

Abstract
Background
Germinal center-derived B cell lymphomas are tumors of the lymphoid tissues representing one of the most heterogeneous malignancies. Here we characterize the variety of transcriptomic phenotypes of this disease based on 873 biopsy specimens collected in the German Cancer Aid MMML (Molecular Mechanisms in Malignant Lymphoma) consortium. They include diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), Burkitt’s lymphoma, mixed FL/DLBCL lymphomas, primary mediastinal large B cell lymphoma, multiple myeloma, IRF4-rearranged large cell lymphoma, MYC-negative Burkitt-like lymphoma with chr. 11q aberration and mantle cell lymphoma.
Methods
We apply self-organizing map (SOM) machine learning to microarray-derived expression data to generate a holistic view on the transcriptome landscape of lymphomas, to describe the multidimensional nature of gene regulation and to pursue a modular view on co-expression. Expression data were complemented by pathological, genetic and clinical characteristics.
Results
We present a transcriptome map of B cell lymphomas that allows visual comparison between the SOM portraits of different lymphoma strata and individual cases. It decomposes into one dozen modules of co-expressed genes related to different functional categories, to genetic defects and to the pathogenesis of lymphomas. On a molecular level, this disease rather forms a continuum of expression states than clearly separated phenotypes. We introduced the concept of combinatorial pattern types (PATs) that stratifies the lymphomas into nine PAT groups and, on a coarser level, into five prominent cancer hallmark types with proliferation, inflammation and stroma signatures. Inflammation signatures in combination with healthy B cell and tonsil characteristics associate with better overall survival rates, while proliferation in combination with inflammation and plasma cell characteristics worsens it. A phenotypic similarity tree is presented that reveals possible progression paths along the transcriptional dimensions. Our analysis provided a novel look on the transition range between FL and DLBCL, on DLBCL with poor prognosis showing expression patterns resembling that of Burkitt’s lymphoma and particularly on ‘double-hit’ MYC and BCL2 transformed lymphomas.
Conclusions
The transcriptome map provides a tool that aggregates, refines and visualizes the data collected in the MMML study and interprets them in the light of previous knowledge to provide orientation and support in current and future studies on lymphomas and on other cancer entities.
Electronic supplementary material
The online version of this article (10.1186/s13073-019-0637-7) contains supplementary material, which is available to authorized users.
Keywords: Tumor heterogeneity, B cell malignancies, Gene regulation, Molecular subtypes, Machine learning
Background
Germinal center-derived B cell lymphomas are tumors of the lymphoid tissues representing one of the most heterogeneous malignancies in terms of their molecular and cellular phenotypes [1]. Frequent B cell lymphomas in adulthood are follicular lymphomas (FL) and diffuse large B cell lymphomas (DLBCL), and, in children, Burkitt’s lymphomas (BL). Especially DLBCL show a very heterogeneous spectrum of phenotypes as revealed by morphological [2], immunohistochemical [3] and metabolic [4] characteristics. Particularly, molecular high-throughput analytics created many ways to disentangle the diversity of this disease into a series of stratification schemes [5–14].
The German Cancer Aid MMML (Molecular Mechanisms in Malignant Lymphoma) consortium collected altogether more than 800 biopsy specimens of mature B cell lymphomas and about 100 samples of tumor cell lines, normal B cell populations and non-neoplastic tonsil tissue serving as different kinds of reference, and recorded their genome-wide transcriptomes by means of microarrays. The B cell lymphomas studied comprise virtually the whole spectrum of this disease. Previous studies published subgroups of samples selected from this cohort to extract a molecular classifier that distinguishes BL from ‘other than BL’ cases [7], to disentangle DLBCL into subclasses [10], to associate DLBCL cases with selected signaling pathway activities [8] and to study other partial aspects of this disease [7, 8, 10, 15–18]. An integrated and comprehensive analysis of all samples including about 200 hitherto unpublished cases is presented here.
We hereby aim at establishing a map of the expression landscape of B cell lymphomas covering the heterogeneity of their molecular expression states. Heterogeneity of lymphomas can be understood as a series of mutually similar molecular states forming a continuum without clear-cut borderlines not only between different DLBCL entities but also with respect to the distinction between DLBCL, FL and, partly, also BL [7, 19]. These in many respects indistinct characteristics of the tumors can reflect overlapping genetic events such as the chromosomal translocation of the MYC gene which represents the genetic hallmark of BL but which also appears in about 5–10% of DLBCL leading to expression phenotypes resembling BL [20] and considered as a separate subtype according to the WHO classification [21]. The continuum of molecular states can also reflect the underlying stages of B cell development affected by cancer initiation and progression, e.g. in the course of histological transformations from FL to DLBCL after the consecutive accumulation of a series of genetic hits [22].
Previously, we have developed an omics ‘portraying’ method using self-organizing map (SOM) machine learning [23, 24] which was applied to a series of data types and diseases [24–29]. SOM portraying takes into account the multidimensional nature of gene regulation and pursues a modular view on co-expression, reduces dimensionality and supports visual perception in terms of individual, case-specific ‘omics’ portraits. By applying SOM portraying on B cell lymphoma transcriptomes, we demonstrate that multidimensional profiling will permit a description of the molecular heterogeneity of this disease in terms of a continuous spectrum of transcriptional states and to visualize them by means of different maps distinguishing lymphoma subtypes and their functional context and to link them to prognosis. The transcriptome map will provide a tool that aggregates, refines and visualizes the data collected in the MMML study and interprets them in the light of previous knowledge to provide orientation and support in current and future studies.
Methods
Lymphoma samples, genetic analyses and expression data
The gene expression data set consists of 913 samples studied by means of Affymetrix HG-U133A GeneChip microarrays. They divide into reference samples (tumor cell lines, sorted B cells, tonsils), mature B cell lymphomas and other tumors collected in the study (see Additional file 1: Table S1 and Additional file 2 for details). One of the lymphoma specimens was measured twice on two arrays. Tumors were diagnosed in panel meetings of the MMML pathology group. Genetic analyses by means of interphase fluorescence in situ hybridization were performed on frozen or paraffin-embedded tissues with the use of probes for IGH, IGK, IGL, MYC, BCL6 and BCL2. Loci in which MYC was fused to IGH, IGK or IGL were referred to as ‘IG-MYC’. Lymphomas with MYC breakpoints without fusion of MYC to an IG locus were called ‘non-IG-MYC’ (see [7] for details). Reference data included different lymphoma cell lines [30, 31], several B cell types isolated either from peripheral blood (pre- and post-germinal center (GC) B cells) or from suspended tonsillar tissue (GC B cells), and tonsillar tissue specimen for comparison of their expression patterns with those of lymphoma as specified in Additional file 1: Table S1.
SOM expression portraying
Gene expression data were preprocessed using hook calibration, quantile normalization and centralization as described in [23, 32]. The preprocessing detects and corrects for possible outlier samples, batch effects and a sample- and transcript-specific background in cancer data [29, 33] (Additional file 1: Figure S1). Preprocessed expression data were then clustered using self-organizing map (SOM) machine learning which translates the expression data matrix consisting of N = 22,283 probe set values covering 13,182 ensemble genes, and M = 913 samples, into a data matrix of reduced dimensionality where the N gene expression profiles are represented by K = 2500 metagene profiles. Hereby, ‘profile’ denotes the vector of M expression values per gene/metagene. The SOM training algorithm distributes the N genes over the K metagenes using the Euclidian distance between the expression profiles as a similarity measure. It ensures that genes with similar profiles cluster together in the same or in closely located metagenes. Each metagene profile can be interpreted as the mean profile averaged over all gene profiles referring to the respective metagene cluster. The metagene expression values of each sample are visualized by arranging them into a two-dimensional 50 × 50 grid and by using maroon to blue colors for maximum to minimum expression values in each of the portraits. The number of genes typically varies from metagene to metagene and ranges from only a few associated single genes to metagenes containing more than a hundred genes (see the population map in Additional file 1: Figure S2a). This way, our approach portrays the transcriptome landscape of each sample in terms of a colored image visualizing its metagene expression values. Group- and subtype-specific mean portraits were generated by averaging the portraits of all cases belonging to one group/subtype. We used the implementation of the method in the Bioconductor R-package ‘oposSOM’ [34].
Sample diversity analyses, spot module detection, gene maps and enrichment analysis
Metagenes of similar profiles cluster together forming ‘spot-like’ red and blue areas of over- and under-expression in the portraits due to the self-organizing properties of the SOM. The spot patterns are characteristic fingerprints of each particular sample enabling us to compare their transcriptomic landscapes by means of diversity analysis using a graph representation called ‘correlation network’ and phylogenetic tree visualization as implemented in ‘oposSOM’ [34]. The spot patterns of the expression portraits reveal clusters of correlated metagenes (Additional file 1: Figure S2d) which collect the associated single genes into modules of co-expressed genes. These modules were defined by segmentation of the map according to an over-expression criterion, collecting adjacent metagenes which exceed 90% of maximum metagene expression in the respective sample class (see also [23, 32] and Additional file 1). The number of spot modules detected represents an intrinsic characteristic of the co-expression network present in the samples. The size of the SOM, K, was chosen to ensure the robust identification of spots by exceeding their number by more than two orders of magnitude as was demonstrated previously [28]. The spots are characterized by their number distributions and by spot co-occurrence networks based on association rules [35]. We additionally performed zoom-in SOM analyses for selected subsets of samples (lymphoma cell lines, B cells and Burkitt’s lymphomas) to validate resolution of the transcriptomic landscape [23].
We applied gene set enrichment analysis to the lists of genes located in each of the spot modules to discover their functional context using right-tailed Fisher’s exact test [36, 37]. The gene set enrichment Z-score (GSZ) was used to evaluate the expression profiles of the gene sets across the samples of the study [32, 38]. Gene maps visualize the position of selected genes within the SOM grid. According to their location in or near a specific spot, one can deduce over- and under-expression characteristics and the potential functional context of the respective gene. Its position is invariant in all expression portraits, which allows for direct comparison.
Pattern types
The sample portraits were stratified into pattern types (PATs), where a PAT is defined by the combination of spot modules over-expressed in the respective samples. Rare PATs found in less than five cases per subtype were rejected from further analysis to focus on recurrent pattern types solely. A sample that shows no expression module activated is still assigned to a PAT if their module expression values correlate with those of a certain PAT with Pearson correlation coefficient r > 0.8. Otherwise, it is assigned to ‘no PAT’ and labeled as ‘∅’. In total, 679 samples (74%) were classified into PATs according to detected spots, 102 (11%) were additionally classified by the correlation step, and 133 (15%) remain unclassified. PAT-specific mean expression portraits are generated as averages over the individual sample portraits of the respective PAT.
Metagene sets of hallmarks of cancer
The hallmarks of cancer constitute a series of biological capabilities commonly acquired by tumors [39]. We assembled eight metagene sets referring to the hallmarks angiogenesis, controlling genomic instability, glucose energetics, inflammation, invasion and metastasis, proliferation and replicative immortality and resisting death according to the hallmark definitions proposed in ref. [40]. Each of these hallmark sets collects from 2 to 12 suited gene sets taken from our repository of gene sets. The lists of gene sets included in each hallmark set are provided in Additional file 1: Table S3.
Cell type and pathway signal flow analyses, and survival analyses
Immune cell composition of the tumor biopsies was estimated from the expression data using the program CIBERSORT based on support vector regression and previous knowledge on purified leukocyte expression profiles [41]. Pathway activity was analyzed using the pathway signal flow method as implemented in oposSOM [42].
Hazard ratios and p values for pairwise comparisons of survival curves were derived utilizing Cox models. The models were additionally adjusted by inclusion of co-factors ‘chemotherapy’ (yes/no) and ‘Rituximab’ (yes/no). Cases without information about therapy were removed from the multivariate model. The prognostic map was generated as follows: For each metagene, lymphoma cases with available survival information were divided into cases showing expression of this metagene above or below the 50% percentile, respectively, and then compared using a Cox model. This way, hazard ratios (HRs) were obtained for all metagenes and visualized in terms of a map using blue to red colors for low to high HRs.
Results
SOM portraits of lymphoma subtypes
The gene expression data set studied here was generated by the German MMML consortium. It consists of biopsy specimens of mature B cell lymphomas, of other tumor cases such as multiple myeloma (MM), of lymphoma cell line specimen (32 samples of 28 different lymphoma cell lines), of sorted B cell populations (30) and of non-neoplastic tonsil tissue samples (10) which were used as reference for comparison of their expression landscapes with that of the lymphomas (see Additional file 1: Table S1). Expression data were complemented by pathological evaluation of tissue samples, genetic and immuno-histochemical analyses and clinical data. The tumor samples were divided into ten major strata based on pathological evaluation, genetic and/or previous gene expression classification criteria (see Additional file 1: Table S1 for details), namely, (i) diffuse large B cell lymphoma (DLBCL, 430 cases), (ii) follicular lymphoma (FL, 145), (iii) intermediate lymphoma according to [7] (81), (iv) prototypic Burkitt’s lymphoma (BL, 74), (v) mixed FL/DLBCL and WHO grade 3b FL (48), (vi) mediastinal large B cell lymphoma (PMBL, 23), (vii) multiple myeloma (MM, 20), (viii) IRF4-rearranged large cell lymphoma (IRF4-LCL, 10), (ix) MYC-negative Burkitt-like lymphomas with a chr. 11q aberration pattern (mnBLL-11q, 6) and (x) mantle cell lymphoma (MCL, 4). DLBCL were further stratified into the germinal center (GCB, 142), activated B cell (ABC, 133), unclassified (97) DLBCL and double-hit (DH, 58) lymphoma and, alternatively, into plasmablastic, centroblastic, anaplastic and immunoblastic DLBCL based on pathological panel diagnosis [43, 44]. FLs were divided according to BCL2-break (positive, negative and NA) and according to tumor grading (1, 2 and 3a). Intermediate lymphomas were split into BL-like (11) and others (70).
The expression data of all samples were used to train a self-organizing map (SOM) which provides ‘portraits’ of the transcriptomic landscape of each individual sample (see Additional file 3 for the whole gallery of the expression portraits), and, after averaging, mean portraits of the different strata considered (Additional file 1: Figure S3). The mean transcriptomic portraits of the lymphoma strata (i)–(x) are shown in Fig. 1a together with the mean portraits of reference samples. The mean portraits reveal unique spot-like patterns of over- (colored in red) or under-expressed (in blue) gene clusters but also partly overlapping spots, e.g. between BL, mnBLL-11q and, partly, intermediate lymphoma and between DLBCL, PMBL and, partly, IRF4-LCL and FL. The correlation network visualizes the heterogeneity of the samples (Fig. 1b): BL cases (red-colored nodes) aggregate into a dense cloud which reflects relatively close similarity between them while the DLBCL cases (blue nodes) form an extended, widely distributed data cloud due to the heterogeneous character of this subtype. It overlaps with the cluster of FL cases (green nodes), thus forming a continuum ranging from BL-related to FL-related expression patterns. The samples of the three reference systems accumulate in localized regions of the similarity network, reflecting relatively homogenous expression patterns contrary to most of the lymphoma subtypes (Fig. 1b). They comprise different lymphoma cell lines and B cell types (Additional file 1: Table S1) showing however relatively similar SOM portraits (Additional file 1: Figure S3). We provided a detailed analysis of these reference systems and of BL in terms of zoom-in SOM analyses and class-related difference portraits in the supplementary text (Additional file 1: Figures S17 - S19). The zoom-in SOM maps partly provide an enhanced resolution of the expression landscapes of the particular subsystems. However, comparison with the results of all samples presented here confirms sufficiently high resolution of this analysis (Additional file 1: Figures S17 - S19). In summary, SOM portraying provides subtype-specific images that visualize their expression landscapes in terms of clusters of over- and under-expressed genes.
Fig. 1.

Expression and sample landscapes of lymphoma subtypes. a Mean expression portraits of the major B cell lymphoma subtypes and of the controls are characterized by red-blue spot patterns which reflect clusters of co-expressed genes up- and downregulated in the subtype on the average, respectively. The complete gallery of individual sample portraits is available in Additional file 3. b The correlation network visualizes the similarity relations between the samples as an undirected graph. The nodes represent the samples and are colored according to their class membership. The edges connect sample pairs whose expression landscapes are mutually correlated with Pearson’s correlation coefficients larger than 0.5. The small networks in the part below highlight each individual class considered. Part of the lymphoma types and of the controls occupy localized areas (e.g. BL and tonsils), while other types distribute over wider regions (e.g. intermediate lymphomas and FL/DLBCL) thus reflecting a more heterogeneous composition of the respective groups
Spot modules partition the expression map
We generated an over-expression-spot map which summarizes all red over-expression spots observed in the single-sample portraits (Fig. 2a, see [23]). In total, 13 spot modules A–M were identified, where each of them represents a module of co-expressed genes with a specific mean expression profile (Additional file 1: Figure S5; for lists of genes, see Additional file 4). Nine of the spots are mainly activated in the lymphomas and four in the controls. The spot-connectivity map in Fig. 2b visualizes the probability of joint spot appearances in the single-sample portraits. Accordingly, BL samples frequently express spots A, B and D together (red circles) while DLBCL tend to co-express E–G (blue circles). The frequency distribution of activated spots and their number distribution in each class show two-to-four recurrently activated modules in BL, cell lines, B cells and tonsils (Fig. 2c, d). For example, tonsils are characterized by ubiquitous presence of the two spots I and J (see also the tonsil portrait in Fig. 1a), which are specifically over-expressed in tonsillar tissue specimen as well as in tumors contaminated with tonsillar tissue this way giving rise to the ‘blue-shift’ of the rest of the portrait (Additional file 1: Figure S3 and S5) [33]. The broader distribution in intermediate lymphoma, DLBCL and FL reflects their more heterogeneous character. No spots were assigned in 133 samples, mainly in DLBCL (77 samples), intermediate lymphoma (24), FL (7), FL/DLBCL (11) and BL (2) due to their relatively flat expression landscapes.
Fig. 2.

Decomposing the expression landscape of lymphomas into spot modules of co-expressed genes. a The overview map collects all differentially expressed modules observed in the subtype-specific portraits into one map. The sample class(es) expressing the respective spot module(s) is/are assigned in the figure, thus segmenting the landscape into regions typically upregulated in certain lymphoma subtypes. Spot modules were labeled by capital letters A–M. Dark red/blue areas refer to over-/under-expression, respectively. b Probabilities of concerted module activation show that diverse sets of spot modules, e.g. A, B and D, are frequently upregulated in concert. Notably, spot A often appears also together with spot I, which is characteristic for double-hit lymphomas (see also Additional file 1: Figure S4). The color of the module labels represents the corresponding lymphoma subtype. c Spot-class association histograms depict the fraction of samples showing a certain spot in each class. It indicates, for example, that spots A, B and D are prevalent in the BL portraits in agreement with the assignments shown in panel a. d The spot number histograms show the fraction of samples with one, two etc. over-expression spots in each class. It reveals, that in most BL samples three spots can be observed, while DLBCL and FL/DLBCL show a broader variability of spots. Only the five most abundant lymphoma strata (i)–(v) are shown
A functional map of the spot modules
Each of the 13 spot clusters is populated typically with a few hundred genes (Additional file 4). Their functional context was analyzed by gene set analysis [32] (Fig. 3a and Additional file 1: Figures S7–S9). Modules activated in BL tumors are related to ‘replication’ and ‘cell cycle’ (spot D, p values < 10− 25 in Fisher’s test) and those in DLBCL to ‘inflammation’ (spot F, < 10− 25) reflecting tumor-infiltrating immune cells [13, 45, 46]. Modules G and I show stromal signatures [9] while module J upregulated in tonsils significantly enriches gene sets related to ‘keratinization’ (< 10− 23), a ‘tonsil signature’ (< 10− 10) [23, 32], and to ‘B cell-mediated adaptive immune response’ (< 10− 11). Genes associated with biological functions of B cells are enriched in modules K (e.g. ‘B cell activation’) and M (‘B cell differentiation’, < 10− 3). For a more detailed assignment of the spot patterns to B cell biology, we estimated enrichment of a series of gene sets taken from literature [47, 48] and from a separate analysis of the B cell samples (Fig. 3a, boxes with blue background). Modules activated in BL accumulate signature genes of the dark zone of the GC whereas modules activated in DLBCL accumulate light zone signature genes. The modules H, K, L and M enrich genes related to ‘plasma cells’ and to ‘pre/post-GC B cells’, respectively. Hence, assigning the functional context of the spot patterns provides a functional map that enables interpretation of the lymphoma portraits in terms of activated cellular programs.
Fig. 3.

Functional analysis of the expression modules. a Enriched gene sets from GO, KEGG and Reactome databases (yellow background; p < 0.05, Fisher’s exact test) and of B cell-related signatures taken from [47–49] and from a separate analysis of our B cell samples (blue background) are assigned to each of the spot modules. For example, spots A and F associate with cell-cycle activity and inflammation, respectively. b Mapping of key genes mutated in lymphomas and multiple myeloma taken from [50–60] into the expression landscape: Most of the genes accumulate in or near the spot modules thus reflecting a subtype-specific modulation of their gene expression. Multiple appearances of gene names refer to different Affymetrix probe sets
Mapping key mutations
Mapping of selected genes with mutations in lymphoma [50, 51, 53–60] into the SOM associates their expression profiles with that of the adjacent expression modules (Fig. 3b). Genes frequently mutated in BL are located in the BL-specific spot A (e.g. ID3, CCND3) and D (e.g. TCF3, SMARCA4, MYC) indicating their increased activity in BL and partly in intermediate lymphomas [50, 61]. Genes frequently mutated in DLBCL, FL and/or multiple myelomas (MM) such as BCL6 and BCL2 are found in or near spot K upregulated in healthy B cells and, to a lesser degree, in FL, and downregulated in BL and DLBCL (Additional file 1: Figure S5). The chromatin-modifying genes CREBBP (mutated in 30% of GCB-DLBCL [11], in early FL stages [62] and shared between primary and transformed FL [63]) and KMT2D (alias MLL2) are located in spots up- or downregulated in part of the FL cases compared with DLBCL suggesting epigenetic deregulation in FL. It presumably also involves HLA class II antigens [64], as supported by genome-wide association study (GWAS) analyses (Additional file 1: Figure S12), and MYD88, CDKN2B and PIK3CD, all affected by mutations preferentially in ABC-DLBCL leading to ‘chronic active’ B cell receptor signaling [11] (see also Additional file 1: Figure S11 for pathway analyses).
Spot H, specifically upregulated in MM and immunoblastic and plasmablastic DLBCL, co-regulates with PRDM1 (alias BLIMP1) promoting plasma cell differentiation by repressing MYC activity [53]. PRDM1 is deactivated in GCB-DLBCL and presumably also other subtypes by mutations, deletions or epigenetic effects [65, 66]. Interestingly, also IRF4 co-regulates with PRDM1 as indicated by its co-location in spot H [11]. The PIM1 oncogene (spot E) is over-expressed in most ABC-DLBCL [63] and in transformed FL (about 50% of patients) with ABC characteristics but it is rarely mutated in primary FL (less than 10%) [65]. Interestingly, both genes, PIM1 (40% in ABC vs 15% in GCB) and PRDM1 (25% vs less than 5%), show high prevalence of activating mutations in ABC-DLBCL [14] as indicated by over-expression of spot modules E and H in the SOM portrait of ABC-DLBCL but not in GCB-DLBCL (see Fig. 4).
Fig. 4.

Expression portraits of B cells and lymphomas, and their relation with respect to the GC biology. See also Additional file 1: Figure S3 for the full gallery of group-related expression portraits. Activated spot combinations are given as letters in the portraits. The particular spot patterns observed for the different lymphoma subtypes can be related to their functional context and associated key genes (compare with Fig. 2). For example, DZ-related types such as BL are proliferative as indicated by upregulation of spot D, which is on a lower expression level in LZ-related DLBCL. ABC-DLBCL and MM activate spot H, which is, in turn, virtually inactive in BL, GCB-DLBCL and FL
We also mapped hereditable risk genes for DLBCL and/or FL which were identified by GWAS (Additional file 1: Figure S12). These genes accumulate near the spots related to the somatic mutations in DLBCL and FL. In summary, mapping of mutations into the expression landscapes directly associates genomic with transcriptional events and allows linking mutations with their possible effects on the different subtypes.
Expression portraits relate to the pathogenesis in the GC
The scheme in Fig. 4 illustrates the relation between the expression portraits of B cells and of lymphoma subtypes and GC biology [52] (see also Additional file 1: Figure S3). B cells simultaneously express the spots J (tonsil signature), and K, L and M as characteristic B cell-specific signatures (Fig. 3a). In contrast to pre- and post-GC B cells, GC B cells over-express spot D that reflects activated proliferation in the dark zone of the GC. Also the portraits of the cancer cell line specimen over-express this proliferation signature (Fig. 1). On the other hand, all cell line systems under-express spot F related to inflammation because of the absence of immunogenic bystander cells. For a more detailed view, we refer to the ‘zoom-in’ SOM analysis provided in the supplementary text (Additional file 1: Figure S17 and S 18).
DLBCL of the GCB and ABC types show common expression of spot F (inflammation), but they differ in the expression of spots containing the key genes MYC (spot D), PIM1 (E) and PRDM1 (H) (see Fig. 4 and previous subsection). The portrait of PMBL closely resembles GCB-DLBCL, which differs from that of ABC-DLBCL. It specifically expresses the plasma cell-related spot H and the proliferation-related spot D. Interestingly, the ABC-type portrait resembles that of plasmablastic and partly also immunoblastic DLBCL while the portraits of anaplastic and centroblastic DLBCL partly agree with that of GCB lymphoma (Additional file 1: Figure S3), where plasmablastic, immunoblastic, anaplastic and centroblastic lymphoma annotate three morphological variants of DLBCL. Spot H shows prominent expression also in multiple myelomas (MM) accompanied by deactivation of BCL6-related transcriptional programs (spot K) as a hallmark of plasma cell maturation which is further paralleled by high expression of spot L reflecting B cell-like characteristics. On the other hand, MM under-express spots D, E and F due to decreased proliferative and inflammatory properties compared with ABC-DLBCL. Interestingly, IRF4-LCL over-express spots D, E and G thus indicating a combination of BL-like (spot D), stromal (spot G) and ABC-DLBCL (spot E) characteristics (Fig. 4). BL-like intermediate lymphomas show over-expression of spot B that accumulates marker genes of BL [7] but also of spot L which is related to post- and pre-GC B cell characteristics. This spot is not observed in prototypic BL and possibly refers to early stages of BL development which is supported by the relatively weak expression of spot D harboring proliferation-related genes such as MYC, TP53 and EZH2 (Fig. 3b). The portrait of mnBLL-11q closely resembles that of intermediate lymphomas and only partly that of prototypic BL [67] which, in turn, resembles that of double-hit lymphoma (DHL, Fig. 4). In the supplemental text, we present a comprehensive analysis of the expression patterns before and after acquiring a second hit combining MYC- with BCL2 or BCL6 translocations (Additional file 1: Figure S4). It illustrates the capability of SOM portraying to identify specific transcriptional patterns. The DZ- (spots D and A) GC signatures were evident in BL, while the LZ-GC signature (spots E–G) was found in GCB-DLBCL, partly FL and also in ABC-DLBCL and intermediate lymphomas in mixed amounts.
FLs of all histological grades express spot I as a transcriptional hallmark of this subtype independent of the presence or absence of the genetic hallmark of FL, namely the t(14;18) translocation (BCL2-break). Spot I partly transforms into spot G with increasing grade of FL paralleled by decreasing gene activities in the regions of other spots which indicates the progressive dominance of FL characteristics over other processes such as DNA processing and B cell characteristics. Grade 3b FL (FL/DLBCL) show a combined pattern of the FL and DLBCL-specific spots I and F, respectively, indicating the continuous transformation from FL into DLBCL. The portrait of double-hit lymphoma resembles that of BL thus reflecting increased transcriptional activity compared with FL (see also Additional file 1: Figure S4 for details). The portrait of MCL shows a unique pattern different from all the other lymphoma groups but sharing similarities with the portraits of B cells especially with strong expression of spot K and, partly, of spot M. MCL split into two subtypes deriving from pre- (type C1) or post-GC memory (C2) B cells, respectively [68]. Both types carry the t(14:18) translation giving rise to over-expression of spot I also found in FL. C1 MCL, in contrast to C2 MCL, express the gene SOX11 near spot A which prevents them from entering the GC. The portrait of tonsils expresses spot J as the unique prominent characteristics.
In summary, stratification of the molecular subtype portraits with respect to histological and genetic diagnosis reveals detailed relations to GC biology such as DZ- and LZ-GC, plasma cell and B cell characteristics. Overall, the criteria used, however, do not provide a consensus with respect to the classification of the tumors.
Pattern types
All subclasses express a combination of spots which makes them suited candidates as landmarks in the expression landscape of lymphoma. To address this multi-dimensionality, we define ‘pattern types’ (PATs) as the combination of spot modules concertedly over-expressed in a sample. We use notations such as ‘A B D’ to annotate cases jointly over-expressing the three modules A, B and D. In total, we identified 35 different PATs where 30 of them refer to lymphomas (Fig. 5a). We further stratified the PATs into 11 PAT groups, where the groups were labeled according to the most characteristic overlapping module(s) of the respective PATs (Fig. 5a). For example, BLs accumulate within five PATs collected into one BL-like group, while DLBCL distribute over four groups with 14 PATs, where one of these groups overlaps with FL. DLBCL were assigned to proliferative PATs with ABC-DLBCL characteristics (E type) or inflammatory and stromal types with GCB-DLBCL characteristics (F and G types, respectively). FL and FL/DLBCL are found in two groups mainly over-expressing spot I and partly also G and F thus forming a continuum between DLBCL and FL expression patterns. Interestingly, a small subgroup of intermediate lymphomas and of FL forms the L type that shares similarities with multiple myeloma (H type), partly expressing plasma cell programs associated with spot H. High expression of spot J indicates contaminations of the lymphoma samples with non-neoplastic tonsillar tissue. They were clustered together with the tonsils showing spot J as a hallmark. B cells divide into two PATs, which accumulate either GC B cells (‘AJ’) or pre/post-GC B cells (‘JKLM’, see also Additional file 1: Figure S3). The samples of each PAT mostly aggregate into compact data clouds in the similarity net which confirms the homogeneous character of their expression landscapes (Fig. 5b).
Fig. 5.

Expression (a) and sample (b) landscapes of the lymphoma pattern types (PATs). PATs were arranged into 11 groups. For each group, number-frequencies of samples diagnosed in the major histological lymphoma subtypes are given as barplot in panel a (see also the enrichment heatmap in Additional file 1: Figure S5). Each group collects similar and largely overlapping spot patterns. They arrange into dense sample clouds in the similarity networks, what is in contrast to the partly heterogeneous subtypes (compare with Fig. 1b)
In summary, PATs and PAT groups provide an expression-driven stratification of lymphoma and reference samples with enhanced resolution and homogeneity compared with the histological subtypes and with reference to activated cellular programs.
Characteristics of the PATs
The plot in Fig. 6a associates selected patient and functional characteristics with the PATs. The BL-related PATs show typical characteristics of this subtype such as the increased incidence in young patients, the presence of an IG-MYC translocation, low expression of BCL2 and a high percentage of KI67-positive highly proliferating cells [7]. DLBCL PATs enrich in older patients with high expression levels of the BCL2 markers and slower proliferation as seen by KI67. Expression modules activated in PATs of BL and FL reflect different transcriptional programs associated with IG-MYC and IG-BCL2 single hits, respectively. The joint appearance of both aberrations in double-hit lymphomas (DHL) specifically activates spot module A (PAT ‘A’) in agreement with recently published DHL expression signatures [69, 70] (Additional file 1: Figure S4c). Hence, the combination of different translocations in double-hit lymphomas does not necessarily combine the spot patterns of the respective single-hit lymphomas, but instead, they can induce new, non-additive expression patterns.
Fig. 6.

Characterization of lymphoma pattern types (PATs). a For each lymphoma patient, the PAT, clinical characteristics, previous molecular classifications, genomic characteristics and immunohistochemical (IHC) phenotypes are indicated in the barplots. Thresholds for classification of IHC markers are described in [17]. b Mapping of cases showing selected characteristics into the correlation network. It shows, e.g. that different previous classifications of lymphomas, such as ABC and GCB-DLBCL, accumulate in different areas of the network, which, in turn, associate with certain PATs. c Percentage of selected leukocyte cells according to their mRNA signatures across the PATs. ‘No PAT’ samples were assigned as ‘∅’ and distributed into PAT groups using a minimum Euclidian distance between sample and mean group portraits
We related the PATs to expression signatures of previous lymphoma classifications schemes [6–8, 10]. As expected, samples of the mBL and non-mBL subtypes [7] show strong correspondence with BL and DLBCL, respectively. The intermediate class (by Hummel et al.) accumulates in the PATs expressing spots A and D but also in the I-type typical for FL which reflects its heterogeneity. This class tends to collect DLBCL with BL resemblance induced, e.g. by IG-BCL2 and IG-MYC translocations, respectively (Additional file 1: Figure S4a). It also collects virtually all double-hit lymphomas, which enrich in PAT ‘A’ as described above. DLBCL tumors with the ABC signature [6] significantly enrich in the PATs ‘E’, ‘F’ and ‘E F’, collecting 75 of all 183 ABC cases (41%, p value < 10− 15; see also the expression portrait of ABC lymphoma in Fig. 4) which associates them with a distinct molecular PAT signature. GCB-DLBCL express predominantly PATs of the G and FIJ types. The classification of Rosolowski et al. [10] shows correspondence with E-, F- and L-type PATs. It reveals enrichment of the HiGA-Pro (high gene activation with proliferative phenotype) class in PATs ‘E’ (p value < 10− 14) and ‘E J’ (p value < 0.005) that also enriches ABC-DLBC (see above), suggesting relevant involvement of spot module E genes in this classifier. LoGA (low gene activity) cases accumulate in PAT ‘L’ which associates with B cell characteristics and thus possibly with early stages of lymphoma development (p values < 0.005, see Fig. 3a). Inflammatory [45] and stromal [9] signatures associate with PATs containing spots F, G or I, respectively (Additional file 1: Figure S8). We also compared our transcriptomic strata with recently established genetic classes of DLBCL [12, 14] by mapping characteristic mutations and chromosomal aberrations into the expression landscape. It turned out that these genetic classes associate with different PAT types covering the expression spectrum ranging from phenotypes of BL resemblance, over ABC and GCB-DLBCL, to FL-like tumors (Additional file 1: Figure S10).
Next, we estimated the percentage of selected immune cells based on their mRNA content in the tumor transcriptomes using CIBERSORT [41] (Fig. 6c). The transcriptomes of BL and partly of intermediate lymphomas (A- and D-type PATs) reflect characteristics of naïve B cells while DLBCL transcriptomes are more related to memory B cells which reflects a higher maturation grade of the B cells upon neoplastic transformation into DLBCL compared with BL. H-type PATs enriching MM show a high abundance of a plasma cell mRNA signature. Tumor-infiltrating macrophages are detected in considerable amounts in DLBCL and FL (F- and G-type PATs) which overall reflects a changing tumor microenvironment with PAT resolution. Previous studies report similar results, however, with lower resolution on a subtype level for BL, DLBCL, FL and MM [71]. Altered B cell receptor signaling in B cell lymphomas [11] will possibly lead to changed immune cell signatures with possible consequences for digital immune cell decomposition. In summary, the PATs can be associated with different functional categories and they show correspondence with previous lymphoma classifications and leukocyte characteristics. The PAT approach thus provides a classification scheme based on a multidimensional understanding of the expression landscape of this disease.
Cancer hallmark types
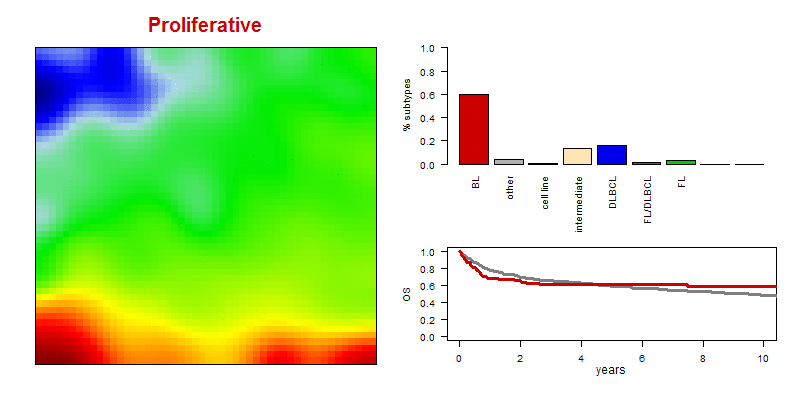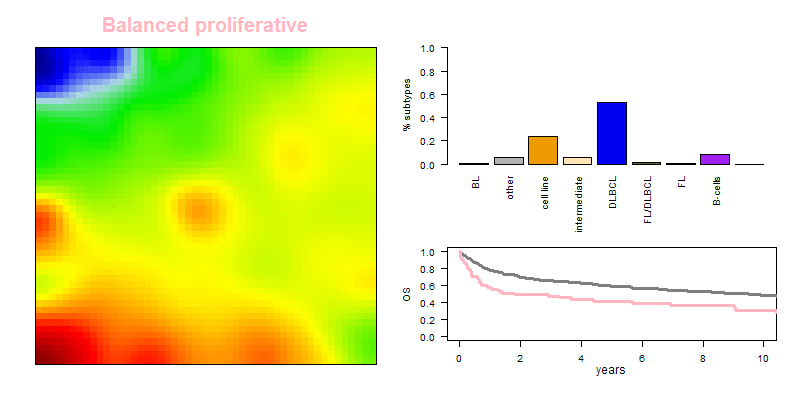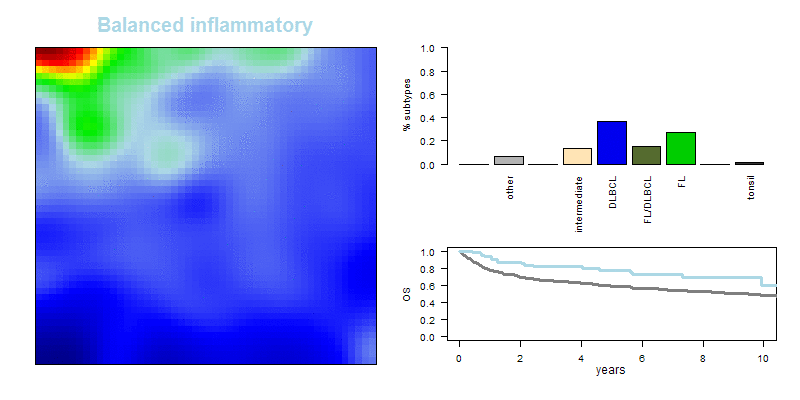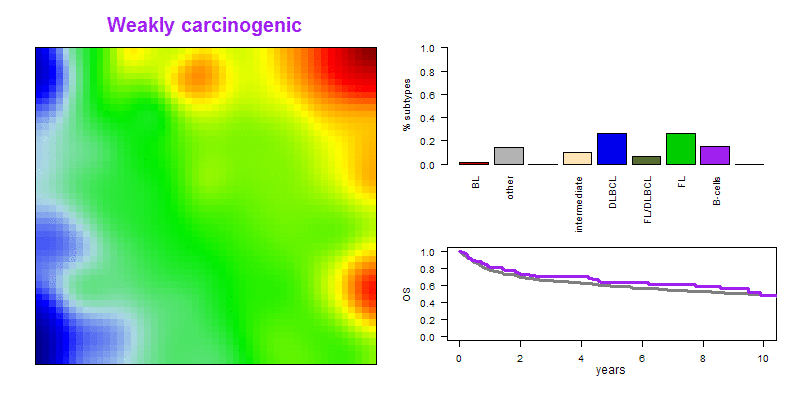
For a more generalized assignment of the PATs, we make use of a cancer hallmark scheme [40]. We defined eight hallmark signatures using GO and literature-gene sets, applied them to each PAT and represented its hallmark signature in terms of a polar diagram (Additional file 1: Figures S13 and S14). The PATs were then grouped into five hallmark types (HTs, see Fig. 7): (i) The proliferative HT with activated hallmark proliferation, controlling genetic instability, invasion and metastasis and, partly, regenerative immortality, collects mainly BL and intermediate lymphoma with over-expressed spots A, B and D. (ii) The balanced proliferative HT with a moderate activation of the hallmark proliferation and a reduced level of invasion and metastasis collects intermediate lymphoma and DLBCL over-expressing spots D, E and H including ABC-DLBCL. (iii) The inflammatory HT with the activated hallmark ‘inflammation’ contains DLBCL especially of the GCB type, FL and, to a lesser degree, DLBCL/FL expressing spots E, F and partly G. (iv) The balanced inflammatory HT with reduced activity of ‘inflammation’ and dominating hallmark ‘angiogenesis’ due to the over-expression of spots G and I collects mainly DLBCL/FL; (v) The weakly carcinogenic HT with generally low overall hallmark activities which collects lymphoma showing partly healthy B cell characteristics. Note that the hallmark ‘angiogenesis’ associates mainly with spot G that enriches stromal [9] and also inflammatory [45] characteristics (Additional file 1: Figure S13c). The samples assigned to each HT occupy almost distinct regions of the similarity net thus reflecting homogeneous expression landscapes (Fig. 7b). Their over-expression spot patterns shift along the edges of the map due to mutual similarities between the HTs (Fig. 7c). Hence, the concept of cancer hallmarks coarsens the expression characteristics and provides a simplified stratification scheme of lymphomas.
Fig. 7.

Cancer hallmark types (HT) were characterized using an expression signature for each of the eight hallmarks and clustering of the lymphoma samples into five HT. a The expression levels of the hallmark signatures were presented in terms of a polar plot (hallmark diagrams) for each of the HTs. Each hallmark is assigned to one polar axis as indicated in the legend. HTs differ markedly regarding the hallmarks ‘inflammation’ on the one hand and ‘proliferation’ and ‘invasion and metastasis’ on the other hand. b Samples assigned to each of the five HTs were colored in the correlation network, where each dot represents one sample. It reveals that the proliferative, inflammatory and weak HTs occupy three different, mutually separated regions while the two balanced HTs fill the transition zones in between them. c Mean expression portraits of the HTs reveal different regions of over- and under-expression, which can be directly compared with the portraits of the subtypes (Fig. 1a) and PATs (Fig. 5)
Prognostic HR map
Next, we generated a prognostic map by associating high expression levels in each of the metagenes of the SOM with the hazard ratio (HR) between the lymphoma patients expressing and not expressing this metagene (Fig. 8a). Red regions of bad prognosis include spots B–D upregulated typically in the proliferative HT and especially the balanced proliferative HT, while blue areas of better prognosis refer mainly to genes upregulated in the balanced inflammatory HT expressing spots G–J predominantly in DLBCL, FL and FL/DLBCL (compare with Fig. 7c). The overall survival (OS) curves of the HTs confirm this observation (Fig. 8c). Inflammation (and stromal) signatures in combination with healthy B cell and tonsil characteristics obviously associate with better survival, while proliferation in combination with inflammation worsens it. Regions of best and worst prognosis near spots K (HR< 0.5) and H (HR > 2), respectively, indeed collect genes that upregulate in the two balanced HTs (compare with Fig. 7c). Interestingly, the respective OS curves (Fig. 8b) resemble that of GCB- and ABC-DLBCL (Fig. 8d), whose portraits show over-expression in the regions of low and high HR around spots K and H, respectively (see Fig. 4). These regions were assigned to B cell development and B cell receptor pathway activity (spot K) and maturation into plasma cells (spot H) harboring the genes BCL6 and PRDM1, respectively, with key roles in lymphomagenesis [72, 73]. The composition of cases from both regions indeed reveals a higher prevalence of ABC-DLBCL and MM with plasma cell characteristics for worse prognosis and of GCB-DLBCL, FL, FL/DLBCL and PMBCL for better prognosis (Fig. 8b). Stratification of the HR map regarding the lymphoma subtypes reveals common prognostic patterns as evident in the overall HR map (Additional file 1: Figure S15).
Fig. 8.

Prognostic map and overall survival (OS) curves for selected groups of tumors. The prognostic map obtained reveals regions of worse prognosis in red and of better prognosis in blue (panel a). The dark blue region near spots K (HR < 0.5) and the maroon region near spot H (HR > 2) associate with best and worst prognosis, respectively. The respective OS curves (b) resemble that of the balanced proliferative HT and of ABC-DLBCL on the one side and that of the balanced inflammatory HT and of GCB-DLBCL on the other side (panels c and d). e–h OS curves of the major subtypes (e) which are further stratified for children and adults for BL (f). OS curves of selected PATs (g) and of DLBCL-related PATs (h) associate spot combinations with prognosis. Hazard ratios (HR) are given for significantly differential curves with p value < 0.01 in Cox model. HRs which are still significant after adjustment for therapy are marked with an asterisk. See also Table S4 and Table S5 for HRs and p values of all pairwise comparisons and of the co-factors
Figure 8e shows OS curves of the major lymphoma subtypes. That of FL tumors reflects the indolent but in most instances incurable character of this disease [74]. In contrast, about 25% of the BL cases die within 2 years after diagnosis, but afterward, the survival curve indicates good prognosis for the survivors. Stratification with respect to age provides a significantly better long-term prognosis for children (p = 0.02, HR = 0.4) in terms of the plateau level (Fig. 8f). Stratification of the OS curves for the PATs further diversifies prognosis (Fig. 8g). The DLBCL cases split into PATs with better (‘G’, ‘E F’ and ‘F G’; HR = 0.5–0.7; HRs refer to all other DLBCL) and worse (‘F’, ‘E’, ‘A’ and ‘none’; HR = 1.3–2.2) prognosis (Fig. 8h, Additional file 1: Table S4). Hence, spot F collecting genes involved in inflammatory response seems to play an ambivalent role, depending if activation is in concert with, e.g., module ‘E’ or sole of spot ‘F’. Sole expression of spot A in double-hit DLBCL drastically worsens prognosis (Fig. 8h). Poor prognosis of DLBCL associates with expression of spot D (see, e.g. the portraits of PATs ‘A’ and ‘E’ in Fig. 5a, and Fig. 8a). These PATs are in correspondence with a recently identified molecular high-grade (MHG) group of DLBC which is characterized by a proliferative and BL-like phenotype which enriches double-hit lymphomas [75].
Overall, it should be taken into account that due to the retrospective nature of the study, patients had been treated with various chemotherapy regimens including rituximab in only a part of cases. Nevertheless, the prognostic map links gene signatures of poor and good prognosis with underlying molecular functions. ABC- and GCB-like transcriptional characteristics associate with worst and best prognosis of DLBCL, respectively. Stratification with respect to PATs associates spot-related molecular programs with the aggressiveness of the disease. GIF animations visualize the mutual relatedness of the PAT- and HT-related SOM portraits (Additional files 5 and 6).
Phenotype similarity and tumor development
SOM portraying further enabled us to establish phenotypic trees of mutual relatedness on three levels of resolution, namely for individual sample portraits, mean subtypes and mean PAT portraits, respectively (Additional file 1: Figure S16). The intermediate PAT level provides the most informative tree structure showing one backbone with two major side branches and well-resolved PAT leaves (Fig. 9). The horizontal backbone describes a series of PATs referring predominantly to lymphomas of the BL, intermediate and DLBCL subtypes (from the left to the right). It is characterized by antagonistic alterations of a dark zone (DZ)-like proliferative signature and more light zone (LZ)-like and inflammatory signatures.
Fig. 9.

The lymphoma phenotype similarity tree. a The PAT-level tree visualizes the similarity relations between the core regions of the subtypes, the mutual transition ranges and their relation to the controls. b Different regions of the landscape associate with different B cell-related expression signatures and changing hallmark characteristics
The left vertical side branch collects mainly DLBCL cases with weak carcinogenic hallmark characteristics and also multiple myeloma showing both similarities of their transcriptomes with healthy B cells. The second side branch on the right contains mainly FL with increasing resemblance with tonsil’s expression signature. On average, the grading of FL increases towards the end of this branch due to gained transcriptional specifics of FL in terms of PATs expressing spot I with increasing grade. On the other hand, FL/DLBCL (FL3b) accumulate along the main backbone as mixed G-type PATs expressing also spot F as the main hallmark of DLBCL which manifests transformation of FL into DLBCL. Hence, FL development splits into two different paths, either reflecting an increasing level of the FL characteristics (spot I) or an increasing contribution of the DLBCL-specific spot-signature F in FL/DLBCL in correspondence with [76]. The expression landscape illustrates also another path of FL progression which is associated with the appearance of a second chromosomal translocation gained in addition to the primary t(14;18) hit [69]. Here, we exemplarily considered a secondary t(8;14) IG-MYC translocation, which induces a jump-like change of the expression phenotype by activating module A. It leads to PATs closely resembling that of IG-MYC-positive single-hit lymphoma with an activated proliferative cellular program (Fig. 9b). Overall, the phenotypic tree establishes similarity relations between the transcriptomes of the major lymphoma subtypes in terms of common and different transcriptional programs; it identifies a distinct branch of lymphomas expressing similarities with healthy B cells, and it reveals possible progression paths, e.g. of FL with increasing grade and composite lymphomas such as DLBCL/FL.
Discussion
We presented a transcriptome map of B cell lymphoma which provides a holistic view on their expression landscape, the heterogeneity of activated gene-regulatory programs and their association with different lymphoma subtypes. The novelty hereby is that the map considers the whole range of variation of mature B cell lymphoma including a series of subtypes and healthy cell references and that it enables modularization of the landscape into expression states, their functional interpretation and visualization in terms of portraits of the different lymphoma strata and individual cases. These states can be grouped into five hallmark types on the coarsest level of stratification with proliferation, inflammation and stroma/angiogenesis as the most relevant hallmark dimensions. Combinatorial pattern types of activated modules stratify the lymphomas with higher resolution. The lymphoma map allows the evaluation of the transcriptome landscape which combines different aspects: (i) subtype-specific over- and under-expression; (ii) biological functions of the related expression modules; (iii) mutations of key genes according to their location in the map and (iv) survival hazard ratios and regions of better and of worse prognosis. Mapping of previous subtyping schemes enables the mutual comparison and characterization of GC-derived B cell lymphomas, of multiple myeloma and mantle cell lymphoma and also of the reference B cells within a unique data landscape. It reflects major aspects of B cell maturation and GC biology.
Exemplarily, our analysis provided a close look on the transition range between FL and DLBCL, on DLBCL with poor prognosis showing expression patterns resembling that of BL, and particularly on ‘double-hit’ MYC and BCL2 transformed lymphomas. In these respects, the definition of clear-cut separating criteria between the different sub-entities of lymphomas is difficult to establish due to the smooth character of their expression landscape that forms rather a continuum of molecular states than distinct clusters. These transition regions have impact regarding tumor development and transformations between different subtypes.
Conclusions
The transcriptome map of lymphomas provides a tool that aggregates, refines, interprets and visualizes previous lymphoma data to provide a reference system in current and future studies. Particularly, it provides a reference landscape which can be utilized to map sets of signature genes and classifiers obtained in new and independent studies for comparison with the MMML cases and strata presented here, and for judging their impact in terms of function and prognosis. It considers the whole spectrum of cases in the MMML cohort thus representing an overview map. Zoom-in maps with enhanced resolution can be generated for more detailed molecular pictures of subsets of cases as demonstrated here for B cells, lymphoma cell lines and BL, and previously for DLBCL and BL [33] and in the context of human tissues [23]. Our analyses demonstrated that consideration of a wide collection of different subtypes into a joint landscape extends the state space of expression phenotypes covered in the map with sufficient resolution and allows for their interpretation in a common context. The map offers the option of extension by adding new cases from other lymphoma studies to further widen the transcriptional landscape and/or to classify and to interpret them according to the classification schemes presented. Tools such as an interactive ‘oposSOM-browser’ are presently under development for potential use in lymphoma diagnostics and molecular interpretation of gene expression patterns. Finally, our multivariate PAT concept provides a nosology scheme for describing heterogeneity also of other cancer types with high granularity.
Additional files
Supplementary text containing Figures S1–S19 and Tables S1–S5. (PDF 6310 kb)
Table of samples, providing histopathological and molecular subtypes. (XLSX 41 kb)
Complete gallery of all 936 sample expression portraits. (PDF 14216 kb)
Lists of genes for each of the spot modules. (XLSX 256 kb)
{kind=link}
Animated expression portraits of the PATs together with survival curves. (GIF 556 kb)
{kind=link}
Animated expression portraits of the HTs together with survival curves. (GIF 249 kb)
Acknowledgements
For a complete list of the members of the MMML consortium, see Additional file 1.
Funding
The Molecular Mechanisms in Malignant Lymphomas Network (MMML) Project was supported by the Deutsche Krebshilfe (FKZ 70-3173-Tr3). The project was further supported by the Federal Ministry of Education and Research (BMBF), project grant No. FKZ 031 6166 (MMML-MYC-SYS) and FKZ 031 5452A (HaematoSYS), the cooperative projects WTZ ARM II-010 (BMBF)/01ZX1304A (AS of RA, to HB and AA), FFE-0034 (BMBF, to HLW) and 16GE-025 (AS of RA, to AA and HB).
Availability of data and materials
The whole expression data set was collected as part of the German MMML consortium (Molecular Mechanisms in Malignant Lymphoma) and is partly available in the GEO repository under accession numbers GSE4475 [7], GSE10172 [15], GSE22470 [16], GSE48184 [18], GSE43677 [10] and GSE103944 [76]. The complete data are available from ‘The Leipzig Health Atlas’ repository under accession number 7VT47TM4GV-1 (https://www.health-atlas.de/index.php/en/lha/7VT47TM4GV-1).
Abbreviations
- ABC
Lymphoma of the activated B cell type
- BL
Burkitt’s lymphoma
- DHL
Double-hit lymphoma
- DLBCL
Diffuse large B cell lymphoma
- DZ
Dark zone of germinal center
- FL
Follicular lymphoma with t(14;18) translocation (BCL2-positive FL)
- GC
Germinal center
- GCB
Lymphoma of the germinal center B cell type
- GSZ
Gene set enrichment Z-score as introduced by [38]
- GWAS
Genome-wide association study
- HiGA-Pro
High gene activity, proliferative phenotype as defined by [10]
- HiGA-Sir
High gene activity, stroma and immune response phenotype as defined by [10]
- IG-MYC
Tumor-biopsy specimens in which MYC was fused to IGH, IGK or IGL
- IHC
Immunohistochemical
- LoGA
Low gene activity phenotype as defined by [10]
- LZ
Light zone of germinal center
- mBL
Molecular Burkitt’s lymphoma subtype according to Hummel et al. [7]
- MM
Multiple myelomas
- MMML
Molecular Mechanisms of Malignant Lymphoma
- non-IG-MYC
Lymphomas with MYC breakpoints without fusion of MYC to an IG locus
- non-mBL
Non-molecular Burkitt’s lymphoma according to Hummel et al. [7]
- PAP
Pathway activation pattern as defined in [8]
- PAT
Pattern types defined in this study
- SOM
Self-organizing map
Authors’ contributions
LT, ML, RS and HS are the steering board of the MMML study. HLW, MK and HB conceived and performed the analysis and are the major contributors to the manuscript. All authors contributed to MS. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was performed as part of the ‘Molecular Mechanisms in Malignant Lymphomas’ Network Project of the Deutsche Krebshilfe for which central (University Hospital, Göttingen), and local ethics approval was obtained. Informed consent was obtained in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. Dido Lenze (now affiliated with AstraZeneca) contributed to the MMML consortium during her employment at Charité Universitätsmedizin, Berlin.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Henry Loeffler-Wirth, Email: wirth@izbi.uni-leipzig.de.
Markus Kreuz, Email: markus.kreuz@imise.uni-leipzig.de.
Lydia Hopp, Email: hopp@izbi.uni-leipzig.de.
Arsen Arakelyan, Email: aarakelyan@sci.am.
Andrea Haake, Email: haake_andrea@web.de.
Sergio B. Cogliatti, Email: sergio.cogliatti@kssg.ch
Alfred C. Feller, Email: feller@haematopathologie-luebeck.de
Martin-Leo Hansmann, Email: m.l.hansmann@em.uni-frankfurt.de.
Dido Lenze, Email: dido.lenze@astrazeneca.com.
Peter Möller, Email: peter.moeller@uniklinik-ulm.de.
Hans Konrad Müller-Hermelink, Email: konrad.mh@mail.uni-wuerzburg.de.
Erik Fortenbacher, Email: fortenbacher@izbi.uni-leipzig.de.
Edith Willscher, Email: willscher@izbi.uni-leipzig.de.
German Ott, Email: German.Ott@rbk.de.
Andreas Rosenwald, Email: rosenwald@uni-wuerzburg.de.
Christiane Pott, Email: c.pott@med2.uni-kiel.de.
Carsten Schwaenen, Email: Carsten.Schwaenen@ortenau-klinikum.de.
Heiko Trautmann, Email: h.trautmann@med2.uni-kiel.de.
Swen Wessendorf, Email: s.wessendorf@klinikum-esslingen.de.
Harald Stein, Email: h.stein@pathodiagnostik.de.
Monika Szczepanowski, Email: m.szczepanowski@med2.uni-kiel.de.
Lorenz Trümper, Email: lorenz.truemper@med.uni-goettingen.de.
Michael Hummel, Email: michael.hummel@charite.de.
Wolfram Klapper, Email: wklapper@path.uni-kiel.de.
Reiner Siebert, Email: reiner.siebert@uni-ulm.de.
Markus Loeffler, Email: markus.loeffler@imise.uni-leipzig.de.
Hans Binder, Email: binder@izbi.uni-leipzig.de.
References
- 1.O'Connor O. A., Tobinai K. Putting the Clinical and Biological Heterogeneity of Non-Hodgkin Lymphoma into Context. Clinical Cancer Research. 2014;20(20):5173–5181. doi: 10.1158/1078-0432.CCR-14-0574. [DOI] [PubMed] [Google Scholar]
- 2.Campo E., Swerdlow S. H., Harris N. L., Pileri S., Stein H., Jaffe E. S. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berglund Mattias, Thunberg Ulf, Amini Rose-Marie, Book Majlis, Roos Göran, Erlanson Martin, Linderoth Johan, Dictor Michael, Jerkeman Mats, Cavallin-Ståhl Eva, Sundström Christer, Rehn-Eriksson Suzanne, Backlin Carin, Hagberg Hans, Rosenquist Richard, Enblad Gunilla. Evaluation of immunophenotype in diffuse large B-cell lymphoma and its impact on prognosis. Modern Pathology. 2005;18(8):1113–1120. doi: 10.1038/modpathol.3800396. [DOI] [PubMed] [Google Scholar]
- 4.Caro Pilar, Kishan Amar U., Norberg Erik, Stanley Illana A., Chapuy Bjoern, Ficarro Scott B., Polak Klaudia, Tondera Daniel, Gounarides John, Yin Hong, Zhou Feng, Green Michael R., Chen Linfeng, Monti Stefano, Marto Jarrod A., Shipp Margaret A., Danial Nika N. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell. 2012;22(4):547–560. doi: 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alizadeh Ash A., Eisen Michael B., Davis R. Eric, Ma Chi, Lossos Izidore S., Rosenwald Andreas, Boldrick Jennifer C., Sabet Hajeer, Tran Truc, Yu Xin, Powell John I., Yang Liming, Marti Gerald E., Moore Troy, Hudson James, Lu Lisheng, Lewis David B., Tibshirani Robert, Sherlock Gavin, Chan Wing C., Greiner Timothy C., Weisenburger Dennis D., Armitage James O., Warnke Roger, Levy Ronald, Wilson Wyndham, Grever Michael R., Byrd John C., Botstein David, Brown Patrick O., Staudt Louis M. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 6.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. [Internet]. 2003 [cited 2012 Nov 15];100:9991–6. Available from: https://www.ncbi.nlm.nih.gov/pubmed/12900505. [DOI] [PMC free article] [PubMed]
- 7.Hummel Michael, Bentink Stefan, Berger Hilmar, Klapper Wolfram, Wessendorf Swen, Barth Thomas F.E., Bernd Heinz-Wolfram, Cogliatti Sergio B., Dierlamm Judith, Feller Alfred C., Hansmann Martin-Leo, Haralambieva Eugenia, Harder Lana, Hasenclever Dirk, Kühn Michael, Lenze Dido, Lichter Peter, Martin-Subero Jose Ignacio, Möller Peter, Müller-Hermelink Hans-Konrad, Ott German, Parwaresch Reza M., Pott Christiane, Rosenwald Andreas, Rosolowski Maciej, Schwaenen Carsten, Stürzenhofecker Benjamin, Szczepanowski Monika, Trautmann Heiko, Wacker Hans-Heinrich, Spang Rainer, Loeffler Markus, Trümper Lorenz, Stein Harald, Siebert Reiner. A Biologic Definition of Burkitt's Lymphoma from Transcriptional and Genomic Profiling. New England Journal of Medicine. 2006;354(23):2419–2430. doi: 10.1056/NEJMoa055351. [DOI] [PubMed] [Google Scholar]
- 8.Bentink S, Wessendorf S, Schwaenen C, Rosolowski M, Klapper W, Rosenwald A, Ott G, Banham A H, Berger H, Feller A C, Hansmann M-L, Hasenclever D, Hummel M, Lenze D, Möller P, Stuerzenhofecker B, Loeffler M, Truemper L, Stein H, Siebert R, Spang R. Pathway activation patterns in diffuse large B-cell lymphomas. Leukemia. 2008;22(9):1746–1754. doi: 10.1038/leu.2008.166. [DOI] [PubMed] [Google Scholar]
- 9.Lenz G., Wright G., Dave S.S., Xiao W., Powell J., Zhao H., Xu W., Tan B., Goldschmidt N., Iqbal J., Vose J., Bast M., Fu K., Weisenburger D.D., Greiner T.C., Armitage J.O., Kyle A., May L., Gascoyne R.D., Connors J.M., Troen G., Holte H., Kvaloy S., Dierickx D., Verhoef G., Delabie J., Smeland E.B., Jares P., Martinez A., Lopez-Guillermo A., Montserrat E., Campo E., Braziel R.M., Miller T.P., Rimsza L.M., Cook J.R., Pohlman B., Sweetenham J., Tubbs R.R., Fisher R.I., Hartmann E., Rosenwald A., Ott G., Muller-Hermelink H.-K., Wrench D., Lister T.A., Jaffe E.S., Wilson W.H., Chan W.C., Staudt L.M. Stromal Gene Signatures in Large-B-Cell Lymphomas. New England Journal of Medicine. 2008;359(22):2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosolowski Maciej, Läuter Jürgen, Abramov Dmitriy, Drexler Hans G., Hummel Michael, Klapper Wolfram, MacLeod Roderick A.F., Pellissery Shoji, Horn Friedemann, Siebert Reiner, Loeffler Markus. Massive Transcriptional Perturbation in Subgroups of Diffuse Large B-Cell Lymphomas. PLoS ONE. 2013;8(11):e76287. doi: 10.1371/journal.pone.0076287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. [Internet]. 2005;87:163–208. Available from: https://www.ncbi.nlm.nih.gov/pubmed/16102574. [cited 2015 Jan 7] [DOI] [PMC free article] [PubMed]
- 12.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. [Internet]. 2018 [cited 2018 Aug 28];24:679–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29713087. [DOI] [PMC free article] [PubMed]
- 13.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell [Internet]. 2017 [cited 2018 Apr 27];171:481–494.e15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28985567. [DOI] [PMC free article] [PubMed]
- 14.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. [Internet]. 2018 [cited 2018 may 17];378:1396–407. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29641966. [DOI] [PMC free article] [PubMed]
- 15.Klapper W, Szczepanowski M, Burkhardt B, Berger H, Rosolowski M, Bentink S, et al. Molecular profiling of pediatric mature B-cell lymphoma treated in population-based prospective clinical trials. Blood [Internet]. 2008 [cited 2014 Apr 9];112:1374–81. Data available from Gene Expression Omnibus (GEO), accession number GSE10172: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10172 [DOI] [PubMed]
- 16.Salaverria I., Philipp C., Oschlies I., Kohler C. W., Kreuz M., Szczepanowski M., Burkhardt B., Trautmann H., Gesk S., Andrusiewicz M., Berger H., Fey M., Harder L., Hasenclever D., Hummel M., Loeffler M., Mahn F., Martin-Guerrero I., Pellissery S., Pott C., Pfreundschuh M., Reiter A., Richter J., Rosolowski M., Schwaenen C., Stein H., Trumper L., Wessendorf S., Spang R., Kuppers R., Klapper W., Siebert R. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood. 2011;118(1):139–147. doi: 10.1182/blood-2011-01-330795. [DOI] [PubMed] [Google Scholar]
- 17.Klapper W., Kreuz M., Kohler C. W., Burkhardt B., Szczepanowski M., Salaverria I., Hummel M., Loeffler M., Pellissery S., Woessmann W., Schwanen C., Trumper L., Wessendorf S., Spang R., Hasenclever D., Siebert R. Patient age at diagnosis is associated with the molecular characteristics of diffuse large B-cell lymphoma. Blood. 2012;119(8):1882–1887. doi: 10.1182/blood-2011-10-388470. [DOI] [PubMed] [Google Scholar]
- 18.Masqué-Soler N, Szczepanowski M, Kohler CW, Spang R, Klapper W. Molecular classification of mature aggressive B-cell lymphoma using digital multiplexed gene expression on formalin-fixed paraffin-embedded biopsy specimens. Blood [Internet]. 2013 [cited 2014 Apr 7];122:1985–6. Data available from Gene Expression Omnibus (GEO), accession number GSE48184: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48184 [DOI] [PubMed]
- 19.Gentles A. J., Alizadeh A. A., Lee S.-I., Myklebust J. H., Shachaf C. M., Shahbaba B., Levy R., Koller D., Plevritis S. K. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood. 2009;114(15):3158–3166. doi: 10.1182/blood-2009-02-202465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aquino Gabriella, Marra Laura, Cantile Monica, De Chiara Annarosaria, Liguori Giuseppina, Curcio Maria, Sabatino Rocco, Pannone Giuseppe, Pinto Antonio, Botti Gerardo, Franco Renato. MYC chromosomal aberration in differential diagnosis between Burkitt and other aggressive lymphomas. Infectious Agents and Cancer. 2013;8(1):37. doi: 10.1186/1750-9378-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beham-Schmid Christine. Aggressive lymphoma 2016: revision of the WHO classification. memo - Magazine of European Medical Oncology. 2017;10(4):248–254. doi: 10.1007/s12254-017-0367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lossos Izidore S., Gascoyne Randy D. Transformation of follicular lymphoma. Best Practice & Research Clinical Haematology. 2011;24(2):147–163. doi: 10.1016/j.beha.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wirth H, Löffler M, von Bergen M, Binder H. Expression cartography of human tissues using self organizing maps. BMC Bioinformatics [Internet]. 2011;12:306–52. Available from: https://www.ncbi.nlm.nih.gov/pubmed/21794127. [cited 2011 Jul 28] [DOI] [PMC free article] [PubMed]
- 24.Binder H, Wirth H. Analysis of Large-Scale OMIC Data Using Self Organizing Maps. In: Khosrow-Pour M, editor. Encycl. Inf. Sci. Technol. [Internet]. 3rd ed. Hershey, PA, USA; 2015 [cited 2014 Sep 17]. p. 1642–53. Available from: http://www.igi-global.com/chapter/analysis-of-large-scale-omic-data-using-self-organizing-maps/112569
- 25.Camp J. Gray, Sekine Keisuke, Gerber Tobias, Loeffler-Wirth Henry, Binder Hans, Gac Malgorzata, Kanton Sabina, Kageyama Jorge, Damm Georg, Seehofer Daniel, Belicova Lenka, Bickle Marc, Barsacchi Rico, Okuda Ryo, Yoshizawa Emi, Kimura Masaki, Ayabe Hiroaki, Taniguchi Hideki, Takebe Takanori, Treutlein Barbara. Multilineage communication regulates human liver bud development from pluripotency. Nature. 2017;546(7659):533–538. doi: 10.1038/nature22796. [DOI] [PubMed] [Google Scholar]
- 26.Binder Hans, Hopp Lydia, Schweiger Michal R, Hoffmann Steve, Jühling Frank, Kerick Martin, Timmermann Bernd, Siebert Susann, Grimm Christina, Nersisyan Lilit, Arakelyan Arsen, Herberg Maria, Buske Peter, Loeffler-Wirth Henry, Rosolowski Maciej, Engel Christoph, Przybilla Jens, Peifer Martin, Friedrichs Nicolaus, Moeslein Gabriela, Odenthal Margarete, Hussong Michelle, Peters Sophia, Holzapfel Stefanie, Nattermann Jacob, Hueneburg Robert, Schmiegel Wolff, Royer-Pokora Brigitte, Aretz Stefan, Kloth Michael, Kloor Matthias, Buettner Reinhard, Galle Jörg, Loeffler Markus. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. The Journal of Pathology. 2017;243(2):242–254. doi: 10.1002/path.4948. [DOI] [PubMed] [Google Scholar]
- 27.Gerber T, Willscher E, Loeffler-Wirth H, Hopp L, Schadendorf D, Schartl M, et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget [Internet]. 2017;8:846–62. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27903987. [cited 2017 Jun 9] [DOI] [PMC free article] [PubMed]
- 28.Binder H, Hopp L, Lembcke K, Wirth H. Personalized disease phenotypes from massive OMICs data. In: Baoying W, Li R, Perrizo W, editors. Big Data Anal. Bioinforma Healthc. [Internet]. Hershey, PA, USA: IGI Global; 2014. p. 359–78. Available from: http://www.igi-global.com/book/big-data-analytics-bioinformatics-healthcare/110030
- 29.Hopp Lydia, Wirth Henry, Fasold Mario, Binder Hans. Portraying the expression landscapes of cancer subtypes. Systems Biomedicine. 2013;1(2):99–121. doi: 10.4161/sysb.25897. [DOI] [Google Scholar]
- 30.Drexler HG. Establishment and culture of leukemia–lymphoma cell lines. Methods Mol. Biol. [Internet]. 2011 [cited 2019 Jan 17]. p. 181–200. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21516408. [DOI] [PubMed]
- 31.Drexler H. The leukemia-lymphoma cell line factsbook [Internet]. Academic Press; 2001 [cited 2019 Jan 24]. Available from: https://books.google.de/books?hl=de&lr=&id=yL5ysmGMGLYC&oi=fnd&pg=PP1&dq=The+Leukemia-Lymphoma+Cell+Line+FactsBook&ots=cg63DvpTa8&sig=zIZtGFzxXKy3SHxb0Trv5O7tBjQ
- 32.Wirth H, von Bergen M, Binder H. Mining SOM expression portraits: feature selection and integrating concepts of molecular function. BioData Min. [Internet]. 2012 [cited 2013 Mar 8];5:18–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23043905. [DOI] [PMC free article] [PubMed]
- 33.Hopp L, Lembcke K, Binder H, Wirth H. Portraying the expression landscapes of B-cell lymphoma - intuitive detection of outlier samples and of molecular subtypes. Biology (Basel) 2013;2:1411–1437. doi: 10.3390/biology2041411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Löffler-Wirth H, Kalcher M, Binder H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on Bioconductor. Bioinformatics [Internet]. 2015 [cited 2015 Jun 14]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/26063839. [DOI] [PubMed]
- 35.Agrawal R, Srikant R. Fast algorithms for minin g association rules in large databases. VLDB ‘94 Proc. 20th Int. Conf. Very Large Data Bases. San Francisco: Morgan Kaufmann Publishers Inc.; 1994. p. 489–99.
- 36.Zhang B., Kirov S., Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Research. 2005;33(Web Server):W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vêncio RZN, Shmulevich I. ProbCD: enrichment analysis accounting for categorization uncertainty. BMC Bioinformatics [Internet]. 2007;8:383. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17935624. [cited 2011 Mar 16] [DOI] [PMC free article] [PubMed]
- 38.Törönen P, Ojala PJ, Marttinen P, Holm L. Robust extraction of functional signals from gene set analysis using a generalized threshold free scoring function. BMC Bioinformatics [Internet]. 2009;10:307. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19775443. [DOI] [PMC free article] [PubMed]
- 39.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell [Internet]. 2000 [cited 2014 Feb 19];100:57–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10647931. [DOI] [PubMed]
- 40.Hanahan Douglas, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Newman Aaron M, Liu Chih Long, Green Michael R, Gentles Andrew J, Feng Weiguo, Xu Yue, Hoang Chuong D, Diehn Maximilian, Alizadeh Ash A. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nersisyan L, Löffler-Wirth H, Arakelyan A, Binder H. Gene set-and pathway-centered knowledge discovery assigns transcriptional activation patterns in brain, blood, and colon cancer: a bioinformatics perspective. Int. J. Knowl. Discov. Bioinforma. [Internet]. 2016 [cited 2018 Apr 6];4. Available from: https://www.igi-global.com/article/gene-set%2D%2Dand-pathway%2D%2Dcentered-knowledge-discovery-assigns-transcriptional-activation-patterns-in-brain-blood-and-colon-cancer/147303
- 43.Feller A, Diebold J. Histopathology of nodal and extranodal non-Hodgkin’s lymphomas (based on the WHO classification) New York: Springer; 2004. [Google Scholar]
- 44.Lennert K, Feller A. Histopathology of non-Hodgkin’s lymphomas. New York: Springer; 1992. [Google Scholar]
- 45.Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. [Internet]. 2004 [cited 2014 Jan 3];351:2159–69. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15548776. [DOI] [PubMed]
- 46.Monti S. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105(5):1851–1861. doi: 10.1182/blood-2004-07-2947. [DOI] [PubMed] [Google Scholar]
- 47.Tarte K, Zhan F, De Vos J, Klein B, Shaughnessy J. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood [Internet]. 2003 [cited 2014 mar 20];102:592–600. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12663452. [DOI] [PubMed]
- 48.Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood [Internet]. 2012;120:2240–8. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22740445. [cited 2014 May 30] [DOI] [PMC free article] [PubMed]
- 49.Haddad R. Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood. 2004;104(13):3918–3926. doi: 10.1182/blood-2004-05-1845. [DOI] [PubMed] [Google Scholar]
- 50.Campo Elias. New pathogenic mechanisms in Burkitt lymphoma. Nature Genetics. 2012;44(12):1288–1289. doi: 10.1038/ng.2476. [DOI] [PubMed] [Google Scholar]
- 51.Küppers Ralf. Mechanisms of B-cell lymphoma pathogenesis. Nature Reviews Cancer. 2005;5(4):251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 52.Schneider Christof, Pasqualucci Laura, Dalla-Favera Riccardo. Molecular pathogenesis of diffuse large B-cell lymphoma. Seminars in Diagnostic Pathology. 2011;28(2):167–177. doi: 10.1053/j.semdp.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Hematol Am Soc Hematol Educ Program. 2013;2013:575–583. doi: 10.1182/asheducation-2013.1.575. [DOI] [PubMed] [Google Scholar]
- 54.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature [Internet] 2012;490:116–120. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tzankov A. Prognostic significance of CD44 expression in diffuse large B cell lymphoma of activated and germinal centre B cell-like types: a tissue microarray analysis of 90 cases. Journal of Clinical Pathology. 2003;56(10):747–752. doi: 10.1136/jcp.56.10.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Jenny, Grubor Vladimir, Love Cassandra L., Banerjee Anjishnu, Richards Kristy L., Mieczkowski Piotr A., Dunphy Cherie, Choi William, Au Wing Yan, Srivastava Gopesh, Lugar Patricia L., Rizzieri David A., Lagoo Anand S., Bernal-Mizrachi Leon, Mann Karen P., Flowers Christopher, Naresh Kikkeri, Evens Andrew, Gordon Leo I., Czader Magdalena, Gill Javed I., Hsi Eric D., Liu Qingquan, Fan Alice, Walsh Katherine, Jima Dereje, Smith Lisa L., Johnson Amy J., Byrd John C., Luftig Micah A., Ni Ting, Zhu Jun, Chadburn Amy, Levy Shawn, Dunson David, Dave Sandeep S. Genetic heterogeneity of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences. 2013;110(4):1398–1403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malek S, Kaminski M, Li H, Ouillette P, Jones S, Fox H, et al. Recurrent STAT6 mutations in follicular lymphoma. Blood [Internet]. 2013 [cited 2014 Jun 16];122. Available from: http://bloodjournal.hematologylibrary.org/content/122/21/503.short
- 58.Pasqualucci Laura, Dominguez-Sola David, Chiarenza Annalisa, Fabbri Giulia, Grunn Adina, Trifonov Vladimir, Kasper Lawryn H., Lerach Stephanie, Tang Hongyan, Ma Jing, Rossi Davide, Chadburn Amy, Murty Vundavalli V., Mullighan Charles G., Gaidano Gianluca, Rabadan Raul, Brindle Paul K., Dalla-Favera Riccardo. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Green M. R., Gentles A. J., Nair R. V., Irish J. M., Kihira S., Liu C. L., Kela I., Hopmans E. S., Myklebust J. H., Ji H., Plevritis S. K., Levy R., Alizadeh A. A. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121(9):1604–1611. doi: 10.1182/blood-2012-09-457283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell [Internet]. 2014;25:91–101. Available from: https://www.ncbi.nlm.nih.gov/pubmed/24434212. [cited 2014 Nov 12] [DOI] [PMC free article] [PubMed]
- 61.Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. [Internet]. 2012 [cited 2015 Jan 8];44:1316–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23143595. [DOI] [PubMed]
- 62.Okosun Jessica, Bödör Csaba, Wang Jun, Araf Shamzah, Yang Cheng-Yuan, Pan Chenyi, Boller Sören, Cittaro Davide, Bozek Monika, Iqbal Sameena, Matthews Janet, Wrench David, Marzec Jacek, Tawana Kiran, Popov Nikolay, O'Riain Ciaran, O'Shea Derville, Carlotti Emanuela, Davies Andrew, Lawrie Charles H, Matolcsy András, Calaminici Maria, Norton Andrew, Byers Richard J, Mein Charles, Stupka Elia, Lister T Andrew, Lenz Georg, Montoto Silvia, Gribben John G, Fan Yuhong, Grosschedl Rudolf, Chelala Claude, Fitzgibbon Jude. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nature Genetics. 2013;46(2):176–181. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasqualucci Laura, Khiabanian Hossein, Fangazio Marco, Vasishtha Mansi, Messina Monica, Holmes Antony B., Ouillette Peter, Trifonov Vladimir, Rossi Davide, Tabbò Fabrizio, Ponzoni Maurilio, Chadburn Amy, Murty Vundavalli V., Bhagat Govind, Gaidano Gianluca, Inghirami Giorgio, Malek Sami N., Rabadan Raul, Dalla-Favera Riccardo. Genetics of Follicular Lymphoma Transformation. Cell Reports. 2014;6(1):130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Green Michael R., Kihira Shingo, Liu Chih Long, Nair Ramesh V., Salari Raheleh, Gentles Andrew J., Irish Jonathan, Stehr Henning, Vicente-Dueñas Carolina, Romero-Camarero Isabel, Sanchez-Garcia Isidro, Plevritis Sylvia K., Arber Daniel A., Batzoglou Serafim, Levy Ronald, Alizadeh Ash A. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proceedings of the National Academy of Sciences. 2015;112(10):E1116–E1125. doi: 10.1073/pnas.1501199112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pasqualucci Laura, Compagno Mara, Houldsworth Jane, Monti Stefano, Grunn Adina, Nandula Subhadra V., Aster Jon C., Murty Vundavally V., Shipp Margaret A., Dalla-Favera Riccardo. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. The Journal of Experimental Medicine. 2006;203(2):311–317. doi: 10.1084/jem.20052204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandelbaum Jonathan, Bhagat Govind, Tang Hongyan, Mo Tongwei, Brahmachary Manisha, Shen Qiong, Chadburn Amy, Rajewsky Klaus, Tarakhovsky Alexander, Pasqualucci Laura, Dalla-Favera Riccardo. BLIMP1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-like Diffuse Large B Cell Lymphoma. Cancer Cell. 2010;18(6):568–579. doi: 10.1016/j.ccr.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salaverria I., Martin-Guerrero I., Wagener R., Kreuz M., Kohler C. W., Richter J., Pienkowska-Grela B., Adam P., Burkhardt B., Claviez A., Damm-Welk C., Drexler H. G., Hummel M., Jaffe E. S., Kuppers R., Lefebvre C., Lisfeld J., Loffler M., Macleod R. A. F., Nagel I., Oschlies I., Rosolowski M., Russell R. B., Rymkiewicz G., Schindler D., Schlesner M., Scholtysik R., Schwaenen C., Spang R., Szczepanowski M., Trumper L., Vater I., Wessendorf S., Klapper W., Siebert R. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood. 2014;123(8):1187–1198. doi: 10.1182/blood-2013-06-507996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Queirós A, Beekman R, Vilarrasa-Blasi R, Duran-Ferrer M, Clot G, Merkel A, et al. Decoding the DNA methylome of mantle cell lymphoma in the light of the entire B cell lineage. Cancer Cell. 2016;30:806–821. doi: 10.1016/j.ccell.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aukema S. M., Siebert R., Schuuring E., van Imhoff G. W., Kluin-Nelemans H. C., Boerma E.-J., Kluin P. M. Double-hit B-cell lymphomas. Blood. 2010;117(8):2319–2331. doi: 10.1182/blood-2010-09-297879. [DOI] [PubMed] [Google Scholar]
- 70.Ennishi Daisuke, Jiang Aixiang, Boyle Merrill, Collinge Brett, Grande Bruno M., Ben-Neriah Susana, Rushton Christopher, Tang Jeffrey, Thomas Nicole, Slack Graham W., Farinha Pedro, Takata Katsuyoshi, Miyata-Takata Tomoko, Craig Jeffrey, Mottok Anja, Meissner Barbara, Saberi Saeed, Bashashati Ali, Villa Diego, Savage Kerry J., Sehn Laurie H., Kridel Robert, Mungall Andrew J., Marra Marco A., Shah Sohrab P., Steidl Christian, Connors Joseph M., Gascoyne Randy D., Morin Ryan D., Scott David W. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. Journal of Clinical Oncology. 2019;37(3):190–201. doi: 10.1200/JCO.18.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gentles Andrew J, Newman Aaron M, Liu Chih Long, Bratman Scott V, Feng Weiguo, Kim Dongkyoon, Nair Viswam S, Xu Yue, Khuong Amanda, Hoang Chuong D, Diehn Maximilian, West Robert B, Plevritis Sylvia K, Alizadeh Ash A. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature Medicine. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vrzalikova Katerina, Woodman Ciaran Bernard John, Murray Paul Gerard. BLIMP1α, the master regulator of plasma cell differentiation is a tumor supressor gene in B cell lymphomas. Biomedical Papers. 2012;156(1):1–6. doi: 10.5507/bp.2012.003. [DOI] [PubMed] [Google Scholar]
- 73.Hatzi Katerina, Melnick Ari. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends in Molecular Medicine. 2014;20(6):343–352. doi: 10.1016/j.molmed.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salles GA. Clinical features, prognosis and treatment of follicular lymphoma. Hematology Am. Soc. Hematol. Educ. Program [Internet]. 2007 [cited 2014 Jun 6];216–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18024633. [DOI] [PubMed]
- 75.Sha Chulin, Barrans Sharon, Cucco Francesco, Bentley Michael A., Care Matthew A., Cummin Thomas, Kennedy Hannah, Thompson Joe S., Uddin Rahman, Worrillow Lisa, Chalkley Rebecca, van Hoppe Moniek, Ahmed Sophia, Maishman Tom, Caddy Josh, Schuh Anna, Mamot Christoph, Burton Catherine, Tooze Reuben, Davies Andrew, Du Ming-Qing, Johnson Peter W.M., Westhead David R. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. Journal of Clinical Oncology. 2019;37(3):202–212. doi: 10.1200/JCO.18.01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horn H, Kohler C, Witzig R, Kreuz M, Leich E, Klapper W, et al. Gene expression profiling reveals a close relationship between follicular lymphoma grade 3A and 3B, but distinct profiles of follicular lymphoma grade 1 and 2. Haematologica [Internet]. 2018 [cited 2018 Apr 27];haematol.2017.181024. Data available from Gene Expression Omnibus (GEO), accession number GSE103944: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103944 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text containing Figures S1–S19 and Tables S1–S5. (PDF 6310 kb)
Table of samples, providing histopathological and molecular subtypes. (XLSX 41 kb)
Complete gallery of all 936 sample expression portraits. (PDF 14216 kb)
Lists of genes for each of the spot modules. (XLSX 256 kb)
Animated expression portraits of the PATs together with survival curves. (GIF 556 kb)
Animated expression portraits of the HTs together with survival curves. (GIF 249 kb)
Data Availability Statement
The whole expression data set was collected as part of the German MMML consortium (Molecular Mechanisms in Malignant Lymphoma) and is partly available in the GEO repository under accession numbers GSE4475 [7], GSE10172 [15], GSE22470 [16], GSE48184 [18], GSE43677 [10] and GSE103944 [76]. The complete data are available from ‘The Leipzig Health Atlas’ repository under accession number 7VT47TM4GV-1 (https://www.health-atlas.de/index.php/en/lha/7VT47TM4GV-1).