

Summary
Aims
It has been demonstrated that neuroinflammation is associated with cardiovascular dysfunction. The phosphoinositide‐3 kinase (PI3K) signaling in the rostral ventrolateral medulla (RVLM), a key region for sympathetic outflow, is upregulated and contributes to increased blood pressure (BP) and sympathetic outflow in hypertension. This study was designed to determine the role of the PI3K signaling in neuroinflammation in the RVLM of hypertension.
Methods
The normotensive WKY rats were performed by intracisternal infusion of lipopolysaccharide (LPS) or angiotensin II (Ang II) for inducing neuroinflammation. Elisa was used to determine the level of proinflammatory cytokines. Western blot was employed to detect the protein expression of PI3K signaling pathway. Gene silencing of PI3K p110δ subunit and overexpression of angiotensin‐converting enzyme 2 (ACE2) were realized by injecting related lentivirus into the RVLM.
Results
In the spontaneously hypertensive rats (SHR), the PI3K signaling in the RVLM was upregulated compared with WKY, gene silencing of PI3K in the RVLM significantly reduced BP and renal sympathetic nerve activity (RSNA), but also decreased the levels of proinflammatory cytokines. In the WKY rats, central infusion of LPS and Ang II significantly elevated BP and RSNA, but also increased the levels of proinflammatory cytokines and PI3K signaling activation in the RVLM. These changes in the Ang II‐induced hypertension were effectively prevented by gene silencing of PI3K in the RVLM. Furthermore, overexpression of ACE2 in the RVLM significantly attenuated high BP and neuroinflammation, as well as decreased the activation of PI3K signaling in hypertensive rats.
Conclusion
This study suggests that the PI3K signaling in the RVLM is involved in neuroinflammation in hypertension and plays an important role in the renin–angiotensin system‐mediated changes in neuroinflammation in the RVLM.
Keywords: ACE2, Hypertension, neuroinflammation, PI3K signaling, RVLM
1. Introduction
It has been documented that inflammation participates in the development of neurogenic hypertension.1 The lipopolysaccharide (LPS)‐induced systemic inflammation leads to a significant increase in blood pressure (BP).2 In the RAG‐1‐/‐ mice lacking T and B cells, hypertension and abnormal vascular function induced by angiotensin II (Ang II) infusion or deoxycorticosterone acetate–salt are significantly attenuated.3 It is reported that low‐grade inflammation has been detected in peripheral cardiovascular system in hypertensive patients4 and animals,5 suggesting that the inflammation is involved in the processing of essential hypertension. The immune inflammatory response in the central nervous system, also called neuroinflammation, is characterized by an increase in the production of proinflammatory cytokines, which is required to activate microglia.6 It is reported that microinjections of the proinflammatory cytokine interleukin‐1β (IL‐1β) or tumor necrosis factor‐α (TNF‐α) into the cardiovascular center paraventricular nucleus (PVN) lead to an increase in BP and renal sympathetic nerve activity (RSNA).7 Preactivated microglia in the brain has prolonged the pressor response induced by intracerebroventricular injection of Ang II.8 The rostral ventrolateral medulla (RVLM) has been recognized as a pivotal region for maintaining basal BP and sympathetic tone.9 The previous evidence indicates that the systemic LPS‐induced neuroinflammation in the RVLM is involved in high BP and sympathetic hyperactivity.2 Therefore, it would be significant to investigate the role of neuroinflammation in the central regulation of cardiovascular function in hypertension, and is helpful for researching for a new strategy against hypertension.
Previous evidence suggests that the phosphoinositide‐3 kinase (PI3K) pathway in the RVLM is involved in regulating BP and sympathetic outflow.10, 11 It has been demonstrated that the PI3K signaling in the RVLM is upregulated in hypertension, and its inactivation leads to reduction in reactive oxygen species (ROS) production and cardiovascular function.10 Moreover, we recently report that gene knockdown of the PI3K subunit p110δ in the RVLM exerts the centrally antihypertensive action.12Interestingly, the PI3K signaling is suggested to be involved in several key events in the inflammatory response to damage and infection.13, 14 Therefore, the major aim of this study was to determine the role of the RVLM PI3K signaling in the neuroinflammation in hypertension.
Overactivity in renin–angiotensin system (RAS) has been shown to play an important role in the pathogenesis of hypertension.15 It is reported that Ang II stimulates the activation of resting microglia and induces the production of proinflammatory cytokines in vivo16 and in vitro.17 Recent evidence indicates that blockade of Ang II receptor (AT1R) with candesartan decreases the brain inflammatory response induced by administration of LPS.16 Therefore, it is suggested that neuroinflammation induced by Ang II acts as a molecular signal for increased sympathetic outflow in hypertension. It has been demonstrated that angiotensin‐converting enzyme 2 (ACE2) plays an important role in compensatory mechanisms to oppose the overactive RAS and catalyzes the conversion from Ang II and Ang‐(1‐9) to Ang‐(1‐7), which exerts physiological functions through combining with Mas receptor.18, 19 The cardiovascular protective effects (eg, vasodilatation and antihypertrophic cardiomyopathy) of ACE2/Ang‐(1‐7)/Mas axis are resulted from increased synthesis of nitric oxide and decreased generation of reactive oxygen species (ROS).20, 21, 22 Cardiovascular neural regulatory regions in the brain‐targeted ACE2 overexpression are reported to reduce BP and sympathetic outflow in hypertension,23 and ameliorate the baroreflex function in chronic heart failure.24 Interestingly, it is reported that Ang‐(1‐7) has a direct antiinflammatory effect on microglia in vitro.25 However, it is not clear whether the effect of RAS in the RVLM on neuroinflammation in hypertension is associated with PI3K signaling.
Hence, this study was designed to determine: (i) whether the PI3K gene silencing in the RVLM inhibits neuroinflammation in hypertensive rats; (ii) whether the PI3K silencing in the RVLM attenuates the Ang II‐induced high BP and neuroinflammation in WKY rats; and (iii) whether the PI3K signaling is involved in the antihypertensive effect of ACE2 overexpression by reducing the level of neuroinflammation in the RVLM in hypertensive rats.
2. Methods
2.1. Animals
Sixteen weeks old male SHRs and WKY rats (260‐300 g) were used for this study and purchased from Sino‐British SIPPR/BK Laboratory Animal Ltd (Shanghai, China). All procedures were obtained approval of the Institutional Animal Care and Use Committee of Second Military Medical University, and all operations in this study were conducted according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
2.2. Construction and production of the human ACE2 and PI3K shRNA lentiviral vector
The PI3K shRNA and human (h) ACE2 lentiviral particles were constructed and produced by Shanghai Innovation Biotech (Shanghai, China), as described in our previous studies.12, 23 In brief, the specific shRNA fragment of rat PI3K p110δ (target sequence: GTAAACGACTTCCGCACTA) was integrated into a pLenR‐GPH vector, and the negative fragment (control sequence: TTCTCCGAACGTGTCACGT) was used for the control shRNA. The hACE2 cDNA was amplified through PCR with the use of specific primer (sense 5′‐ATGCTGCGCGCCGCACTCAGCAC‐3′ and antisense 5′‐TTACGAG TTCTTCTGTGGCACTT‐3′). Amplified hACE2 cDNA was integrated into pLenO‐DCE lentiviral cloning vector, and the pLenO‐DCE vector carried a green fluorescent protein (GFP) reporter gene, which acts as control. These two kinds of lentiviral particles were harvested and tested through transfecting 293T cell, and the infectious titer was 5×109 transducing units/mL.
2.3. Injections of lentiviral particles into the RVLM
Procedures for microinjection of the lentivirus into the bilateral RVLM were based on the previous study.23 In brief, rats were fixed in a stereotaxic instrument (Shanghai Alcott Biotech) after anesthetized by inhaling 3% isofluorane. The skull was exposed, and two symmetrical small holes were drilled on the skull surface. According to the atlas of rats,26 lentiviral particles (250 nL) were slowly injected into the RVLM (2.0 mm lateral to the midline, 3.0 mm posterior to the Lamda, and 10 mm deep to the skull surface) with a 32‐gauge Hamilton syringe (5 μL) in 5 minutes. After viral injections, rats were treated with 1000 units of penicillin through muscle injection to prevent infection.
2.4. Intracisternal infusion
To induce neuroinflammation in normotensive WKY rats, intracisternal (fourth ventricle) infusion of Ang II (24 μg/d, 1 week) or LPS (6 μg/d, 2 weeks) was performed by the osmotic minipump, and the infusion doses of agents were based on previous studies.12, 27 In brief, rats were anesthetized through inhaling 3% isofluorane and placed on the stereotaxic frame. The atlantooccipital membrane was exposed, and the dura was punctured with a needle. After observation of leakage of cerebrospinal fluid (CSF) from the hole, a PE‐10 catheter was advanced for 3 mm into the cisterna magna (fourth ventricle). The infusion catheter was sealed to the dura with tissue glue, and the incision was closed with layered sutures. The outer end of the catheter was connected to an osmotic minipump (Model 1002 or 1007D, Alzet, USA) containing LPS or Ang II, which was fixed in the neck subcutaneous region.
2.5. Measurements of BP, HR, and RSNA recording
In conscious rats, the noninvasive tail‐cuff system was performed to monitor the systolic BP, diastolic BP, mean arterial pressure (MAP), and heart rate (HR), as described previously.28 Briefly, conscious rats were restricted by a recording chamber for 15 minutes before measurement of BP to make the rat acclimate the holding device, and to keep rat quiet and comfortable during the whole process. In addition, retaining the ambient temperature of 30°C in chamber with controlled warming plate under the rats is beneficial to vasodilate the tail artery. Systolic BP (SBP), diastolic BP, mean arterial pressure (MAP), and HR were simultaneously monitored by noninvasive tail‐cuff system (ALC‐NIBP, Shanghai Alcott Biotech). The values resulted from averaging of at least six consecutive measurements, and BP and HR were measured every week or every 3 days after treatments.
As described previously,29 levels of BP, HR, and baseline RSNA were also examined in the anesthetized rats after completion of intracisternal infusion. In anaesthetized rats (urethane 800 mg/kg ip and a‐chloralose 40 mg/kg ip), trachea was cannulated and the right femoral artery was catheterized for monitoring BP and HR with the PowerLab system. The left renal sympathetic nerve was isolated retroperitoneally and placed on a pair of silver recording electrodes. The RSNA signal was amplified, integrated, and recorded with the PowerLab system. Based on previous studies,29 the maximum of RSNA was measured when rat was euthanized with an overdose of pentobarbital sodium (200 mg/kg). Baseline RSNA was expressed as a percentage of maximal RSNA.
2.6. Western blot analysis
As previously described30 in brief, after the rats were euthanized with an overdose of pentobarbital sodium (200 mg/kg), the brain was removed quickly and stored in −80°C. The RVLM tissues were punched on a freezing microtome according to the rat atlas,26 lysed with lysate, and sonicated. The supernatants were collected after centrifugation. The protein concentration of supernatants was determined by BCA kit, and protein samples were denatured with loading buffer in proportion heating to 100°C for 10 minutes. Protein sample (30 μg) was used to run on a 8% or 12% SDS‐PAGE gel and transferred to PVDF membrane, and 5% milk was dissolved in Tris‐buffered saline Tween and used to block the membrane; the blocked membranes were incubated with anti‐ACE2 (no.3583‐1, Epitomics), anti‐PI3K110δ (no.3295‐1, Epitomics), anti‐phospho‐AKT (Ser473) (no.4060, CST), anti‐AKT (no.9272, CST), and anti‐GAPDH (no.sc‐47724, Santa Cruz) antibody overnight at 4°C. After three times of washes, species‐specific secondary antibody‐conjugated horseradish peroxidase was used to interact with epitope‐specific primary antibodies in room temperature for 2 hours, and then, the membranes were subjected to chemiluminescent agent for detecting the target protein binding by GeneTools software (Gene Company). GAPDH served as control to normalize the expression of target proteins.
2.7. Immunofluorescence
As described previously,23 the fluorescence staining for GFP was detected by laser confocal microscopy. In brief, after the rats were euthanized with excessive pentobarbital sodium (200 mg/kg, ip), 0.9% NaCl solution and 4% paraformaldehyde in 0.1M phosphate buffer solution (PBS) were used to perfuse the aorta through a constant flow pump. The rat brain was removed and fixed with 4% paraformaldehyde in 0.1M PBS overnight at 4°C. Sucrose solution (20%) was used to dehydrate the brain tissues until it sank to the bottom. Sections of 20 μm thickness made by a cryostat were frozen at −20°C floated in 0.01M PBS (pH 7.4); these sections were then mounted on slides after three 5‐min washes and cover‐slipped with antifade medium. In addition, the GFP fluorescence was detected by a laser confocal microscopy (Leica, TCS‐SP5).
2.8. Detection of proinflammatory cytokines in the RVLM
According to the rat atlas,26 RVLM tissues were punched, lysed, and sonicated. The supernatants were extracted after centrifugation for detecting proinflammatory cytokines, Ang II/Ang‐(1‐7), in the RVLM tissues. Elisa kits for determining the level of IL‐1β (no.F15810), IL‐6 (no.F15870), TNF‐α (no.F16960), Ang II (no.F15050), and Ang‐(1‐7) (no.F15051) were purchased from Shanghai Westang Bio‐tech Co., LTD, according to the manufacturer's instructions. In brief, the protein samples were added to coated wells for 40 minutes at 37°C. Then wells were washed 3 times, and added biotinylated antibody for 20 minutes at 37°C. Next, wells were washed 3 times again and added sequentially with horseradish peroxidase conjugated secondary antibody and substrates (TMB Solution). Finally, the reactions in wells were stopped with Stop Solution. The level of proinflammatory cytokines, Ang II and Ang‐(1‐7), and the absorbance at 450 nm with an automated microplate reader were determined and were calculated according to the standard curve.
2.9. Data analysis
Data are expressed as means±SEM. Statistical differences between the WKY rats treated with LPS or Ang II alone were analyzed by a Student's t test. Comparisons of MAP and HR obtained by tail‐cuff system in conscious rats among groups were analyzed by repeated measurement ANOVA followed by Tukey's post hoc test. Two‐way ANOVA followed by Bonferroni's post hoc test was used for the other multiple comparisons. Differences were considered to be significant for P<.05.
3. Results
3.1. Gene silencing of the PI3K signaling in the RVLM attenuated neuroinflammation and cardiovascular activity in SHR
As shown in Figure 1A, GFP immunofluorescence staining for lentivirus transfer was expressed restrictedly in the RVLM. Furthermore, transfer efficacy of lentivirus containing specific PI3K p110δ shRNA fragment injected into the RVLM was confirmed by a decrease in expression level of PI3K p110δ protein (Figure 1B). It was also observed that the expression level of the RVLM PI3K p110δ was significantly upregulated in SHR compared with WKY rats. As indicated in Figure 1B, compared with WKY, SHR showed a significant increase in the content of proinflammatory cytokines (IL‐1β: 55.9±18.2 vs 141.8±7.54 pg/mg, P<.05; IL‐6: 309±18.2 vs 598±26.0 pg/mg, P<.05; TNF‐α: 56.3±3.76 vs 98.0±15.4 pg/mg, P<.05), MAP (125±2.50 vs 183±8.05 mm Hg), HR (363±10.6 vs 441±11.4 bpm), and basal RSNA (11.3±0.514 vs 38.0±2.03% Max). Importantly, the levels of IL‐1β, IL‐6, and TNF‐α in the RVLM (Figure 1C) and cardiovascular activity under anesthesia (Table 1) and conscious state (Figure 1D) were significantly decreased in SHR 4 weeks after knockdown of PI3K p110δ in the RVLM. In additional, injection of lenti‐PI3K shRNA into the RVLM had no influence on BP and HR in WKY rats.
Figure 1.
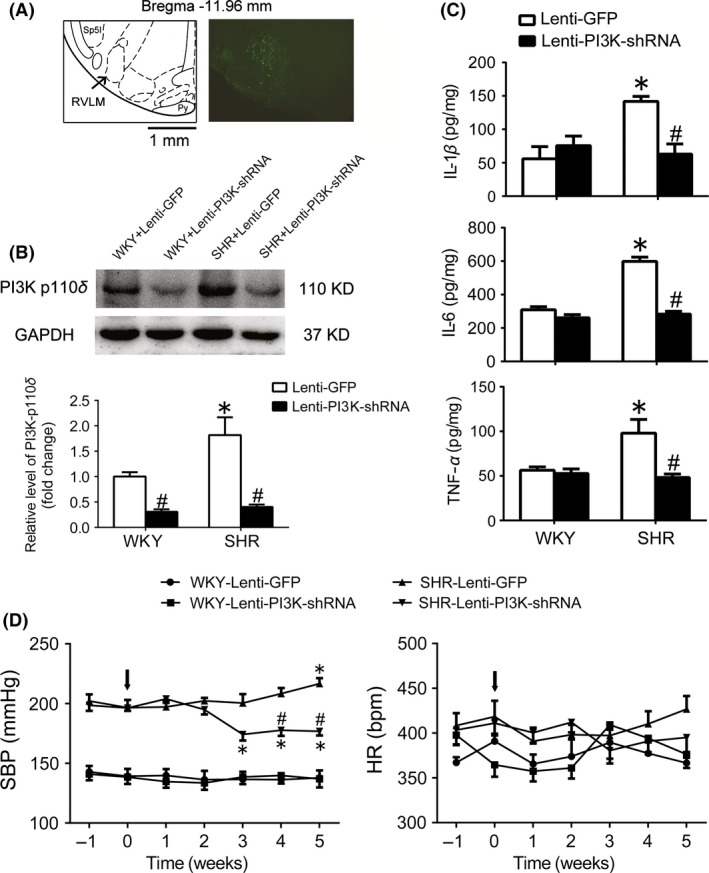
Effect of PI3K knockdown in the RVLM on the levels of neuroinflammation and cardiovascular activity in SHR. A, Location of silencing PI3K in the RVLM. According to the rat's atlas (left), green fluorescent protein (GFP) was expressed in the RVLM area. B, Representative gel bands (top) and quantification bar graph (bottom) show the protein levels of PI3K p110δ in the RVLM in WKY and SHR after treatment with lenti‐PI3K shRNA injected into the RVLM. C, The levels of IL‐1β, IL‐6, and TNF‐α in the RVLM of WKY and SHR treated with lenti‐PI3K shRNA injections. Values are expressed as mean±SEM, n=5/group. *P<.05 vs WKY; # P<.05 vs lenti‐GFP. D, Time courses of systolic blood pressure (SBP) and HR in the WKY rats and SHR treated with lenti‐GFP or lenti‐PI3K‐shRNA injected into the RVLM (arrows). Values are expressed as mean±SEM, n=5/group, *P<.05 vs 0 wk, #P<.05 vs lenti‐GFP
Table 1.
Levels of BP, HR, and RSNA in the anesthetized WKY and SHR rats 4 wk after PI3K shRNA injected into the RVLM
| WKY | SHR | |||
|---|---|---|---|---|
| Lenti‐GFP | Lenti‐PI3K | Lenti‐GFP | Lenti‐PI3K | |
| MAP (mm Hg) | 125±2.50 | 126±1.99 | 183±8.05* | 153±7.92# |
| HR (bpm) | 363±10.6 | 368±6.62 | 441±11.4* | 399±16.3 |
| RSNA (% Max) | 11.3±0.514 | 9.70±0.742 | 38.0±2.03* | 24.2±1.28# |
Values are expressed as mean±SEM, n=5/group,*P<.05 vs WKY+Lenti‐GFP; #P<.05 vs SHR+Lenti‐GFP.
3.2. Gene silencing of PI3K signaling in the RVLM attenuated the Ang II‐induced neuroinflammation and cardiovascular excitation
As indicated in Figure 2, intracisternal infusion of Ang II (24 μg/d) significantly increased approximately 2.5‐fold in the expression level of p110δ subunit of PI3K, and caused an approximately 1.5‐fold increase in AKT phosphorylation at Ser473 in the RVLM of WKY rats.
Figure 2.
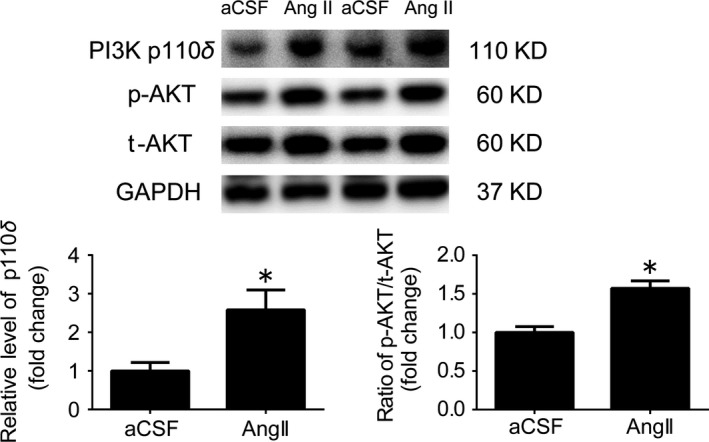
Intracisternal infusion of Ang II significantly upregulated the PI3K signaling in the RVLM. Representative gel bands (top) and quantification results (bottom) of PI3K p110δ expression, and AKT phosphorylation in the RVLM of WKY rats received Ang II infusion. The results of Western blots are shown as mean±SEM, n=5/group, *P<.05 vs aCSF
In order to confirm the role of PI3K in the neuroinflammation induced by Ang II, PI3K shRNA lentivirus was injected into the RVLM before intracisternal infusion of Ang II in WKY rats. As shown in Table 2, it was found that intracisternal infusion (1 week) of Ang II in the WKY rats significantly enhanced cardiovascular activity (MAP: 148±1.89 vs 116±2.75 mm Hg; HR: 430±10.6 vs 367±14.6 bpm; and basal RSNA: 24.6±2.72 vs 10.2±1.00% Max), but also increased levels of proinflammatory cytokines in the RVLM (IL‐1β: 188±17.7 vs 126±3.76 pg/mg; IL‐6: 686±35.5 vs 544±5.39 pg/mg; and TNF‐α: 94.5±5.94 vs 66.0±3.55 pg/mg) compared with infusion of aCSF (Figure 3B). Interestingly, the Ang II‐induced increases in levels of proinflammatory cytokines (IL‐1β, IL‐6, and TNF‐α) in the RVLM and cardiovascular activity (BP and HR) were significantly attenuated after pretreatment with silencing of PI3K in the RVLM.
Table 2.
Effects of intracisternal infusion of Ang II on BP, HR, and RSNA in the anesthetized WKY rats after PI3K shRNA injected into the RVLM
| Lenti‐GFP | Lenti‐PI3K shRNA | |||
|---|---|---|---|---|
| aCSF | Ang II | aCSF | Ang II | |
| MAP (mm Hg) | 116±2.75 | 148±1.89* | 126±2.68 | 135±2.71# |
| HR (bpm) | 367±14.6 | 430±10.6* | 367±13.1 | 383±6.98# |
| RSNA (% Max) | 10.2±1.00 | 24.6±2.72* | 10.9±0.461 | 16.5±0.868# |
Values are expressed as mean±SEM, n=5/group,*P<.05 vs Lenti‐GFP+aCSF; # P<.05 vs Lenti‐GFP+Ang II.
Figure 3.
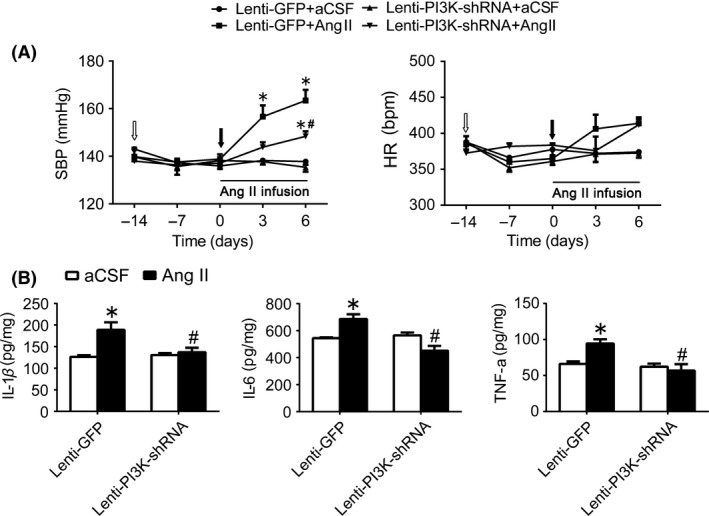
Effects of PI3K knockdown in the RVLM on changes in cardiovascular function and proinflammatory cytokines induced by intracisternal infusion of Ang II (1 wk) in WKY rats. A: time courses of systolic blood pressure (SBP) and HR in the WKY rats treated with intracisternal infusion of Ang II in response to pretreatment with lenti‐GFP or lenti‐PI3K shRNA injected into the RVLM (arrows). Values are expressed as mean±SEM, n=5/group, *P<.05 vs 0 d, #P<.05 vs lenti‐GFP. B, the Ang II‐induced changes in IL‐1β, IL‐6, and TNF‐α in the RVLM in response to pretreatment with lenti‐PI3K shRNA injections. Values are expressed as mean±SEM, n=5/group. *P<.05 vs aCSF, # P<.05 vs lenti‐GFP
3.3. Overexpression of ACE2 inhibited neuroinflammation in the RVLM
As shown in Figure 4A, transfer efficacy of lentivirus containing ACE2 gene (lenti‐ACE2) injected into the RVLM was confirmed by an increase in ACE2 protein expression and the ratio of Ang‐(1‐7) to Ang II in the RVLM. As indicated in Figure 4B, in the GFP group, levels of MAP and HR were significantly (P<.05) increased in conscious WKY rats after treatment with intracisternal infusion of LPS (2 weeks) compared with artificial cerebrospinal fluid (aCSF) infusion. The WKY rats treated with LPS infusion showed a significant increase in baseline RSNA compared with aCSF infusion (Table 3). Furthermore, the LPS‐induced increases in the levels of proinflammatory cytokines (IL‐1β, IL‐6, and TNF‐α) in the RVLM and cardiovascular activity (BP and HR) were significantly attenuated in WKY rat after pretreatment with ACE2 overexpression in the RVLM. Moreover, intracisternal infusion of LPS (2 weeks) significantly upregulated the expression level of p110δ subunit of PI3K and AKT phosphorylation at Ser473 in the RVLM of WKY rats, which was prevented by overexpression of ACE2.
Figure 4.
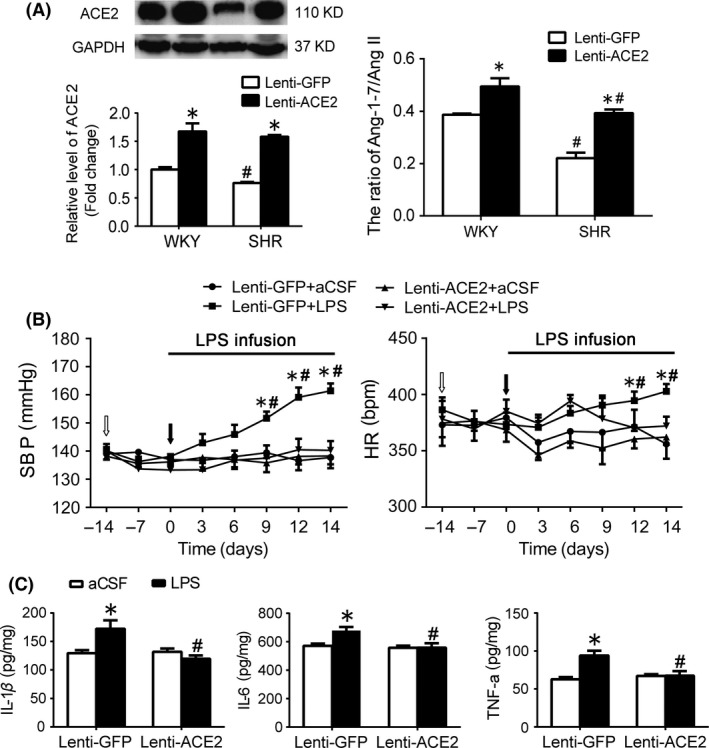
Effects of ACE2 overexpression in the RVLM on changes in cardiovascular function and proinflammatory cytokines induced by intracisternal infusion of LPS (2 wk) in WKY rats. A, Efficacy of overexpression of ACE2 in the RVLM. Bar graphs show the protein levels of ACE2 and the ratio of Ang‐(1‐7) to Ang II in the RVLM in WKY rats and SHR after treatment with lenti‐ACE2 injected into the RVLM. B, time courses of systolic blood pressure (SBP) and HR in the WKY rats treated with intracisternal infusion of LPS in response to pretreatment with lenti‐GFP or lenti‐ACE2 injected into the RVLM (arrows). Values are expressed as mean±SEM, n=5/group, *P<.05 vs 0 d, #P<.05 vs lenti‐GFP. C, the LPS‐induced changes in IL‐1β, IL‐6, and TNF‐α in the RVLM of WKY rats after pretreatment with ACE2 overexpression in the RVLM. Values are expressed as mean±SEM, n=5/group, *P<.05 vs aCSF; # P<.05 vs lenti‐GFP
Table 3.
Effects of intracisternal infusion of LPS on BP, HR, and RSNA in anesthetized WKY rats 4 wk after ACE2 overexpression in the RVLM
| Lenti‐GFP | Lenti‐ACE2 | |||
|---|---|---|---|---|
| aCSF | LPS | aCSF | LPS | |
| MAP (mm Hg) | 121±4.39 | 146±1.61* | 125±3.33 | 123±4.19# |
| HR (bpm) | 364±15.4 | 421±10.3* | 385±6.53 | 380±11.9 |
| RSNA (% Max) | 10.4±1.54 | 24.4±4.70* | 9.82±1.27 | 11.5±1.45# |
Values are expressed as mean±SEM, n=5/group,*P<.05 vs Lenti‐GFP+aCSF; #P<.05 vs Lenti‐GFP+LPS.
In SHR, it was observed that SHR showed a significant decrease in BP, HR, and basal RSNA 4 weeks after ACE2 overexpression in the RVLM (Table 4). Compared with WKY, SHR showed a significant increase in the content of proinflammatory cytokines (IL‐1β: 92.9±13.3 vs 136±9.62 pg/mg, P<.05; IL‐6: 428±83.8 vs 582±51.5 pg/mg, P<.05; TNF‐α: 40.6±4.0 vs 65.4±11.1 pg/mg, P<.05), all of which were attenuated by pretreatment with overexpression of ACE2 in the RVLM (Figure 5). Overexpression of ACE2 in the RVLM caused a significant decrease in the expression level of p110δ subunit of PI3K and AKT phosphorylation in SHR. However, overexpression of ACE2 in the RVLM in WKY rats had no influence on BP and the level of PI3K signaling (Figure 6).
Table 4.
Levels of BP, HR, and RSNA in the anesthetized WKY and SHR rats 4 wk after ACE2 overexpression in the RVLM
| WKY | SHR | |||
|---|---|---|---|---|
| Lenti‐GFP | Lenti‐ACE2 | Lenti‐GFP | Lenti‐ACE2 | |
| MAP (mm Hg) | 120±1.27 | 112±4.07 | 170±6.04* | 152±3.45# |
| HR (bpm) | 345±22.0 | 349±14.0 | 397±12.6* | 352±12.3# |
| RSNA (% of Max) | 15.9±1.49 | 13.2±2.00 | 38.3±1.33* | 24.1±1.53# |
Values are expressed as mean±SEM, n=5/group,*P<.05 vs WKY+Lenti‐GFP; #P<.05 vs SHR+Lenti‐GFP.
Figure 5.
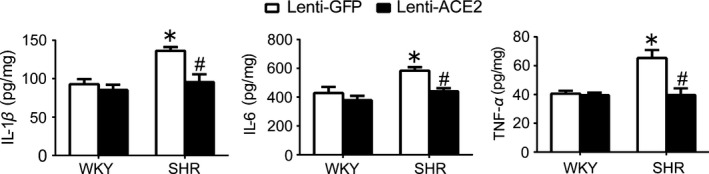
Effect of ACE2 overexpression in the RVLM on neuroinflammation in SHR. A, Levels of IL‐1β, IL‐6, and TNF‐α in the RVLM in SHR 4 wk after ACE2 overexpression. Values are expressed as mean±SEM, n=5/group. *P<.05 vs WKY; # P<.05 vs lenti‐GFP
Figure 6.
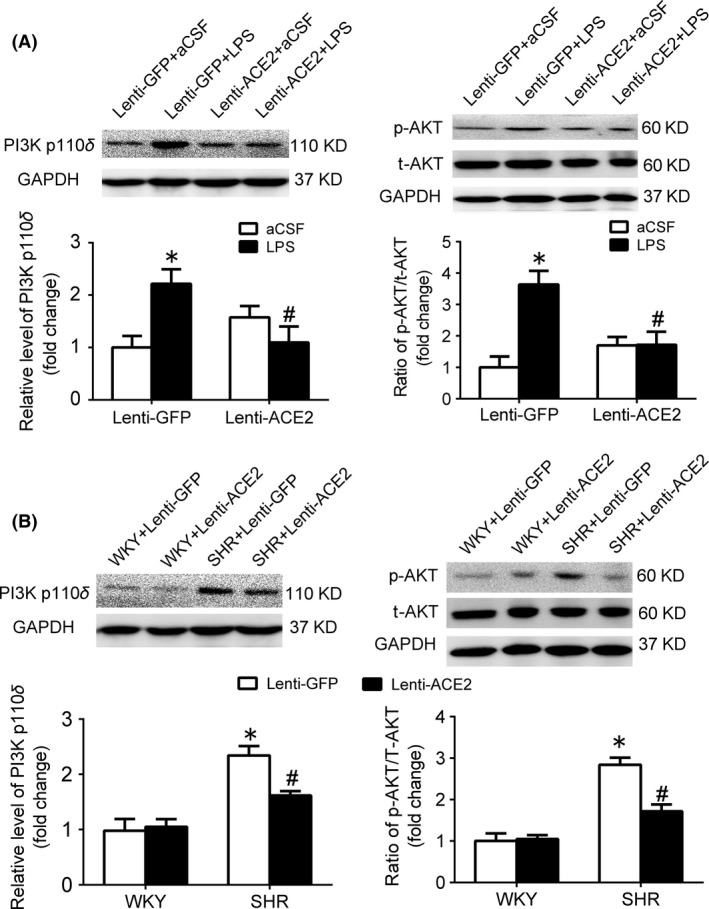
Effects of ACE2 overexpression in the RVLM on changes in PI3K signaling in the LPS‐induced hypertensive rats and SHR. Representative gel bands (top) and quantification data (bottom) of PI3K p110δ and AKT in the RVLM in the LPS‐treated WKY rats (A) and SHR (B) after pretreatment with overexpression of ACE2. Values are shown as mean±SEM, n=5/group, *P<.05 vs aCSF; # P<.05 vs lenti‐GFP
4. Discussion
In this study, the major findings are that (i) gene silencing of PI3K in the RVLM attenuated the levels of neuroinflammation and cardiovascular function in SHR; (ii) silencing of PI3K in the RVLM inhibited the cardiovascular excitation and neuroinflammation induced by central Ang II; and (iii) ACE2 overexpression in the RVLM attenuated the increased neuroinflammation, cardiovascular excitation, and upregulation of PI3K/AKT signaling in the LPS‐induced hypertensive rats and SHR. Taken together, these findings indicate that the PI3K signaling in the RVLM is involved in the pathogenesis of neuroinflammation in hypertension. It is also suggested that inactivation of the PI3K signaling mediates the antineuroinflammation effect of ACE2 overexpression in the RVLM in hypertensive rats.
It has been demonstrated that the proinflammatory cytokines produced by microglia in the brain increase sympathetic outflow, which is relative to the development of hypertension.31 In this study, we confirmed that the level of proinflammatory cytokines in the RVLM was increased in SHR; silencing PI3K signaling in the RVLM attenuated the neuroinflammation in SHR, suggesting a link between neuroinflammation and PI3K signaling in the central cardiovascular regulatory region in SHR. We also found that intracisternal infusion of Ang II produced a significant increase in the level of proinflammatory cytokines, as well as an increased activation of PI3K signaling in the RVLM. To avoid the inflammatory reaction in the RVLM induced by the microinjection‐induced tissue damage, therefore, intracisternal infusion was selected as an approach for inducing neuroinflammation. Although neuroinflammation induced by intracisternal infusion may be unselective in other brain regions, the inflammatory reaction in the RVLM was significantly observed. Furthermore, pretreatment with PI3K shRNA lentivirus in the RVLM in WKY rats could reduce the elevated BP and neuroinflammation induced by Ang II infusion. Based on these observations, it is suggested that the neuroinflammation in the RVLM is associated with high levels of BP and sympathetic outflow in hypertension. Moreover, the present study found that activation of PI3K signaling is capable of increasing the inflammatory response at the level of RVLM in hypertension. This supports the view that PI3K signaling may be an important pathway for inducing neuroinflammation in hypertension. It has been documented that central Ang II system is significantly increased and contributes to sympathetic overactivity in hypertension and chronic heart failure.32, 33 The previous study has suggested that the Ang II activates the T‐cell RAS leading to amplified low‐grade inflammation in hypertensive patients.34 Blockade of AT1R inhibits the acute inflammatory response induced by LPS treatment in vitro and in vivo.16 Therefore, these observations lead to a possibility that PI3K signaling is involved in neuroinflammation, which mediates the sympathoexcitation and hypertension in central Ang II‐induced hypertensive rats and SHR.
Another finding in the present study is that overexpression of ACE2 in the RVLM inhibits neuroinflammation in SHR or the LPS‐induced hypertension. The previous studies from our and other laboratories have demonstrated that overexpression of ACE2 in the RVLM significantly reduces BP and sympathetic nerve activity in hypertensive rats.23, 35 Previous studies have indicated a possible relationship between the ACE2/Ang‐(1‐7)/MasR axis and inflammatory response. For example, it is reported that Ang‐(1‐7) prevents the radiation‐induced inflammation in rat primary astrocyte via inhibiting the activation of MAP kinase signaling.36 In a vitro study, Ang‐(1‐7) decreases significantly the basal level of the proinflammatory cytokines in primary microglial cells, suggesting that Ang‐(1‐7) exerts a direct antiinflammatory effect on microglia.25 In this work, gene transfer of ACE2 in the RVLM was confirmed by increases in ACE2 protein expression and ratio of Ang1‐7/AngII content in the RVLM. In addition, it is found that GPF is expressed in both neurons and microglia in the RVLM after gene transfer. Therefore, it is possible that functional state of both neurons and microglia contributes to beneficial effect of the RVLM ACE2 on hypertension. Importantly, the current data revealed that pretreatment with ACE2 overexpression in the RVLM significantly attenuated the LPS‐induced increase in BP, RSNA, and proinflammatory cytokines. Moreover, enhancement in proinflammatory cytokines in the RVLM of SHR is significantly reduced by pretreatment with ACE2 overexpression. These findings suggest that antiinflammation plays an important role in mediating the beneficial effects of central ACE2 on hypertension.
In this study, the major finding was to also confirm the importance of the PI3K signaling in mediating the ACE2‐mediated antineuroinflammation in hypertension. First, we found that expression levels of PI3K and phosphorylated AKT in the RVLM were upregulated in WKY rats treated by intracisternal infusion of LPS. Second, pretreatment with overexpression of ACE2 in the RVLM significantly attenuated the LPS‐induced increase in expression levels of PI3K and phosphorylated AKT in the RVLM, and inhibited the activation of PI3K signaling in the RVLM in SHR. More importantly, gene transfer of PI3K p110δ shRNA in the RVLM reduced levels of cardiovascular activity, but also inhibits neuroinflammation in Ang II‐induced hypertensive rats and SHR. The subunit p110δ belongs to the catalytic subunit of PI3K, and its blockade effectively prevents activity of PI3K in the RVLM.37 In this study, there is a limitation that we did not directly observe the effect of PI3K knockdown on the LPS‐induced neuroinflammation. It has been widely demonstrated that increased Ang II plays an important role in central regulation of cardiovascular activity in hypertension. Instead of LPS, therefore, the Ang II‐induced neuroinflammation was subjected in response to gene transfer of PI3K shRNA in the RVLM. We confirmed that gene knockdown of PI3K in the RVLM prevented increased proinflammatory cytokines induced by Ang II. These observations lead to a conclusion that inactivation of the PI3K signaling plays an important role in mediating the ACE2‐induced antiinflammation in the RVLM of hypertension.
It is reported that the activity of the PI3K/AKT pathway is enhanced in the RVLM of SHR,10 and the phosphorylation of AKT in the PVN of hypertensive rats is also increased.38 Recently, we also confirmed that an inactivation of PI3K signaling in the RVLM is involved in antioxidative stress induced by central antihypertensive mechanism.12 However, the present data does not address the question how ACE2 reduces the production of proinflammatory cytokines via an inactivation of PI3K signaling. It is well known that nuclear factor‐kappa B (NF‐κB) plays an important role in production of proinflammatory cytokines. Chronic Ang‐(1‐7) treatment induces an inactivation of NF‐κB pathway in the epididymal fat tissue of experimental rats for the metabolic syndrome model.39 In additional, Ang‐(1‐7) can attenuate the Ang II‐induced dysfunction in human brain microvascular endothelial cells via inhibiting PI3K pathways.40 Interestingly, it also has been reported that ACE2 prevents the LPS‐induced inflammation via inhibiting MAPK and NF‐κB pathways in human retinal pigment epithelium.41 Therefore, it is possible that increase in endogenous Ang‐(1‐7) by ACE2 overexpression inhibits the PI3K/AKT‐mediated NF‐κB activation, in turn which reduces the production of proinflammatory cytokines and neuroinflammation.
5. Conclusion
The current results suggest that both neuroinflammation and cardiovascular excitation induced by central infusion of Ang II are prevented by pretreatment with RVLM PI3K knockdown. In addition, silencing of PI3K signaling in the RVLM attenuates neuroinflammation and cardiovascular excitation in SHR. Overexpression of ACE2 significantly reduces neuroinflammation in the RVLM of SHR and LPS‐treated WKY rats, as well as inhibits PI3K signaling. This work provides new evidence to be helpful for our understanding of neuroinflammation mediated by PI3K involved in neural control of cardiovascular activity in hypertension.
Conflict of Interests
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81370363, 81470534 and 81570385) and grants from the key Laboratory of Medical Electrophysiology, Ministry of Education of China (KeyME‐2014‐04).
Tan X, Jiao P‐L, Wang Y‐K, et al. The phosphoinositide‐3 kinase signaling is involved in neuroinflammation in hypertensive rats. CNS Neurosci Ther. 2017;23:350–359. 10.1111/cns.12679
The first two authors contribute equally to this work.
Contributor Information
Miao‐Ling Li, Email: limiaolingcc@163.com.
Wei‐Zhong Wang, Email: wangwz68@hotmail.com.
References
- 1. Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409. [DOI] [PubMed] [Google Scholar]
- 2. Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Salles GF, Fiszman R, Cardoso CR, Muxfeldt ES. Relation of left ventricular hypertrophy with systemic inflammation and endothelial damage in resistant hypertension. Hypertension. 2007;50:723–728. [DOI] [PubMed] [Google Scholar]
- 5. El‐Bassossy HM, Shaltout HA. Allopurinol alleviates hypertension and proteinuria in high fructose, high salt and high fat induced model of metabolic syndrome. Transl Res. 2015;165:621–630. [DOI] [PubMed] [Google Scholar]
- 6. Block ML. Neuroinflammation: modulating mighty microglia. Nat Chem Biol. 2014;10:988–989. [DOI] [PubMed] [Google Scholar]
- 7. Shi Z, Gan XB, Fan ZD, et al. Inflammatory cytokines in paraventricular nucleus modulate sympathetic activity and cardiac sympathetic afferent reflex in rats. Acta Physiol (Oxf). 2011;203:289–297. [DOI] [PubMed] [Google Scholar]
- 8. Shen XZ, Li Y, Li L, et al. Microglia participate in neurogenic regulation of hypertension. Hypertension. 2015;66:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. [DOI] [PubMed] [Google Scholar]
- 10. Wu KL, Wu CA, Wu CW, Chan SH, Chang AY, Chan JY. Redox‐sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid Redox Signal. 2013;18:36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai CY, Chang AY, Chan JY, Chan SH. Activation of PI3K/Akt signaling in rostral ventrolateral medulla impairs brain stem cardiovascular regulation that underpins circulatory depression during mevinphos intoxication. Biochem Pharmacol. 2014;88:75–85. [DOI] [PubMed] [Google Scholar]
- 12. Wang YK, Yu Q, Tan X, et al. Centrally acting drug moxonidine decreases reactive oxygen species via inactivation of the phosphoinositide‐3 kinase signaling in the rostral ventrolateral medulla in hypertensive rats. J Hypertens. 2016;34:993–1004. [DOI] [PubMed] [Google Scholar]
- 13. Xiao Z, Peng J, Yang L, Kong H, Yin F. Interleukin‐1beta plays a role in the pathogenesis of mesial temporal lobe epilepsy through the PI3K/Akt/mTOR signaling pathway in hippocampal neurons. J Neuroimmunol. 2015;282:110–117. [DOI] [PubMed] [Google Scholar]
- 14. Min KJ, Kim JH, Jou I, Joe EH. Adenosine induces hemeoxygenase‐1 expression in microglia through the activation of phosphatidylinositol 3‐kinase and nuclear factor E2‐related factor 2. Glia. 2008;56:1028–1037. [DOI] [PubMed] [Google Scholar]
- 15. Dupont AG, Brouwers S. Brain angiotensin peptides regulate sympathetic tone and blood pressure. J Hypertens. 2010;28:1599–1610. [DOI] [PubMed] [Google Scholar]
- 16. Benicky J, Sanchez‐Lemus E, Honda M, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodriguez‐Pallares J, Rey P, Parga JA, Munoz A, Guerra MJ, Labandeira‐Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH‐derived ROS. Neurobiol Dis. 2008;31:58–73. [DOI] [PubMed] [Google Scholar]
- 18. Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin‐converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin‐converting enzyme 2. Circulation. 2005;111:2605–2610. [DOI] [PubMed] [Google Scholar]
- 19. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1‐7 Axis of the Renin‐Angiotensin System in Heart Failure. Circ Res. 2016;118:1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giani JF, Mayer MA, Munoz MC, et al. Chronic infusion of angiotensin‐(1‐7) improves insulin resistance and hypertension induced by a high‐fructose diet in rats. Am J Physiol Endocrinol Metab. 2009;296:E262–E271. [DOI] [PubMed] [Google Scholar]
- 21. Giani JF, Gironacci MM, Munoz MC, Pena C, Turyn D, Dominici FP. Angiotensin‐(1 7) stimulates the phosphorylation of JAK2, IRS‐1 and Akt in rat heart in vivo: role of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol. 2007;293:H1154–H1163. [DOI] [PubMed] [Google Scholar]
- 22. Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin‐converting enzyme 2 and angiotensin‐(1–7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289:H2281–H2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang YK, Shen D, Hao Q, et al. Overexpression of angiotensin‐converting enzyme 2 attenuates tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;307:H182–H190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao L, Gao L, Lazartigues E, Zucker IH. Brain‐selective overexpression of angiotensin‐converting enzyme 2 attenuates sympathetic nerve activity and enhances baroreflex function in chronic heart failure. Hypertension. 2011;58:1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu M, Shi P, Sumners C. Direct anti‐inflammatory effects of angiotensin‐(1‐7) on microglia. J Neurochem. 2016;136:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates, 3rd edn New York: Academic Press; 1998. [Google Scholar]
- 27. Porter JP. Chronic intracerebroventricular infusion of angiotensin II increases brain AT1 receptor expression in young rats. Brain Res Dev Brain Res. 1999;112:293–295. [DOI] [PubMed] [Google Scholar]
- 28. Zha YP, Wang YK, Deng Y, et al. Exercise training lowers the enhanced tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. CNS Neurosci Ther. 2013;19:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hao F, Gu Y, Tan X, et al. Estrogen Replacement Reduces Oxidative Stress in the Rostral Ventrolateral Medulla of Ovariectomized Rats. Oxid Med Cell Longev. 2016;2016:2158971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng JF, Wu ZT, Wang YK, et al. GABAergic mechanism in the rostral ventrolateral medulla contributes to the hypotension of moxonidine. Cardiovasc Res. 2011;89:473–481. [DOI] [PubMed] [Google Scholar]
- 31. Winklewski PJ, Radkowski M, Wszedybyl‐Winklewska M, Demkow U. Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation. 2015;12:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanderford MG, Bishop VS. Central mechanisms of acute ANG II modulation of arterial baroreflex control of renal sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2002;282:H1592–H1602. [DOI] [PubMed] [Google Scholar]
- 33. Zhu GQ, Gao L, Patel KP, Zucker IH, Wang W. ANG II in the paraventricular nucleus potentiates the cardiac sympathetic afferent reflex in rats with heart failure. J Appl Physiol. 1985;2004:1746–1754. [DOI] [PubMed] [Google Scholar]
- 34. Coppo M, Bandinelli M, Berni A, et al. Ang II Upregulation of the T‐lymphocyte renin‐angiotensin system is amplified by low‐grade inflammation in human hypertension. Am J Hypertens. 2011;24:716–723. [DOI] [PubMed] [Google Scholar]
- 35. Yamazato M, Yamazato Y, Sun C, Diez‐Freire C, Raizada MK. Overexpression of angiotensin‐converting enzyme 2 in the rostral ventrolateral medulla causes long‐term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension. 2007;49:926–931. [DOI] [PubMed] [Google Scholar]
- 36. Moore ED, Kooshki M, Metheny‐Barlow LJ, Gallagher PE, Robbins ME. Angiotensin‐(1‐7) prevents radiation‐induced inflammation in rat primary astrocytes through regulation of MAP kinase signaling. Free Radic Biol Med. 2013;65:1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu ZT, Ren CZ, Yang YH, et al. The PI3K signaling‐mediated nitric oxide contributes to cardiovascular effects of angiotensin‐(1‐7) in the nucleus tractus solitarii of rats. Nitric Oxide. 2016;52:56–65. [DOI] [PubMed] [Google Scholar]
- 38. Sriramula S, Xia H, Xu P, Lazartigues E. Brain‐targeted angiotensin‐converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase‐mediated inflammation. Hypertension. 2015;65:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marcus Y, Shefer G, Sasson K, et al. Angiotensin 1‐7 as means to prevent the metabolic syndrome: lessons from the fructose‐fed rat model. Diabetes. 2013;62:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao X, Zhang C, Ma X, et al. Angiotensin‐(1‐7) counteracts angiotensin II‐induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp Cell Res. 2015;336:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao L, Qiu Y, Fu X, et al. Angiotensin‐converting enzyme 2 activator diminazene aceturate prevents lipopolysaccharide‐induced inflammation by inhibiting MAPK and NF‐kappaB pathways in human retinal pigment epithelium. J Neuroinflammation. 2016;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]