

Summary
Aim
To study whether inhibiting microglia migration to the ischemic boundary zone (IBZ) at the early phase could improve neurological outcomes after stroke.
Methods
The transient middle cerebral artery occlusion (tMCAO) was induced in adult male Sprague‐Dawley rats. AMD3100, a highly selective CXC‐chemokine receptor 4 (CXCR4) antagonist, was used to inhibit microglia migration. Microglia was evaluated by immunofluorescence in vivo, and their migration was tested by transwell assay in vitro. Expressions of cytokines were detected by real‐time PCR. Infarct volume was determined by triphenyltetrazolium chloride (TTC) staining. Functional recovery of tMCAO rats was evaluated by behavior tests.
Results
M1 microglia in the IBZ was rapidly increased within 3 days after tMCAO, accompanied with enhanced expression of CXCR4. Chemokine CXC motif chemokine ligand 12 (CXCL12) was also increased in the IBZ. And AMD3100 could obviously decline M1 microglia migration induced by CXCL12 and secretion of related inflammatory cytokines in the IBZ after stroke. This was accompanied by significant attenuated infarct volume and improved neurological outcomes.
Conclusion
This study confirms the protective efficacy of inhibiting microglia migration at the hyperacute phase as a therapeutic strategy for ischemic stroke in tMCAO model of rats, and its therapeutic time window could last for 24 h after cerebral ischemia reperfusion.
Keywords: AMD3100, CXCL12/CXCR4, Hyperacute phase, M1 microglia, Migration, tMCAO
Introduction
Stroke is envisioned to be a pathophysiological multiphase process. It is known that cerebral infarction expands slowly over 24 h from the core to the peripheral area after ischemic stroke, and the peripheral area which is also called ischemic boundary zone (IBZ) is a potential therapeutic target 1, 2. Nowadays, massive studies support the conception that inflammation is a major cause in propagating tissue damage after cerebral ischemia 3, 4. Within the first hour after stroke, inflammatory response was rapidly initiated and various inflammatory cells, especially microglia, flocked to the ischemic cerebral parenchyma 3, 5. Then, considerable proinflammatory cytokines were released from microglia and contributed to severe brain damage 6. Thus, it is very important and beneficial to patients to intervene in the inflammatory responses at the early phase after stroke.
Microglia, which is the resident immune cell of the central nervous system (CNS), plays a critical role in the progress of inflammatory response after cerebral ischemia 7. In rodent models, microglia began to produce proinflammatory cytokines very early after stroke. It is reported that microglia started to increase at 12 h after ischemia and proliferated largely after 24 h in the IBZ 3, 8.Microglia is categorized into a classic proinflammatory (M1) or an alternative antiinflammatory (M2) polarization 9, 10. M1 microglia mainly expresses proinflammatory molecules, such as TNF‐α, IL‐1β, IL‐18 and CCL2 and iNOS, which can aggravate inflammation 7, 11. M2 microglia can secrete TGF‐β, BDNF, NGF, IL‐10, and IL‐4 which shows antiinflammatory and neuroprotective effects 11, 12. At present, accumulative evidences show that M1 microglia contributes to the inflammatory cascade and further propagates cell death beyond the initial ischemic region 13. In this study, we also found the number of microglia, especially the M1 phenotype was rapidly increased in the IBZ several hours after transient middle cerebral artery occlusion (tMCAO), accompanied with increased expression of proinflammatory cytokines. This may be the primary cause of brain damage at the hyperacute period after ischemic stroke. However, the pathogenesis that induced the M1 microglia stream into the IBZ during the hyperacute phase is yet to be characterized.
Early work demonstrated that rodent microglia express CXC‐chemokine receptor 4 (CXCR4) 14, 15, 16. CXCR4 and its ligand CXC motif chemokine ligand 12 (CXCL12) are key regulators of microglia migration and recruitment 14, 17. Emerging evidences showed that the expression of CXCL12 was increased in the IBZ after ischemia 18, 19. Our previous study also confirmed that CXCL12 expression in the IBZ was increased as early as 3 h after permanent MCAO of rats 20. So it naturally reminds us to wonder whether inhibiting CXCR4 at the very early phase after ischemic stroke could attenuate the inflammatory response initiated rapidly after ischemia.
In the present study, we used a rat tMCAO model to investigate (1) whether CXCL12 could induce M1 microglia migration and proinflammatory cytokine production, and (2) whether inhibiting the migration of M1 microglia could diminish the infarct volume and improve long‐term outcome after transient focal ischemia. We aimed to provide the available intervention time window of CXCR4 antagonist and a feasible stroke treatment strategy.
Materials and Methods
Animals
Adult (7–8 weeks old) male Sprague‐Dawley rats weighing 250–280 g were used and housed individually under standard conditions of temperature and humidity and a 12 h of light/dark cycle (lights on at 08:00) with free access to food and water. Adequate measures were taken to minimize pain or discomfort during surgeries. All the experiments were carried out in accordance with the Institutional Guidelines of the Animal Care and Use Committee (Huazhong University of Science and Technology).
Establishment of tMCAO Model
Rats were anesthetized with 10% chloral hydrate (300 mg/kg, intraperitoneal injection) and subjected to MCAO as described previously 21. Briefly, the right common carotid artery, external carotid artery, and internal carotid artery were exposed via a midline pretracheal incision. A poly‐L‐lysine‐coated 4/0 monofilament nylon suture with a rounded tip was inserted from the lumen of right external carotid artery to the internal carotid artery until a mild resistance was felt. Therefore, the origin of right MCA was occluded. The filament was left in place for 2 h and then withdrawn. The rectal temperature was maintained at 37.0 ± 0.5°C with a feedback‐regulated heating pad during surgery. Sham‐operated rats underwent the same anesthesia and surgical procedures except MCAO.
Intracerebroventricular Injection of AMD3100
AMD3100, which was widely applied as CXCL12/CXCR4 blocker 22, 23, 24, was chosen in this study to antagonize CXCR4. AMD3100 (Abcam, Cambridge, MA, USA) was dissolved with normal saline to a concentration of 3 μg/μL. Five microliters of AMD3100 was injected into the right lateral ventricle at 1.0 mm posteriorly to bregma, 2.5 mm laterally from midline, and 3.5 mm vertically from the skull surface. The same amount of normal saline solution was used in the vehicle groups. Rats were divided into four groups: 12‐h‐treated group, rats received twice (6‐h interval) AMD3100 or vehicle at 12 h after tMCAO; 24‐h‐treated group, rats received twice (6‐h interval) AMD3100 or vehicle at 24 h after tMCAO; 48‐h‐treated group, rats received twice (6‐h interval) AMD3100 or vehicle at 48 h after tMCAO; 72‐h‐treated group, rats received twice (6‐h interval) AMD3100 or vehicle at 72 h after tMCAO.
Evaluation of Infarct Volume
Twenty‐four hours after the second injection, rat brain tissues were rapidly removed and sectioned into six slices along the coronal plane. The brain slices were then incubated with 2% triphenyltetrazolium chloride (TTC; Sigma‐Aldrich, St. Louis, MO, USA) at 37°C for 20 min in the dark as described previously 25. Infarct brain was identified as an area of unstained tissue. Then, the TTC‐stained sections were photographed. Infarct volumes were calculated using Image J (National Institutes of Health, Bethesda, MD, USA) and expressed as the percentage of infarction in an ipsilateral hemisphere.
Behavioral Tests
In another group of independent experiments, rats were allowed for recovery after AMD3100 or vehicle injection. Then, each rat was subjected to a series of behavioral tests to evaluate various aspects of neurological functions. Measurements were performed at 24 h before operation, 24 h, 3, 7, 14, and 28 days postischemic injury by an investigator who was blinded to the experimental groups.
Neurological Scores
Neurological performance was scored using modified neurological severity scores (mNSS) 26, 27. Scores were ranked from 0 to 18 at an interval of 1. As shown in Table 1, the mNSS is a composite of motor, sensory, reflex, and balance tests. In the severity scores of injury, one score point is awarded for the inability to perform the test or for the lack of a tested reflex; thus, the higher the score, the more severe the injury is.
Table 1.
Modified neurological severity score points
| Tests | Points |
|---|---|
| Motor tests | |
| Raising the rat by the tail | |
| Flexion of forelimb | 1 |
| Flexion of hindlimb | 1 |
| Head moving more than 10° (vertical axis) | 1 |
| Placing the rat on the floor | |
| Inability to walk straight | 1 |
| Circling toward the paretic side | 1 |
| Falling down to the paretic side | 1 |
| Sensory tests | |
| Visual and tactile placing | 1 |
| Proprioceptive test (deep sensory) | 1 |
| Beam balance tests | |
| Grasps side of beam | 1 |
| Hugs the beam and one limb falls down from the beam | 2 |
| Hugs the beam and two limbs fall down from the beam or spins on beam (>60 s) | 3 |
| Attempts to balance on the beam but falls off (>40 s) | 4 |
| Attempts to balance on the beam but falls off (>20 s) | 5 |
| Falls off: no attempt to balance or hang on to the beam (<20 s) | 6 |
| Reflexes (blunt or sharp stimulation) absent of: | |
| Pinna reflex (a head shake when touching the auditory meatus) | 1 |
| Corneal reflex (an eye blink when lightly touching the cornea with cotton) | 1 |
| Startle reflex (a motor response to a brief loud paper noise) | 1 |
| Seizures, myoclonus, myodystony | 1 |
| Maximum points | 18 |
One point is awarded for the inability to perform the tasks or for the lack of a tested reflex; 13–18 indicates severe injury; 7–12, moderate injury; 1–6, mild injury.
Rotarod Test
Motor coordination of the animals was measured using a rotarod treadmill for rats under the accelerating rotor mode (speed increased from 4 to 40 rpm within 5 min) 27. The trial will be ended if the rats fell off the rungs, or gripped and spun around for two complete revolutions without attempting to walk on the rungs. The time that the rat remained on the rung was recorded as the retention time. The mean duration on the rod was recorded with three rotarod measurements 1 day before surgery. Performance on the rotarod test was measured three times a day in the following 4 weeks after ischemic injury and AMD3100 administration. Motor test data are expressed as a percentage of mean duration per day on the rotarod compared with the presurgery control value.
M1 and M2 Microglia Polarization and Oxygen–Glucose Deprivation (OGD)
The murine microglial cell line BV2 was cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured at 37°C in a saturated humidified incubator with 5% CO2 and were passaged every 3 days. BV2 cells were distributed into six‐well plates at the density of 1 × 106 cells/well, and then lipopolysaccharides (LPS, 1 μg/mL, Sigma‐Aldrich) or IL‐4 (20 ng/mL, Sangon Biotech, Shanghai, China) were used to polarize microglia to M1 or M2 phenotype 28.
For exposure to OGD, M1, and M2 microglia were washed with glucose‐free phosphate‐buffered saline (PBS) twice and were placed in glucose‐free medium. Then, they were incubated for 3 h at 37°C in an anaerobic chamber (5% CO2, 10% H2, and 85% N2 atmosphere). After 3 h of OGD exposure, they were reoxygenated with glucose‐containing Dulbecco's modified Eagle's medium (DMEM) and incubated under normoxic conditions for transwell assay.
Primary Culture of Astrocytes and Cortical Neurons
Brain tissue from Sprague‐Dawley rats was used for primary culture of astrocytes as previously described 29. Briefly, dissociated cerebral cortices from neonatal rats were digested and cultured in the high‐glucose DMEM supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). After 14 days, cells were gently shaken at 260 rpm to remove the loosely adhered oligodendrocytes and microglial cells. Then astrocytes were cultured in 24‐well plate (5 × 105/well), subjected to OGD for 60 min and then incubated under normoxic conditions for transwell assay.
Neonatal rats were used for the culture of cortical neurons. Rat cortices were dissected and then dissociated in Neurobasal‐A medium (Gibco) containing 2% B27 (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). The cell suspension (1 × 104 cells/cm2) was then plated on 96‐well plate coated with poly‐D‐lysine (0.1 mg/mL, Sigma) for about 8 days. Then, they were prepared to perform the following experiments.
Coculture of Microglia‐Conditioned Medium and Neurons
BV2 microglia treated with LPS or IL‐4 for 24 h to polarize into M1 or M2 phenotype, and the culture medium (CM) was collected. To generate microglia‐conditioned medium and neurons cocultures, 8‐day‐old neurons cultured in 96‐well plate were subjected to OGD for 60 min, and then, their medium was changed with the CM from M1 or M2 microglia. Twenty‐four hours later, the cell viability assay and quantitative enzyme‐linked immunosorbent assay (ELISA) for MAP2 were performed.
Cell Viability Assay
Cell viability assay was performed as previously reported with some modifications 30. The neurons were seeded onto 96‐well plates at a density of approximate 1 × 104 cells/well. After the treatment, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT, Sigma‐Aldrich) was added to each well to reach a final concentration of 0.5 mg/mL. After 4‐h incubation at 37°C, the medium was removed. Then, 100 μL dimethyl sulfoxide was added to each well and left to stand for 10 min. The absorbance at 570 nm was measured with a microplate reader (iMarKTM, Bio‐Rad Laboratories, Inc., Hercules, CA, USA). Results were expressed as the percentage of control.
Quantitative ELISA for MAP2
Quantitative ELISA for MAP2 was performed as previously described 31. After fixed with 4% paraformaldehyde and blocked with 10% bovine serum albumin, neurons were incubated with MAP2 primary antibody (1:1000; Abcam) overnight at 4°C. Alkaline–phosphatase‐labeled secondary antibody was then applied (rabbit anti‐mouse IgG, 1:1000; Jackson, West Grove, PA, USA) for 2 h at room temperature. P‐nitrophenyl phosphate substrate was used to detect the expression of MAP2. The absorbance was measured at 405 nm using a microplate reader, and the enzyme activity was calculated according to p‐nitrophenol standards.
Transmigration Assay
The transwell system with an aperture of 8 μm was purchased from Millipore Inc. (Billerica, MA, USA). A total of 200 μL M1 or M2 phenotype cells with the concentration of 1.5 × 105 cells/mL containing 2.5 μg/mL AMD3100 or vehicle were placed in the upper chamber and 600 μL medium containing 200 ng/mL CXCL‐12 (Millipore) or vehicle was placed in the lower chamber. In another independent experiment, post‐OGD astrocytes were seeded in the lower chamber. Cells were fixed for 1 h with 4% paraformaldehyde after incubation for 16 h at 37°C. Then, un‐migrated cells were removed from the top by cotton swabs, and the migrated cells on the bottom surface were stained with 0.1%crystal violet (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 10 min and counted with Olympus LCX100 Imaging system (Olympus Corporation, Tokyo, Japan). All experiments were performed at least in triplicates.
Immunofluorescence
Rats were deeply anesthetized and transcardially perfused with 0.9% saline followed by ice‐cold 4% paraformaldehyde in 0.1 M PBS. A tissue block for standard paraffin‐embedding was obtained and cut into 4‐μm‐thick sections serially. After deparaffinization, rehydration, and antigen retrieval, the sections were blocked with 10% donkey serum (Jackson Immunoresearch, West Grove, PA, USA) for 30 min. For fluorescence imaging, the sections were incubated with goat anti‐Iba1 (1:200, Abcam), rabbit anti‐iNOS (1:100, Abcam), rabbit anti‐CD206 (1:100, Abcam), or rabbit anti‐CXCR4 (1:100, Abcam) at 4°C overnight, then with fluorescence‐labeled secondary antibodies for 2 h. DAPI (1:1000, Sigma‐Aldrich) was used to label cell nuclei. Then mounted with Fluorescence ProLong Gold antifade reagent (Beyotime, Jiangsu, China), cover‐slipped, and examined under a TCS SP5 multiphoton laser scanning confocal microscope (Nikon, Tokyo, Japan).
The cell number calculation was determined by counting three randomly selected microscopic fields across five slides in the IBZ of the ipsilateral cortex. Data were expressed as mean numbers of cells per square millimeter.
Real‐time Polymerase Chain Reaction (PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen), and 5 μg was used to synthesize cDNA using M‐MLV reverse transcriptase following the manufacturer's protocol (Invitrogen). Real‐time PCR reactions were performed using SYBR® Green Realtime Master Mix and ABI PRISM® 7700 (Applied Biosystems, Waltham, MA, USA). Threshold cycle values were used to calculate the fold change in the transcript levels using the 2−ΔΔCt method. The relative mRNA expression levels were normalized to the β‐actin gene. Primer sequences for the genes analyzed are summarized in Table 2.
Table 2.
Primers for real‐time polymerase chain reaction (PCR)
| Gene | Primer (5′‐3′) |
|---|---|
| Rats | |
| TNF‐α | SENS: TGCCTCAGCCTCTTCTCATT |
| REVS: GCTTGGTGGTTTGCTACGAC | |
| iNOS | SENS: ACCTCTATGTTTGTGGCGATG |
| REVS: TCAACCTGCTCCTCACTCAA | |
| IL‐1β | SENS: CCCTGCAGCTGGAGAGTGTGG |
| REVS: TGTGCTCTGCTTGAGAGGTGCT | |
| IL‐6 | SENS: CGAGCCCACCAGGAACGAAAGTC |
| REVS: CTGGCTGGAAGTCTCTTGCGGAG | |
| IL‐10 | SENS: CAACTGCATAGAAGCCTACGTG |
| REVS: GGGAACTGAGGTATCAGAGGTAA | |
| CD206 | SENS: CAAGGAAGGTTGGCATTTGT |
| REVS: CCTTTCAGTCCTTTGCAAGC | |
| Arg‐1 | SENS: GGCGTTGACCTTGTCTTGTT |
| REVS: CTGTTCGGTTTGCTGTGATG | |
| TGF‐β | SENS: TGCGCCTGCAGAGATTCAAG |
| REVS: AGGTAACGCCAGGAATTGTTGCTA | |
| Mouse | |
| TNF‐α | SENS: AGCCCACGTCGTAGCAAACCAC |
| REVS: AGGTACAACCCATCGGCTGGCA | |
| iNOS | SENS: CAAGCACCTTGGAAGAGGAG |
| REVS: AAGGCCAAACACAGCATACC | |
| IL‐1β | SENS: CCTGCAGCTGGAGAGTGTGGAT |
| REVS: TGTGCTCTGCTTGTGAGGTGCT | |
| CD86 | SENS: ACGATGGACCCCAGATGCACCA |
| REVS: GCGTCTCCACGGAAACAGCA | |
Statistical Analysis
All variance values are represented as mean ± SEM. Unpaired t‐tests or one‐way Analysis of Variance (ANOVA) were performed for two groups or for multiple group comparisons (with a post hoc Student–Newman–Keuls test), respectively. Two‐way ANOVA were performed for group comparisons with time course analysis. Statistical processing was performed using SPSS 16.0 (IBM, Armonk, NY, USA). All tests were two‐sided. Statistically significant differences were defined as P < 0.05.
Results
M1 Microglia Increased Over Time in the IBZ within the First 72 h after Stroke
To evaluate the change of microglia in the IBZ (Figure 1A and Figure S1) after tMCAO, we investigated M1 and M2 phenotypes by detecting coexpression of iNOS (M1 marker) or CD206 (M2 marker) with microglia marker Iba‐1 within the first 72 h after ischemic injury (Figure 1B). At 12 h after tMCAO, the number of M1 microglia (iNOS+/Iba‐1+ cells) increased obviously in the IBZ (152.6 ± 20.8/mm2 vs. 5.7 ± 1.5/mm2; Figure 1C). Then, it enhanced abundantly (224.9 ± 32.3/mm2 at 24 h, 526.2 ± 38.6/mm2 at 48 h and 575.6 ± 52.7/mm2 at 72 h after tMCAO, respectively; Figure 1C). Inconsistently, the number of the M2 microglia (CD206+/Iba‐1+ cells) changed slightly at 12 h after tMCAO (22.5 ± 5.5/mm2 vs. 3.6 ± 3.7/mm2; Figure 1C). It began to progressively increase 24 h later (54.0 ± 17.5/mm2 at 24 h, 93.3 ± 21.1/mm2 at 48 h and 159.2 ± 39.1/mm2 at 72 h, respectively; Figure 1C). Importantly, the count of M1 phenotype increased more dramatically than M2 phenotype at each point in time, implying the dominant presence of M1 microglia in the IBZ during the early 72 h after ischemia.
Figure 1.
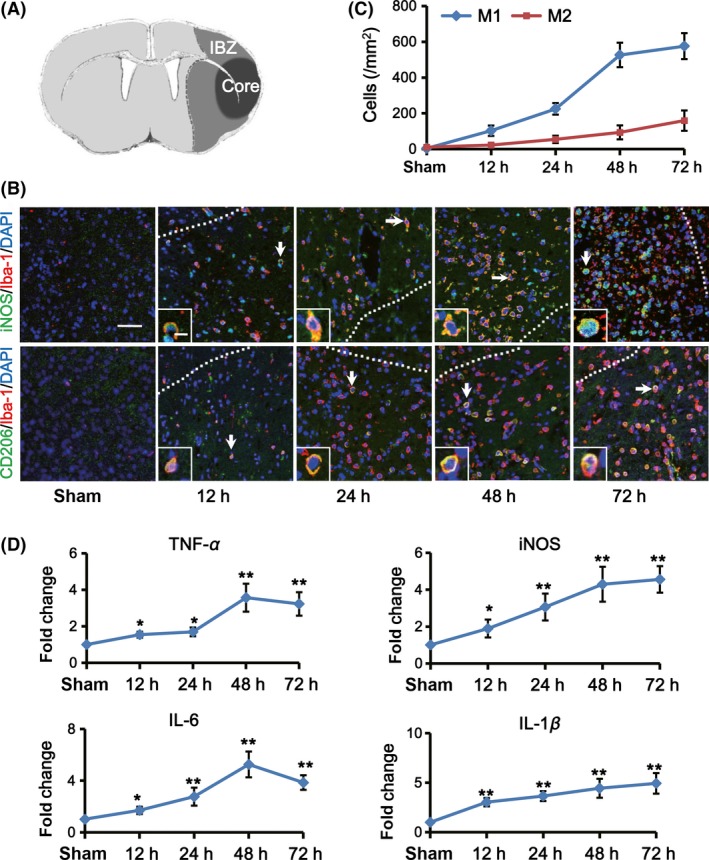
M1 microglia increased over time in the IBZ within the first 72 h after ischemic stroke. (A) Sketch picture illustrates the ischemic core (black) and the IBZ (gray) after tMCAO. (B) Representative images of the IBZ after 12, 24, 48, and 72 h of reperfusion double‐stained with Iba‐1 (red) and M1 marker iNOS (green) or M2 marker CD206 (green) antibodies. DAPI staining (blue) was used to label the nucleus. Double positive cells indicated by arrows are shown at higher magnification in the left‐bottom panels. Scare bar: 40 and 10 μm, respectively. (C) Quantification of iNOS+/Iba‐1+ (M1 microglia) and CD206+/Iba‐1+ (M2 microglia) cells per mm2 in the IBZ (n = 5 per group). (D) Quantification of M1‐associated inflammatory cytokine expression in the IBZ by real‐time PCR at indicated time points after tMCAO or sham treatment (n = 5 per group). Data are expressed as fold change versus sham. *P < 0.05, ** P < 0.01 versus sham.
Then, we detected the expression of M1‐associated proinflammatory factors and M2‐associated antiinflammatory cytokines by real‐time PCR. We found that the expression of TNF‐α, iNOS, IL‐6, and IL‐1β, which were mainly secreted by M1 phenotype, enhanced as early as 12 h after tMCAO (*P < 0.05, ***P < 0.001, Figure 1D). However, M2‐associated antiinflammatory cytokines including Arg‐1, IL‐10, CD206, and TGF‐β had no significant increase until 72 h after ischemia (Figure S2). It is known that some of the M1 and M2 signature genes are expressed not only in microglia but also in other brain cells or infiltrating immune cells. But as shown in the immunofluorescence results, iNOS+/iba1+ cells were in the majority of iNOS+ cells in the IBZ. Taken together, these data suggested that microglia initially flocking into the site of IBZ exhibited mainly M1 phenotype. They increased over time and outnumbered M2 phenotype largely within the first 72 h poststroke.
CXCR4 and CXCL12 were Upregulated after tMCAO
Previous studies showed that rodent microglia expressed CXCR4. CXCR4 and its ligand CXCL12 are key regulators of microglia migration and recruitment. In tMCAO rats, we analyzed the expression of CXCR4 in microglia in the IBZ by RT‐PCR after 12, 24, 48, and 72 h of reperfusion. Results showed that CXCR4 increased significantly over time from 12 to 72 h after tMCAO (*P < 0.05, **P < 0.01 and ***P < 0.001; Figure 2A). Then, we did double immunofluorescence staining with anti‐CXCR4 and anti‐iNOS and found that most of the CXCR4 were coexpressed with iNOS, a marker of M1 microglia (Figure 2B). In addition, the number of CXCR4+/iNOS+ cells increased progressively in the IBZ over time within 72 h after tMCAO (12 h: 94.3 ± 10.8/mm2, 24 h: 191.7 ± 15.2/mm2, 48 h: 551.2 ± 53.5/mm2 and 72 h: 567.2 ± 58.1/mm2, respectively; ***P < 0.001; Figure 2C) compared with sham group (3.7 ± 1.4/mm2). Concurrently, our results revealed the expression of CXCL12, the ligand of CXCR4, increased and peaked as early as 12 h after tMCAO and maintained at high levels within 72 h compared to the sham group (***P < 0.001; Figure 2D).
Figure 2.
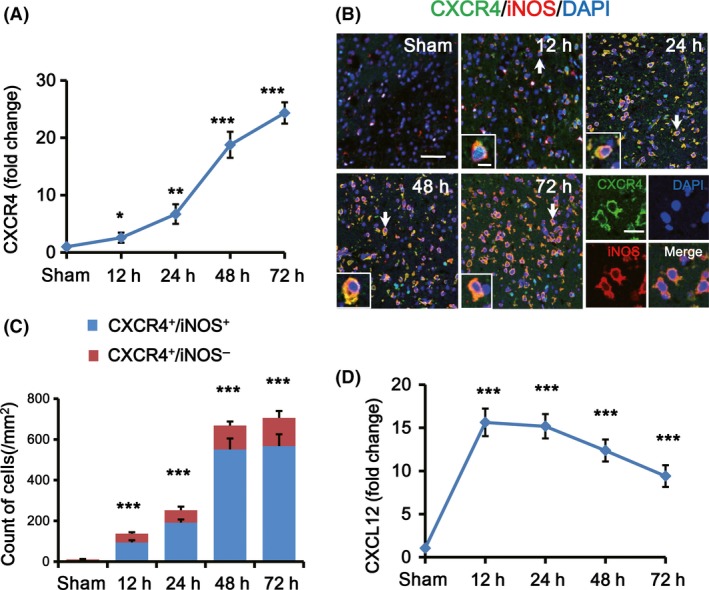
CXCR4 and CXCL12 were increased after ischemic stroke. (A) Quantification of CXCR4 mRNA expression in the IBZ by real‐time PCR after 12, 24, 48, and 72 h of reperfusion (n = 5 per group). Data are expressed as fold change versus sham. (B) Representative images of the IBZ at indicated time points after tMCAO double‐stained with CXCR4 (green) and iNOS (red) antibodies. DAPI staining (blue) was used to label the nucleus. Double positive cells indicated by arrows are shown at higher magnification in the left‐bottom panels. Scare bar: 40, 10, and 20 μm, respectively. (C) Quantification of CXCR4+/iNOS+ and CXCR4+/iNOS− cells per mm2 in the IBZ at indicated time points after tMCAO or sham treatment. (D) Quantification of CXCL12 mRNA expression in the IBZ by real‐time PCR after 12, 24, 48, and 72 h of reperfusion (n = 5 per group). Data are expressed as fold change versus sham. *P < 0.05, **P < 0.01 and ***P < 0.001 versus sham.
Migration of M1 Microglia was Induced via CXLC12/CXCR4 Pathway
To confirm the effect of CXCL12/CXCR4 pathway on M1 microglia, we used the transwell assay in vitro to assess whether inhibiting CXCR4 could disturb the chemotaxis of M1 microglia to CXCL12. Firstly, BV2 microglia was treated by LPS for 24 h to polarize them into M1 phenotype. Double immunostaining for Iba‐1 and iNOS showed that most of BV2 microglia was induced to M1 phenotype (Figure 3A). Additionally, the mRNA expression of M1‐associated inflammatory factors (IL‐1β, TNF‐α, iNOS, and CD86) also increased after LPS treatment (Figure 3B), which further revealed that LPS‐treated BV2 microglia acquired M1 phenotype characteristics.
Figure 3.
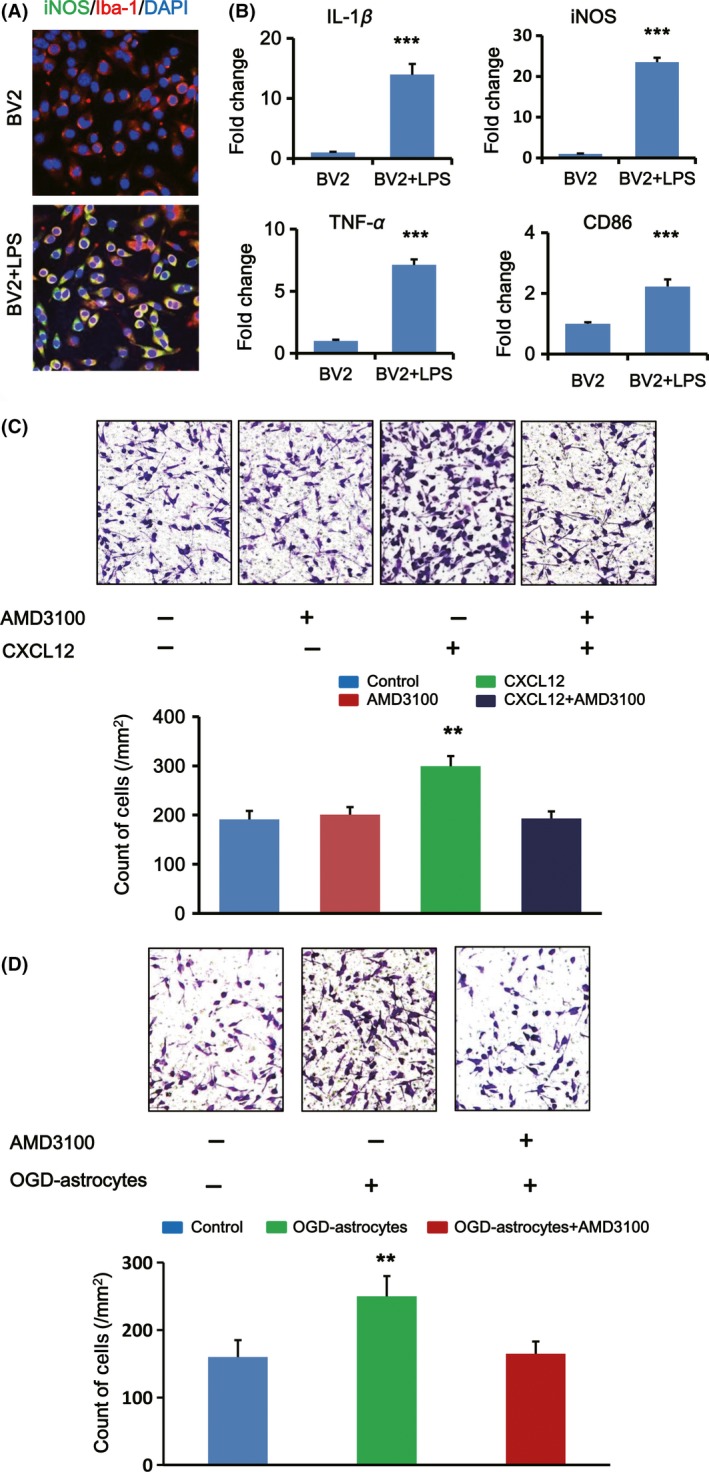
Migration of M1 microglia in vitro was induced via CXLC12/CXCR4 pathway. (A) Immunofluorescent double staining for iNOS (green) and Iba‐1 (red) of BV2 microglia in the absence or presence of LPS. The nuclei were stained with DAPI. (B) Quantification for mRNA expression of M1‐associated inflammatory cytokine (IL‐1β, iNOS, TNF‐α, and CD86) in BV2 microglia stimulated by LPS (n = 3 per group). Data are expressed as fold change versus un‐stimulated control. (C) Representative images showing the migration of M1 microglia on a transwell system affected by AMD3100 under OGD condition with or without CXCL12 (upper panel). Quantification of migrated M1 microglia count on the opposite surface of the upper chamber per mm2 (lower panel, n = 5 per group). (D) Representative images showing the migration of M1 microglia on a transwell system affected by AMD3100 under OGD condition with or without post‐OGD astrocytes (upper panel). Quantification of migrated M1 microglia count on the opposite surface of the upper chamber per mm2 (lower panel, n = 5 per group). ** P < 0.01, ***P < 0.001 versus control.
These LPS‐treated BV2 microglia (M1 microglia) were exposed under ODG condition for 3 h to imitate the ischemia‐reperfusion model. Then, we did the transwell assay with AMD3100 or vehicle in the upper chamber and CXCL12 or vehicle in the lower chamber. AMD3100 was used in this study as a highly selective CXCR4 antagonist. When adding vehicle to both chambers, only a few M1 microglia could transmigrate to the opposite surface of the upper chamber, while more M1 microglia migrated to the lower chamber when CXCL12 was added to the lower chamber (**P < 0.001; Figure 3C). However, after adding AMD3100 to the upper chamber, the migration effect of M1 microglia induced by CXCL12 was suppressed (Figure 3C).
Previous studies confirm that in stroke animal models, CXCL12 was released primarily by activated astrocytes and endothelial cells 18, 32. Considered the abundant activated astrocytes in the IBZ, post‐OGD astrocytes were cultured and seeded in the lower chambers of transwell system to imitate the in vitro ischemic environment. Results showed that OGD astrocytes could induce more M1 microglia to migrate to the opposite surface and AMD3100 could inhibit this phenomenon significantly compared with the control group (Figure 3D).
Disturbing CXLC12/CXCR4 Pathway at the Hyperacute Phase after Stroke could Inhibit M1 Microglia Migration and Inflammatory Responses in the IBZ
Then, we confirmed the effect of CXCL12/CXCR4 pathway on M1 microglia migration to the IBZ after ischemic stroke. Rats started to receive twice (6‐h interval) AMD3100 or vehicle injection at 12, 24, 48, and 72 h after tMCAO and were sacrificed 24 h after the last injection (Figure S3). The number of M1 microglia in the IBZ was determined by double immunofluorescence staining of anti‐Iba‐1 and anti‐iNOS (Figure 4A). Results showed that AMD3100 treatment at the hyperacute phase (12 and 24 h) after stroke could significantly reduce the count of M1 microglia in the IBZ compared to vehicle groups (12‐h‐treated group: 30.5 ± 6.2/mm2 vs. 75.1 ± 10.0/mm2, 24‐h‐treated group: 162.8 ± 27.2/mm2 vs. 359.6 ± 22.7/mm2; **P < 0.01; Figure 4B). While no obvious differences were observed in 48‐h‐ and 72‐h‐treated groups compared to vehicle groups (48‐h‐treated group: 578.9 ± 50.7/mm2 vs. 582.8 ± 39.4/mm2, 72‐h‐treated group: 552.6 ± 37.9/mm2 vs. 564.2 ± 26.5/mm2; Figure 4B).
Figure 4.
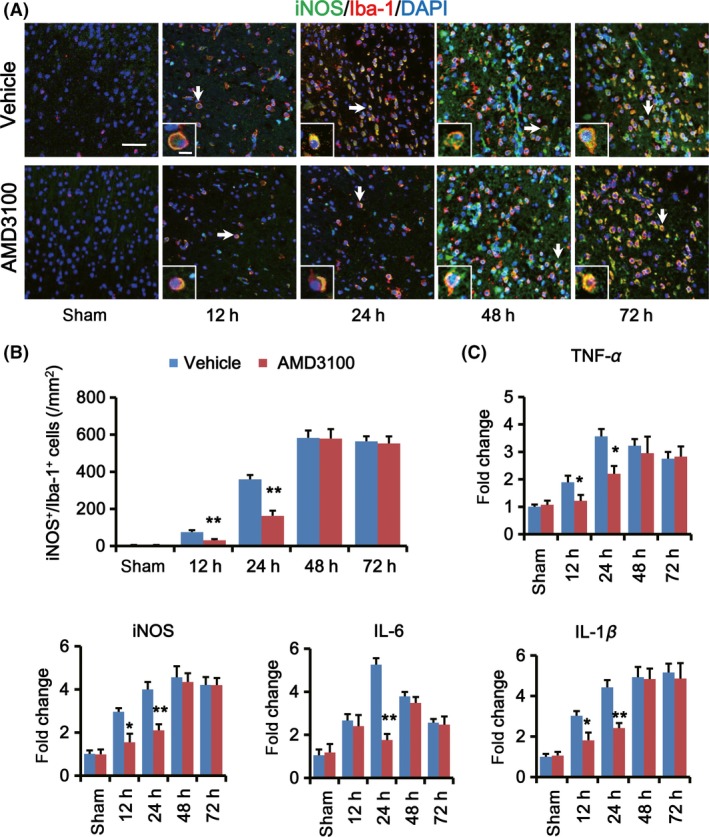
Disturbing CXLC12/CXCR4 pathway at the hyperacute phase after stroke could inhibit M1 microglia migration and inflammatory responses in the IBZ. (A) Representative images of the IBZ double‐stained with iNOS (green) and Iba‐1 (red) antibodies. DAPI staining (blue) was used to label the nucleus. Rats received AMD3100 or vehicle treatment twice with 6‐h interval at 12, 24, 48, and 72 h after tMCAO, and brain slices were prepared 24 h after last treatment. Double positive cells indicated by arrows are shown at higher magnification in the bottom‐left panels. Scare bar: 40 and 10 μm, respectively. (B) Quantification of the M1 microglia cells (iNOS+/Iba‐1+ cell) in the IBZ per mm2 at indicated time points after AMD3100 or vehicle treatment (n = 5 per group). (C) Quantification of mRNA expression for TNF‐α, iNOS, IL‐6, and IL‐1β in the IBZ at indicated time points after AMD3100 or vehicle treatment. Data are expressed as fold change versus sham (n = 5 per group). *P < 0.05, **P < 0.01, AMD3100 versus vehicle.
Further, the mRNA levels of M1‐associated inflammatory cytokines in the IBZ were examined by RT‐PCR. After AMD3100 treatment, the expression of TNF‐α, iNOS, and IL‐1β decreased significantly both in 12‐h‐ and 24‐h‐treated groups and IL‐6 expression decreased in 24‐h‐treated group (*P < 0.05, **P < 0.01; Figure 4C). However, there were no obvious changes in 48‐h‐ and 72‐h‐treated groups compared to vehicle‐treated groups (Figure 4C).
Disturbing CXLC12/CXCR4 Pathway at the Hyperacute Phase after Stroke could Improve Outcome of tMCAO Rats
We found that CM from M1 microglia significantly reduced the cell viability and MAP2 expression of post‐OGD neurons, while CM from M2 phenotype improved the cell viability in vitro, suggesting that M1 microglia could impede neuronal survival after ischemia (Figure S4). Then, we detected the effect of AMD3100 on infarct volume and neurological outcomes of tMCAO rats. As shown in Figure S3, rats were sacrificed 24 h after the last injection for TTC staining to quantify the infarct sizes (Figure 5A). Results showed that infarct volumes of tMCAO rats in the 12‐h‐treated group were attenuated significantly compared to the vehicle rats (*P < 0.05; Figure 5B). For 24‐h‐treated rats, the infarct size slightly decreased but had no significant differences compared to the vehicle rats; for 48‐h‐ and 72‐h‐treated groups, there were no significant differences (Figure 5B).
Figure 5.
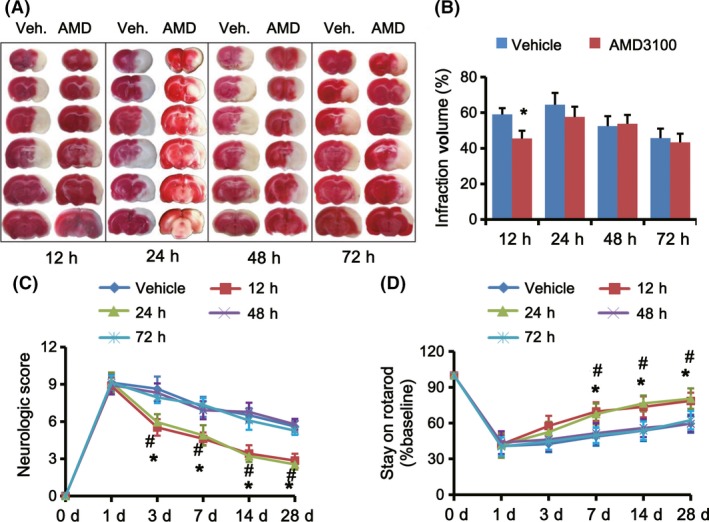
Disturbing CXLC12/CXCR4 pathway at the hyperacute phase after stroke could attenuate infarct volume and improve neurological outcome. (A) TTC staining for brain coronal sections of rats received AMD3100 (AMD) or vehicle (Veh.) treatment twice with 6‐h interval at 12, 24, 48, and 72 h after tMCAO shows the area sizes of cerebral infarct. Brain slices were prepared 24 h after the last treatment. (B) Percentage of the brain infarct volumes of tMCAO rats received AMD3100 or vehicle treatment (n = 10 per group). *P < 0.05 AMD3100 group versus vehicle group. Overall neurological severity scores (C) and the performance on rotarod tests (D) were analyzed throughout the indicated period of tMCAO rats treated with AMD3100 or vehicle at different time points (n = 10 per group). *P < 0.05, 12‐h‐treated group versus vehicle group; #P < 0.05, 24‐h‐treated group versus vehicle group.
Ischemia‐induced neurological deficits were evaluated by modified neurological severity scores and an accelerating rotarod test. We observed that among the AMD3100‐treated groups, 12‐h‐ and 24‐h‐treated rats had lower neurological severity scores at 3, 7, 14, and 28 days after stroke and a better rotarod performance at 7, 14, and 28 days after stroke compared to vehicle‐treated rats (*P < 0.05, 12‐h‐treated group vs. vehicle‐treated group; #P < 0.05, 24‐h‐treated group vs. vehicle‐treated group; Figure 5C,D). However, 48‐h‐treated and 72‐h‐treated groups did not exhibit better performance than vehicle‐treated rats.
Discussion
Inflammation induced by stroke is regarded as a major factor contributing to tissue damage 4. Cerebral ischemia causes a robust neuroinflammatory response that includes phenotyping of various endogenous CNS cell types (astrocytes, neurons, and microglia) and influx of leukocytic cells (neutrophils, macrophages, and T‐cells) from the periphery 33. As the resident immune cells in the CNS, microglia has versatile effectors under ischemic conditions, which is known to secrete proinflammatory cytokines (M1 phenotype) contributing to tissue damage and phagocytose debris (M2 phenotype) involving tissue repair 34. Selective modulation of microglia phenotype function could be a promising strategy to improve recovery after stroke.
In this study, we found that microglia, mainly the M1 phenotype in the IBZ, began to increase rapidly several hours after tMCAO. The main sources of M1 microglia cells in the IBZ within 48 h after stroke include both the activation of resident microglia and chemotaxis from surrounding brain tissue 35. Proliferation of original microglia and infiltration of blood‐derived macrophages were observed 48 h after ischemia, in which period the number of M2 microglia began to increase 36, 37, 38. Therefore, it might be taking measures to inhibit the increase of M1 microglia in the early phase of stroke in order to alleviate the subsequent inflammatory injury of stroke.
Our previous study demonstrated that the expression of chemokine CXCL12 in the IBZ was obviously increased over time after permanent MCAO 20. In this study, we also observed the same phenomenon in tMCAO model. Most importantly, we found CXCL12 could induce the migration of M1 microglia in vitro and this effect could be inhibited effectively by the antagonist of CXCR4, AMD3100. It reminded us that the migration of M1‐polarized microglia was carried out via CXCL12/CXCR4 signaling pathway. Then, to determine whether disturbing CXCL12/CXCR4 pathway could inhibit M1 microglia recruitment to the IBZ in vivo, we delivered AMD3100 to rats at different time points after tMCAO. We discovered that only AMD3100 treatment at the hyperacute phase (within 24 h after ischemia) could obviously inhibit the migration of M1 phenotype to the IBZ and suppress the production of inflammatory cytokines. Consistently, AMD3100 administered at the hyperacute phase could attenuate infarct volume and improve neurological outcomes of tMCAO rats. However, when AMD3100 was used later than 24 h, around 48 or 72 h after ischemia, M1 microglia had been abundantly recruited into the IBZ, and the benefits could not be achieved. Zhao et al. 39 even found that AMD3100 abrogated the enhanced neurogenesis and behavioral recovery by infusing of AMD3100 for 14 consecutive days. AMD3100 could also inhibit the migration of M2 microglia (shown in supplementary materials 5 and 6) which began to increase obviously 48 h after ischemia, as we previously mentioned 36, 37, 38. When AMD3100 was given after the hyperacute phase (for example 48 h and 72 h after tMACO), the count of M2 microglia could also be reduced; however, it had no significant change for AMD3100 treatment at hyperacute phase (for example, 12 h and 24 h after tMACO; shown in Figure S6). It indicated that inhibiting CXCR4/CXCL12 pathway after the hyperacute phase could inhibit M2 microglia migration. Maybe this is the reason why rats had no better neurological outcomes if we inhibited CXCR4/CXCL12 pathway after the hyperacute phase of stroke. Therefore, it highlights the importance of therapeutic time window which could last for 24 h after cerebral ischemia reperfusion.
The effects of AMD3100 in ischemic stroke had been studied previously 40, 41, 42. By blocking CXCL12/CXCR4 signaling, AMD3100 could contribute to attenuation of microglia activation, immune cells infiltration, and leukocyte migration. Ruscher et al. confirmed that AMD3100 treatment in mice 2 days after induction of photothrombosis could enhance recovery of lost neurological function through attenuating microglia activation 40, 41. Huang et al. 42 found that AMD3100 treatment for three consecutive days could reduce blood–brain barrier disruption and infarct volume in pMCAO mice by suppressing leukocyte migration, infiltration, and proinflammatory cytokine expression. In this study, we underscored the effects of AMD3100 on microglia migration at the hyperacute phase after tMCAO. The various mechanisms might be due to the different doses, concentrations, and ways of delivery of AMD3100 used. Another reason might be dependent on the experimental model (permanent vs. transient model) and species, including specific pharmacokinetics in mice and rats. Therefore, further studies are needed for elucidating the exact roles of AMD3100 in ischemic stroke.
In addition, we found that there were a few CXCR4+ cells in the IBZ which did not costain with microglia markers. Previous studies demonstrated that CXCR4 expression was increased in neural progenitor cells, neuroblasts, neurons, and endothelial progenitor cells after ischemic stroke 43, 44. In our study, which kind of cells these noncollocated CXCR4+ cells belonged to and what functions AMD3100 have on these cells during the hyperacute phase after stroke still need further studies. However, considering M1 microglia accounted for the majority percentage of CXCR4+ cells (above 80% shown in figure 2C), it is reasonable to think that AMD3100 mainly affects the M1 microglia at the hyperacute phase after stroke.
Collectively, our study provides evidence that inhibiting the migration of M1 microglia to the IBZ at the hyperacute phase of stroke could significantly reduce the infarct size and promote recovery of neurological function. Our results support the idea that CXCR4 antagonist AMD3100 might be exploited in future approaches to improve rehabilitative treatment for stroke patients, especially for thrombolysis patients with reperfusion injury.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. H&E staining of coronal section of rat brain in low magnification showing the area of the infarct core (IC) and the ischemic boundary zone (IBZ) after tMCAO.
Figure S2. Expression of M2‐associated anti‐inflammatory cytokines in the IBZ within the first 72 h after ischemic stroke (A‐D) Quantification of M2‐associated anti‐inflammatory cytokine (Arg‐1, IL‐10, CD206 and TGF‐β) expression in the IBZ by real‐time PCR at 12, 24, 48 and 72 h after tMCAO or sham treatment (n = 5 per group).
Figure S3. AMD3100 treatment diagram.
Figure S4. M1‐polarized microglia impeded post‐OGD neuronal survival.
Figure S5. Migration of M2 microglia in vitro was induced via CXLC12/CXCR4 pathway.
Figure S6. The effects of disturbing CXLC12/CXCR4 pathway on M2 microglia migration.
Acknowledgments
The authors thank the support of National Natural Science Foundation of China (Grants: 81571119, and 81371311 to BH, 81301165 to JHJ), National Key Research and Development Program of China (2016YFC1300600), and New Century Excellent Talents in University (Grants: NCET‐10‐0406 to BH).
The first two authors contributed equally to this work.
Contributor Information
Hui‐Juan Jin, Email: jinhuijuan1983@163.com.
Bo Hu, Email: hubo@mail.hust.edu.cn.
References
- 1. Mabuchi T, Kitagawa K, Ohtsuki T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000;31:1735–1743. [DOI] [PubMed] [Google Scholar]
- 2. Ginsberg MD, Pulsinelli WA. The ischemic penumbra, injury thresholds, and the therapeutic window for acute stroke. Ann Neurol 1994;36:553–554. [DOI] [PubMed] [Google Scholar]
- 3. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 4. Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron 2010;67:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol 2006;66:232–245. [DOI] [PubMed] [Google Scholar]
- 6. Doll DN, Barr TL, Simpkins JW. Cytokines: Their role in stroke and potential use as biomarkers and therapeutic targets. Aging Dis 2014;5:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol 2013;2013:746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Durham JT, Surks HK, Dulmovits BM, Herman IM. Pericyte contractility controls endothelial cell cycle progression and sprouting: Insights into angiogenic switch mechanics. Am J Physiol Cell Physiol 2014;307:C878–C892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J Neuroinflammation 2014;11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation 2011;8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi K, Imagama S, Ohgomori T, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 2013;4:e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS‐derived interleukin‐4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci 2007;27:10714–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Denker SP, Ji S, Dingman A, et al. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J Neurochem 2007;100:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanabe S, Heesen M, Yoshizawa I, et al. Functional expression of the CXC‐chemokine receptor‐4/fusin on mouse microglial cells and astrocytes. J Immunol 1997;159:905–911. [PubMed] [Google Scholar]
- 15. Ohtani Y, Minami M, Kawaguchi N, et al. Expression of stromal cell‐derived factor‐1 and CXCR4 chemokine receptor mRNAs in cultured rat glial and neuronal cells. Neurosci Lett 1998;249:163–166. [DOI] [PubMed] [Google Scholar]
- 16. Mennicken F, Maki R, de Souza EB, Quirion R. Chemokines and chemokine receptors in the CNS: A possible role in neuroinflammation and patterning. Trends Pharmacol Sci 1999;20:73–78. [DOI] [PubMed] [Google Scholar]
- 17. Lipfert J, Odemis V, Wagner DC, Boltze J, Engele J. CXCR4 and CXCR7 form a functional receptor unit for SDF‐1/CXCL12 in primary rodent microglia. Neuropathol Appl Neurobiol 2013;39:667–680. [DOI] [PubMed] [Google Scholar]
- 18. Hill WD, Hess DC, Martin‐Studdard A, et al. SDF‐1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J Neuropathol Exp Neurol 2004;63:84–96. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Li C, Chen Y, et al. Hypoxia enhances CXCR4 expression favoring microglia migration via HIF‐1alpha activation. Biochem Biophys Res Commun 2008;371:283–288. [DOI] [PubMed] [Google Scholar]
- 20. Mao L, Huang M, Chen SC, et al. Endogenous endothelial progenitor cells participate in neovascularization via CXCR4/SDF‐1 axis and improve outcome after stroke. CNS Neurosci Ther 2014;20:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 22. Hendrix CW, Flexner C, MacFarland RT, et al. Pharmacokinetics and safety of AMD‐3100, a novel antagonist of the CXCR‐4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother 2000;44:1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liles WC, Broxmeyer HE, Rodger E, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 2003;102:2728–2730. [DOI] [PubMed] [Google Scholar]
- 24. Devine SM, Flomenberg N, Vesole DH, et al. Rapid mobilization of CD34 + cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non‐Hodgkin's lymphoma. J Clin Oncol 2004;22:1095–1102. [DOI] [PubMed] [Google Scholar]
- 25. Elvington A, Atkinson C, Zhu H, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol 2012;189:4640–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen LH, Li Y, Chen J, et al. Intracarotid transplantation of bone marrow stromal cells increases axon‐myelin remodeling after stroke. Neuroscience 2006;137:393–399. [DOI] [PubMed] [Google Scholar]
- 27. Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 2001;32:1005–1011. [DOI] [PubMed] [Google Scholar]
- 28. Liu HC, Zheng MH, Du YL, et al. N9 microglial cells polarized by LPS and IL4 show differential responses to secondary environmental stimuli. Cell Immunol 2012;278:84–90. [DOI] [PubMed] [Google Scholar]
- 29. Li Y, Xia Y, Wang Y, et al. Sonic hedgehog (Shh) regulates the expression of angiogenic growth factors in oxygen‐glucose‐deprived astrocytes by mediating the nuclear receptor NR2F2. Mol Neurobiol 2013;47:967–975. [DOI] [PubMed] [Google Scholar]
- 30. Gerlier D, Thomasset N. Use of MTT colorimetric assay to measure cell activation. J Immunol Methods 1986;94:57–63. [DOI] [PubMed] [Google Scholar]
- 31. Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063–3070. [DOI] [PubMed] [Google Scholar]
- 32. Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF‐1 induction of SDF‐1. Nat Med 2004;10:858–864. [DOI] [PubMed] [Google Scholar]
- 33. Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Future Neurol 2010;5:227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 2000;20:53–65. [DOI] [PubMed] [Google Scholar]
- 35. Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol 2015;6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2005;196:290–297. [DOI] [PubMed] [Google Scholar]
- 37. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003;183:25–33. [DOI] [PubMed] [Google Scholar]
- 38. Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007;27:1941–1953. [DOI] [PubMed] [Google Scholar]
- 39. Zhao S, Qu H, Zhao Y, et al. CXCR4 antagonist AMD3100 reverses the neurogenesis and behavioral recovery promoted by forced limb‐use in stroke rats. Restor Neurol Neurosci 2015;33:809–821. [DOI] [PubMed] [Google Scholar]
- 40. Walter HL, van der Maten G, Antunes AR, Wieloch T, Ruscher K. Treatment with AMD3100 attenuates the microglial response and improves outcome after experimental stroke. J Neuroinflammation 2015;12:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruscher K, Kuric E, Liu Y, et al. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J Cereb Blood Flow Metab 2013;33:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. CXCR4 antagonist AMD3100 protects blood‐brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013;44:190–197. [DOI] [PubMed] [Google Scholar]
- 43. Li Y, Huang J, He X, et al. Postacute stromal cell‐derived factor‐1alpha expression promotes neurovascular recovery in ischemic mice. Stroke 2014;45:1822–1829. [DOI] [PubMed] [Google Scholar]
- 44. Ardelt AA, Bhattacharyya BJ, Belmadani A, Ren D, Miller RJ. Stromal derived growth factor‐1 (CXCL12) modulates synaptic transmission to immature neurons during post‐ischemic cerebral repair. Exp Neurol 2013;248:246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. H&E staining of coronal section of rat brain in low magnification showing the area of the infarct core (IC) and the ischemic boundary zone (IBZ) after tMCAO.
Figure S2. Expression of M2‐associated anti‐inflammatory cytokines in the IBZ within the first 72 h after ischemic stroke (A‐D) Quantification of M2‐associated anti‐inflammatory cytokine (Arg‐1, IL‐10, CD206 and TGF‐β) expression in the IBZ by real‐time PCR at 12, 24, 48 and 72 h after tMCAO or sham treatment (n = 5 per group).
Figure S3. AMD3100 treatment diagram.
Figure S4. M1‐polarized microglia impeded post‐OGD neuronal survival.
Figure S5. Migration of M2 microglia in vitro was induced via CXLC12/CXCR4 pathway.
Figure S6. The effects of disturbing CXLC12/CXCR4 pathway on M2 microglia migration.