

Summary
Aims
We previously demonstrated that intrathecal IL‐1β upregulated phosphorylation of p38 mitogen‐activated protein kinase (P‐p38 MAPK) and inducible nitric oxide synthase (iNOS) in microglia and astrocytes in spinal cord, increased nitric oxide (NO) release into cerebrospinal fluid, and induced thermal hyperalgesia in rats. This study investigated the role of spinal glutamatergic response in intrathecal IL‐1β‐induced nociception in rats.
Methods
The pretreatment effects of MK‐801 (5 μg), minocycline (20 μg), and SB203580 (5 μg) on intrathecal IL‐1β (100 ng) in rats were measured by behavior, Western blotting, CSF analysis, and immunofluorescence studies.
Results
IL‐1β increased phosphorylation of NR‐1 (p‐NR1) subunit of N‐methyl‐D‐aspartate receptors in neurons and microglia, reduced glutamate transporters (GTs; glutamate/aspartate transporter by 60.9%, glutamate transporter‐1 by 55.0%, excitatory amino acid carrier‐1 by 39.8%; P<.05 for all), and increased glutamate (29%‐133% increase from 1.5 to 12 hours; P<.05) and NO (44%‐101% increase from 4 to 12 hours; P<.05) levels in cerebrospinal fluid. MK‐801 significantly inhibited all the IL‐1β‐induced responses; however, minocycline and SB203580 blocked the IL‐1β‐downregulated GTs and elevated glutamate but not the upregulated p‐NR1.
Conclusion
The enhanced glutamatergic response and neuron‐glia interaction potentiate the intrathecal IL‐1β‐activated P‐p38/iNOS/NO signaling and thermal hyperalgesia.
Keywords: glutamate, N‐methyl‐D‐aspartate receptor, spinal cord, transporter
1. INTRODUCTION
Glutamate functions as neurotransmitter and its extracellular concentration is normally low in the central nervous system (CNS). It activates glutamate receptors and plays a significant role in nociception. Glutamate transporters (GTs), including glutamate/aspartate transporter (GLAST), glutamate transporter‐1 (GLT‐1), and excitatory amino acid carrier‐1 (EAAC1), remove glutamate from the synapses and maintain glutamatergic homeostasis. Elevated glutamate concentration caused by either excessive release or decreased uptake associates with pain1 and glutamate administration produces pain.2 The N‐methyl‐D‐aspartate (NMDA) receptors are glutamate receptors composed of NR1 subunits, and activation of NMDA receptor (NMDAR) by phosphorylating NR1 (p‐NR1) is important in nociceptive processing and correlates with pain.3, 4, 5, 6 Animal studies on nociception have revealed upregulation of p‐NR1 in dorsal root ganglion (DRG) neurons and primary afferent fibers,7 postsynaptic neurons,8, 9 microglia,8 and astrocytes9 in the spinal cord dorsal horn (SCDH). In addition, activation of presynaptic NMDARs increases glutamate release and excitatory postsynaptic currents (EPSC) in DRG neurons of rats with ligated spinal nerves.10 However, p‐NR1 expression in rat DRG neurons decreases following intraplantar injection with complete Freund's adjuvant (CFA).11
Activation of astrocytes and microglia is associated with increased TNF‐α expression, reduced expression of GLAST, GLT‐1, and EAAC1 in SCDH, and elevated glutamate level in CSF in rats with sciatic nerve injury.12 GLT‐1 downregulation occurs in activated astrocytes in rat models of morphine‐tolerant pain, peripheral neuropathic pain, and experimental autoimmune encephalomyelitis, whereas intrathecal ceftriaxone and MK‐801 abolish these responses.13, 14 In contrast, inhibition of GLT‐1 and GLAST reduces nociception and glutamate concentrations in CSF in rats receiving intraplantar injection of formalin.15, 16
IL‐1β involves in the initiation and maintenance of pain, and its levels in CSF and SCDH significantly increase 2‐3 hours after intraplantar injection of CFA and lipopolysaccharide in rodents.17, 18 Our previous studies showed that intrathecal IL‐1β increases p38 mitogen‐activated protein kinase phosphorylation (P‐p38 MAPK) and inducible nitric oxide synthase (iNOS) in astrocytes and microglia in SCDH, releases nitric oxide (NO) in CSF, and induces thermal hyperalgesia in rats.19, 20 In contrast, we previously had reported that pretreatment with inhibitors of microglia (minocycline), astrocytes (fluorocitrate), IL‐1 receptor (IL‐1ra), and p38 MAPK (SB203580) suppresses the IL‐1β‐induced P‐p38/iNOS/NO signaling and hyperalgesia.19, 20 As endogenous IL‐1β upregulates p‐NR1 that colocalize with IL‐1 receptor type I (IL‐1RI) in spinal neurons, IL‐1 receptor antagonist (IL‐1ra) inhibits the expression of p‐NR1 and hyperalgesia in rat model of inflammatory pain.21 However, the roles of glutamate, NMDAR, and GTs in intrathecal IL‐1β‐induced thermal hyperalgesia are not well characterized.
This study is a continuation of our preceding ones in a rat model of intrathecal IL‐1β‐induced thermal hyperalgesia,19, 20 and we hypothesized that exogenous IL‐1β may activate NMDAR and inhibit GTs in the spinal cord (SC) and this glutamatergic response participates in intrathecal IL‐1β‐induced nociceptive signaling. The aim of this study was 3‐fold: to investigate (i) the spinal glutamatergic response after IL‐1β injection; (ii) the interaction between glutamatergic response and P‐p38/iNOS/NO signaling; and (iii) the intercellular interactions in SCDH related to IL‐1β‐induced nociception.
2. MATERIALS AND METHODS
2.1. Animal preparation and intrathecal drug delivery
All experiments were approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital, and the use of animals conformed to the Guiding Principles for the Care and Use of Animals published by the American Physiological Society. All efforts were made to minimize the number of animals used and their suffering.
Preparation of the intrathecal catheter, microdialysis probe, and animals was as previously described.19, 20 Adult male Wistar rats (260‐320 g; BioLASCO Taiwan Co., Taipei, Taiwan) were anesthetized with 2%‐3% inhaled isoflurane, and an intrathecal catheter was implanted with or without a microdialysis probe. Postoperative care included subcutaneous amoxicillin (150 mg/kg) and lidocaine infiltration. Rats were allowed to recover for 4 days before experimentation, housed individually, given food and water ad libitum, and maintained on a standard 12‐hours light/dark schedule at room temperature. The intrathecal catheter and microdialysis tubes were perfused with artificial CSF (aCSF) every other day to prevent obstruction. Rats with locomotor dysfunction were excluded from the study.
On day 5 after intrathecal catheterization, all drugs were administered through the intrathecal catheter, followed by a 10‐μL flush with aCSF. Rats were randomly assigned to one of six groups: (i) control rats received 5 μL of saline; (ii) IL rats received 100 ng of IL‐1β; (iii) MK rats received 5 μg of MK‐801 (non‐competitive NMDAR antagonist); (iv) MK+IL rats received 5 μg MK‐801 pretreatment and 100 ng of IL‐1β 1 hour later; (v) SB+IL rats received 5 μg of SB203580 (p38 MAPK inhibitor) pretreatment and 100 ng of IL‐1β 1 hour later; and (vi) Mino+IL group rats received 20 μg of minocycline pretreatment and 100 ng of IL‐1β 1 hour later. The doses of IL‐1β, MK‐801, minocycline, and SB203580 were based on previous studies.19, 20, 22 All experimenters were blinded to the group allocations except the one who injected the drugs.
2.2. Nociceptive behavioral assessment
Paw withdrawal latency (PWL) to radiant heat applied to the right hindpaw (Ugo Basile Biological Instruments, Comerio, Italy) was assessed as previously described.19, 20 The heat intensity was adjusted to obtain an average PWL of 17.6±0.4 seconds, and the cutoff time was set at 22±0.4 seconds to prevent tissue damage. Paw withdrawal latency was assessed at baseline and different times after drug injection.
2.3. Analysis of nitric oxide and glutamate in spinal CSF dialysates
A CMA102 microinfusion pump with a gastight Hamilton syringe was used to continuously perfuse aCSF through one end of the microdialysis probe at a rate of 5 μL/min, and the other end was used to collect CSF dialysates. Dialysates were collected in polypropylene tubes on ice prior to drug administration (baseline) and every 30 minutes for 12 hours after drug administration. Samples were frozen at −80°C until analysis. Nitric oxide concentrations were measured by chemiluminescence (NOA 280; Sievers Instruments Inc., Boulder, CO, USA) as described previously,19, 20 and the glutamate concentration was determined by high‐performance liquid chromatography (Agilent 1200 HPLC System; Agilent Technologies, Palo Alto, CA, USA) using a fluorescence detector (340 nm excitation, 450 nm emission) as described previously.23 The concentrations of NO and glutamate in the dialysates are shown as percentage change relative to the basal concentration.
2.4. Western blotting
The dorsal part of lumbar SC enlargement was collected by exsanguination under isoflurane anesthesia at 30 minutes or 6 hours after drug injection. The Western blotting analysis was performed as described previously in refs.19, 20 The polyvinylidene difluoride membranes were incubated with either one of the primary antibodies including polyclonal rabbit anti‐phospho‐NR1 Ser896 antibody (1:1000; EMD Millipore, Temecula, CA, USA), polyclonal rabbit anti‐GLAST antibody (1:1000; Abcam, Cambridge, MA, USA), polyclonal guinea pig anti‐GLT‐1 antibody (1:500; EMD Millipore), monoclonal mouse anti‐EAAC1 antibody (1:1000; EMD Millipore), monoclonal mouse anti‐iNOS antibody (1:1000; Transduction Laboratories, Lexington, KY, USA), polyclonal rabbit p38 or phospho‐p38 MAPK antibodies (1:1000; PhosphoPlus® p38 MAP Kinase Antibody Kit; Cell Signaling Technology®, Beverly, MA, USA), or monoclonal mouse anti‐β‐actin antibody (1:5000; Sigma‐Aldrich®, St Louis, MO, USA), re‐incubated with horseradish peroxidase‐conjugated secondary antibodies, and then measured by chemiluminescence. Densitometry was used to evaluate the density of bands relative to background. β‐actin was used as the internal control for protein loading. The densities of P‐p38 MAPK bands were calculated and normalized to the total amount of p38 MAPK of each sample. Western blotting experiments were conducted in triplicate for semi‐quantification of p‐NR1 (n=4 per group), glutamate transporters (n=4 per group), P‐p38 MAPK/p38 MAPK (n=6 per group), and iNOS (n=6 per group).
2.5. Immunohistochemical assay
Lumbar SC enlargement was subjected to immunohistochemical analysis. Cell markers for neurons (NeuN), astrocytes (glial fibrillary acidic protein, GFAP), and microglia (CD11b) were used to determine cellular colocalization with p‐NR1. p‐NR1 expression was assessed at 30 minutes (four rats per group) and 6 hours (four rats per group), and GTs expression was assessed at 6 hours (four rats per group).
The immunohistochemical analysis was performed as described previously in ref.20 The sample was cut into 20‐μm sections on a cryostat at −25°C, mounted serially onto microscope slides, and processed for immunofluorescence studies. All primary antibodies were purchased from EMD Millipore: mouse monoclonal anti‐NeuN antibody (1:600), mouse monoclonal anti‐GFAP antibody (1:200), mouse monoclonal anti‐CD11b antibody (1:200), rabbit polyclonal anti‐phospho‐NR1 Ser897 antibody (1:50), guinea pig polyclonal anti‐GLAST antibody (1:1000), guinea pig polyclonal anti‐GLT‐1 antibody (1:1000), and goat polyclonal anti‐EAAC1 antibody (1:1000). After washing in phosphate‐buffered saline, sections were incubated with either Alexa Fluor 488‐labeled chicken anti‐mouse IgG antibody (1:400; Life Technologies Corporation, USA; green fluorescence) or DyLight 549‐conjugated donkey anti‐rabbit secondary antibody (1:400; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA; red fluorescence). The sections were examined under a Leica DM‐6000B fluorescence microscope (Leica, Wetzlar, Germany) equipped with a CCD Spot Xplorer integrating camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA) and analyzed by SPOT software (Diagnostic Instruments Inc.). Confocal double‐immunostaining images were captured with a Leica TCS SP5 II confocal microscope equipped with a Leica HyD (Hybrid Detector). Image J software (National Institutes of Health, Bethesda, MD, USA) was used for pixel measurement and analysis. For quantification of immunofluorescence acquired from lamina I to lamina IV in SCDH, four randomly selected SC sections were measured in each rat, and the means of 4 rats in each group were calculated. The sizes and conditions of the image captures and views were kept constant at ipsilateral SCDH, and pixel measurement and analysis was performed with MetaVue Imaging software (Molecular Devices Corporation, Sunnyvale, CA, USA).
2.6. Drug preparation
Recombinant rat IL‐1β (R&D Systems, Minneapolis, MN, USA), MK‐801 (Dizocilpine hydrogen maleate; Sigma‐Aldrich®), minocycline (Sigma‐Aldrich®), and SB203580 (Calbiochem, La Jolla, CA, USA) were dissolved in sterile 0.9% physiological saline at a concentration of 50 ng/μL, 1 μg/μL, 2 μg/μL, and 1 μg/μL, respectively. Each drug was freshly prepared on the morning of the experiment.
2.7. Statistical analysis
Data are expressed as means±SEMs. Changes in PWL, and in NO and glutamate concentrations are expressed relative to baseline values. Protein expression, immunofluorescence intensity, and ratio of cell colocalization are analyzed by one‐way or two‐way analysis of variance (ANOVA), followed by the Student‐Newman‐Keuls post hoc test. In addition, repeated‐measures ANOVA followed by the Student‐Newman‐Keuls post hoc test, with time (before and after intrathecal injection) as the within‐subjects variable and grouping as the between‐subjects variable, is used to analyze changes in PWL, NO, and glutamate concentrations. Mauchly's sphericity test is used to check the equal variance assumption of the repeated‐measures ANOVA, and the Huynh‐Feldt correction is applied if the assumption of sphericity is violated. A P‐value <.05 is considered statistically significant. Statistical analyses are performed with the Statistical Package for the Social Sciences (SPSS), version 17.0 for Windows (SPSS Inc., Chicago, IL, USA).
3. RESULTS
3.1. Effect of MK‐801 pretreatment on IL‐1β‐induced thermal hyperalgesia
We initially examined the role of the spinal glutamatergic response in IL‐1β‐induced nociception by examining the pretreatment effect of MK‐801 before IL‐1β injection (Figure 1). The results indicate a significant difference among the four groups (F[3,32]=43.313, P<.001) and over time (F=14.244, df=4.568, P<.001), and a significant interaction between groups and time (F=5.363, df=13.705, P<.001). Saline had no effect on PWL, but IL‐1β decreased the PWL (28.0%‐48.7% reduction from 2 to 12 hours) with a nadir at 6 hours. MK‐801 alone only slightly reduced PWL at 12 hours (11.8% reduction; P=.046). Pretreatment with MK‐801 partially blocked the effect of IL‐1β at 2‐8 hours and 12 hours (P<.01 for all), but not at 10 hours (P=.076). These results demonstrate that MK‐801 pretreatment suppresses IL‐1β‐induced thermal hyperalgesia, and suggest the NMDAR involves in IL‐1β‐induced nociception.
Figure 1.
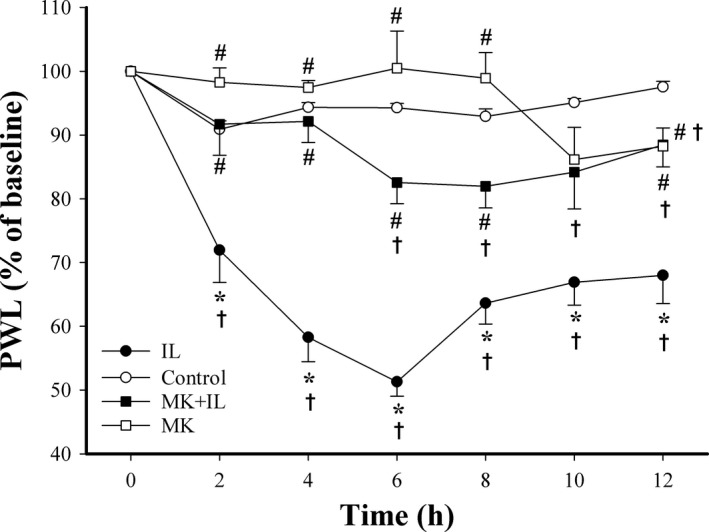
Effect of IL‐1β, MK‐801, and MK‐801+IL‐1β on thermal hyperalgesia in rats. Paw withdrawal latency (PWL) was measured in rats given intrathecal saline (control, n=8), IL‐1β (IL, n=8), MK‐801 alone (MK, n=8), or MK‐801 at 1 h before IL‐1β (MK+IL, n=12). All values were normalized to the baseline (100%) and expressed as means±SEMs. *P<.05 relative to controls; #P<.05 relative to IL‐1β; †P<.05 relative to baseline
3.2. IL‐1β upregulates NR1 phosphorylation in neurons and microglia, and SB203580 and minocycline pretreatment abolish the effect of IL‐1β on p‐NR1
Immunohistochemistry experiments showed that control rats had very low expression of p‐NR1 in SCDH at 30 minutes (Figure 2A). IL‐1β dramatically upregulated NR1 phosphorylation from 30 minutes to 6 hours compared to controls (15.6‐ and 31.9‐fold, respectively; both P<.05) (Figure 2A,d). Double immunofluorescent staining showed that p‐NR1‐immunofluorescence in SCDH was not totally colocalized with neurons, microglia, and astrocytes in this study (Figure 2B). We focused on the effect of IL‐1β on the p‐NR1 expression among neurons, microglia, and astrocyte in this study, and double‐immunostaining showed that p‐NR1 immunoreactivity colocalized in both NeuN‐positive neurons and CD11b‐positive microglia, but not GFAP‐positive astrocytes (yellow, with arrows; Figure 2B). The sum of the percentages of neurons and microglia expressing p‐NR1 to the total number of p‐NR1‐positive cells was 47.8%, 42.8%, and 78.9% among rats injected with either saline or IL‐1β for 30 minutes and 6 hours, respectively (Figure 2B,d,i,n). It suggests that oligodendrocytes might be the other possible cellular source of p‐NR1 expression in spinal cord and further investigation is required. We further examined the percentage of p‐NR1‐positive neurons or microglia to the total number of neurons or microglia to evaluate the effect of IL‐1β on p‐NR1 expression in neurons and microglia. The percentage of p‐NR1‐positive neurons (42.3%) to total neurons in SCDH was significantly higher than that of the percentage of p‐NR1‐positive microglia (1.6%), and the majority of p‐NR1‐positive cells were neurons in saline‐injected control rat spinal cord (Figure 2B,e). Although IL‐1β significantly increased the intensity of p‐NR1 immunoreactivity, the percentage of p‐NR1‐positive neurons was similar among rats injected with either saline or IL‐1β for 30 minutes and 6 hours (42.3%, 40.4%, and 46.7%, respectively; Figure 2B,e,j,o). In contrast, IL‐1β markedly increased the percentage of p‐NR1‐positive microglia to 11.0% at 30 minutes and 29.9% at 6 hours compared to the control rat spinal cord (1.6%, P<.05; Figure 2B,e,j,o).
Figure 2.
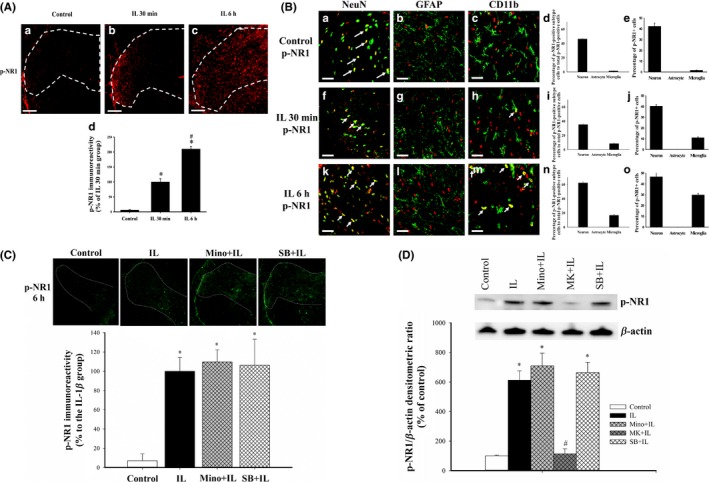
Effects of IL‐1β, SB203580, and minocycline on the level of p‐NR1 in the rat lumbar spinal cord dorsal horn (SCDH) and colocalization of p‐NR1 with other proteins. (A) Representative immunohistochemical images of p‐NR1 (red) in control rats (a), and at 30 min (b) and 6 h (c) after intrathecal injection of IL‐1β (magnification: 100×; scale bars: 200 μm). Quantification of these results (d, 4 sections per rat, 4 rats per group). *P<.05 relative to control. #P<.05 relative to 30 min after IL‐1β injection. (B) Representative confocal immunofluorescence microscopy images (20‐μm sections) showing the localization of p‐NR1 (red in all images) and NeuN in neurons (green in left row), GFAP in astrocytes (green in middle row), and CD11b in microglia (green in right row) in three groups (magnification: 630×; scale bar: 25 μm). Colocalization is indicated by yellow and arrows (a, f, h, k, and m). The percentage of neurons, astrocytes, and microglia expressing p‐NR1 to the total number of p‐NR1‐positive cells are depicted in histograms (d, i, and n). The percentages of p‐NR1‐positive cells in neurons, astrocytes, and microglia populations are depicted in histograms (e, j, and o). (C) Representative immunohistochemistry results show the distribution of p‐NR1 in SCDH (top, magnification: 25×) and quantification of these results (bottom) in the control, IL, Mino+IL, and SB+IL groups. *P<.05 relative to controls. #P<.05 relative to IL‐1β‐injected rats. (D) Representative Western blotting results of p‐NR1 in SCDH at 6 h after drug treatment (top) and quantification of these results (bottom). Values indicate means±SEMs, with 4 sections per rat and 4 rats for each group. *P<.05 vs saline‐injected controls. #P<.05 vs IL‐1β‐injected rats
At 6 hours after drug injection, there were significant differences in the level of p‐NR1 among the groups (P<.001) (Figure 2C,D). IL‐1β increased p‐NR1 at 6 hours after injection, but minocycline and SB203580 pretreatment did not inhibit this potentiation response as revealed in immunohistochemical (Figure 2C) and Western blotting (Figure 2D) analyses. However, MK‐801 pretreatment prevented the IL‐1β‐upregulated p‐NR1 expression (97.3% reduction; P<.001) (Figure 2D). These results suggest that IL‐1β primarily upregulates p‐NR1 expression in neurons of SCDH.
3.3. Effect of drug pretreatments on IL‐1β‐induced downregulation of GLAST, GLT‐1, and EAAC1
Additional immunohistochemistry studies indicated that the immunoreactivities of GLAST, GLT‐1, and EAAC1 were abundantly located in the control rats but decreased at 6 hours after IL‐1β injection in SCDH (Figure 3A). These results are consistent with the results of Western blotting, which showed that IL‐1β decreased GLAST by 60.9%, GLT‐1 by 55.0%, and EAAC1 by 39.8% (P<.05 for all; Figure 3B‐D). The Western blotting experiments also showed that MK‐801 alone had no significant effect on these GTs expression, but pretreatment with MK‐801, minocycline, and SB203580 blocked the effect of IL‐1β (P<.001 for all; Figure 3B). Similar to the effect of MK‐801 on glutamate transporters, both minocycline and SB203580 alone did not affect the protein expression of GLAST, GTL‐1, and EAAC1 as compared to the saline‐injected controls in our studies (data not show).
Figure 3.
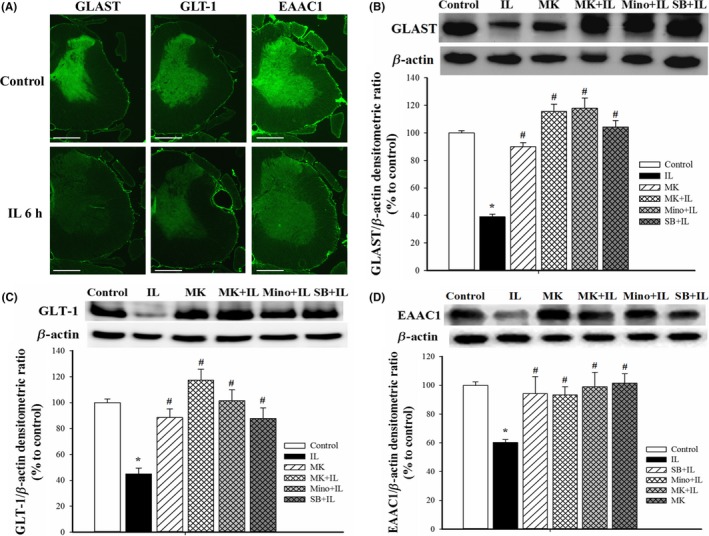
Effects of different treatments on the levels of glutamate transporters in SCDH. (A) Representative immunofluorescence images show the expression of GLAST, GLT‐1, and EAAC1 in the gray matter of spinal cord in controls (top row) and 6 h after IL‐1β injection (bottom row) (magnification: 25×; scale bar: 1000 μm). (B) Representative Western blotting results of GLAST (top), and quantification of these results in the five groups (bottom). Here and below, β‐actin was the loading control, quantification was performed with 4 rats per group, and values are expressed as means±SEMs. *P<.05 relative to controls; #P<.05 relative to rats given IL‐1β. (C). Representative Western blotting of GLT‐1 (top) and quantification of these results in the five groups (bottom). (D) Representative Western blotting of results of EAAC1 (top) and quantification of these results in the five groups (bottom)
3.4. Effect of drug pretreatments on IL‐1β‐induced glutamate release into the CSF
IL‐1β injection markedly increased glutamate concentration in CSF dialysates starting at 2 hours (57% increase), with a maximum at 5.5‐6 hours (132%‐133% increase), and an increase of 113% till 12 hours (P<.05 for all; Figure 4A). MK‐801 alone did not affect the glutamate concentration, but MK‐801 pretreatment prevented the IL‐1β‐induced glutamate release from 1.5‐12 hours (P<.05 for all; Figure 4A). Although minocycline and SB203580 pretreatment also prevented IL‐1β‐induced glutamate release, minocycline did not reduce the glutamate level within 1.5‐3.5 hours after IL‐1β injection (Figure 4B).
Figure 4.
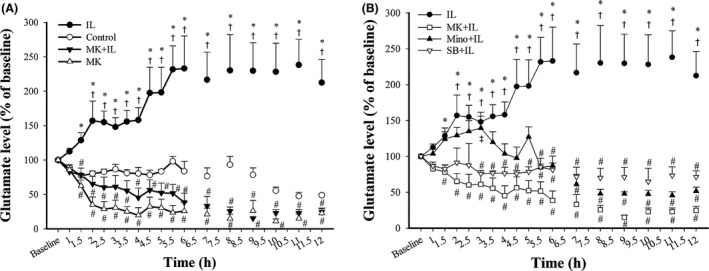
Effect of different treatments on changes in the glutamate concentrations in CSF dialysates. (A) Glutamate in the control, IL, MK+IL, and MK groups. (B) Glutamate in the IL, MK+IL, Mino+IL, and SB+IL groups. All values were normalized to the baseline (100%) and expressed as means±SEMs (n=4 in the control group, n=9 in the IL group, n=4 in the MK group, n=6 in the MK+IL group, n=4 in the Mino+IL group, n=4 in the SB+IL group); *P<.05 vs controls. #P<.05 vs IL‐1β‐injected rats; †P<.05 vs baseline. ‡P<.05 vs MK‐801‐pretreated rats
3.5. Effect of MK‐801 on IL‐1β‐induced P‐p38/iNOS/NO signaling
We also examined the effect of MK‐801 on IL‐1β‐induced NO release into the CSF. The results indicate that IL‐1β increased the NO level in CSF dialysates starting at 4 hours (44% increase), with a maximum at 6 hours (156% increase), and an increase of 101% at 12 hours (P<.05 for all; Figure 5A). MK‐801 alone had no effect, but MK‐801 pretreatment significantly attenuated the IL‐1β‐induced NO release from 4 to 12 hours (P<.005 for all; Figure 5A). Western blotting analysis confirmed that IL‐1β increased the level of P‐p38 MAPK by 2.4‐fold at 30 minutes and of iNOS by 78.4‐fold at 6 hours (Figure 5B‐D). Pretreatment with MK‐801, minocycline, or SB203580 significantly blocked the IL‐1β‐induced p38 MAPK phosphorylation and iNOS expression (Figure 5B‐D), but SB20358019 had no effect and minocycline had little effect when given alone (data not shown).
Figure 5.
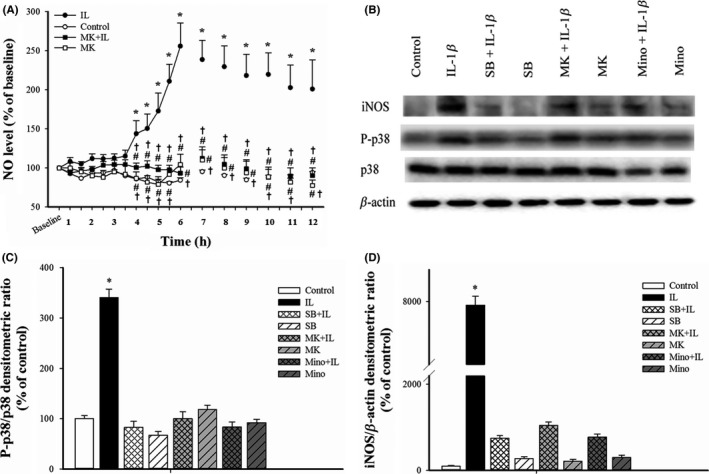
Effect of MK‐801 on the IL‐1β‐activated P‐p38/iNOS/NO nociceptive pathway in the rat spinal cord. (A) Change of NO concentration in CSF dialysates after intrathecal injection with saline, IL‐1β, MK‐801+IL‐1β, and MK‐801 (n=6 in the control group; n=6 in the IL‐1β group, n=5 in the MK‐801 group, and n=9 in the MK801+IL‐1β group). Here and below, all values were normalized to the baseline (100%) and expressed as means±SEMs. (B) Representative Western blotting results showing the effects of various treatments on p38 MAPK phosphorylation and iNOS expression. (C) Quantitative analysis of Western blotting experiments (n=6 per group) showing the effect of various treatments on the P‐p38 MAPK/p38 MAPK ratio at 30 min after drug treatment. (D) Quantitative analysis of Western blotting experiments (n=6 per group) showing the effect of various treatments on expression of iNOS relative to β‐actin in SCDH at 6 h after drug treatment (n=6 rats per group). *P<.05 vs controls; #P<.05 vs IL‐1β‐injected rats; †P<.05 vs baseline
4. DISCUSSION
This study demonstrated that IL‐1β activates spinal neurons and microglia with upregulated p‐NR1 expression, downregulates GTs, and increases extracellular glutamate level. MK‐801 pretreatment not only reverses these glutamatergic responses but also inhibits the IL‐1β‐activated P‐p38/iNOS/NO signaling and thermal hyperalgesia. Pretreatment with minocycline or SB203580 abolishes the effect of IL‐1β on glutamate release and GTs expression, but not the upregulated p‐NR1. Taken together, these results indicate that both NMDAR/GTs/glutamate and the P‐p38/iNOS/NO signaling play pivotal roles in IL‐1β‐induced thermal hyperalgesia. IL‐1β upregulates p‐NR1 expression majorly in neurons and minorly in microglia and activates P‐p38/iNOS/NO signaling in microglia and astrocytes. An earlier NMDAR‐dependent followed by a later P‐p38 MAPK‐dependent nociceptive signaling in neuron‐glia interaction in SCDH may be responsible for the intrathecal IL‐1β‐induced thermal hyperalgesia.
Significant upregulation of endogenous IL‐1β has been demonstrated in many models of nociception,17, 21 and application of exogenous IL‐1β produces nociception.19, 20 IL‐1β binds to the IL‐1RI to produce its effect,23 and IL‐1ra completely reverses the IL‐1β‐induced nociceptive signaling and behaviors in rats.19 IL‐1RIs ubiquitously express in gray and white matters of SC and in neurons, astrocytes, microglia, and oligodendrocytes.24 We previously demonstrated that intrathecal IL‐1β activated microglia and astrocytes in SCDH with upregulated expression of P‐p38 MAPK and iNOS,20 and the present study further showed that IL‐1β upregulates p‐NR1 expression in activated neurons and microglia and downregulates GTs in SCDH. We propose that the nociceptive effect of IL‐1β is mediated by IL‐RI located on neurons, microglia, and astrocytes; however, the cellular localization of IL‐1RI in response to intrathecal IL‐1β needs to be determined.
N‐methyl‐D‐aspartate receptor activation involves in central nociceptive sensitization4, 21, 25 and NR1 knockout mice experience less pain.11, 26, 27 Immunofluorescent and electrophysiological studies show that IL‐1RI and p‐NR1 are colocalized in primary afferent fibers and spinal neurons, and that IL‐1ra and NMDAR antagonist inhibit both nociceptive behavior and NR1 phosphorylation in many models of pain including peripheral injection of IL‐1β.21, 22, 25, 28, 29, 30 IL‐1β enhances spontaneous EPSC‐ and NMDA‐induced inward current but reduces spontaneous inhibitory postsynaptic currents on rat spinal neurons in whole‐cell patch‐clamp study,31, 32 while IL‐1ra pretreatment abolishes the effect of IL‐1β on NMDA‐induced currents and minocycline pretreatment inhibit the NMDA‐induced currents.32 Molecular mechanisms underlying the IL‐1β‐induced NMDAR activation involve protein kinase A and C, neutral sphingomyelinase/ceramide, inositol triphosphate receptor, and phospholipase A2 signaling.25, 29, 30, 33, 34, 35, 36 NMDA infusion in the nucleus tractus solitarius increases glutamate level in rats, while MK‐801 prevents glutamate release during hypoglycemic coma in piglet.37, 38 These data suggest that NMDAR activation increases the release of glutamate. Neurons,10, 29 astrocytes,39, 40, 41, 42 and microglia43, 44 have been shown to release glutamate. Additionally, IL‐1β activates presynaptic NMDAR to enhance glutamate release from the primary afferents in whole‐cell recording study on DRG and spinal slice of spinal nerve‐ligated rats.29 These might indicate that IL‐1β activates NMDAR and enhance glutamate release in an IL‐1RI‐dependent manner in primary afferents, spinal neurons, and microglia, and is responsible for the IL‐1β‐induced thermal hyperalgesia.
The rate of glutamate uptake and amount of GTs are important for maintaining glutamate homeostasis in CNS. Increased glutamate level in CSF and reduced GTs expression in SCDH occur in rats with peripheral nerve injury and morphine‐induced hyperalgesia.12, 14, 23, 45, 46 Pharmacological inhibition of GTs induces hypersensitivity to nociceptive stimulus, while increasing GT expression relieves nociception.12, 13, 15, 16, 47, 48, 49 Cervical nerve root compression downregulates GLT‐1 expression in SCDH in rats but restoring GLT‐1 expression alleviates pain.50 Both GLAST and GLT‐1 expressions in SCDH decreased in morphine‐tolerant rats and increased trafficking of GTs onto glial cell surface preserves the morphine's antinociceptive effect.14, 44 EAAC1 localizes in spinal neurons and DRG neurons.51 Additionally, inhibition of GTs with DL‐threo‐β‐benzyloxyaspartate impairs glutamate uptake that leads to NMDAR activation and extrasynaptic glutamate spillover in rats with partial sciatic nerve ligation.52, 53 Taken together, our results suggest that decreased GTs increases glutamate level, enhances NMDAR activation, and participates in IL‐1β‐induced hyperalgesia.
Multiple pathways downstream of NMDAR have roles in nociception.32, 54, 55 IL‐1β enhances NMDAR‐mediated Ca2+ inward flow through phosphorylated NMDAR in rat neurons,44, 56 and this elevated intracellular Ca2+ concentration activates the Ca2+/calmodulin‐dependent protein kinase II and the MAPK kinase‐MAPK pathways.57 NR1 knockout prohibits nociceptive behavior and attenuates the upregulation of phospho‐PKCγ and phosphorylated extracellular signal‐regulated kinase in spinal neurons in mouse with CFA‐induced pain.26 NMDA upregulates P‐p38 MAPK expression in spinal microglia and neurons and induces hyperalgesia through a P‐p38‐dependent way in rats with streptozotocin‐induced diabetes, while MK‐801 inhibits them.58
Our present study shows that MK‐801, minocycline, and SB203580 all reduce the IL‐1β‐induced glutamatergic response; MK‐801 is the most potent followed by SB203580 and minocycline in descending order. MK‐801 inhibits NMDAR and increases GTs expression, and this provides evidence that IL‐1β binds to IL‐1RI to upregulate neighboring NMDAR and glutamate release. MK‐801 pretreatment abolishes the IL‐1β‐induced P‐p38/iNOS/NO signaling and glutamatergic response, while SB203580 inhibits P‐p38 upregulation and reverses GTs downregulation without inhibiting effect on NMDAR activation. Therefore, it suggests that the upregulated NMDAR expression is upstream of p38 and contributes to the IL‐1β‐activated P‐p38/iNOS/NO signaling. In addition, the P‐p38/iNOS/NO pathway retrogradely enhances glutamate release. In contrast, minocycline inhibits microglia and astrocytes but not neurons. Although minocycline reverses both the downregulated GTs and activated P‐p38/iNOS/NO signaling, it does not reduce the glutamate level within 1.5‐3.5 hours after IL‐1β injection. It may indicate that intrathecal IL‐1β activates neurons, microglia, and astrocytes and/or the primary afferents to release glutamate in a time‐dependent manner, with a prominent role of neurons in the early phase (within 3.5 hours) and of microglia in the late phase. Furthermore, the IL‐1β‐induced NMDAR activation and glutamate release potentiate the P‐p38/iNOS/NO signaling.
Our present and previous studies showed that intrathecal IL‐1β activated p‐NR1 in neurons and microglia and upregulated P‐p38 MAPK and iNOS in microglia and astrocytes.20 The percentage of p‐NR1‐positive neurons was similar in saline or IL‐1β‐injected rat spinal cord at 30 minutes and 6 hours, while IL‐1β markedly increased the percentage of p‐NR1‐positive microglia at 30 minutes and 6 hours as compared to the control group. This finding further supports the important role of microglia in the intrathecal IL‐1β‐induced thermal hyperalgesia in rats. In addition, MK‐801 pretreatment not only reverses these glutamatergic responses but also inhibits the IL‐1β‐activated P‐p38/iNOS/NO signaling. However, pretreatment with either minocycline or SB203580 abolishes the effect of IL‐1β on glutamate release and GTs expression, but not p‐NR1 upregulation. Taken together, these results indicate that both NMDAR/GTs/glutamate and P‐p38/iNOS/NO signaling play pivotal roles in IL‐1β‐induced thermal hyperalgesia. In addition, IL‐1β upregulates p‐NR1 expression mainly in neurons and less in microglia, and activates P‐p38/iNOS/NO signaling in microglia and astrocytes. Furthermore, activation of microglial p‐NR1 receptor initiates glutamate response that leads to activation of P‐p38 MAPK‐dependent nociceptive signaling in microglia.
Based on the timeline of the measured responses, it appears that IL‐1β‐induced thermal hyperalgesia is initially NMDAR‐dependent, followed by a P‐p38 MAPK‐dependent pathway. IL‐1β activates spinal microglia through both the NMDAR‐ and p38 MAPK‐dependent pathways. In contrast, it activates neurons through the NMDAR‐dependent pathway but activates astrocytes through the p38 MAPK‐dependent pathway.
5. CONCLUSIONS
A complex neuron‐glia interaction and distinct molecular mechanisms in SCDH underlie the intrathecal IL‐1β‐evoked thermal hyperalgesia in rats. We suggest that pharmacological intervention that disrupts these specific targets may be suitable for clinical pain management.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This research was supported by the Ministry of Science and Technology, Taiwan (NSC95‐2314‐B‐075‐096‐MY2 and NSC102‐2314‐B‐075‐024), Taipei Veterans General Hospital, Taiwan (V95C1‐119 and V96C1‐116), and Anesthesiology Research and Development Foundation, Taiwan (ARDF9703‐Y3). The authors thank the Clinical Research Core Laboratory of Taipei Veterans General Hospital, Taipei, Taiwan for experimental support.
Sung C‐S, Wen Z‐H, Feng C‐W, et al. Potentiation of spinal glutamatergic response in the neuron‐glia interactions underlies the intrathecal IL‐1β‐induced thermal hyperalgesia in rats. CNS Neurosci Ther. 2017;23:580–589. 10.1111/cns.12705
REFERENCES
- 1. Harris RE, Sundgren PC, Craig AD, et al. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009;60:3146‐3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beirith A, Santos AR, Calixto JB. Mechanisms underlying the nociception and paw oedema caused by injection of glutamate into the mouse paw. Brain Res. 2002;924:219‐228. [DOI] [PubMed] [Google Scholar]
- 3. Dauch JR, Yanik BM, Hsieh W, Oh SS, Cheng HT. Neuron‐astrocyte signaling network in spinal cord dorsal horn mediates painful neuropathy of type 2 diabetes. Glia. 2012;60:1301‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garraway SM, Xu Q, Inturrisi CE. siRNA‐mediated knockdown of the NR1 subunit gene of the NMDA receptor attenuates formalin‐induced pain behaviors in adult rats. J Pain. 2009;10:380‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan PH, Chia YY, Chow LH, et al. Gene knockdown of the N‐methyl‐D‐aspartate receptor NR1 subunit with subcutaneous small interfering RNA reduces inflammation‐induced nociception in rats. Anesthesiology. 2010;112:1482‐1493. [DOI] [PubMed] [Google Scholar]
- 6. Yang X, Yang HB, Xie QJ, Liu XH, Hu XD. Peripheral inflammation increased the synaptic expression of NMDA receptors in spinal dorsal horn. Pain. 2009;144:162‐169. [DOI] [PubMed] [Google Scholar]
- 7. Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N‐methyl‐D‐aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci U S A. 1994;91:8383‐8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daulhac L, Maffre V, Mallet C, et al. Phosphorylation of spinal N‐methyl‐D‐aspartate receptor NR1 subunits by extracellular signal‐regulated kinase in dorsal horn neurons and microglia contributes to diabetes‐induced painful neuropathy. Eur J Pain. 2011;15:169. [DOI] [PubMed] [Google Scholar]
- 9. Moon ES, Karadimas SK, Yu WR, Austin JW, Fehlings MG. Riluzole attenuates neuropathic pain and enhances functional recovery in a rodent model of cervical spondylotic myelopathy. Neurobiol Dis. 2014;62:394‐406. [DOI] [PubMed] [Google Scholar]
- 10. Yan X, Jiang E, Gao M, Weng HR. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J Physiol. 2013;591:2001‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Zhang RX, Wang R, Qiao JT. Decreased expression of N‐methyl‐D‐aspartate (NMDA) receptors in rat dorsal root ganglion following complete Freund's adjuvant‐induced inflammation: an immunocytochemical study for NMDA NR1 subunit. Neurosci Lett. 1999;265:195‐198. [DOI] [PubMed] [Google Scholar]
- 12. Chen NF, Huang SY, Chen WF, et al. TGF‐β1 attenuates spinal neuroinflammation and the excitatory amino acid system in rats with neuropathic pain. J Pain. 2013;14:1671‐1685. [DOI] [PubMed] [Google Scholar]
- 13. Ramos KM, Lewis MT, Morgan KN, et al. Spinal upregulation of glutamate transporter GLT‐1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience. 2010;169:1888‐1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen N, Mo LQ, Hu F, Chen PX, Guo RX, Feng JQ. A novel role of spinal astrocytic connexin 43: mediating morphine antinociceptive tolerance by activation of NMDA receptors and inhibition of glutamate transporter‐1 in rats. CNS Neurosci Ther. 2014;20:728‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niederberger E, Schmidtko A, Rothstein JD, Geisslinger G, Tegeder I. Modulation of spinal nociceptive processing through the glutamate transporter GLT‐1. Neuroscience. 2003;116:81‐87. [DOI] [PubMed] [Google Scholar]
- 16. Niederberger E, Schmidtko A, Coste O, Marian C, Ehnert C, Geisslinger G. The glutamate transporter GLAST is involved in spinal nociceptive processing. Biochem Biophys Res Commun. 2006;346:393‐399. [DOI] [PubMed] [Google Scholar]
- 17. Samad TA, Moore KA, Sapirstein A, et al. Interleukin‐1β‐mediated induction of COX‐2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471‐475. [DOI] [PubMed] [Google Scholar]
- 18. Wang C, Song S, Zhang Y, et al. Inhibition of the Rho/Rho kinase pathway prevents lipopolysaccharide‐induced hyperalgesia and the release of TNF‐α and IL‐1β in the mouse spinal cord. Sci Rep. 2015;5:14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sung CS, Wen ZH, Chang WK, et al. Inhibition of p38 mitogen‐activated protein kinase attenuates interleukin‐1β‐induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J Neurochem. 2005;94:742‐752. [DOI] [PubMed] [Google Scholar]
- 20. Sung CS, Cherng CH, Wen ZH, et al. Minocycline and fluorocitrate suppress spinal nociceptive signaling in intrathecal IL‐1β–induced thermal hyperalgesic rats. Glia. 2012;60:2004‐2017. [DOI] [PubMed] [Google Scholar]
- 21. Zhang RX, Li A, Liu B, et al. IL‐1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR‐1 subunit in rats. Pain. 2008;135:232‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wen ZH, Chang YC, Cherng CH, Wang JJ, Tao PL, Wong CS. Increasing of intrathecal CSF excitatory amino acids concentration following morphine challenge in morphine‐tolerant rats. Brain Res. 2004;995:253‐259. [DOI] [PubMed] [Google Scholar]
- 23. Chen WF, Sung CS, Jean YH, et al. Suppressive effects of intrathecal granulocyte colony‐stimulating factor on excessive release of excitatory amino acids in the spinal cerebrospinal fluid of rats with cord ischemia: role of glutamate transporters. Neuroscience. 2010;165:1217‐1232. [DOI] [PubMed] [Google Scholar]
- 24. Wang XF, Huang LD, Yu PP, et al. Upregulation of type I interleukin‐1 receptor after traumatic spinal cord injury in adult rats. Acta Neuropathol. 2006;111:220‐228. [DOI] [PubMed] [Google Scholar]
- 25. Guo W, Wang H, Watanabe M, et al. Glial‐cytokine‐neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006‐6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng HT, Suzuki M, Hegarty DM, et al. Inflammatory pain‐induced signaling events following a conditional deletion of the N‐methyl‐D‐aspartate receptor in spinal cord dorsal horn. Neuroscience. 2008;155:948‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quintero GC, Erzurumlu RS, Vaccarino AL. Decreased pain response in mice following cortex‐specific knockout of the N‐methyl‐D‐aspartate NR1 subunit. Neurosci Lett. 2007;425:89‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ji XT, Qian NS, Zhang T, et al. Spinal astrocytic activation contributes to mechanical allodynia in a rat chemotherapy‐induced neuropathic pain model. PLoS One. 2013;8:e60733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan X, Weng HR. Endogenous interleukin‐1β in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N‐methyl‐D‐aspartate acid receptors. J Biol Chem. 2013;288:30544‐30557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim MJ, Lee SY, Yang KY, et al. Differential regulation of peripheral IL‐1β‐induced mechanical allodynia and thermal hyperalgesia in rats. Pain. 2014;155:723‐732. [DOI] [PubMed] [Google Scholar]
- 31. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin‐1β, interleukin‐6, and tumor necrosis factor‐α in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189‐5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu T, Jiang CY, Fujita T, Luo SW, Kumamoto E. Enhancement by interleukin‐1β of AMPA and NMDA receptor‐mediated currents in adult rat spinal superficial dorsal horn neurons. Mol Pain. 2013;9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin‐1β enhances NMDA receptor‐mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692‐8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Balosso S, Maroso M, Sanchez‐Alavez M, et al. A novel non‐transcriptional pathway mediates the proconvulsive effects of interleukin‐1β. Brain. 2008;131:3256‐3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao X, Kim HK, Chung JM, Chung K. Enhancement of NMDA receptor phosphorylation of the spinal dorsal horn and nucleus gracilis neurons in neuropathic rats. Pain. 2005;116:62‐72. [DOI] [PubMed] [Google Scholar]
- 36. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383‐400. [DOI] [PubMed] [Google Scholar]
- 37. Ichord RN, Johnston MV, Traystman RJ. MK801 decreases glutamate release and oxidative metabolism during hypoglycemic coma in piglets. Brain Res Dev Brain Res. 2001;128:139‐148. [DOI] [PubMed] [Google Scholar]
- 38. Matsuo I, Hirooka Y, Hironaga K, et al. Glutamate release via NO production evoked by NMDA in the NTS enhances hypotension and bradycardia in vivo. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1285‐R1291. [DOI] [PubMed] [Google Scholar]
- 39. Bardoni R, Ghirri A, Zonta M, et al. Glutamate‐mediated astrocyte‐to‐neuron signalling in the rat dorsal horn. J Physiol. 2010;588:831‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bezzi P, Carmignoto G, Pasti L, et al. Prostaglandins stimulate calcium‐dependent glutamate release in astrocytes. Nature. 1998;391:281‐285. [DOI] [PubMed] [Google Scholar]
- 41. Kanno T, Nishizaki T. A2a adenosine receptor mediates PKA‐dependent glutamate release from synaptic‐like vesicles and Ca2+ efflux from an IP3‐ and ryanodine‐insensitive intracellular calcium store in astrocytes. Cell Physiol Biochem. 2012;30:1398‐1412. [DOI] [PubMed] [Google Scholar]
- 42. Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci U S A. 2000;97:8629‐8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noda M, Nakanishi H, Akaike N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid‐beta peptide. Neuroscience. 1999;92:1465‐1467. [DOI] [PubMed] [Google Scholar]
- 44. Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L‐glutamate released from activated microglia downregulates astrocytic L‐glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L‐glutamate concentration in neuroinflammation. J Neuroinflammation. 2012;9:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tai YH, Wang YH, Wang JJ, Tao PL, Tung CS, Wong CS. Amitriptyline suppresses neuroinflammation and up‐regulates glutamate transporters in morphine‐tolerant rats. Pain. 2006;124:77‐86. [DOI] [PubMed] [Google Scholar]
- 46. Yang CP, Cherng CH, Wu CT, Huang HY, Tao PL, Wong CS. Intrathecal ultra‐low dose naloxone enhances the antinociceptive effect of morphine by enhancing the reuptake of excitatory amino acids from the synaptic cleft in the spinal cord of partial sciatic nerve‐transected rats. Anesth Analg. 2011;113:1490‐1500. [DOI] [PubMed] [Google Scholar]
- 47. Liaw WJ, Stephens RL Jr, Binns BC, et al. Spinal glutamate uptake is critical for maintaining normal sensory transmission in rat spinal cord. Pain. 2005;115:60‐70. [DOI] [PubMed] [Google Scholar]
- 48. Schroeder JA, Quick KF, Landry PM, Rawls SM. Glutamate transporter activation enhances nicotine antinociception and attenuates nicotine analgesic tolerance. NeuroReport. 2011;22:970‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006;138:1351‐1360. [DOI] [PubMed] [Google Scholar]
- 50. Nicholson KJ, Gilliland TM, Winkelstein BA. Upregulation of GLT‐1 by treatment with ceftriaxone alleviates radicular pain by reducing spinal astrocyte activation and neuronal hyperexcitability. J Neurosci Res. 2014;92:116‐129. [DOI] [PubMed] [Google Scholar]
- 51. Tao F, Liaw WJ, Zhang B, et al. Evidence of neuronal excitatory amino acid carrier 1 expression in rat dorsal root ganglion neurons and their central terminals. Neuroscience. 2004;123:1045‐1051. [DOI] [PubMed] [Google Scholar]
- 52. Nie H, Weng HR. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol. 2009;101:2041‐2051. [DOI] [PubMed] [Google Scholar]
- 53. Nie H, Weng HR. Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J Neurophysiol. 2010;103:2570‐2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu LJ, Zhuo M. Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics. 2009;6:693‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bradley J, Carter SR, Rao VR, Wang J, Finkbeiner S. Splice variants of the NR1 subunit differentially induce NMDA receptor‐dependent gene expression. J Neurosci. 2006;26:1065‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin‐1β enhances NMDA receptor‐mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034:172‐179. [DOI] [PubMed] [Google Scholar]
- 57. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, et al. Diabetes‐induced mechanical hyperalgesia involves spinal mitogen‐activated protein kinase activation in neurons and microglia via N‐methyl‐D‐aspartate‐dependent mechanisms. Mol Pharmacol. 2006;70:1246‐1254. [DOI] [PubMed] [Google Scholar]