

Summary
Aims
To analyze brain functional connectivity in the somatomotor and default‐mode networks (DMNs) of patients with Huntington disease (HD), its relationship with gray matter (GM) volume loss, and functional changes after pridopidine treatment.
Methods
Ten patients and ten untreated controls underwent T1‐weighted imaging and resting‐state functional magnetic resonance imaging (fMRI); four patients were also assessed after 3 months of pridopidine treatment (90 mg/d). The seed‐based functional connectivity patterns from the posterior cingulate cortex and the supplementary motor area (SMA), considered cortical hubs of the DMN and somatomotor networks, respectively, were computed. FMRIB Software Library voxel‐based morphometry measured GM volume.
Results
Patients had GM volume decrease in all cortical and subcortical areas of the somatomotor network with preservation of the SMA, and increased somatomotor and DMN connectivity. In DMN structures, functional connectivity impairment preceded volume loss. Pridopidine reduced the intensity of these aberrant connections.
Conclusion
The abnormal connectivity of the somatomotor and DMN observed in HD patients may represent an early dysfunction marker, as it preceded volume loss in DMN. Pridopidine reduced connectivity of these networks in all four treated patients, suggesting that connectivity is sensitive to treatment response.
Keywords: biomarker, Huntington disease, magnetic resonance imaging, pridopidine
1. Introduction
Huntington disease (HD), an autosomal‐dominant disease determined by a cytosine‐adenine‐guanine (CAG) expansion mutation in the HTT gene encoding the protein huntingtin, results in progressive neuronal degeneration, motor and behavioral disturbances, and cognitive deficits. More CAG repeats result in earlier disease onset.1
Reliable individual‐level markers of brain changes would allow evaluation of treatment efficacy and monitoring of disease progression. Current structural imaging studies have described reduced caudate and putamen volumes, cortical thinning, and early impairment of white matter fibers.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15
Network‐sensitive neuroimaging techniques have shown that spatial pattern of atrophy of a disease relates to functional connectivity, as mapped by resting‐state functional magnetic resonance imaging (fMRI).16 fMRI measures rates of spontaneous fluctuations in the blood oxygen level‐dependent (BOLD) signal, allowing the exploration of brain regions showing temporal coherence in slow spontaneous fluctuations (<0.1 Hz), giving rise to resting‐state networks (RSNs). These interconnected cerebral areas, called nodes, are described by their degree (number of connections), centrality, and modularity17 A node occupying a central position in the overall network organization is called a hub. Mapping these cortical hubs and plasticity of RSNs may help explain why some lesions are particularly disruptive,18 how the neurodegenerative process spreads across functionally interconnected brain regions,16 and how a potential drug may modify these connections.
Literature on multimodal (fMRI, electroencephalography, and magnetoencephalography) imaging19, 20, 21, 22 supports evidence of RSNs, among which the somatomotor network (SMN) and default‐mode network (DMN) are typically very reproducible. DMN is characterized by greater activation during rest than during goal‐directed tasks, suggesting network activity reflects a default state of neuronal activity of the human brain18, 23, 24, 25, 26 It comprises the posterior cingulate cortex (PCC), medial frontal and inferior parietal regions, and plays a fundamental role in integration across different functional domains.27 We previously identified the PCC and supplementary motor area (SMA) as core regions for the DMN and SMN, respectively.21 DMN is a backbone around which other cortical networks amalgamate with the PCC, acting as a major transit hub for exchange of information.21 Furthermore, the SMA has been associated with planning, preparation, and execution of voluntary movements. It is involved in the corticospinal tract and has anatomical connections with the primary motor cortex and basal ganglia,21 areas known to be impaired in HD.
It is unclear whether the DMN is impaired during the presymptomatic stage28, 29, 30, 31, 32 or from manifest HD. Some studies have shown reduced functional connectivity in DMN‐related areas, such as the PCC, ventromedial prefrontal cortex (vmPFC), and angular gyrus,33 while others have highlighted the functional connectivity increase.34, 35 Regarding the SMN, functional connectivity increase has been described,35, 36 suggesting a compensatory process due to striatal or cortical degeneration, or a generalized spreading of activity due to reduced specialization of function.37 Longitudinal studies in presymptomatic subjects showed no differences compared with controls over time.38, 39
Alterations in dopamine and dopamine‐receptor levels in the brain contribute to clinical symptoms of HD.40 Pridopidine, a new dopamine‐ligand compound, is thought to stabilize dysregulated psychomotor functions, by modulating hyper‐ or hypo‐active functioning in areas of the brain receiving dopamine input.41 Pridopidine showed potential neuroprotective properties through interaction with the sigma‐1 receptor.41, 42 In a Phase III study, pridopidine improved total motor score (TMS) on the Unified Huntington's Disease Rating Scale (UHDRS)43 without worsening cognitive or psychiatric symptoms after 1 year.44 To date, pharmacological effects of pridopidine on functional connectivity, mapped by fMRI, have not been reported in patients with HD.
The aim of our study is to analyze brain functional connectivity in the SMN and DMN in HD patients, its relationship with gray matter (GM) volume loss, and functional changes after short‐term treatment with pridopidine 90 mg/d.
2. Methods
2.1. Patients
Ten HD patients with CAG repeat expansions ranging from 42 to 66, and 10 age‐ and gender‐matched controls were enrolled. All patients were clinically examined by the same neurologist (F.S.) using the UHDRS motor, cognitive, behavioral, and functional subscales.45 Patient age at onset was established on the basis of neurological manifestations by interviews with patients and relatives, and analysis of clinical files. Disease stage was determined according to previously published standards with slight modifications12: HD stage‐I (total functional capacity [TFC] 11‐13); stage‐II (TFC 7‐10); stage‐III (TFC<7). The TFC is a standardized scale to assess capacity to work, handle finances, perform domestic chores, self‐care tasks, and live independently. It ranges from 13 (normal) to 0 (severe disability). The UHDRS cognitive subscale includes a phonetic verbal fluency test, the symbol digit test, the Stroop test, and the Mini‐Mental State Examination (MMSE), which is a global index of cognitive deterioration, ranging from 30 (no impairment) to 0 (maximum impairment). In the phonetic verbal fluency test, a single letter is provided and the subject has to generate as many words starting with the specified letter as he or she can within a specified period of time. The symbol digit test measures the time to pair abstract symbols with specific numbers. It evaluates attention, visuoperceptual processing, working memory, and psychomotor speed. The score is the number of correctly coded items from 0 to 110 in 90 seconds. The Stroop test consists of three conditions: color naming requires naming the colors of blocks presented horizontally. Word reading requires reading color words printed in black ink, and the interference condition, naming the ink color of color words while inhibiting word reading. The number of correct responses in 45 seconds determines the score in each condition. The three conditions permit separation of the probable contribution of psychomotor speed from the executive function of response inhibition. Furthermore, the behavioral scale included in the UHDRS was also administered. This scale explores mood, apathy, psychosis, and obsessive‐compulsive symptoms. Controls were selected from a database of volunteers in our Institute. Demographic and clinical characteristics of the sample are shown in Table S1. Our local ethics committee approved the study according to the Declaration of Helsinki, and written informed consent was obtained from all the participants.
All patients were free of medication when assessed at baseline. Four of ten patients were assessed after 3 months of pridopidine treatment (90 mg/d). Treatment was administered within the context of the ACR16CU014/NeuroSearch study, approved by local ethics committee.43
2.2. MRI acquisition
All participants were examined using a 3T Allegra scanner with a standard‐quadrature coil (Siemens Medical Solutions). We acquired a gradient echo planar imaging (EPI) sequence with 38 axial slices (repetition time [TR]=65, echo time [TE]=30, field of view [FoV]=192 mm2, voxel size=2.5 mm3) during two resting sessions of 200 volumes. Participants were instructed to visually fix on the cross and not to fall asleep. Additional high‐resolution T1‐weighted images were obtained by a modified driven‐equilibrium Fourier transform (MDEFT) sequence (TR=7.92 ms, TE=2.4 ms, FoV=256 mm2, voxel size=1 mm3).
2.3. MRI data processing
2.3.1. fMRI preprocessing
The preprocessing pipeline was performed using a combination of FMRIB Software Library tools (FSL 4.1.9, www.fmrib.ox.ac.uk/fsl/) and Physiologic Estimation by Temporal Independent Component Analysis (PESTICA) software. Data were initially motion‐corrected using the FSL‐MCFLIRT tool. Physiological respiration and cardiac artifacts were removed by means of PESTICA, a data‐driven estimator of cardiac and respiratory effects that uses the Infomax algorithm with enforced temporal independence (www.nitrc.org/plugins/mwiki/index.php/pestica:MainPage). This approach identifies four cardiac and two respiratory time‐series regressors, based on spatial weighting maps of cardiac and respiratory effects and manual selection of the artifact temporal power‐spectrum band. A detailed description can be found in de Pasquale et al.21
2.3.2. Computation of functional connectivity maps
The adopted measure of functional connectivity is based on the Pearson correlation coefficient computed among BOLD time series of brain voxels. To reduce the computational burden, data were interpolated to a final voxel size of 4×4×4 mm using a second‐order nearest‐neighbor interpolation (FSL interpolation tool). We standardized brains in Montreal Neurological Institute (MNI) space and selected a common brain space including only voxels present in all the subjects of a group. The brains in MNI space consisted of 25 806 common voxels for the control group and 25 921 common voxels for the patient's sample. To obtain the DMN and SMN connectivity maps, we used a seed‐based approach by adopting the PCC and SMA as seeds corresponding to the MNI coordinates reported previously.21 To obtain a more robust estimate, we computed the average BOLD time course within a cube of 3 voxels centered on each seed, which we correlated with the rest of the brain. Correlation matrices of the same patient were averaged before any group‐level analyses. After the PESTICA step, no further regression to remove a global mean signal was performed as the fluctuations typically associated with nuisance variables (eg, respiration) were already accounted for. Additionally, it has been argued that global signal regression could have controversial effects on correlation values.46 Each averaged cross‐correlation matrix of every patient was Fisher‐transformed to obtain normally distributed values. From DMN and SMN connectivity maps, a statistical test was performed to identify significant connections with the seed47 We adopted false discovery rate (FDR) correction48 at the significance level of P=.05 to account for multiple comparisons.
For the computation of average values within different RSNs and graph theory‐based measures, we used a set of nodes within six RSNs reported in the literature.22 The complete list of nodes and their labeling is reported in Table S2. On this set of nodes, we determined node centrality, as measured by the degree (number of incident links of each node)17
2.3.3. Whole‐brain analysis of gray matter volume
The structural stability of the main nodes of both networks was evaluated using structural data (T1 images) by an optimized voxel‐based morphometry (VBM) protocol49 carried out with FSL tools50 First, structural images were brain‐extracted and GM‐segmented before being registered to the MNI 152 standard space using a nonlinear registration, yielding a study‐specific GM template. Second, all native GM images were nonlinearly registered to this template and modulated to correct for local expansion/contraction due to the nonlinear component of spatial transformation, and then smoothed with an isotropic Gaussian kernel of 4 mm. Finally, a voxel‐wise general linear model was applied using a permutation‐based nonparametric test with a threshold for the cluster‐based corrections (t<2.3), correcting for multiple comparisons across space (P <.05). The result is a statistical parametric map highlighting regions in which GM concentrations differ significantly between groups.
2.3.4. FreeSurfer analysis
To perform a correlation analysis between functional connectivity and GM volume, we estimated the absolute GM volume of the considered nodes using FreeSurfer (X86_64 RedHat running in a stable Linux GNU version, v5.2. [http://ftp.nmr.mgh.harvard.edu/]) and the Desikan atlas.51
3. Results
Demographic data are reported in Table S1. There were no significant differences in age or gender distribution between groups (P<.05).
3.1. Resting‐state networks
3.1.1. Gray matter analysis
The analysis of GM showed structural impairment in all cortical and subcortical areas involved in SMN, with preservation of SMA (Figure 1, yellow arrows). There was global structural preservation of all cortical areas of the DMN (Figure 1, red arrows). The most significant GM loss occurred in the caudate, putamen, globus pallidus, and thalamus bilaterally; the primary somatomotor and somatosensory areas bilaterally (pre‐ and postcentral gyri, supramarginal gyrus), the left precuneus, and right lingual gyrus.
Figure 1.
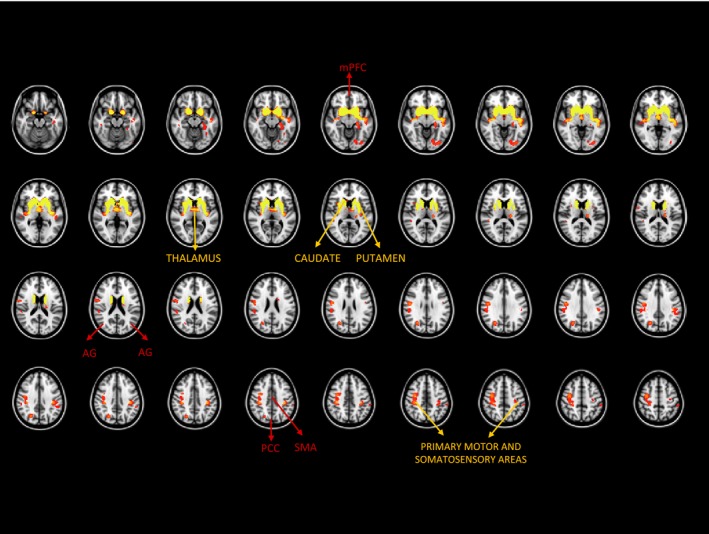
Significant GM volume loss in HD patients (FWE‐corrected, P<.05). The main components of the somatomotor and the default‐mode networks are noted. AG, angular gyrus; FWE, family‐wise error; mPFC, medial prefrontal cortex; PCC, posterior cingulate cortex; SMA, supplementary motor area
3.1.2. Somatomotor network functional topography
SMN functional connectivity is impaired in HD patients compared with controls. Figure 2 shows the spatial topography of the SMN obtained when the seed is the SMA. The SMA is connected with the pre‐SMA and primary somatomotor and somatosensory areas in controls, whereas in patients, there is an aberrant spread of significant connections between the SMA and premotor, medial prefrontal, anterior cingulate, and primary somatomotor and somatosensory areas. We computed the percentage overlap between patients’ and controls’ connectivity maps and percentage difference in overall connectivity between groups. After calculating the total number of significant connections with SMA as seed (P<.05, FDR‐corrected; Figure 2), obtaining 5789 connections for the patients and 827 connections for controls; we computed a mask of the common connections across patients and controls, namely 599 common connections, and normalized the number of common connections by the total number of connections observed in controls (599/827=0.72). Accordingly, there was an overlap of 72% between patients and controls SMN maps. However, those profiles were not correlated (r=.26; P=.2), meaning that although we observed a good overlap between networks across the two groups (72%), the large amount of additional (non in common) connections found in patients leads to a poor similarity between the network topographies. Finally, we computed the percentage difference between the total amount of significant connections between groups, obtaining an increase in the number of connections by 85% in patients (ie, (5789‐827)/5789=0.85).
Figure 2.
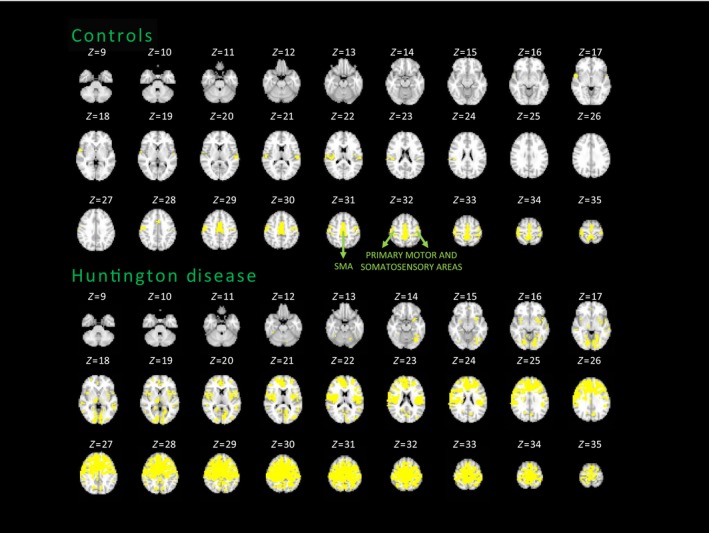
Average somatomotor network maps of the HD and control groups. FDR‐corrected, P<.05. SMA, supplementary motor area
Four patients were also assessed after 3 months of pridopidine treatment. Figure 3 shows individual maps of functional connectivity in the SMN, with the SMA as seed, in patients before and after treatment. Percentage change in the amount of connectivity was calculated as the difference between number of connections at time 1 and time 2 normalized by the same quantity at time 1, that is, [(#conn at t2 ‐ #conn at t1)/(#conn at t1)] (Table S3). We also calculated the intensity of significant connections at time 1 and time 2 and its percentage change in time [(intensity connections at t2 ‐ intensity of connections at t1)/(intensity of connections at t1)]. We observed a reduction in the intensity of significant connections in all patients after pridopidine treatment. In two patients (Pat01 and Pat03), we also observed a reduction of 64% and 74% in number of connections of the SMN from baseline to follow‐up, consistent with the decrease in intensity (1%‐15%), that is, reduced aberrant spread of connectivity leading to a SMN topography more similar to controls. The remaining patients (Pat02 and Pat04) showed an increase in number of connections after treatment (55% and 177%), corresponding to a decrease in intensity of significant connections by 3% and 11%, respectively.
Figure 3.
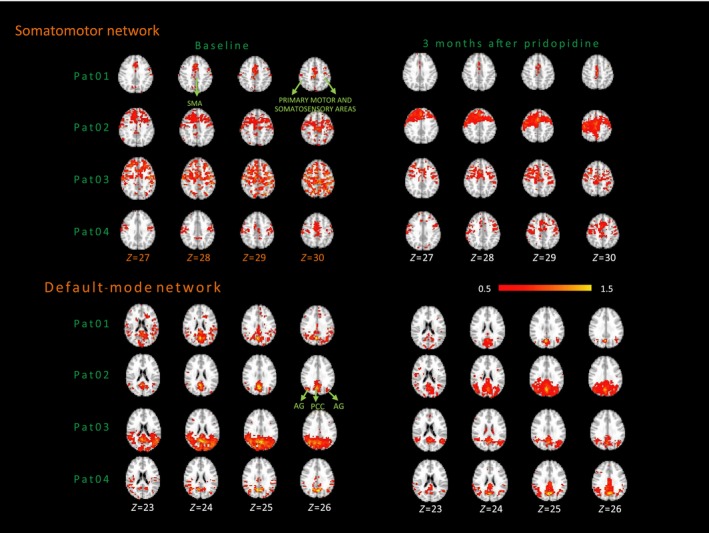
Individual SMN and DMN maps of patients before and after pridopidine treatment. The color bar indicates the Z values (mean+2SD). AG, angular gyrus; PCC, posterior cingulate cortex; SMA, supplementary motor area
3.1.3. Default‐mode network functional topography
Figure 4 shows spatial topography of the DMN obtained using PCC as the seed in both groups (P<.05, FDR‐corrected). Controls show PCC connections with all nodes that compose the DMN, that is, the vmPFC and angular gyrus bilaterally. In patients, PCC connectivity over‐recruits extensive parieto‐occipital regions. As described for the SMN, here we obtained DMN maps overlapped at 67% between patients and controls (2883 and 974 significant connections for patients and controls, respectively, with 651 common connections, thus 651/974=0.67), but both profiles were not correlated (r=.3; P=.3), showing an increase in number of connections by 66% in patients ((2883‐974)/2883=0.66).
Figure 4.
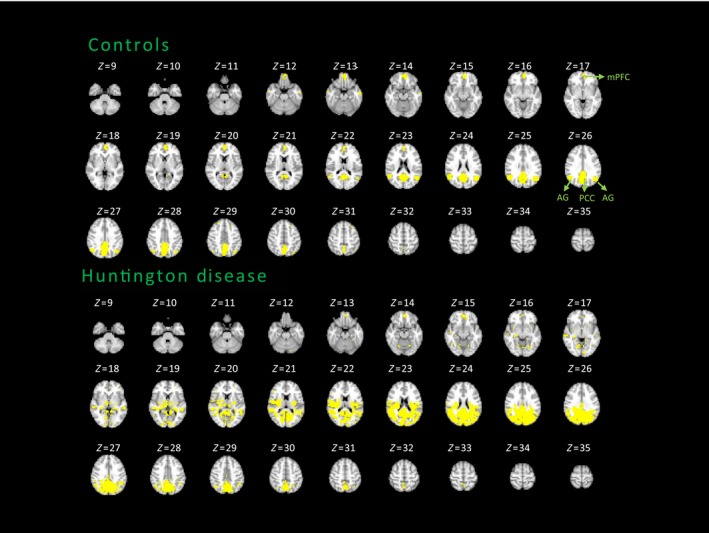
Average DMN maps of the HD and control groups. FDR‐corrected, P<.05. AG, angular gyrus; mPFC, medial prefrontal cortex; PCC, posterior cingulate cortex; SMA, supplementary motor area
Further, Figure 3 displays individual functional DMN connectivity maps, with the PCC as the seed, for patients who received pridopidine 90 mg/d at baseline and after 3 months of treatment. Table S3 shows reduction in the intensity of connections among three of the four patients (ranging from −2% to −15%), while in one patient there was a slight increase (8%). Pat01 and Pat03 decreased the number of connections after 3 months of pridopidine treatment (ranging from −46% to −78%), whereas in Pat02 and Pat04, the number of connections increased (14% and 63%). The increased connectivity observed in Pat02 might be explained by an increase in the coupling intensity (approximately 8%) between nodes that compose the network. The other three patients had a reduction in the intensity of connections, with two showing decreases in the noisy connections at baseline (Pat01 and Pat03), while the third (Pat04) showed a strengthened number of connections. All showed a topographically better‐organized network at time 2 (as shown in Figure 3), more closely resembling the maps of controls. Thus, in the DMN we also observed a general trend of recovery of functional connectivity, in terms of network focusing, which may be attributed to pridopidine.
3.2. Resting‐state networks: functional architecture
3.2.1. Nodal degree of connectivity
To test whether the centrality of a hub influences its preservation during the neurodegenerative process, we adopted a graph measure of centrality: the nodal degree. We evaluated the mean degree obtained, for every node, by averaging its degree of connectivity across the whole brain. We defined nodes showing a high degree as those whose degree exceeded the threshold [mean+two standard deviations (2SD) of estimated degree values] (dashed line in Figure 5A), which were PCC for the DMN, and the SMA and primary motor and somatosensory areas for the SMN (bars in Figure 5A).
Figure 5.
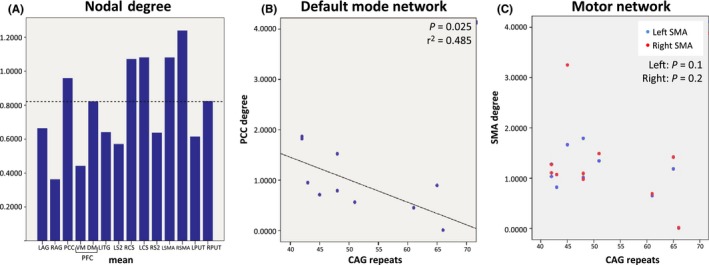
A) Bars indicate the mean degree of connectivity of each node in patients (σ=2 mm). The dashed line represents the mean+2SD. B) PCC correlation with the number of CAG repeats. C) Right and left SMA correlations with the CAG repeat length . LAG, leU angular gyrus; LCS, leU central sulcus; LDIFG, leU dorsal inferior frontal gyrus; LITG, leU inferior temporal gyrus; LS2, leU secondary somatosensory area; LPUT, leU putamen; LSMA, leU supplementary motor area; DMPFC, dorsal‐medial prefrontal cortex; PCC, posterior cingulate; RAG, right angular gyrus; RCS, right central sulcus; RS2, right secondary somatosensory area; RPUT, right putamen; RSMA, right supplementary motor area; VMPFC, ventral‐medial prefrontal cortex
3.2.2. Correlation between node centrality and clinical and genetic data
The number of CAG repeats significantly correlates with degree of connectivity in PCC (P=.02; r=−.696; Figure 5B), the main hub of the DMN. For SMA, the correlation was not significant, but a negative trend was observed (left SMA: P=.1; right SMA: P=.2; Figure 5C). No other nodes showed any significant correlation with the number of CAG repeats, suggesting that node centrality in the PCC and SMA is inversely related to number of CAG repeats.
Among the remaining DMN nodes, the vmPFC degree correlated negatively with TMS (r=−.878; P=.001), and positively with the symbol digit score (r=.715; P=.04) and TFC (r=.618; P=.05) (Fig. S1A). In the SMN, however, the right putamen degree correlated positively with the Stroop word test (r=.708; P=.049), Stroop color test (r=.788; P=.02) and negatively with obsessive symptoms (r=−.805; P=.005) (Fig. S1B).
3.2.3. Relationship between functional connectivity and local gray matter volume
We observed a significant correlation between the SMA connectivity and the volume of the superior frontal gyri, where this area is located (r=.959; P=.04). No other nodes showed any significant correlation with GM volume.
4. Discussion
The aim of our study was to investigate changes in fMRI of the SMN and DMN in HD patients and their correlation with the genetic burden, clinical scores, and GM loss.
Our findings show the following: (i) increased functional connectivity in both the SMN and DMN in HD patients that preceded the GM volume loss in the DMN; (ii) the centrality value (degree) of the PCC and SMA showed an inverse relationship with number of CAG repeats; and the centrality value of the vmPFC and right putamen correlated with patients’ clinical and genetic data.
Structural data showed HD patients have increased GM loss compared with controls in the majority of nodes of the SMN network, apart from the SMA. Areas of significant GM loss were the caudate, putamen, globus pallidus, thalamus, and primary somatomotor and somatosensory areas, consistent with previous published studies.6, 7, 8, 11, 12 Interestingly, SMA volume is preserved in our sample and correlates to its increased functional connectivity. We speculate it might be linked to the central role of SMA as a hub, which allows the SMN to integrate with other functional circuits. These results seem to suggest a pattern of damage from local to more central nodes.
The functional connectivity of the SMN showed HD patients having a 85% increase in functional connectivity compared with controls. The degree of functional connectivity in the right putamen, a critical SMN node affected early in HD, correlated with cognitive and behavioral measures in our cohort. Interestingly, the higher the connectivity was in the right putamen, the better the cognitive performance and less severe the obsessive‐compulsive behavior, a frequently occurring psychiatric manifestation in HD52 Together, these results suggest increased functional connectivity in the SMN in HD patients could represent a compensatory mechanism to maintain motor and cognitive performance.36 Consistent with our results, previous studies showed increased functional connectivity in areas related to motor function in HD, which was correlated with motor and cognitive functioning.35, 36 Increased connectivity in the putamen, together with other striatal‐prefrontal structures, has been related to improved performance in a Stroop task in different cohorts (ie, Parkinson disease patients, cocaine‐dependent subjects, and middle‐aged healthy subjects).53, 54, 55 Thus, stronger putaminal connectivity reflects compensatory strategies to meet task demands and improve performance levels. Furthermore, the putamen underlies obsessive‐compulsive traits.56, 57, 58 Obsessive‐compulsive disorder patients have reduced functional connectivity between the putamen and prefrontal areas56, 58 and the volume and shape of the putamen correlate with obsessive‐compulsive symptoms in healthy subjects.57 Previous task‐related fMRI studies have also shown enhanced activation in HD patients as a compensatory process due to striatal or cortical degeneration.38, 39, 40 Thus, in HD patients, the functional connectivity will be increased by the regulatory role of dopamine that the striatal impairment exerts in this circuit; and by the cumulative effect of diffuse cortical structural alterations, altogether causing the characteristic deterioration in motor and cognitive functions.24
Another important aim of our study was to analyze how short‐term administration of pridopidine at 90 mg/d could modify brain functional connectivity in a small subgroup of patients, as this dose has been shown to improve TMS in early and moderate HD patients.43 Dopaminergic and glutamatergic signaling pathways act synergistically to enhance the sensitivity of striatal neurons to mutated HTT toxicity and may represent crucial therapeutic targets for HD treatment. Our results show pridopidine modulated the functional connectivity of both SMN and DMN, namely the intensity of significant connections, in the majority of patients. Regarding the SMN connectivity, the observed decrease in connection intensity and changes in the number of connections suggest the drug induces a network focusing around the typical topography observed in controls, which may improve network efficiency. Recent evidence suggesting that pridopidine exerts protective properties by binding sigma‐1 receptor, a critical receptor in signal transmission, supports the hypothesis that pridopidine may reinforce synaptic connections. Theoretically, if confirmed in a larger population, our data may support the hypothesis that pridopidine has potential antidysfunctional and neuroprotective effects.41, 42 We hoped that a further drug trial with new pridopidine dosages will document positive long‐term effects.
Furthermore, patients have increased functional connectivity of the DMN in parieto‐occipital areas, as compared with controls, but show no structural changes in GM. The degree of connectivity of its hub, the PCC, correlated with the number of CAG repeats suggests that the genetic burden might influence functional architecture of the DMN. Also, increased degree of connectivity of the vmPFC, a salient node in the DMN, significantly correlated with better motor and cognitive functioning.
The DMN is active when individuals are engaged offline, internally focused tasks.59 There is evidence that despite progressive GM volume loss, clinical measures do not necessarily deteriorate in HD gene‐mutation carriers. This fact supports that neural activity reorganizes to maintain normal behavior despite the loss of brain volume, as a “neural compensation”.36 Our results support this hypothesis by showing a relationship between vmPFC connectivity and specific cognitive functions in HD. Previous studies reported contradictory results regarding DMN connectivity in HD patients. These differences might be due to different methodologies used, as well as clinical differences (our subjects have more CAG repeats and are younger). Dumas et al.33 used networks of interest obtained by an Independent Component Analysis as reference for the connectivity estimation. In our work, we adopted a seed‐based approach, thus using a region as reference. This may lead to differences in the change in connectivity. In particular, our results show connectivity changes with respect to a given region (PCC), while other results assume an identical signal belonging to each identified network between regions. In addition, the regression steps that remove the ventricles and global signal contributions are known to induce negative connectivity values and thus affect the number of significant connections.50 Notably, there are other studies supporting the finding in HD patients of increased connectivity, which has been discussed as a compensatory process34, 35, 36 or as a generalized spreading of activity due to reduced specialization of function.37
Our study is limited by the overall small population and small sample of patients treated with pridopidine. Although the purpose of this study was only to investigate fMRI as a valuable technique for monitoring pharmacological effects in HD patients, preliminary data open the possibility of extending the sample in a longitudinal, multicentric, clinical, imaging study. If treatment goal is to improve cerebral function, both structural and functional imaging may predict whether an individual will respond to a particular treatment. However, therapeutic conclusions cannot be extracted.
In conclusion, our preliminary data describe, to our knowledge for the first time, an abnormal functional connectivity of the SMN and DMN in HD, which precedes brain atrophy in the DMN, is sensitive to changes driven by a drug, and may represent a biomarker of HD dysfunction. These results may have the potential, once confirmed in larger data sets, to be transferred into clinical practice to monitor response to experimental therapies.
Disclosures
The authors report no conflicts of interest.
Supporting information
Acknowledgements
This work was supported by the European Commission, through a Marie Curie Fellowship for career development to C.S.‐C. (FP7‐PEOPLE‐2011‐IEF), by the Catalan Government, through a Beatriu Pinós fellowship awarded to C.S.‐C. (2013BP‐B 00269) (COFUND 7th Marie Curie Actions), and by the Italian Ministry for Health (IMA) GRANT 204/GR‐2009‐1606835 (to M.P). This study was made possible thanks to the constant support of LIRH Foundation (Lega Italiana Ricerca Huntington e malattie correlate onlus, www.lirh.it) and to the collaboration of patients and families. We thank NeuroSearch for drug supplies. Editorial support was provided by Arianna Grove, PhD (Chameleon Communications International with funding from Teva Pharmaceutical Industries).
Sánchez‐Castañeda C, de Pasquale F, Caravasso CF, et al. Resting‐state connectivity and modulated somatomotor and default‐mode networks in Huntington disease. CNS Neurosci Ther. 2017;23:488–497. 10.1111/cns.12701
Funding information
This work was supported by the European Commission, through a Marie Curie Fellowship for career development to C.S.‐C. (FP7‐PEOPLE‐2011‐IEF), by the Catalan Government, through a Beatriu Pinós fellowship awarded to C.S.‐C. (2013BP‐B 00269) (COFUND 7th Marie Curie Actions), and by the Italian Ministry for Health (IMA) GRANT 204/GR‐2009‐1606835 (to M.P). This study was made possible thanks to the constant support of LIRH Foundation (Lega Italiana Ricerca Huntington e malattie correlate onlus, www.lirh.it) and to the collaboration of patients and families. We thank NeuroSearch for drug supplies. Editorial support was provided by Arianna Grove, PhD (Chameleon Communications International with funding from Teva Pharmaceutical Industries).
Contributor Information
Umberto Sabatini, Email: sabatini@unicz.it.
Ferdinando Squitieri, Email: f.squitieri@css-mendel.it.
References
- 1. Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398‐403. [DOI] [PubMed] [Google Scholar]
- 2. Ciarmiello A, Cannella M, Lastoria S, et al. Brain white‐matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med. 2006;47:215‐222. [PubMed] [Google Scholar]
- 3. Aylward EH, Nopoulos PC, Ross CA, et al. Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry. 2011;82:405‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aylward EH, Liu D, Nopoulos PC, et al. Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiatry. 2012;71:822‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745‐747. [DOI] [PubMed] [Google Scholar]
- 6. Rosas HD, Lee SY, Bender AC, et al. Altered white matter microstructure in the corpus callosum in Huntington's disease: implications for cortical “disconnection”. NeuroImage. 2010;49:2995‐3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the Predict‐HD study. Arch Neurol. 2006;63:883‐890. [DOI] [PubMed] [Google Scholar]
- 8. Paulsen JS, Nopoulos PC, Aylward E, et al. Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull. 2010;82:201‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jurgens CK, van de Wiel L, van Es AC, et al. Basal ganglia volume and clinical correlates in ‘preclinical’ Huntington's disease. J Neurol. 2008;255:1785‐1791. [DOI] [PubMed] [Google Scholar]
- 10. Klöppel S, Draganski B, Golding CV, et al. White matter connections reflect changes in voluntary‐guided saccades in pre‐symptomatic Huntington's disease. Brain. 2008;131:196‐204. [DOI] [PubMed] [Google Scholar]
- 11. Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol. 2011;10:31‐42. [DOI] [PubMed] [Google Scholar]
- 12. Tabrizi SJ, Reilmann R, Roos RA, et al. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK‐HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11:42‐53. [DOI] [PubMed] [Google Scholar]
- 13. Sánchez‐Castañeda C, Cherubini A, Elifani F, et al. Seeking Huntington disease biomarkers by multimodal, cross‐sectional basal ganglia imaging. Hum Brain Mapp. 2013;34:1625‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Phillips O, Sanchez‐Castaneda C, Elifani F, et al. Tractography of the corpus callosum in Huntington's disease. PLoS One. 2013;8:e73280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Phillips O, Squitieri F, Sanchez‐Castaneda C, et al. Deep white matter in Huntington's disease. PLoS One. 2014;9:e109676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou J. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73:1216‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van den Heuvel MP, Sporns O. Network hubs in the human brain. Trends Cogn Sci. 2013;17:683‐696. [DOI] [PubMed] [Google Scholar]
- 18. Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009;29:1860‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Pasquale F, Della Penna S, Snyder AZ, et al. Temporal dynamics of spontaneous MEG activity in brain networks. Proc Natl Acad Sci USA. 2010;107:6040‐6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Pasquale F, Della Penna S, Snyder AZ, et al. A cortical core for dynamic integration of functional networks in the resting human brain. Neuron. 2012;74:753‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Pasquale F, Sabatini U, Della Penna S, et al. The connectivity of functional cores reveals different degrees of segregation and integration in the brain at rest. NeuroImage. 2013;69:51‐61. [DOI] [PubMed] [Google Scholar]
- 22. de Pasquale F, Della Penna S, Sporns O, Romani GL, Corbetta M. A dynamic core network and global efficiency in the resting human brain. Cereb Cortex. 2016;26:4015‐4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andrews‐Hanna JR, Reidler JS, Huang C, Buckner RL. Evidence for the default network's role in spontaneous cognition. J Neurophysiol. 2010;104:322‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quarantelli M, Salvatore E, Giorgio SM, et al. Default‐mode network changes in Huntington's disease: an integrated MRI study of functional connectivity and morphometry. PLoS One. 2013;8:e72159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di X, Biswal BB. Identifying the default mode network structure using dynamic causal modeling on resting‐state functional magnetic resonance imaging. NeuroImage. 2014;86:53‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fox KC, Spreng RN, Ellamil M, Andrews‐Hanna JR, Christoff K. The wandering brain: meta‐analysis of functional neuroimaging studies of mind‐wandering and related spontaneous thought processes. NeuroImage. 2015;111:611‐621. [DOI] [PubMed] [Google Scholar]
- 27. Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA. 2003;100:253‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harrington DL, Rubinov M, Durgerian S, et al. Network topology and functional connectivity disturbances precede the onset of Huntington's disease. Brain. 2015;38:2332‐2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koenig KA, Lowe MJ, Harrington DL, et al. Functional connectivity of primary motor cortex is dependent on genetic burden in prodromal Huntington disease. Brain Connect. 2014;4:535‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poudel GR, Egan GF, Churchyard A, Chua P, Stout JC, Georgiou‐Karistianis N. Abnormal synchrony of resting state networks in premanifest and symptomatic Huntington disease: the IMAGE‐HD study. J Psychiatry Neurosci. 2014;39:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wolf RC, Sambataro F, Vasic N, et al. Default‐mode network changes in preclinical Huntington's disease. Exp Neurol. 2012;237:191‐198. [DOI] [PubMed] [Google Scholar]
- 32. Unschuld PG, Joel SE, Liu X, et al. Impaired cortico‐striatal functional connectivity in prodromal Huntington's Disease. Neurosci Lett. 2012;514:204‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dumas EM, van den Bogaard SJ, Hart EP, et al. Reduced functional brain connectivity prior to and after disease onset in Huntington's disease. Neuroimage Clin. 2013;2:377‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paulsen JS. Functional imaging in Huntington's disease. Exp Neurol. 2009;216:272‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Werner CJ, Dogan I, Saß C, et al. Altered resting‐state connectivity in Huntington's disease. Hum Brain Mapp. 2014;35:2582‐2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolf RC, Sambataro F, Vasic N, et al. Abnormal resting‐state connectivity of motor and cognitive networks in early manifest Huntington's disease. Psychol Med. 2014;44:3341‐3356. [DOI] [PubMed] [Google Scholar]
- 37. Rajah MN, D'Esposito M. Region‐specific changes in prefrontal function with age: A review of PET and fMRI studies on working and episodic memory. Brain. 2005;128:1964‐1983. [DOI] [PubMed] [Google Scholar]
- 38. Odish OF, van den Berg‐Huysmans AA, van den Bogaard SJ, et al. Longitudinal resting state fMRI analysis in healthy controls and premanifest Huntington's disease gene carriers: a three‐year follow‐up study. Hum Brain Mapp. 2015;36:110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seibert TM, Majid DS, Aron AR, Corey‐Bloom J, Brewer JB. Stability of resting fMRI interregional correlations analyzed in subject‐native space: a one‐year longitudinal study in healthy adults and premanifest Huntington's disease. NeuroImage. 2012;59:2452‐2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gardoni F, Bellone C. Modulation of the glutamatergic transmission by dopamine: a focus on Parkinson, Huntington and addiction diseases. Front Cell Neurosci. 2015;9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Squitieri F, de Yebenes JG. Profile of pridopidine and its potential in the treatment of Huntington disease: the evidence to date. Drug Des Devel Ther. 2015;9:5827‐5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Squitieri F, Di Pardo A, Favellato M, Amico E, Maglione V, Frati L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J Cell Mol Med. 2015;19:2540‐2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Yebenes JG, Landwehrmeyer B, Squitieri F, et al. Pridopidine for the treatment of motor function in patients with Huntington's disease (MermaiHD): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2011;10:1049‐1057. [DOI] [PubMed] [Google Scholar]
- 44. Squitieri F, Landwehrmeyer B, Reilmann R, et al. One‐year safety and tolerability profile of pridopidine in patients with Huntington disease. Neurology. 2013;80:1086‐1094. [DOI] [PubMed] [Google Scholar]
- 45. Huntington Study Group . The Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord. 1996;11:136‐142. [DOI] [PubMed] [Google Scholar]
- 46. Murphy K, Birn RM, Handwerker DA, Jones TB, Bandettini PA. The impact of global signal regression on resting state correlations: are anti‐correlated networks introduced? NeuroImage. 2009;44:893‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaufman B, Rousseeuw PJ. Finding Groups in Data: An Introduction to Cluster Analysis. New York, NY: Wiley; 1990. [Google Scholar]
- 48. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289‐300. [Google Scholar]
- 49. Good CD. A voxel‐based morphometric study of ageing in 465 normal adult human brains. NeuroImage. 2001;14:21‐36. [DOI] [PubMed] [Google Scholar]
- 50. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage. 2004;23(suppl 1):S208‐S219. [DOI] [PubMed] [Google Scholar]
- 51. Desikan RS. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage. 2006;31:968‐980. [DOI] [PubMed] [Google Scholar]
- 52. van Duijn E, Craufurd D, Hubers AA, et al. Neuropsychiatric symptoms in a European Huntington's disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry. 2014;85:1411‐1418. [DOI] [PubMed] [Google Scholar]
- 53. Müller‐Oehring EM, Sullivan EV, Pfefferbaum A, et al. Task‐rest modulation of basal ganglia connectivity in mild to moderate Parkinson's disease. Brain Imaging Behav. 2015;9:619‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mitchell MR, Balodis IM, Devito EE, et al. A preliminary investigation of Stroop‐related intrinsic connectivity in cocaine dependence: associations with treatment outcomes. Am J Drug Alcohol Abuse. 2013;39:392‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zysset S, Schroeter ML, Neumann J, et al. Stroop interference, hemodynamic response and aging: an event‐related fMRI study. Neurobiol Aging. 2007;28:937‐946. [DOI] [PubMed] [Google Scholar]
- 56. Bernstein GA, Mueller BA, Schreiner MW, et al. Abnormal striatal resting‐state functional connectivity in adolescents with obsessive‐compulsive disorder. Psychiatry Res. 2016;247:49‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kubota Y, Sato W, Kochiyama T, et al. Putamen volume correlates with obsessive compulsive characteristics in healthy population. Psychiatry Res. 2016;249:97‐104. [DOI] [PubMed] [Google Scholar]
- 58. Vaghi MM, Vértes PE, Kitzbichler MG, et al. Specific frontostriatal circuits for impaired cognitive flexibility and goal‐directed planning in obsessive‐compulsive disorder: evidence from resting‐state functional connectivity. Biol Psychiatry. 2016;S0006–3223:32670‐32671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buckner RL, Andrews‐Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1‐38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials