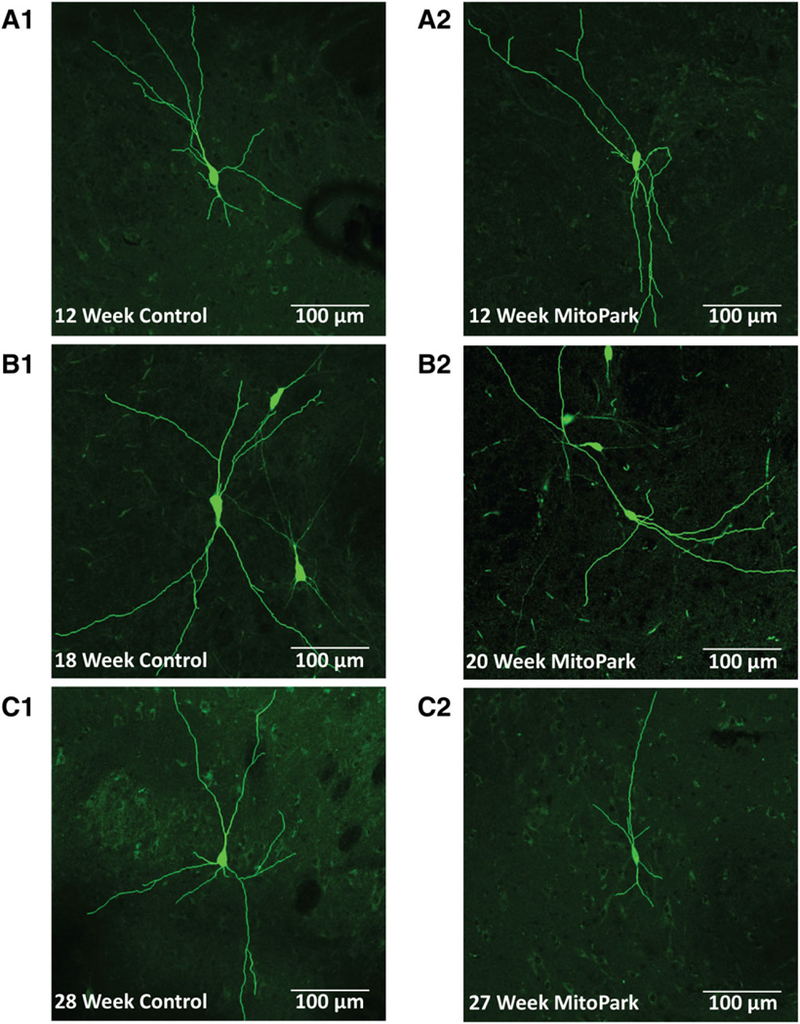
As age increases, somatodendritic morphology in dopamine neurons from MitoPark mice becomes disrupted. Substantia nigra dopamine neurons from 12-week-old MitoPark mice (A2) exhibited a similar appearance to neurons from littermate controls (A1). In contrast, the number of branches appeared to be reduced in 16–20-week old MitoPark mice (B2 vs B1). Furthermore, branch number and length as well as soma size were dramatically reduced in 27- to 31-week-old MitoPark mice (C2 vs C1). Neurons from control mice exhibited consistent morphology throughout all age groups.
We next sought to quantify deficits in somatodendritic morphology of dopamine neurons using multiple methods of analysis. Dopamine neurons from 12-week-old MitoPark mice exhibited a similar number of neurites as age-matched littermate controls (P = .895; Fig. 2A). Cumulative neurite length was also not significantly different between genotypes at 12 weeks (P = .690; Fig. 2B). Sholl analysis was next used to explore neurite arborization with greater precision. Using flattened 2-dimensional skeletons containing neurite traces, ImageJ software was used to create a series of concentric circles 20-μm apart starting from the soma center and extending up to 200 μm (Fig. 2C). When the number of circle-neurite intersections was analyzed in neurons from 12-week-old mice, there was no main effect of genotype (F1,32 = 0.473, P = .497) and no distance–genotype interaction (F9,288 = 0.228, P = .990; Fig. 2D). We next estimated soma volume by measuring the area of the soma in each plane within a z-stack and then multiplying by the distance between each plane of the stack. Somas from 12-week-old MitoPark mice appeared similar to those from age-matched littermate controls (Fig. 2E), consistent with the quantitative analysis of multiple neurons (P = .146; Fig. 2F). Overall, these data indicate that somatodendritic morphology of dopamine neurons was unaltered in 12-week-old MitoPark mice.
FIG. 2.
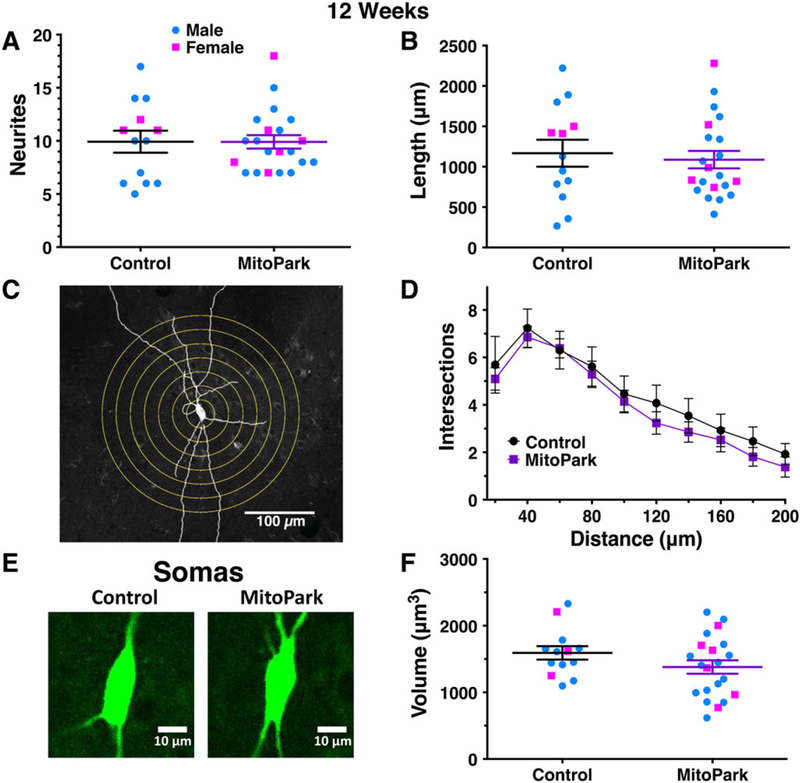
Dopamine neurons from MitoPark mice exhibit normal morphology at 12 weeks of age. Morphological parameters were quantified for individual dopamine neurons from 12-week-old male (blue circles) and female (pink squares) MitoPark mice and littermate controls. Neurite number (A) and cumulative neurite length (B) were not different between genotypes. Sholl analysis was performed by digitally drawing concentric circles at 20-μm intervals around the soma and counting neurite intersections (C). The number of intersections did not vary between genotypes (D). Soma size (E) was estimated by measuring the two-dimensional area through multiple z-planes. Soma volumes were not significantly different in MitoPark mice at this age (F).
We next compared dopamine neurons from mice that were 16 to 20 weeks of age, which is when locomotor decrements become more evident but the mice are still predominantly healthy. In this age range, neurite number was reduced in MitoPark mice when compared with littermate controls (P = .0011; Fig. 3A), as was cumulative neurite length (P = .0006; Fig. 3B). Sholl analysis indicated that neurite arbors were less extensive in MitoPark mice (F1,71 = 12.4, P = .0007) with no genotype–distance interaction (F9,639 = 0.8 1 7, P = .601; Fig. 3C). Soma volume was also reduced in neurons from 16- to 20-week-old MitoPark mice (P = .0016; Fig. 3D). Together these data indicate that neurite number and branching as well as somatic morphology are significantly impaired by 16 to 20 weeks of age in MitoPark mice.
FIG. 3.
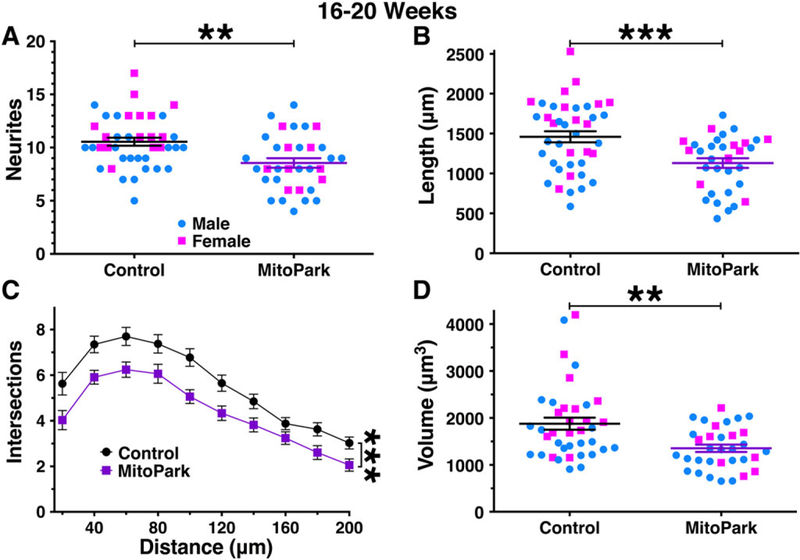
Morphological parameters are mildly reduced in dopamine neurons from 16- to 20-week-old MitoPark mice. By 16 to 20 weeks of age, dopamine neurons from male (blue circles) and female (pink squares) MitoPark mice exhibited a measurable impairment in somatodendritic morphology. This was observed for neurite count (A), cumulative neurite length (B), number of intersections/Sholl analysis (C), and soma volume (D). **P < .01; ***P < .001 between genotypes.
We then compared the morphology of dopamine neurons from 27- to 31-week-old MitoPark mice, an age at which dopamine neuron loss is severe and the mice have lost weight and appear in poor health.10 Indeed, despite our extensive experience performing patch clamp in the substantia nigra of aged mice,14,26 dopamine neurons were few in number and difficult to successfully patch in the MitoPark group. Regardless of the limited sample size, dopamine neurons from MitoPark mice exhibited a clear reduction in neurite number (P < .0001; Fig. 4A) and cumulative neurite length (P < .0001; Fig. 4B) when compared with age-matched littermate controls. Sholl analysis indicated a reduction in neurite arborization (F1,43 = 27.8, P < .0001), with a significant genotype–distance interaction (F9,387 = 6.34, P < .0001; Fig. 4C). In addition, neurite branching in the dopamine neurons from 27- to 31-week-old MitoPark mice did not increase 20 to 60 μm from the soma, as indicated by a significant genotype–distance interaction specifically within that range (F1,86 = 13.24, P = .0001). Finally, soma volume was also dramatically reduced in the neurons from MitoPark mice (P < .0001; Fig. 4D). Together these data indicate that, in addition to substantial death of dopamine neurons that has occurred in MitoPark mice by 27 to 31 weeks, the neurons that are still alive exhibit a severe impairment in somatodendritic morphology.
FIG. 4.
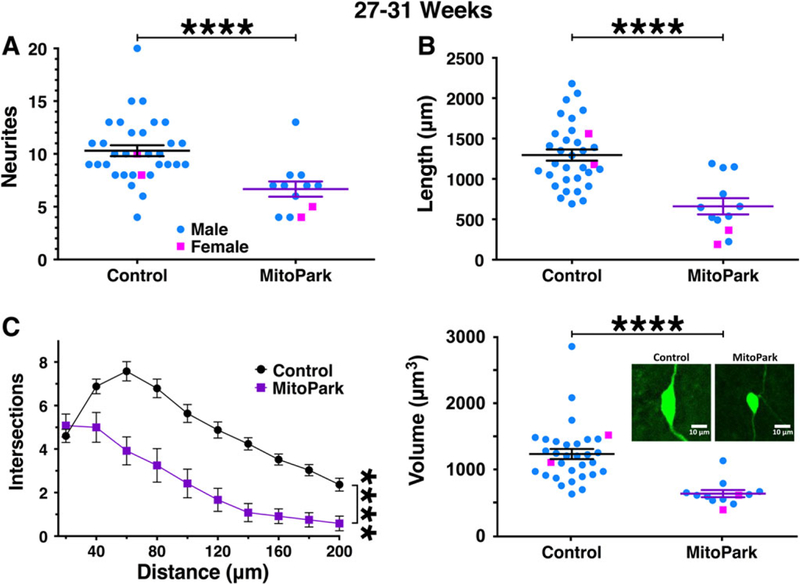
Morphological parameters are severely reduced in dopamine neurons from 27- to 31-week-old MitoPark mice. By 27 to 31 weeks of age, dopamine neurons from male (blue circles) and female (pink squares) MitoPark mice exhibited a severe impairment in somatodendritic morphology. This was observed for all parameters tested including neurite count (A), cumulative neurite length (B), and number of intersections/Sholl analysis (C). Soma volume was also severely reduced (D) with the inset showing representative 2-dimensional images from this age group. ****P < .0001 between genotypes.
To further explore the data we next analyzed morphology based on anatomical location. Cells were segregated into two separate analyses. All cells were divided along both dorsal-ventral and medial-lateral planes, with the caveat that the cells were all located in the medial substantia nigra27 (see Supplementary Fig. 1). As expected, morphology across anatomical location was consistent at 12 weeks and universally disrupted in 27- to 31-week-old MitoPark mice (not shown). An analysis of 16- to 20-week-old mice indicated that neurites from lateral cells may be more susceptible to damage than medial cells (Supplementary Fig. 3). Furthermore, there appeared to be a group of ventromedial neurons with large somas that were not present in MitoPark mice at this age.
We next explored the relationship between electrophysiological and cellular parameters from the oldest and youngest groups. As expected, morphological parameters generally showed a negative correlation with input resistance and a positive correlation with cell capacitance (Table 1). Soma size exhibited a strong positive association with H-current amplitude. When broken apart by genotype, neurons from the control mice showed a negative association between input resistance and neuritic parameters that was not present in MitoPark mice. In contrast, altered soma volumes were associated with all 3 physiological parameters in neurons from MitoPark mice.
TABLE 1.
Correlations between electrophysiological and morphological parameters in single neurons,
| All cells |
Control only |
MitoPark only |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Comparison | r | P | N | r | P | N | r | P | N |
| Input resistance vs neurite count | –0.265 | .0318* | 66 | –0.412 | .0082** | 40 | –0.207 | .31 | 26 |
| Input resistance vs neurite length | –0.371 | .0022** | 66 | –0.447 | .0038** | 40 | –0.3 | .136 | 26 |
| Input resistance vs soma volume | –0.361 | .0031** | 65 | 0.0208 | .9 | 39 | –0.498 | .0097** | 26 |
| Cell capacitance vs neurite count | 0.266 | .0306* | 66 | 0.125 | .444 | 40 | 0.383 | .054 | 26 |
| Cell capacitance vs neurite length | 0.269 | .0291* | 66 | 0.08 | .624 | 40 | 0.38 | .056 | 26 |
| Cell capacitance vs soma volume | 0.558 | <.0001**** | 65 | 0.3 | .064 | 39 | 0.777 < | .0001**** | 26 |
| H-current vs neurite count | 0.161 | .236 | 56 | 0.0564 | .75 | 34 | 0.281 | .21 | 22 |
| H-current vs neurite length | 0.193 | .154 | 56 | 0.0651 | .715 | 34 | 0.31 | .16 | 22 |
| H-current vs soma volume | 0.58 | <.0001**** | 55 | 0.415 | .016* | 33 | 0.724 | .0001*** | 22 |
P <.05,
P <.01,
P <.001,
P <.0001
Although not the primary goal of the study, we next determined if decrements in dopamine neuron morphology in MitoPark mice proceeded similarly between sexes by analyzing Sholl analysis/intersection data (Supplementary Fig. 4). As expected, there was no significant effect of genotype on neurite arborization at 12 weeks of age in either males or females. In contrast, by 16 to 20 weeks, significant arborization deficits were evident in both males (F1,45 = 4.74, P = .0348) and females (F1,24 = 7.70, P = .0105). Similarly, by 27 to 31 weeks, dopamine neurons from males exhibited severe deficits in arborization (F1,39 = 15.66, P = .0003), whereas in females this analysis was not significant, likely because of the small sample size in the MitoPark group (F1,2 = 16.2, P = .0567). Overall, these data indicate no obvious effect of sex on the decline of neuritic branching in dopamine neurons from MitoPark mice. In an independent experiment, we also observed no difference in dopamine neuron morphology between 4-month-old male and female wild-type C57Bl/6 mice (Supplementary Fig. 5). Our interpretation at this time is that there is no convincing effect of sex on dopamine neuron morphology in MitoPark mice and/or control mice at any age tested.
Discussion
Wholesale loss of substantia nigra dopamine neurons is a cardinal pathophysiological finding in PD and is responsible for the motor symptoms of the disease.3,4 Diagnosis often occurs with the appearance of overt motor impairment, when dopamine neuron loss reaches 50% to 70%.6,7 This suggests that compensatory mechanisms may mask disease pathology for years before diagnosis28 and may even contribute to the disease process itself. Current PD treatments do not alter disease progression but primarily focus on the relief of symptomatology. Establishing novel disease-modifying treatments will first require a better knowledge of the anatomical and pathophysiological changes that occur in the early stages of PD.
Of the models currently available for PD studies in rodents, MitoPark mice are particularly well-suited for studying dopamine neurons at multiple time points during the neurodegenerative process. MitoPark mice were engineered to lack the Tfam gene specifically in dopamine neurons, which is vitally important for mitochondrial function and cell health.10,13 Behaviorally, MitoPark mice exhibit an adult onset and progressive phenotype similar to PD in humans but on the timescale of a mouse’s life.10 The MitoPark model faithfully recapitulates most of the key characteristics of PD including age-dependent loss of dopamine neurons, motor impairment, development of inclusion bodies, and positive response to L-dopa treatment that worsens with time.10–12 Models of parkinsonism based on neurotoxins (such as 6-hydroxydopamine and MPTP) develop neuronal degeneration much faster than the progression of the disease in humans, thus obscuring the contribution of putative compensatory processes that may eventually become part of the pathophysiology of the disease. Similarly, models of PD based on human genetic studies, broadly speaking, have not been as effective at mimicking the mass neurodegeneration of dopamine neurons and motor arrestment associated with the human condition. Thus, the MitoPark mouse model is comparably suited for delineating the sequence of cellular events that occurs in the early stages of neurodegeneration.
Morphology in Relation to Physiology and Behavior
Our findings demonstrate a striking, progressive decline in neurite count, neurite length, and soma volume in MitoPark mice, beginning sometime between 12 and 20 weeks of age and becoming severe by 27 to 31 weeks. Alterations in somatic and dendritic structure are likely to have direct effects on dopamine-dependent motor function and reward learning. In these neurons, dendrites serve important roles as sites for synaptic termination and contribute to many determinants of cell excitability.29,30 Voltage-gated ion channels distributed throughout the somatodendritic compartment directly determine intrinsic firing, which is thought to maintain extracellular dopamine levels that are crucial for voluntary movement.31,32 In addition, dopamine neurons communicate with each other through somatodendritic dopamine release and dendrodendritic neurotransmission.33,34 The timing of the appearance of morphological deficits is coincident with a disruption of basal physiological parameters in MitoPark mice, including decreasing cell capacitance (an indirect measure of membrane surface area), elevated input resistance, and decreased spike width14 as well as increasing size of intracellular protein aggregates.10 Although synaptic input to dopamine neurons has not been extensively studied in MitoPark mice, our previous work suggests that dopamine release and receptor signaling are both disrupted prior to the appearance of morphological deficits.14 Behaviorally, 12 to 20 weeks of age in MitoPark mice is associated with decreased acoustic startle (i.e., enhanced prepulse inhibition35 as well as decreased body weight.12 This age range is also associated with altered ion channel gene expression that is consistent with an adaptive mechanism to increase cell excitability in the face of declining dopamine function.14
This study is, to our knowledge, the first to quantify in vivo decrements in dopamine neuron morphology by filling single cells in brain slices acutely isolated from a progressive model of PD. The findings are consistent with previous work suggesting a role for somatodendritic morphology in the development of parkinsonian syndromes. For example, dendrites from nonidentified substantia nigra neurons appear to be shorter in postmortem tissue from PD patients.20 Diffusion tensor imaging indicates that fractional anisotropy is reduced in the substantia nigra during the early stages of PD,36 which could indicate a reduced dendritic arbor.37 Reduced soma volumes have also been reported in PD patients.21 In rodents, the overexpression of alpha-synuclein, which is believed to have a major role in Parkinson’s development,38,39 initially reduces neurite length within cultured midbrain dopamine neurons while somas remained unaffected.40 Similarly, rats injected with a viral vector driving overexpression of alpha-synuclein exhibit widespread loss of dopaminergic dendrites while cell bodies are only mildly affected.41 Mutated leucine-rich repeat kinase 2, also thought to play a role in certain types of PD, disrupts neuronal branching in cultured cells in an autophagy-dependent manner.42–44 Toxin-based models of PD also exhibit reductions of neurite length and branching in nigral dopamine cells, as quantified from midbrain cultures.45,46 In the current study, the raw numbers obtained for neurite number and branching were almost certainly underestimates because of the limited thickness (200 μm) of our midbrain slices. However, these measurements of dopamine neuron morphology allow a straightforward interpretation of the time course of neuronal decline in vivo and the degree to which it proceeds in the presence of neuroprotective elements such as vasculature, extracellular matrices, and glia. Our findings demonstrate that dendritic branching and soma size, although intact in dopamine neurons from 12-week-old MitoPark mice, are progressively and severely disrupted in a manner that undoubtedly impairs cellular function in advance of neuronal death.
A secondary aim of this study was to analyze possible sex differences within the MitoPark model, as PD afflicts human males more predominately than females.47,48 Although evidence from toxin models indicates a greater loss of nigral dopamine neurons in males,49–51 sex differences have not been extensively studied in MitoPark mice. Overall, the results indicate that the decline in somatodendritic morphology in MitoPark mice proceeds similarly in both sexes. This complements the previously published claim that 8- to 12-week-old MitoPark mice do not exhibit sex differences in regard to various motor and cognitive tasks.12
Mitochondrial Mechanisms of Neurodegeneration
The current findings could provide insight into how dopamine neurons degenerate when faced with mitochondrial/energetic impairment. Dopamine neurons are autonomous pacemakers whose firing is associated with large intracellular calcium oscillations.52 The large energetic demand along with dopamine metabolism generate high basal levels of reactive oxygen species, suggesting that added oxidative stress from mitochondrial dysfunction could be especially damaging.53–55 Indeed, dopamine neurons from MitoPark mice exhibit increased markers of oxidative stress as well as altered morphology in nigral microglia indicative of neuroinflammation.56 Furthermore, dopamine neurons in MitoPark mice exhibit structurally altered mitochondria that are associated with the intracellular protein aggregates observed with this model.10 Tfam is necessary for transcription initiation at mitochondrial promoters,57 and its ablation leads to mitochondrial respiratory chain failure through the disruption of mitochondrial DNA.13,58–61 Growing evidence suggests that mitochondria could represent a crucial crossroad in PD pathogenesis, as many genes implicated in familial forms of PD also regulate mitochondrial function.62–64 Moreover, mitochondrial respiratory chain dysfunction in dopamine neurons appears to be a convergence point for most if not all rodent models of PD.55,65,66
The initial locomotor deficits and disruption of ion channel physiology in dopamine neurons precede the morphological decrements observed in MitoPark mice, suggesting that mitochondrial impairment could produce physiological deficits prior to anatomical degeneration. We observed no significant effect of genotype on morphological measures in dopamine neurons from 12-week-old mice; however, the first mild locomotor decrements are already observable at this age,12 and in one study were observed as early as 8 weeks.35 Furthermore, MitoPark mice also exhibit impaired learning in an escape task and a decline in novel object recognition prior to 12 weeks of age.12 This suggests that frank morphological alterations in the somatodendritic compartment may not be involved in the initial motor and cognitive impairments. Indeed, previous work suggests that neurodegeneration of dopamine neurons in PD likely begins at axonal terminals of the caudate/putamen and progresses caudally back to the cell bodies, sometimes referred to as “dying back.”67–69 In MitoPark mice, dopamine neurons exhibit the beginnings of both elevated input resistance and mildly decreased cell capacitance at this age as well as deficits in both striatal innervation and evoked dopamine release.10,14,15 However, decrements in somatodendritic ion channel and synaptic physiology can also be observed prior to 12 weeks, including decreased pacemaker firing fidelity, H-current, amphetamine-induced dopamine efflux, and synaptic transmission mediated by dendritic release of endogenous dopamine.14,15 This suggests that physiological determinants of neuronal excitability, driven by both intrinsic conductances and synaptic transmission, could be responsible for the subtle, initial decrements in behavior seen prior to 16 weeks in MitoPark mice. Thus, morphological decline could represent an effect of mitochondrial impairment that is more resistant to insults than neuronal excitability.
In summary, we have shown that the somatodendritic compartment in substantia nigra dopamine neurons from MitoPark mice exhibits a progressive decline in morphological parameters that follows the initial decrements in ion channel physiology but precedes widespread neuronal death. This decline progresses at similar rates in both sexes and is concurrent with many of the published physiological and behavioral observations of this model. This work could help identify specific therapeutic treatments as well as an ideal time window to target initial nigral dopamine degeneration to avert the debilitating motor impairments that are a hallmark of PD.
Supplementary Material
Acknowledgments:
We would like to thank Drs. Exing Wang and Jim Lechleiter from the imaging core facility at University of Texas Health, San Antonio and Julie Crane and Ben Fowler from the imaging core facility at the Oklahoma Medical Research Foundation for their assistance with confocal imaging. We also thank Dr. Bill Freeman and the Oklahoma Medical Research Foundation Quantitative Analysis Core for helpful suggestions.
Funding agencies: Support was provided by the National Institute on Aging Grant R01 AG052606 (to M.J.B.) as well as funds from the William & Ella Owens Medical Research Foundation, the Presbyterian Health Foundation, and the Oklahoma Center for Adult Stem Cell Research.
Footnotes
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
Relevant conflicts of interests/financial disclosures: Nothing to report.
References
- 1.Goetz CG. The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb Perspect Med 2011;1:a008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalia LV, Lang AE. Parkinson’s disease. The Lancet 2015;386: 896–912. [DOI] [PubMed] [Google Scholar]
- 3.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39:889–909. [DOI] [PubMed] [Google Scholar]
- 4.Hornykiewicz O Parkinson’s disease and its chemotherapy. Bio-chem Pharmacol 1975;24:1061–1065. [DOI] [PubMed] [Google Scholar]
- 5.Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013;136:2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. Brain 1999;122:1437–1408. [DOI] [PubMed] [Google Scholar]
- 7.Marsden CD. Parkinson’s disease. J Neurol Neurosurg Psychiatr 1994;57:672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terzioglu M, Galter D. Parkinson’s disease: genetic versus toxin-induced rodent models. FEBS J 2008;275:1384–1391. [DOI] [PubMed] [Google Scholar]
- 9.Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 2009;15(suppl 3): S185–S188. [DOI] [PubMed] [Google Scholar]
- 10.Ekstrand MI, Terzioglu M, Galter D, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A 2007;104:1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galter D, Pernold K, Yoshitake T, et al. MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson’s disease. Genes Brain Behav 2010;9:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Redus L, Chen C, et al. Cognitive dysfunction precedes the onset of motor symptoms in the MitoPark mouse model of Parkinson’s disease. PLoS ONE 2013. 8:e71341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsson NG, Wang J, Wilhelmsson H, et al. Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat Genet 1998;18:231–236. [DOI] [PubMed] [Google Scholar]
- 14.Branch SY, Chen C, Sharma R, Lechleiter JD, Li S, Beckstead MJ. Dopaminergic neurons exhibit an age-dependent decline in electrophysiological parameters in the MitoPark Mouse model of Parkinson’s disease. J Neurosci 2016;36:4026–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Good CH, Hoffman AF, Hoffer BJ, et al. Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. FASEB J 2011;25:1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci 2009;29: 3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Liu W, Oo TF, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci 2009;12:826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morfini G, Pigino G, Opalach K, et al. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A 2007;104:2442–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tagliaferro P, Burke RE. Retrograde axonal degeneration in parkinson disease. J Parkinsons Dis 2016;6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patt S, Gertz HJ, Gerhard L, Cervós-Navarro J. Pathological changes in dendrites of substantia nigra neurons in Parkinson’s disease: a Golgi study. Histol Histopathol 1991;6:373–380. [PubMed] [Google Scholar]
- 21.Rudow G, O’Brien R, Savonenko AV, et al. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol 2008;115:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Branch SY, Beckstead MJ. Methamphetamine produces bidirectional, concentration-dependent effects on dopamine neuron excitability and dopamine-mediated synaptic currents. J Neurophysiol 2012;108:802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 2011;27:2453–2454. [DOI] [PubMed] [Google Scholar]
- 24.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira TA, Blackman AV, Oyrer J, et al. Neuronal morphometry directly from bitmap images. Nat Methods 2014;11 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Branch SY, Sharma R, Beckstead MJ. Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J Neurosci 2014;34:9310–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates, Fourth Edition New York: Elsevier Academic Press; 2013. [Google Scholar]
- 28.de la Fuente-Fernández R, Schulzer M, Kuramoto L, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol 2011;69:803–810. [DOI] [PubMed] [Google Scholar]
- 29.Henny P, Brown MT, Northrop A, et al. Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat Neurosci 2012;15:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blythe SN, Wokosin D, Atherton JF, Bevan MD. Cellular mechanisms underlying burst firing in substantia nigra dopamine neurons. J Neurosci 2009;29:15531–15541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carli M, Evenden JL, Robbins TW. Depletion of unilateral striatal dopamine impairs initiation of contralateral actions and not sensory attention. Nature 1985;313:679–682. [DOI] [PubMed] [Google Scholar]
- 32.Tepper JM, Creese I, Schwartz DH. Stimulus-evoked changes in neostriatal dopamine levels in awake and anesthetized rats as measured by microdialysis. Brain Res 1991;559:283–292. [DOI] [PubMed] [Google Scholar]
- 33.Kalivas PW, Duffy P. Effects of daily cocaine and morphine treatment on somatodendritic and terminal field dopamine release. J Neurochem 1988;50:1498–1504. [DOI] [PubMed] [Google Scholar]
- 34.Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 2004;42:939–946. [DOI] [PubMed] [Google Scholar]
- 35.Grauer SM, Hodgson R, Hyde LA. MitoPark mice, an animal model of Parkinson’s disease, show enhanced prepulse inhibition of acoustic startle and no loss of gating in response to the adenosine A (2A) antagonist SCH 412348. Psychopharmacology (Berl) 2014; 231:1325–1337. [DOI] [PubMed] [Google Scholar]
- 36.Vaillancourt DE, Spraker MB, Prodoehl J, et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology 2009;72:1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bock AS, Olavarria JF, Leigland LA, Taber EN, Jespersen SN, Kroenke CD. Diffusion tensor imaging detects early cerebral cortex abnormalities in neuronal architecture induced by bilateral neonatal enucleation: an experimental model in the ferret. Front Syst Neurosci 2010;4:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron 2013;79:1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 40.Koch JC, Bitow F, Haack J, et al. Alpha-synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis 2015;6:e1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Decressac M, Mattsson B, Lundblad M, Weikop P, Björklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of α-synuclein in midbrain dopamine neurons. Neurobiol Dis 2012;45:939–953. [DOI] [PubMed] [Google Scholar]
- 42.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006;52:587–593. [DOI] [PubMed] [Google Scholar]
- 43.Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 2008;105:1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramonet D, Daher JP, Lin BM, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 2011;6:e18568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cisbani G, Drouin-Ouellet J, Gibrat C, et al. Cystamine/cysteamine rescues the dopaminergic system and shows neurorestorative properties in an animal model of Parkinson’s disease. Neurobiol Dis 2015;82:430–444. [DOI] [PubMed] [Google Scholar]
- 46.Gómez FJ, Aguirre P, Gonzalez-Billault C, Núñez MT. Iron mediates neuritic tree collapse in mesencephalic neurons treated with 1-methyl-4-phenylpyridinium (MPP+). J Neural Transm 2011;118: 421–431. [DOI] [PubMed] [Google Scholar]
- 47.Elbaz A, Bower JH, Maraganore DM, et al. Risk tables for parkinsonism and Parkinson’s disease. J Clin Epidemiol 2002;55:25–31. [DOI] [PubMed] [Google Scholar]
- 48.Smith KM, Dahodwala N. Sex differences in Parkinson’s disease and other movement disorders. Exp Neurol 2014;259:44–56. [DOI] [PubMed] [Google Scholar]
- 49.Liu B, Dluzen DE. Oestrogen and nigrostriatal dopaminergic neurodegeneration: animal models and clinical reports of Parkinson’s disease. Clin Exp Pharmacol Physiol 2007;34:555–565. [DOI] [PubMed] [Google Scholar]
- 50.McArthur S, McHale E, Gillies GE. The size and distribution of midbrain dopaminergic populations are permanently altered by perinatal glucocorticoid exposure in a sex- region- and time-specific manner. Neuropsychopharmacology 2007;32:1462–1476. [DOI] [PubMed] [Google Scholar]
- 51.Murray HE, Pillai AV, McArthur SR, et al. Dose- and sex-dependent effects of the neurotoxin 6-hydroxydopamine on the nigrostriatal dopaminergic pathway of adult rats: differential actions of estrogen in males and females. Neuroscience 2003;116:213–222. [DOI] [PubMed] [Google Scholar]
- 52.Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol 2000;83: 3084–3100. [DOI] [PubMed] [Google Scholar]
- 53.Chan CS, Guzman JN, Ilijic E, et al. “Rejuvenation” protects neurons in mouse models of Parkinson’s disease. Nature 2007;447: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 54.Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2:a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 2013; 106–107:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langley M, Ghosh A, Charli A, et al. Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in MitoPark transgenic mice. Antioxid Redox Signal 2017;27:1048–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 2002;31:289–294. [DOI] [PubMed] [Google Scholar]
- 58.Silva JP, Köhler M, Graff C, et al. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 2000;26:336–340. [DOI] [PubMed] [Google Scholar]
- 59.Sörensen L, Ekstrand M, Silva JP, et al. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci 2001;21:8082–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Wilhelmsson H, Graff C, et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet 1999;21:133–137. [DOI] [PubMed] [Google Scholar]
- 61.Wredenberg A, Wibom R, Wilhelmsson H, et al. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 2002;99:15066–15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen J, Cookson MR. Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron 2004;43:301–304. [DOI] [PubMed] [Google Scholar]
- 63.Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006;441: 1157–1161. [DOI] [PubMed] [Google Scholar]
- 64.Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006;441:1162–1166. [DOI] [PubMed] [Google Scholar]
- 65.Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays 2002;24:308–318. [DOI] [PubMed] [Google Scholar]
- 66.Schober A Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res 2004;318:215–224. [DOI] [PubMed] [Google Scholar]
- 67.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 1973;20:415–455. [DOI] [PubMed] [Google Scholar]
- 68.Cheng H-C, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67: 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scherman D, Desnos C, Darchen F, Pollak P, Javoy-Agid F, Agid Y. Striatal dopamine deficiency in Parkinson’s disease: role of aging. Ann Neurol 1989;26:551–557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.