

Summary
Aims
To evaluate the therapeutic effects of n‐butylidenephthalide (BP) in SOD1G93A mouse model of amyotrophic lateral sclerosis and explore the possible mechanisms.
Methods
The SOD1G93A mice were treated by oral administration of BP (q.d., 400 mg/kg d) starting from 60 days of age and continuing until death. The rotarod test was performed to assess the disease onset. The expression levels of apoptosis‐related proteins, inflammatory molecules, and autophagy‐associated proteins were determined. The number of apoptotic motor neurons and the extent of microglial and astroglial activation were also assessed in the lumbar spinal cords of BP‐treated mice. Grip strength test, hematoxylin‐eosin staining, nicotinamide adenine dinucleotide hydrogen staining, and malondialdehyde assay were conducted to evaluate the muscle function and pathology.
Results
Although BP treatment did not delay the disease onset, it prolonged the life span and thereafter extended the disease duration in SOD1G93A mouse model of ALS. BP treatment also reduced the motor neuron loss through inhibiting apoptosis. We further demonstrated that the neuroprotective effects of BP might be resulted from the inhibition of inflammatory, oxidative stress, and autophagy.
Conclusion
Our study suggests that BP may be a promising candidate for the treatment of ALS.
Keywords: amyotrophic lateral sclerosis, apoptosis, inflammation, n‐butylidenephthalide, oxidative stress
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the selective degeneration of upper motor neurons and lower motor neurons, resulting in the progressive paralysis.1 Approximately 90% of ALS cases are sporadic (sALS), while the remaining 10% cases were familial (fALS).2 In addition, over 20% of fALS cases are caused by missense mutations in Cu/Zn superoxide dismutase (SOD1) and several other gene defects related to RNA processing and protein clearance.2, 3, 4, 5 Although the underlying etiological and pathogenic mechanisms of ALS are still unknown, several pathogenic factors have been proposed, such as misfolding proteins accumulation, oxidative stress, inflammation, excitotoxicity, and apoptosis.6
Current treatments of ALS rely on relieving symptoms and extending life expectancy. Symptomatic treatments include the inhibition of salivation, ease of muscle cramps, alleviations of fatigue, relief of spasticity and pain. In the late stage of ALS, ventilatory assistance and anti‐infective therapy were often used to prevent the occurence of respiratory failure. Other alternative treatments such as physical therapy may also provide some benefits to some of the clinical symptoms of patients with ALS, such as easing the pain, delaying the loss of strength, and preventing the complications. Additionally, although a few novel therapeutic strategies such as cell and gene therapy have been tested for the better treatment of ALS, there are still no definitive and conclusive outcome yet.7, 8, 9 Riluzole, the only drug approved by Food and Drug Administration (FDA) of United States, can moderately prolong ALS patients’ life span by 2‐3 months through the inhibition of the glutamate release, but it may only benefit a subset of patients with ALS.10, 11 Thus, it is indispensable to develop novel therapeutic drugs for this devastating disease.
n‐butylidenephthalide (BP) is one of the main components derived from the volatile oil of Danggui (also referred as Angelica sinensis) which is widely used in the composite formula to treat gynecological disease and cardiovascular disease as a traditional Chinese medicine.12, 13 BP has been reported to have multiple pharmacological effects including antitumor, anti‐angina, anti‐atherosclerosis, and antiplatelet agglutination activities.14, 15, 16 As for neurodegenerative diseases, BP has been found to exert anti‐apoptosis, anti‐inflammation, and neuroprotective effects in the models of Parkinson's disease and Alzheimer's disease.17, 18 Recently, BP has been tested in SOD1G93A mouse model of ALS to show some neuroprotective effects.19 However, the detailed molecular mechanisms for its neuroprotective impacts are still not fully demonstrated. In addition, dl‐3‐n‐butylphthalide, one chemical analogue sharing similar chemical structure with BP, has been recently reported to exert antineuroinflammation activity and may have neuroprotective effects in SODG93A mouse model of ALS.20, 21 In this study, we used SOD1G93A mouse model of ALS to systematically investigate the neuroprotective properties of BP and to explore its possible underlying mechanisms.
2. Materials and Methods
2.1. Animals and treatments
Transgenic mice carrying mutant human SOD1 with a substitution of glycine to alanine in position 93 (SOD1G93A) develop a neuromuscular disease very similar to human ALS with the clinical phenotypes and histopathological features. SOD1G93A mice have been widely used in ALS research.22 We purchased the SOD1G93A mice from Jackson laboratory (B6SJL‐Tg‐SOD1*G93A‐1Gur/J, stock no. 002726). The colony was maintained by breeding transgenic male mice to wild‐type (WT) females from the same background. The genotypes of offspring were determined by PCR of DNA extracted from tail tissues according to our previous protocol.23 All experiments and animals care procedures were conducted in accordance with the Laboratory Animals Care Guidelines approved by the Animal Committee of Shanghai Jiao Tong University School of Medicine. All mice were kept in constant temperature and controlled lighting/dark cycle (light/dark 12/12 hours).
BP was purchased from Alfa Aesar (A10353, Tewksbury, MA, USA). The chemical structure of BP was shown in Figure 1A. BP was dissolved in olive oil (40 mg/mL) before oral administration. Sixty SOD1G93A mice were randomly divided into three groups (20 mice per group): (i) Vehicle group, treated with 10 mL/kg·d olive oil; (ii) BP group, treated with 400 mg/kg d BP; (iii) Riluzole group, treated with 30 mg/kg d riluzole (Rilutek, Sanofi, Paris, France) immersed in double distilled water. Twenty WT littermates were given 10 mL/kg d olive oil as normal WT control. Half of the SOD1G93A mice received the treatment by gavage once a day starting from 60 days of age to the day of their death. Ten WT mice were treated with vehicle from 60 to 160 days of age. Animal's body weight was recorded every 2 days. In addition, the rest half of SOD1G93A mice and WT littermates were also treated from 60 days of age and sacrificed at the age of 120 days. After treatments, five mice of each group were perfused with paraformaldehyde (PFA) for histological analysis of the spinal cords, and the rest of animals were used to obtain fresh muscles and spinal cords.
Figure 1.
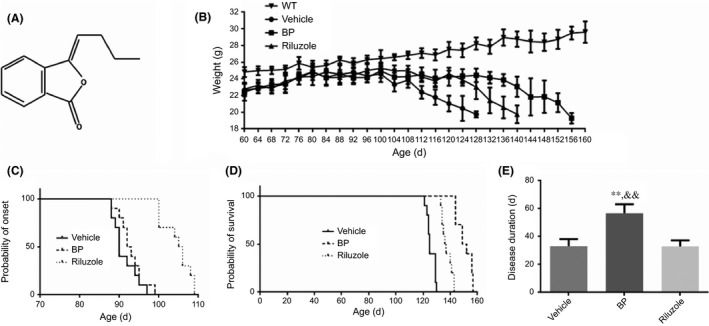
Chemical structure of BP and the effects of BP on body weight, disease onset, life span, and disease duration in SOD1G93A mice. (A) The chemical structure of BP; (B) the body weight curves of four groups; (C) the probability of disease onset analyzed by Kaplan‐Meier survival analysis; (D) the probability of survival analyzed by Kaplan‐Meier survival analysis; (E) the comparing result of disease duration. The values were presented as mean±SD. **P<.01 vs vehicle group; && P<.01 vs riluzole‐treated mice. n=5 in each group
2.2. Rotarod test
The disease onset of SOD1G93A mice was assessed by rotarod test. From the age of 63 days, the mice were subjected to a 7‐day training session (5 min/day) to adapt to the rotarod test apparatus (4 cm diameter, 20 rpm). Starting from 70 days of age, the rotarod test was performed with three trials (5 min/trial) every day. The behavioral performance was qualitatively recorded to check whether the ALS mice could complete one of all three 5‐minute trials. When the animal could not insist for 5 minutes on the apparatus for all three trials, a “×” (but not the exact duration) will be recorded to characterize the disease onset. As described in our previous studies,23, 24 once the mouse could not complete all three trials, we considered it as the disease onset.
2.3. Grip strength test
The hindlimb grip strength was measured at the age of 90 and 120 days. The hindlimb of mice caught the grasping force flatbed when their forelimb griped the steel wire nets. The measurements were recorded when the hindlimb separated from the flatbed by pulling their tails, and they were repeated for three times and the highest numerical record was taken.
2.4. Assessment of life span
The life span was determined as the duration between the day of birth and the day of death that was defined as an animal's inability to right itself within 30 seconds after being placed on its back.25 The age of death was recorded for the assessment of life span.
2.5. Nissl staining
Mice were anesthetized with chloral hydrate and given transcardiac perfusion firstly by cold 100 mmol/L phosphate buffered saline (PBS, PH 7.4) and then by 4% PFA at the age of 120 days. The spinal cords were removed and postfixed in 4% PFA overnight at 4°C. The tissues were then transferred to 15% and 30% sucrose dissolved in PBS, respectively, for 24 hours to be embedded by optimal cutting temperature (OCT). The embedded lumbar spinal cords (L4‐L5) were sectioned to 10 μm thickness with a Leica cryostat and frozen at −80°C until needed. Every forth section from 200 slices of each mouse was selected for Nissl staining. After being stained by 1% cresyl violet (Sigma, C5042, Saint Louis, MO, USA) for 10 minutes, the slices were dehydrated in gradient alcohol and cleared in xylol. Sample slides were observed and photographed with a microscope (Olympus, BX51, Tokyo, Japan). The motor neurons in the anterior horns of both sides were counted by a technician who was blind to the experimental design and animals grouping. The counting criteria was as follows: (i) the motor neurons in anterior horns and before the central canal; (ii) the neurons with a maximum diameter>20 μm; (iii) the neurons with a distinct nucleus.26 The average number of motor neurons was calculated based on 250 slices from five mice in each group.
2.6. Immunofluorescent staining
The spinal cords for immunofluorescent staining were obtained at the age of 120 days. The sections mounted on gelatin‐coated slides were roasted at 55°C and washed in PBS for three times. After being incubated in immunologic staining blocking buffer (Beyotime, Shanghai, China) for 1 hour, the slides were then incubated at 4°C overnight with the following primary antibodies: ionized calcium binding adapter molecule (Iba1) (1:500; Wako, 019‐19741, Tokyo, Japan), glial fibrillary acidic protein (GFAP) (1:500, Dako, Z0334, Glostrup, Denmark). The primary antibodies were removed and the slides were washed with PBS, incubated with Alexa Fluor dyes conjugated IgG secondary antibody (1:500, Invitrogen, Waltham, MA, USA) for 2 hours. The slides were then sealed with antifade reagent (Life technologies, Waltham, MA, USA) and were visualized with a fluorescent microscope (BX51, Olympus).
2.7. TdT‐mediated dUTP Nick‐End labeling (TUNEL) assay
TUNEL assays were performed to detect the apoptosis of motor neurons in the anterior horn of spinal cords according to the instructions of one‐step TUNEL apoptosis assay kit (Beyotime, C1089). After being washed by PBS twice, the sections were incubated for 2 minutes at 4°C by PBS containing 0.1% Triton X‐100. Then, sections were washed again by PBS and incubated with the prepared working solution for 60 minutes at 37°C. Afterward, the slides were sealed by antifade reagent with DAPI (Life technologies) and were visualized with a fluorescent microscope (BX51, Olympus). Ten sections from each mouse were randomly selected for TUNEL staining. The percentage of apoptotic motor neurons were calculated as the number of TUNEL‐positive motor neurons among total motor neurons.
2.8. Western blot
The acquired fresh spinal cords at the age of 120 days were lysed with radio‐immunoprecipitation assay (RIPA) lysis buffer (Beyotime, P0013B) containing 1% protease inhibitor phenylmethanesulfonyl fluoride (Beyotime, ST506). The lysates were centrifuged at 13 000 g for 30 minutes at 4°C with an high‐speed centrifuge (Beckman coulter, Brea, CA, USA) to obtain the supernatant fluid. The proteins were quantitated with a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, 23225, Waltham, MA, USA). Forty‐microgram proteins were loaded in 8% or 12% SDS gel and then transferred to 0.22 or 0.45 μm polyvinylidenefloride membranes as required. The membranes were blocked in 5% nonfat milk dissolved in TBST (50 mmol/L Tris, 150 mmol/L NaCl, 0.1% Tween‐20) buffer for 1 hour, and then incubated overnight at 4°C with the following primary antibodies: cleaved caspase‐3 (1:1000, Cell signaling technology, 9664, Danvers, MA, USA), Bax (1:1000, Cell signaling technology, 2772), Bcl‐2 (1:1000, Cell signaling technology, 3498), PARP (1:1000, Cell signaling technology, 9542), iNOS (1:1000, Cell signaling technology, 13120), TNF‐α (1:1000, Cell signaling technology, 11948), LC3 (1:1000, Sigma, L7543), P62 (1:1000, Cell signaling technology, 5114), Beclin‐1 (1:1000, Cell signaling technology, 3738), p‐AKT (1:1000, Cell signaling technology, 4060), AKT (1:1000, Abcam, ab8805, Cambridge, UK), p‐mTOR (1:1000, Cell signaling technology, 2983), mTOR (1:1000, Cell signaling technology, 5536), p‐p70s6k (1:1000, Cell signaling technology, 9204), p70s6k (1:1000, Cell signaling technology, 2708), β‐actin (1:20000, Abcam, ab49900). The anti‐rabbit (1:2000, Cell signaling technology, 7076) or anti‐mouse (1:2000, Cell signaling technology, 7074) HRP‐linked secondary antibodies were used, and the protein band images were visualized after reacting with a chemiluminescent horseradish peroxidase substrate (ECL, Pierce, 34075, Waltham, MA, USA). Band density was analyzed by Image Lab software version 5.2 (Bio‐Rad, Hercules, CA, USA).
2.9. Histopathology of skeletal muscles
After PBS perfusion, at the age of 120 days, the fresh gastrocnemius muscles (5×5×10 mm3) were dissected and immersed immediately in isopentane cooled in liquid nitrogen and embedded with bassora gum. The muscle tissues were cross‐sectioned at 10 μm thickness; then, hematoxylin‐eosin (H&E) staining or nicotinamide adenine dinucleotide hydrogen (NADH) staining was performed. Both the area of gastrocnemius muscle fibers and the percent of type I fiber were measured in 10 visual fields of each mouse (n=5 in each group). The dissected fresh gastrocnemius muscles were weighed, and 0.9% normal saline was added at a ratio of 1:9 (w/v). The muscles homogenate was prepared with ultrasonic homogenizer and then centrifuged at 1000 g at 4°C for 10 minutes. The malondialdehyde (MDA) level was determined with commercial kit (Jiancheng, A003‐2, Nanjing, China) according to the manufacturer's instruction. Briefly, the MDA level was measured by fluorescence assay of thiobarbituric acid reaction substance formation, detected at 532 nm, and quantified with a microplate reader (Biotek, Synergy H4, Winooski, VT, USA).
2.10. Statistical analysis
All statistical analyses were performed using SPSS 19.0 software (IBM, Armonk, NY, USA). Statistical analyses of disease onset and life span were performed by Kaplan‐Meier method. The other data were assessed by one‐way ANOVA with significance reported at the level of P<.05. While the variances between groups were equal (P>.05) or not equal, LSD test or Dunnett t‐test was performed, respectively. All data were presented as mean±SD.
3. Results
3.1. Effects of BP on disease onset and life span in SOD1G93A mice
BP treatment significantly slowed down the body weight loss in SOD1G93A mice. As shown in Figure 1B, while body weight loss can be easily observed during disease progression in three groups of SOD1G93A mice, the body weight of WT mice was gradually increased. In addition, compared with those vehicle‐treated SOD1G93A mice, the BP treatment significantly ameliorated body weight loss.
Moreover, the rotarod test was conducted to identify the disease onset. As shown in Figure 1C, BP treatment showed no significant delay on disease onset compared with vehicle‐treated SOD1G93A mice (94.1±3.07 vs 92.8±3.49, P>.05), and the disease onset of riluzole‐treated mice was significantly delayed (105±3.62, P<.01) vs BP‐ or vehicle‐treated SOD1G93A mice. However, BP treatment significantly prolonged the life span of SOD1G93A mice (Figure 1D) when compared with that of vehicle‐ or riluzole‐treated mice (151±5.55 vs 126±3.11 or 138±3.84, P<.01). Consequently, the disease duration of BP‐treated SOD1G93A mice was significantly extended (Figure 1E), as compared with that of vehicle‐ and riluzole‐treated SOD1G93A mice (56.7±6.48 vs 33.1±5.22 or 32.9±4.38, P<.01).
3.2. Effects of BP on motor neuron survival and apoptosis
The Nissl staining was performed to investigate the effects of BP on the motor neuron survival in SOD1G93A mice (Figure 2A). As shown in Figure 2B, there were fewer motor neurons in the anterior horn of vehicle‐treated SOD1G93A mice at 120 days of age compared with their age‐matched WT littermates (numbers of motor neurons in WT mice 24.9±2.02 vs in vehicle‐treated SOD1G93A mice 5.24±2.25, P<.01). More surviving motor neurons were found after BP and riluzole treatment (numbers of motor neurons in vehicle‐treated SOD1G93A mice 5.24±2.25, in BP‐treated SOD1G93A mice 16.9±2.08, in riluzole‐treated SOD1G93A mice 13.9±2.00, P<.01). BP‐treated SOD1G93A mice appeared to have more motor neuron survival than riluzole‐treated SOD1G93A mice (P<.01, Figure 2B). These results suggested that BP administration significantly ameliorated the motor neurons loss and further indicated a better neuroprotective activity than riluzole.
Figure 2.
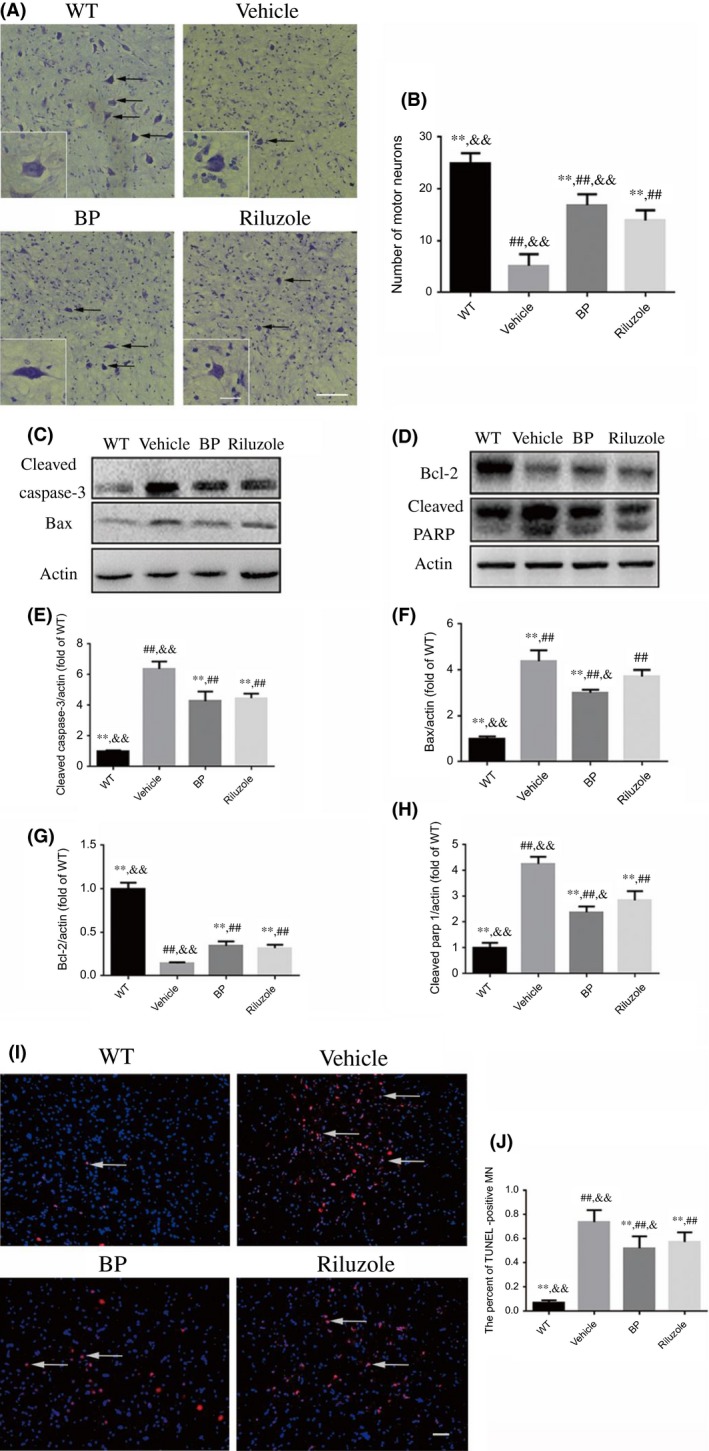
Effects of BP on the motor neuron survival, and the expression of apoptosis‐related proteins in the lumbar sections of the spinal cords of SOD1G93A mice. (A) The photomicrographs of motor neurons (black arrows) in the anterior horn of L4‐L5 spinal cord of four groups of mice by Nissl staining; scale bar=100 μm; the small inset scale bar=25 μm; (B) the number of motor neurons in the anterior horn of L4‐L5 spinal cord of four groups, n=5 in each group (50 slices in each mouse). Western blot assays of (C) cleaved caspase‐3 and Bax, (D) Bcl‐2 and cleaved PARP; the quantitative analysis of the expression levels of (E) cleaved caspase‐3, (F) Bax, (G) Bcl‐2, and (H) cleaved PARP in four groups; n=5 in each group. (I) BP treatment decreased the number of apoptotic motor neurons (DAPI: blue, TUNEL: red) in the spinal cords. White arrows point to the TUNEL‐positive cells. Scale bar=50 μm; (J) the comparing results of the percent of TUNEL‐positive motor neurons (MN) in four groups, n=5 in each group (10 slices in each mouse). **P<.01 vs vehicle group; ## P<.01 vs WT group; & P<.05 and && P<.01 vs riluzole group
We then examined the effects of BP on apoptosis by immunoblotting of cleaved caspase‐3, Bax, Bcl‐2, and cleaved PARP (Figure 2C,D). Consistent with our previous studies,23, 24 the quantitative analysis showed that the protein levels of cleaved caspase‐3 (Figure 2E), Bax (Figure 2F), and cleaved PARP (Figure 2H) in the vehicle‐treated SOD1G93A mice were increased by 3‐, 1.5‐, and 4.2‐fold, respectively, as compared with WT mice, while the protein level of Bcl‐2 in the vehicle group of SOD1G93A mice was decreased by 53% (Figure 2G). Moreover, BP treatment decreased the expression levels of cleaved caspase‐3 (Figure 2E), Bax (Figure 2F), and cleaved PARP (Figure 2H) by 33%, 32%, and 48%, respectively, and increased the expression of Bcl‐2 (Figure 2G) by 1.4‐fold as compared with vehicle‐treated SOD1G93A mice. Riluzole treatment can also decrease the expression of cleaved caspase‐3, Bax, and cleaved PARP as well as increase expression of Bcl‐2.
Consistent with Western blot data, TUNEL assay suggested that the number of TUNEL‐positive motor neurons in the spinal cords of vehicle‐treated SOD1G93A mice was significantly increased as compared with WT mice (Figure 2I,J). Both BP and riluzole treatments reduced the number of TUNEL‐positive motor neurons in the SOD1G93A mice (Figure 2I,J).
3.3. Effects of BP on neuroinflammation
Inflammatory responses and glial cells activation have been reported to be accompanied by motor neuron degeneration in ALS animal models and patients.27 Here, we used Iba‐1 and GFAP immunostainings to detect the activation of microglia and astrocytes, respectively. As shown in Figure 3A, the immunofluorescence staining indicated that the microglia and astrocytes were highly activated in vehicle‐treated SOD1G93A mice compared with their WT littermates. The BP and riluzole treatment significantly ameliorated the activation of microglia and astrocyte (Figure 3A‐C).
Figure 3.
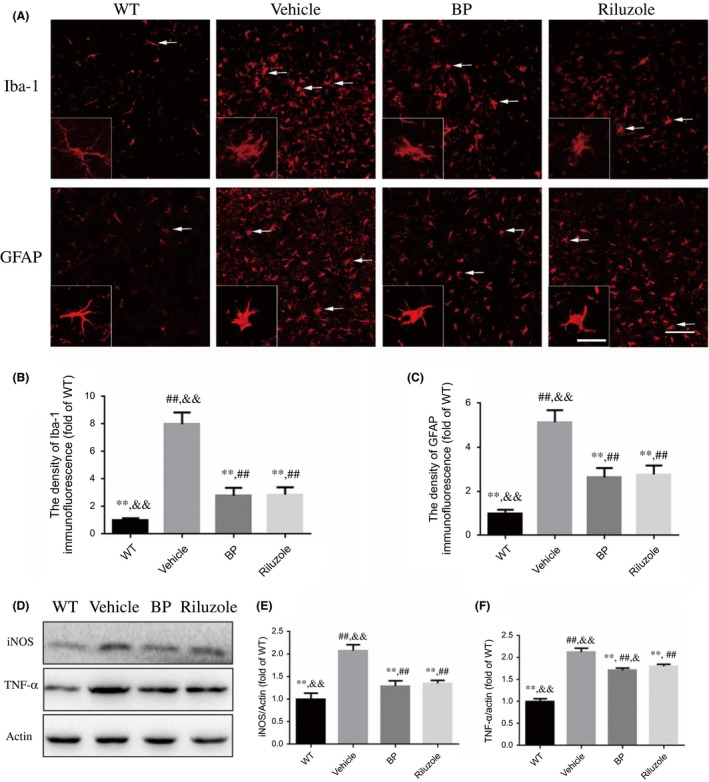
Effects of BP on the activation of glial cells and the expression of inflammation‐related proteins. The photomicrographs of immunofluorescence of the lumbar spinal cords in four groups at the age of 120 days. (A) Anti‐Iba1 and GFAP antibodies were applied as markers of microglia and astrocytes. BP treatment significantly inhibited the activation of microglia and astrocytes (white arrows); Scale bar=100 μm; inset scale bar=50 μm; (B) the quantitative analysis of Iba‐1 immunofluorescence, n=5 in each group (10 slices in each mouse); (C) the quantitative analysis of GFAP immunofluorescence, n=5 in each group (10 slices in each mouse); (D) Western blot assays of inflammation‐related proteins; the quantitative analysis of the expression levels of (E) iNOS and (F) TNF‐α, n=5 in each group. **P<.01 vs vehicle‐treated mice; ## P<.01 vs WT mice; and & P<.05 and && P<.01 vs riluzole‐treated mice
The data from Western blot analysis further demonstrated that vehicle‐treated SOD1G93A mice showed higher levels of iNOS and TNF‐α, two inflammatory markers, than WT mice, while these changes can be significantly suppressed by BP treatment (Figure 3D‐F).
3.4. Effects of BP on hindlimb grip strength and histopathology of gastrocnemius muscles
As shown in Figure 4A, the hindlimb grip strength of all SOD1G93A mice was declined when compared with WT mice, although there was no significance among three groups of SOD1G93A mice at the age of 90 days (WT mice 106±7.57, vehicle‐treated SOD1G93A mice 87.4±4.17, BP‐treated SOD1G93A mice 90.9±6.20, riluzole‐treated SOD1G93A mice 91.7±6.54). Nevertheless, the grip strength of BP‐treated SOD1G93A mice was much better than that of riluzole‐ and vehicle‐treated SOD1G93A mice at the age of 120 days (WT mice 110±7.01, vehicle‐treated SOD1G93A mice 20.5±6.26, BP‐treated SOD1G93A mice 72.1±6.14, riluzole‐treated SOD1G93A mice 52.2±4.60, P<.01). These results indicated that BP could strengthen hindlimb muscles greater than riluzole.
Figure 4.
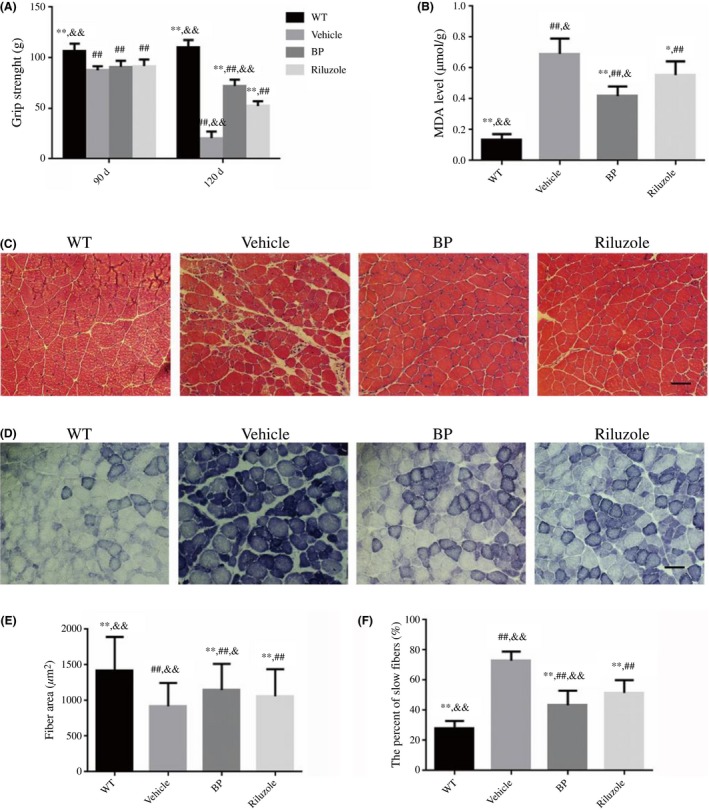
The effects of BP on the gastrocnemius muscles of SOD1G93A mice. (A) The hindlimb grip strength in four groups of mice; (B) the comparing results of MDA levels in four groups; (C) the photomicrographs of gastrocnemius muscle sections by H&E staining in SOD1G93A mice; and (D) the NADH staining of gastrocnemius muscle sections; scale bar=50 μm; (E) the comparing results of muscle fiber area, n=5 in each group; (F) the percent of slow fibers in four groups, n=5 in each group. The values were expressed as mean±SD. *P<.05 and **P<.01 vs vehicle‐treated mice; ## P<.01 vs WT mice; & P<.05 and && P<.01 vs riluzole‐treated mice
The structures and function of muscles fibers apparently changed in the vehicle‐treated SOD1G93A mice as compared with WT mice. H&E staining showed the typical atrophic features of gastrocnemius muscles such as atrophic fibers, central nuclei, and hematoxylin inclusions in the vehicle‐treated SOD1G93A mice (Figure 4C). BP and riluzole administration ameliorated the atrophy of muscles, as evidenced by larger myofiber, less central nuclei, and less aggregated nucleus in BP‐ and riluzole‐treated SOD1G93A mice than vehicle‐treated SOD1G93A mice (Figure 4C). Figure 4E showed that the fiber area of BP‐treated SOD1G93A mice was significantly larger than both vehicle‐ and riluzole‐treated SOD1G93A mice (P<.05). The oxidative metabolism of muscles can be visualized by NADH staining (dark blue). As shown in Figure 4D, there were more dark blue areas and grouped myofibers in the vehicle‐treated SOD1G93A mice than WT mice. The dark blue areas and grouped myofibers in BP‐ and riluzole‐treated SOD1G93A mice decreased as compared with the vehicle‐treated SOD1G93A mice. The data further indicated that BP‐treated SOD1G93A mice had significantly lower percentage of type I fibers than both vehicle‐ and riluzole‐treated SOD1G93A mice (P<.05).
Furthermore, we measured MDA content to identify the oxidative stress level in the muscle of SOD1G93A mice and the possible effect of BP. Consistent with NADH staining, as shown in Figure 4B, the level of MDA in vehicle‐treated SOD1G93A mice was significantly higher than that in WT mice (0.69±0.10 vs 0.14±0.04, P<.01). Additionally, the BP treatment reduced MDA level by 40% (0.42±0.06 vs 0.69±0.10, P<.01), while riluzole induced less decrease on MDA than BP (0.55±0.09 vs 0.42±0.06, P<.05, Figure 4B).
3.5. Effects of BP on autophagy
To investigate the effect of BP treatment on autophagy, we examined the expression levels of LC3‐II, Beclin‐1, and P62 (Figure 5A). The quantitative analysis of Western blot bands showed that the expression levels of LC3‐II (Figure 5B), P62 (Figure 5C), and Beclin‐1 (Figure 5D) in vehicle‐treated SOD1G93A mice were significantly increased as compared with WT mice (P<.01). All these changes can be ameliorated by BP treatment (63%, 74%, and 78% of LC3‐II, P62, and Beclin‐1, respectively, in vehicle‐treated SOD1G93A mice). To explore the involvement of AKT/mTOR signaling pathway in BP‐modulated autophagy, we also measured the expression levels of mTOR, p‐mTOR, AKT, p‐AKT, P70s6k, and p‐P70s6k. Our data revealed that the expression ratios of p‐AKT/AKT (Figure 5F), p‐mTOR/mTOR (Figure 5G), and p‐P70s6k/p‐P70s6k (Figure 5H) in vehicle‐treated SOD1G93A mice were significantly reduced as compared with WT mice. Moreover, all these changes could be partially reversed by BP treatment, indicating that BP may influence the autophagy flux via AKT/mTOR‐related pathway.
Figure 5.
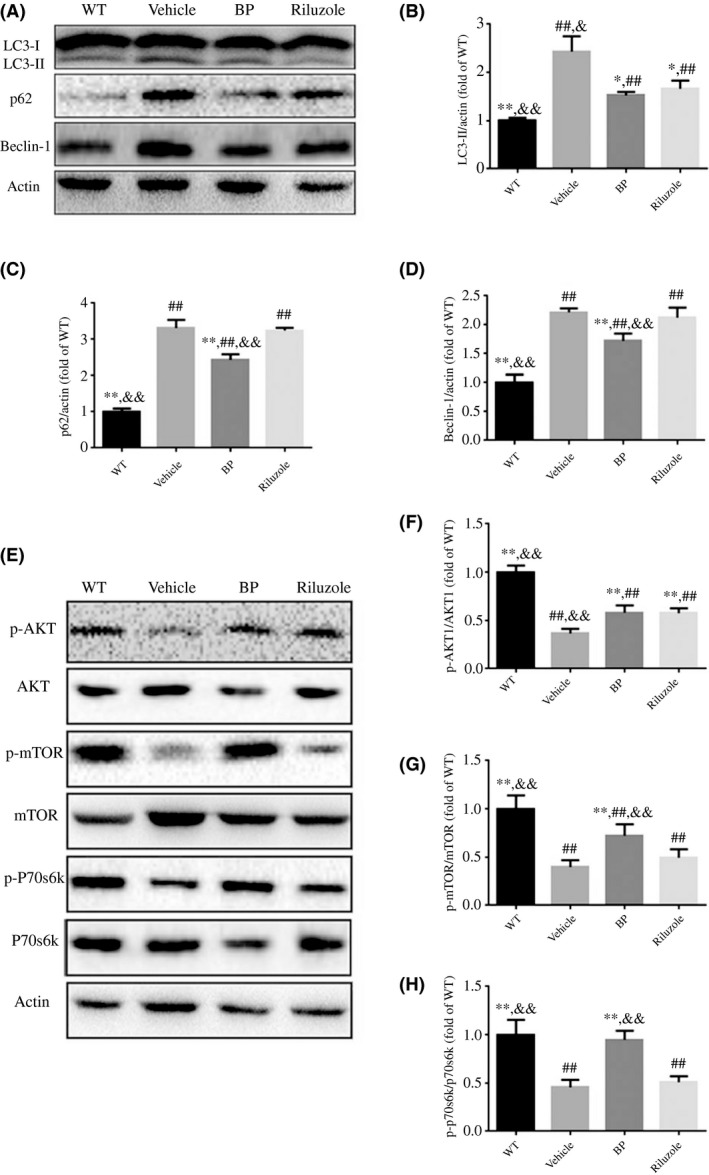
Effects of BP on the autophagy‐related proteins in lumbar spinal cords. (A) Western blot analysis of LC3‐I, LC3‐II, P62, and Beclin‐1 in four groups; quantitative analysis of the expression levels of (B) LC3‐II, (C) P62, and (D) Beclin‐1 in four groups. (E) Western blot analysis of mTOR pathway‐related proteins (p‐AKT, AKT, p‐mTOR, mTOR, p‐p70s6k, and p70s6k). And the quantitative analysis of the expression levels of p‐AKT (F), p‐mTOR (G), and p‐p70s6k (H). The values were expressed as mean±SD. *P<.05 and **P<.01 vs vehicle‐treated mice; ## P<.01 vs WT mice. & P<.05 and && P<.01 vs riluzole‐treated mice. n=5 in each group
4. Discussion
Our present data showed that BP treatment could slow disease progression, prolong the life span, and extend disease duration in SOD1G93A mouse model of ALS. The overall anti‐ALS effect of BP is even better than riluzole to some certain extent. Furthermore, we demonstrated here that BP treatment could increase the motor neuron survival in the anterior horn of spinal cords and protect the gastrocnemius by restoring their function in SOD1G93A mouse model of ALS. The neuroprotective effects of BP treatment may be resulted from its anti‐apoptosis, anti‐inflammation, antioxidative stress, and autophagy inhibition activities.
It has been reported that BP possesses multiple pharmacological properties against several neurological and non‐neurological diseases, and it seems that its regulation on autophagy is only part of protective actions against ALS pathologies.19 Thus, in this study, we have further investigated several possible neuroprotective mechanisms of BP on ALS.
The motor neuron degeneration of the spinal cord is the main feature of ALS. Therefore, it is very important to determine whether any treatment for ALS can prevent motor neuron loss. We found that BP treatment significantly attenuated motor neuron loss in the spinal cord of SOD1G93A mice. Accumulating evidences support the motor neuron degeneration is partially mediated by the activation of apoptosis, which is an ongoing process and a contributor to the pathogenesis of ALS.24, 28, 29 We measured the apoptosis level by detecting the expression of apoptosis‐related proteins and by TUNEL staining. As expected, SOD1G93A mice at the age of 120 days showed increased levels of cleaved caspase‐3, pro‐apoptotic protein Bax and cleaved PARP, and a decreased level of antiapoptotic protein Bcl‐2, together with the increased apoptotic motor neurons in the lumbar spinal cords. Previous study reported that BP can protect against dopaminergic neuron degeneration in Caenorhabditis elegans model of Parkinson's disease by inhibiting apoptosis pathways.18 Similarly, our study demonstrated that BP treatment can inhibit the apoptosis of motor neurons in the spinal cords. In mammalian cells, the Bcl‐2 homology domain 3 (BH3)‐only proteins (Bad, Bid, Bim, Bik, Puma, Noxa et al.) are the important upstream activators of apoptosis.30 BH3‐only proteins can inhibit the antiapoptotic effect of Bcl‐2 by interacting with Bcl‐2 and promote the activation of Bax.30, 31 The expression of BH3‐only proteins was found higher in patients with ALS and ALS mouse model.32, 33 And it was reported that the deletion of the BH3‐only protein Puma could delay the disease onset and protect motor neurons from apoptosis.32 Based on these previous research reports, BP may decrease the expression of BH3‐only proteins to counter against the apoptosis of motor neurons.18 Consistent with the previous studies, we found that riluzole augmented the level of Bcl‐2 and suppressed the levels of cleaved caspase‐3, Bax, and cleaved PARP, and exhibit antiapoptosis activity.34, 35 Interestingly, we documented that the expression levels of cleaved PARP and Bax in BP‐treated SOD1G93A mice were significantly lower than that in riluzole‐treated SOD1G93A mice (Figure 2F,H), indicating that regulating apoptosis may become an important action for BP to protect the motor neurons in this mouse model of ALS.
Inflammatory response is considered to be related with ALS pathogenesis.27 The expression levels of iNOS and TNF‐α usually increase in the spinal cord of SOD1G93A mouse model of ALS.24, 36 iNOS is the marker of macrophage activation while TNF‐α plays an essential role in inflammatory response. In the same way, we found the activation of microglia and astrocytes increased greatly in the anterior horn of spinal cords of vehicle‐treated SOD1G93A mice, but not in WT mice. In addition, the levels of inflammatory factors such as iNOS and TNF‐α were elevated in vehicle‐treated SOD1G93A mice than WT mice. Previous study reported that dl‐3‐n‐butylphthalide, one chemical analogue of BP, exerted its neuroprotective effects by reducing the activation of microglia and astrocytes to inhibit the inflammation response.21 BP can provide neuroprotective effect by inhibiting the release of various proinflammatory molecules and suppressing microglial activation.37 Consistent with these previous findings, in the present study, we found that both the activation of glial cells and the levels of inflammation‐related factors (iNOS and TNF‐α) could be significantly suppressed by BP treatment. BP treatment may attenuate the inflammation response by suppressing the nuclear factor κB pathway. It may directly decrease the activity of inhibitor of nuclear factor κB (IKK), which downregulates the activation of NF‐κB signal pathway.38
Muscle atrophy is another important feature in ALS. Consistent with previous studies, our data demonstrated that SOD1G93A mice exhibited atrophic myofibers, central nuclei, and hematoxylin inclusions in gastrocnemius muscles.23, 24 Our data further showed that BP treatment not only ameliorated the atrophy but also restored the muscular function (elevating the hindlimb grip strength). NADH staining is usually applied to evaluate the muscle metabolic pattern. The type I fibers relying on the oxidative metabolism have the dark staining of NADH, while type II fibers relying on the glycolytic metabolism have the light staining.39, 40 In ALS, the type II fibers tend to either atrophy or convert to the type I fibers. Here, we found that BP treatment inhibited the trend of transition from “type II” to “type I” as evidenced by attenuating the dark staining of NADH in the gastrocnemius muscles of the ALS mice. Consistent with previous reports in which BP can protect cells against oxidative stress, our present study found that BP treatment reversed the high level of MDA production in the spinal cords of ALS mice, indicating an increased level of reactive oxygen species.41, 42 Therefore, BP may protect cells against oxidative stress by activating the nuclear factor erythroid 2‐related factor 2 (Nrf2) pathway,41 which participates in the transcription activation of many antioxidative genes, such as SOD1, quinone 1, and NADH dehydrogenase.41
More and more evidences have suggested that autophagy plays an important role in ALS pathogenesis. Autophagy‐lysosome system is one of the major systems participates in the intracellular protein degradation.43 The mutant SOD1 protein accumulation can activate autophagy in SOD1G93A mice.44 Our previous study demonstrated that the LC3‐II and P62 expressions were increased in the spinal cords of SOD1G93A mice, suggesting the activation of autophagy in ALS model.45 Recently, Hsueh et al. also reported the therapeutic potential of BP in SOD1G93A mice possibly via a down‐regulated autophagy.19 In the present study, we found that the levels of LC3‐II, Beclin‐1, and P62 were significantly decreased in BP‐treated SOD1G93A mice, while the levels of p‐AKT, p‐mTOR, and p‐p70 were increased as compared with vehicle‐treated SOD1G93A mice, suggesting that BP could inhibit autophagy through AKT/mTOR signaling pathway.
In conclusion, our data support the therapeutic potential of BP for ALS. BP could slow the disease progression, prolong the life span, and extend disease duration in SOD1G93A mouse model of ALS. BP could reduce the motor neuron loss, attenuate the muscles atrophy, and restore the gastrocnemius muscles function. Collectively, the therapeutic effects of BP may be resulted from its antiapoptosis, anti‐inflammation, antioxidative stress activities, as well as its inhibition on mTOR‐dependent autophagy.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Sciences Foundation of China (81430021, 81370470 and 81671241), and Shanghai New Youth Science and technology Project (15QA1403000).
Zhou Q‐M, Zhang J‐J, Li S, Chen S, Le W‐D. n‐butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci Ther. 2017;23:375‐385. 10.1111/cns.12681
Contributor Information
Sheng Chen, Email: mztcs@163.com.
Wei‐Dong Le, Email: wdle@sibs.ac.cn.
References
- 1. Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46. [DOI] [PubMed] [Google Scholar]
- 2. Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 2012;8:13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirby J, Halligan E, Baptista MJ, et al. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–1706. [DOI] [PubMed] [Google Scholar]
- 4. Gianforcaro A, Hamadeh MJ. Vitamin D as a potential therapy in amyotrophic lateral sclerosis. CNS Neurosci Ther. 2014;20:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ravits J, Appel S, Baloh RH, et al. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(S1):5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Redler RL, Dokholyan NV. Chapter 7–The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog Mol Biol Transl Sci. 2012;107:215–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abdul Wahid SF, Law ZK, Ismail NA, Azman AR, Lai NM. Cell‐based therapies for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2016;11:CD011742. doi: 10.1002/14651858.CD011742.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mcbean GJ, López MG, Wallner FK. Redox‐based therapeutics in neurodegenerative disease. Br J Pharmacol. 2016;. doi: 10.1111/bph.13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mceachin ZT, Donsante A, Boulis N. Gene therapy for the treatment of neurological disorders: amyotrophic lateral sclerosis. Methods Mol Biol. 2016;1382:399–408. [DOI] [PubMed] [Google Scholar]
- 10. Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17:4–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mu X, Azbill RD, Springer JE. Riluzole improves measures of oxidative stress following traumatic spinal cord injury. Brain Res. 2000;870:66–72. [DOI] [PubMed] [Google Scholar]
- 12. Yim TK, Wu WK, Pak WF, Mak DHF, Liang SM, Ko KM. Myocardial protection against ischaemia‐ reperfusion injury by a Polygonum multiflorum extract supplemented ‘Dang‐Gui decoction for enriching blood’, a compound formulation, ex vivo. Phytother Res. 2000;14:195–199. [DOI] [PubMed] [Google Scholar]
- 13. Yi L, Liang Y, Wu H, Yuan D. The analysis of Radix Angelicae sinensis (Danggui). J Chromatogr A. 2009;1216:1991–2001. [DOI] [PubMed] [Google Scholar]
- 14. Tsai NM, Chen YL, Lee CC, et al. The natural compound n ‐butylidenephthalide derived from Angelica sinensis inhibits malignant brain tumor growth in vitro and in vivo 3. J Neurochem. 2006;99:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan SK, Choi OK, Jones RL, Lin G. Mechanisms underlying the vasorelaxing effects of butylidenephthalide, an active constituent of Ligusticum chuanxiong, in rat isolated aorta. Eur J Pharmacol. 2006;537:111–117. [DOI] [PubMed] [Google Scholar]
- 16. Liu PY, Sheu JC, Lin PC, et al. Expression of Nur77 induced by an n ‐butylidenephthalide derivative promotes apoptosis and inhibits cell growth in oral squamous cell carcinoma. Invest New Drugs. 2012;30:79–89. [DOI] [PubMed] [Google Scholar]
- 17. Chang CY, Chen SM, Lu HE, et al. N‐butylidenephthalide attenuates Alzheimer's disease‐like cytopathy in Down syndrome induced pluripotent stem cell‐derived neurons. Sci Rep. 2015;5:8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu RH, Harn HJ, Liu SP, et al. n‐Butylidenephthalide protects against dopaminergic neuron degeneration and α‐synuclein accumulation in Caenorhabditis elegans Models of Parkinson's disease. PLoS One. 2014;9:e85305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsueh KW, Chiou TW, Chiang SF, et al. Autophagic down‐regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology. 2016;108:152–160. [DOI] [PubMed] [Google Scholar]
- 20. Zhang HL, Liu YL, Neurology DO. Effects of dl‐3n‐butylphthalide on lifespan and CDK5 and P35 expression in mice model of amyotrophic lateral sclerosis. Chinese J New Drug Clin Rem. 2014;33:524–528. [Google Scholar]
- 21. Feng X, Peng Y, Liu M, Cui L. dl ‐3‐n‐butylphthalide extends survival by attenuating glial activation in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology. 2011;62:1004–1010. [DOI] [PubMed] [Google Scholar]
- 22. Ludolph AC, Bendotti C, Blaugrund E, et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop, Amyotrophic Lateral Sclerosis, Informa healthcare. Amyotroph Lateral Scler. 2007;8:217–223. [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Chen S, Song L, et al. MTOR‐independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song L, Gao Y, Zhang X, Le W. Galactooligosaccharide improves the animal survival and alleviates motor neuron death in SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2013;246:281–290. [DOI] [PubMed] [Google Scholar]
- 25. Koh SH, Kim Y, Kim HY, Hwang S, Chang HL, Kim SH. Inhibition of glycogen synthase kinase‐3 suppresses the onset of symptoms and disease progression of G93A‐SOD1 mouse model of ALS. Exp Neurol. 2007;205:336–346. [DOI] [PubMed] [Google Scholar]
- 26. Manabe Y, Nagano I, Gazi MSA, et al. Glial cell line‐derived neurotrophic factor protein prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Neurol Res. 2003;25:195–200. [DOI] [PubMed] [Google Scholar]
- 27. Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. [DOI] [PubMed] [Google Scholar]
- 28. Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis. J Neuropathol Exp Neurol. 1999;58:459–471. [DOI] [PubMed] [Google Scholar]
- 29. Kumar V, Islam A, Hassan MI, Ahmad F. Therapeutic progress in amyotrophic lateral sclerosis‐beginning to learning. Eur J Med Chem. 2016;121:903–917. [DOI] [PubMed] [Google Scholar]
- 30. Tuzlak S, Villunger A. BH3‐Only Proteins. eLS: John Wiley & Sons, Ltd; 2016. doi: 10.1002/9780470015902.a0021569.pub2 [DOI] [Google Scholar]
- 31. Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl‐xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19:341–352. [DOI] [PubMed] [Google Scholar]
- 32. Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3‐only protein puma protects motoneurons from ER stress‐induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci USA. 2007;104:20606–20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shinoe T, Wanaka A, Nikaido T, et al. Upregulation of the pro‐apoptotic BH3‐only peptide harakiri in spinal neurons of amyotrophic lateral sclerosis patients. Neurosci Lett. 2001;313:153–157. [DOI] [PubMed] [Google Scholar]
- 34. Hassanzadeh K, Habibi‐Asl B, Roshangar L, Nemati M, Ansarin M, Farajnia S. Intracerebroventricular administration of riluzole prevents morphine‐induced apoptosis in the lumbar region of the rat spinal cord. Pharmacol Rep. 2010;62:664–673. [DOI] [PubMed] [Google Scholar]
- 35. Hassanzadeh K, Roshangar L, Habibi‐Asl B, et al. Riluzole prevents morphine‐induced apoptosis in rat cerebral cortex. Pharmacol Rep. 2011;63:697–707. [DOI] [PubMed] [Google Scholar]
- 36. Graves M, Fiala M, Lu AD, et al. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph Lateral Scler Other Motor Neuron Disord. 2009;5:213–219. [DOI] [PubMed] [Google Scholar]
- 37. Nam KN, Kim KP, Cho KH, et al. Prevention of inflammation‐mediated neurotoxicity by butylidenephthalide and its role in microglial activation. Cell Biochem Funct. 2013;31:707–712. [DOI] [PubMed] [Google Scholar]
- 38. Fu RH, Hran HJ, Chu CL, et al. Lipopolysaccharide‐stimulated activation of murine DC2.4 cells is attenuated by n‐butylidenephthalide through suppression of the NF‐κB pathway. Biotechnol Lett. 2011;33:903–910. [DOI] [PubMed] [Google Scholar]
- 39. Pilehvarian AA. An ultrastructural and histochemical study of the flexor tibialis muscle fiber types in male and female stick insects (Eurycantha calcarata, L). J Exp Zool A Ecol Genet Physiol. 2015;323:527–539. [DOI] [PubMed] [Google Scholar]
- 40. López‐García K, Mariscal‐Tovar S, Castelán F, Jiménez‐Estrada I. Fiber type composition of pubococcygeus and bulbospongiosus striated muscles is modified by multiparity in the rabbit. Neurourol Urodyn. 2016. doi: 10.1002/nau.23143. [DOI] [PubMed] [Google Scholar]
- 41. Saw CLL, Wu Q, Su ZY, et al. Effects of natural phytochemicals in Angelica sinensis (Danggui) on Nrf2‐mediated gene expression of phase II drug metabolizing enzymes and anti‐inflammation. Biopharm Drug Dispos. 2013;34:303–311. [DOI] [PubMed] [Google Scholar]
- 42. Zhao B, Zhang Q, Lin H, et al. Effect of co‐existent components in CO2 supercritical fluid extract of Angelica sinensis Radix on metabolism of Z‐ligustilide after oral administration in rats. J Trad Chin Med Sci. 2015;51:126–134. [Google Scholar]
- 43. Rubinsztein DC. The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature. 2006;443:780–786. [DOI] [PubMed] [Google Scholar]
- 44. Wootz H, Weber E, Korhonen L, Lindholm D. Altered distribution and levels of cathepsin D and cystatins in amyotrophic lateral sclerosis transgenic mice: possible roles in motor neuron survival. Neuroscience. 2006;143:419–430. [DOI] [PubMed] [Google Scholar]
- 45. Liang L, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy. 2008;4:290–293. [DOI] [PubMed] [Google Scholar]