

Summary
Aims
Although oxidized low‐density lipoprotein (ox‐LDL) in the brain induces neuronal death, the mechanism underlying the damage effects remains largely unknown. Given that the ultimate outcome of a cell is depended on the balance between autophagy and apoptosis, this study was performed to explore whether ox‐LDL induced HT‐22 neuronal cell damage via autophagy impairment and apoptosis enhancement.
Methods
Flow cytometry and transmission electron microscopy (TEM) were used to evaluate changes in cell apoptosis and autophagy, respectively. The protein expression of LC3‐II, p62, Bcl‐2, and Bax in HT‐22 cells was measured by Western bolt analysis.
Results
Our study confirmed that 100 μg/mL of ox‐LDL not only promoted TH‐22 cell apoptosis, characterized by elevated cell apoptosis rate and Bax protein expression, decreased Bcl‐2 protein expression, and damaged cellular ultrastructures, but also impaired autophagy as indicated by the decreased LC3‐II levels and the increased p62 levels. Importantly, all of these effects of ox‐LDL were significantly aggravated by cotreatment with chloroquine (an inhibitor of autophagy flux). In contrast, cotreatment with rapamycin (an inducer of autophagy) remarkably reversed these effects of ox‐LDL.
Conclusions
Taken together, our results indicated that ox‐LDL‐induced shift from autophagy to apoptosis contributes to HT‐22 cell damage.
Keywords: apoptosis, autophagy, cell damage, HT‐22 cell, oxidized low‐density lipoprotein
1. Introduction
Neurodegenerative diseases, such as Alzheimer's disease (AD) and Parkinson's disease (PD), are associated with substantial neuronal loss, which predicts the severity of clinical symptoms.1, 2 Numerous studies show that hypercholesterolemia is involved in neuronal loss and neurodegenerative disorders.3, 4 Recently, growing attention has been focused on the role of oxidized low‐density lipoprotein (ox‐LDL) in the development of neurodegenerative diseases. As we know, the LDL in the brain is prone to oxidative modifications and formation of ox‐LDL.5, 6 ox‐LDL is recognized by scavenger receptors (SRs), such as SR‐A and SR‐B, which are present on neuronal cells.7 Although high dose of ox‐LDL in the brain induced neuronal death and loss, the mechanism by which ox‐LDL contributes to neuronal loss in AD is not clearly understood.
Among three types of cell death, the effect of apoptosis on the pathogenesis of neuronal loss in neurodegenerative diseases has been extensively studied.8 Neuronal apoptosis is featured morphologically by condensed chromatin balls, damaged cytoplasmic organelles, and apoptotic bodies.9, 10 However, apoptosis is not the only factor that determines a neuron's fate. Currently, increasing studies have focused on the effect of autophagy on the loss of neurons. Autophagy is a conserved procedure that delivers intracellular contents to lysosomes for degradation.9, 11 It is important for maintaining neuronal homeostasis through degrading the aggregate‐prone proteins such as ox‐LDL and may protect neuronal cells from apoptosis. Accumulating evidence indicates that autophagy can protect neuronal cell damage, and autophagy dysfunction may be involved in neurodegenerative diseases such as AD, PD, Huntington's disease.12, 13, 14 Recent studies have showed that the cross talk between apoptosis and autophagy is mediated by the interconnected molecular regulators such as mTOR, Bcl‐2, and Bax.15, 16, 17 For an instance, Bcl‐2/Bax pathway regulates the cross talk between both.18 Therefore, the ultimate outcome of a cell is determined by the balance of autophagy and apoptosis.
Several studies have revealed that ox‐LDL inhibits autophagy and promotes apoptosis in human umbilical vein endothelial cells.19, 20 Our previous studies showed that THP‐1 cell exposure to ox‐LDL (100 μg/mL) for 24 hours exhibited autophagy flux impairment, intracellular ox‐LDL accumulation, and cell injury.21 These effects of ox‐LDL were rescued by pretreatment with the autophagy inducer rapamycin (RAP) and aggravated by autophagy flux inhibitor chloroquine (CQ). However, the association between autophagy and apoptosis in HT‐22 cells exposed to ox‐LDL remains poorly understood. Therefore, we hypothesize that ox‐LDL promotes HT‐22 cell apoptosis via autophagy inhibition and apoptosis enhancement. To confirm this hypothesis, we first explored the effects of ox‐LDL on cell viability, apoptosis, and autophagy of HT22 cells. Then, we assessed the effects of autophagy flux inhibitor CQ and autophagy inducer RAP on the expression of autophagy‐related proteins and apoptosis‐related proteins, and apoptosis rate in HT22 cells exposed to ox‐LDL. Our results establish apoptosis enhancement and autophagy impairment in ox‐LDL‐treated HT22 cells and suggest a link between apoptosis and autophagy in ox‐LDL‐induced neurotoxicity.
2. Materials and methods
2.1. Materials
Rapamycin and CQ were supplied by Sigma Chemical Co. (St. Louis, MO, USA). ox‐LDL was purchased from Yi‐yuan Biotechnology (Guangdong, China). Cell counter kit‐8 (CCK‐8) was bought from Dojindo Laboratories (Kumamoto, Japan). Annexin V Apoptosis Detection Kit was obtained from eBioscience (San Diego, CA, USA). Specific monoclonal anti‐LC3 I/II, SQSTM1/P62, Bcl‐2, and Bax antibody were purchased from Cell Signaling Technology, Inc (Beverly, MA, USA). High‐glucose Dulbecco's modified Eagle's medium (DEME) and fetal bovine serum (FBS) were supplied by Gibco BRL (Ground Island, NY, USA).
2.2. Cell culture and treatment
HT‐22 cells, a mouse hippocampal neuronal cell line,22 were supplied by the Experimental Animal Center of Sun Yat‐sen University (Guangzhou, China). Cells were cultured in DMEM medium (Sigma) supplemented with 10% FBS in 10‐cm2 dishes and incubated at 37°C in atmosphere containing 5% CO2. Cells at 60% confluence were treated with ox‐LDL. Native LDL (nLDL), 100 μg/mL, was used as a control. For CQ and RAP treatment, CQ and RAP were prepared before use in 3 mM and 4 μM stocks, respectively, and diluted in medium to the indicated final concentrations. HT‐22 cells were treated with 100 μg/mL of ox‐LDL in the presence CQ (30 μM) or RAP (400 nM) for 24 hours. ox‐LDL treatment groups were supplemented with an equal amount of DMSO as a control. After 24‐hour co‐incubation, cells were detected as described later.
2.3. Determination of cell viability
The viability of HT‐22 cells was determined by CCK‐8 assay according to the manufacturer's instructions. Briefly, HT‐22 cells were plated in 96‐well plates at a concentration of 5 × 103 cells/well and cultured in DMEM medium containing 2% FBS. When HT‐22 cells were almost 60% confluent, the cells were treated with indicated agents. After treatment with specific agents, 5 μL of CCK‐8 solutions was added to each well and incubated at 37°C for an additional 2 hours. Subsequently, the optical density (OD) of each well was measured at 450 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
2.4. Flow cytometry analysis of apoptosis with PI and annexin V double staining
HT‐22 cells were digested with 0.05% trypsin‐EDTA (Gibco Life Technologies) and collected in Eppendorf tubes. All cells were washed twice with cold PBS by centrifugation at 168 g for 5 minutes. After discarding the supernatants, the collected HT‐22 cells were resuspended in 400 μL binding buffer at a concentration of 10 × 105 cells/mL. Then, PI and annexin V double‐staining apoptosis assay kit was used to quantify apoptotic cells. Apoptosis rate was assessed by counting the apoptotic cell number per 1 × 104 cells using flow cytometry (FCM, BD Bioscience).
2.5. Transmission electron microscopy
After washing twice with ice‐cold PBS, HT‐22 cells were fixed with 2.5% glutaraldehyde in 0.15 mM sodium cacodylate at 4°C overnight. The cells were postfixed in 2% osmium tetroxide. All samples were dehydrated in ethanol and embedded in epoxy resin. Then, ultrathin sections (70 nm) of adherent cells were performed on an ultramicrotome. The sections were counterstained with uranyl acetate and lead citrate and observed using a Jeol JEM SX 100 electron microscope (Jeol, Tokyo, Japan) with images captured.
2.6. Western blot analysis
Western blot analyses were performed as described previously.21 Briefly, HT‐22 cells were harvested after treatment with indicated agents and washed twice with cold PBS. Total proteins were extracted with a lysis buffer [20 mM Tris‐HCL, pH 7.5, 150 mM NaCl, 1% Triton X‐100, 1 mM phenylmethylsulphonylfluoride (PMSF), 1 mM Na3VO4, leupeptin, and EDTA] according to the manufacturer's indications. The samples were centrifuged at 2016 g for 10 minutes at 4°C, and the supernatant was collected for Western blots. Protein concentration was assessed using a BCA protein assay kit (ComWin Biotech, Beijing, China). Samples were denatured for 5 minutes in boiling buffer. Equal amounts of the boiled proteins (20‐30 μg per lane) were separated by 10% SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE). And then, the proteins were transferred to a PVDF membrane and blocked in TBS‐T buffer (50 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20) containing 5% bovine serum albumin (BSA, Sigma) for 2 hours. Subsequently, membranes were incubated overnight at 4°C with primary antibodies (anti‐LC3‐I/II, 1:1000; anti‐SQSTML/P62, 1:1000; anti‐Bcl‐2, 1:1000; anti‐Bax, 1:1000; anti‐β‐actin, 1:2000). After washing with TBST for three times, the membranes were incubated with anti‐rabbit secondary antibody conjugated to horseradish peroxidase (1:5000) for 2 hours. The bands were visualized using an enhanced chemiluminescence system (ECL, Millipore, Boston, MA, USA). The quantitative analysis of each bolt was carried out by Sigma Scan Pro5 software (San Jose, CA, USA) and normalized to that of β‐actin.
2.7. Statistical analysis
Data were expressed as mean ± SEM, and the significance of intergroup differences was evaluated by one‐way analysis of variance (ANOVA: least‐significant difference's test for post hoc comparisons). Differences were considered statistically significant at P<.05.
3. Results
3.1. ox‐LDL decreased cell viability of HT‐22 cells
Considerable researches indicated that ox‐LDL can cause neuronal cell death.23, 24, 25 To understand the neurotoxicity of ox‐LDL on HT‐22 cells, we incubated the HT‐22 cells with different concentrations of ox‐LDL (0, 12.5, 25, 50, 100 μg/mL) or 100 μg/mL of nLDL for 24 hours and then detected the cell viability by CCK‐8 kit. CCK‐8 assay indicated that 100 μg/mL of nLDL had no remarkable influence on HT‐22 cell viability (Figure 1). However, HT22 cell activity was significantly decreased by ox‐LDL in a dose‐dependent manner (at the range of 12.5‐100 μg/mL) with the maximal effect at 100 μg/mL (Figure 1). Therefore, 100 μg/mL of ox‐LDL was used to treat HT‐22 cells for 24 hours in subsequent experiments. Taken together, our data suggested that treatment with ox‐LDL (100 μg/mL) for 24 hours notably lowered the cell viability of HT‐22 cells.
Figure 1.
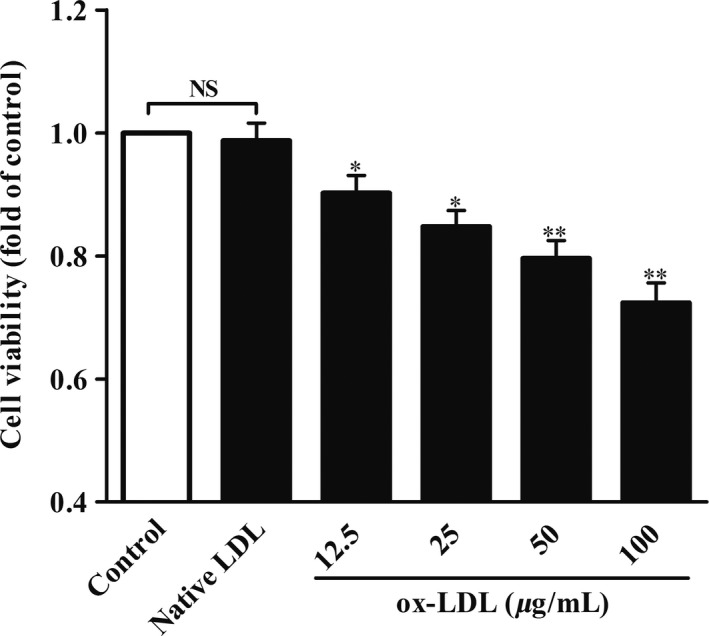
Effects of ox‐LDL on cell viability in HT‐22 cells. HT‐22 cells were treated with different concentrations of native LDL (100 μg/mL) or ox‐LDL (0, 12.5, 25, 50, 100 μg/mL) for 24 hours. Cell viability was determined by CCK‐8 assay. All the data were shown as mean ± SEM of three independent experiments. NS, no significant difference. *P<.05, **P<.01
3.2. Autophagy flux was impaired in ox‐LDL‐induced HT‐22 cells
To investigate the contribution of autophagy to HT‐22 cell injury, we first characterized the autophagy changes between control group and ox‐LDL‐treated group of HT‐22 cells by comparing LC3‐II and p62 protein levels using Western blot assay. Impaired autophagy can be identified by decreased expression of LC3‐II and increased expression of p62 protein. Compared with the control group, LC3‐II levels (Figure 2A,C) in ox‐LDL‐treated HT‐22 cells were obviously declined, while p62 levels were significantly elevated (Figure 2B,D). These findings indicated that ox‐LDL suppressed autophagy. To further confirm whether autophagy flux impairment occurred in ox‐LDL‐treated HT‐22 cells, CQ (CQ, an inhibitor of autophagy flux) and rapamycin (Rap, an inducer of autophagy) were utilized to block and activate autophagy in HT‐22 cells, respectively. Our results revealed that cotreatment with CQ slightly increased LC3‐II (Figure 2A,C) and p62 levels (Figure 2B,D) in ox‐LDL‐treated HT‐22 cells, indicating that ox‐LDL treatment caused a defective autophagy flux in HT‐22 cells. However, cotreatment with RAP could rescue the impaired autophagy flux in ox‐LDL‐treated HT‐22 cells, as characterized by elevated LC3‐II and lowered p62 levels.
Figure 2.
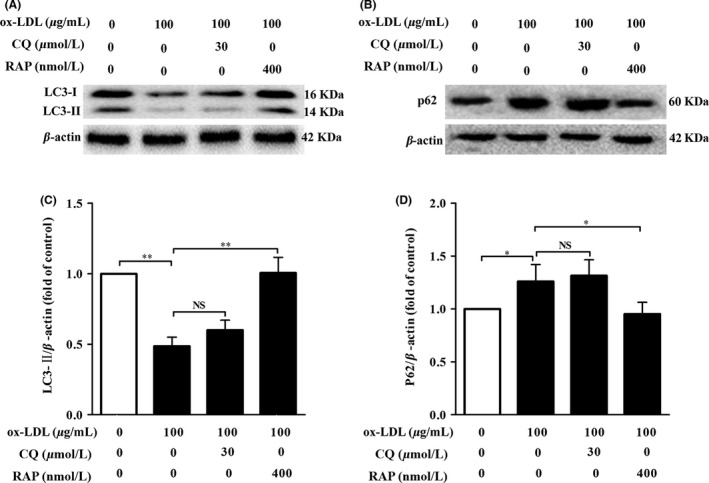
ox‐LDL caused impairment of autophagy flux in HT‐22 cells. HT‐22 cells were treated with ox‐LDL (100 μg/mL) in the presence of chloroquine (30 μM) or rapamycin (400 nM) for 24 hours. Cell lysates were harvested and analyzed by Western blotting assay for LC3‐I (16 kDa) and LC3‐II (14 kDa) (A), and p62 (60 kDa) (B) protein levels. Each lane was loaded with 20 μg of proteins in all experiments. (C) and (D) LC3‐II, and p62 levels were quantified with Sigma Scan Pro5 software. Values are the mean ± SEM. (n=3). NS, no significant difference. *P<.05, **P<.01
3.3. ox‐LDL damaged the ultrastructures of HT‐22 cells
To explore whether ox‐LDL‐induced autophagy flux defect was associated with HT‐22 cell damage, we observed the morphological‐ultrastructural changes under the transmission electron microscope (TEM). Our results indicated that the number of autophagosome (AP), a double‐membrane vesicle enclosing a portion of cytoplasm and organelles, in ox‐LDL group was less than that in control group (Figure 3A,B). Meanwhile, those cells in ox‐LDL group exhibited the features of damaged cells, characterized by cell shrinkage, mitochondria swelling, and chromatin condensation. Compared with ox‐LDL group, cotreatment with CQ caused a slight increase in the number of APs and further exacerbated morphological‐ultrastructural alterations. In contrast, cotreatment with RAP markedly improved the ultrastructural features of TH‐22 cells as compared with those of ox‐LDL group. These results suggested that autophagy flux impairment was involved in 100 μg/mL of ox‐LDL‐induced HT‐22 neuronal cell injury.
Figure 3.
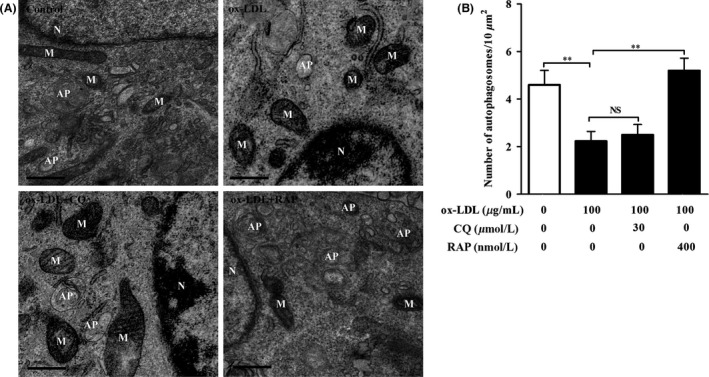
ox‐LDL resulted in the ultrastructure damage to HT‐22 cells. TEM was used to observe ultrastructure alteration in ox‐LDL‐treated HT‐22 cells. (A) HT‐22 cells were treated with ox‐LDL (100 μg/mL) in the presence of chloroquine (30 μM) or rapamycin (400 nM) for 24 hours. Mitochondria (M), nucleus (N), and autophagosome (AP) were indicated. (B) Average number of APs was quantified (n=10 cells/group). Values are the mean ± SEM. NS, no significant difference. **P<.01. Scale bar, 500 nm
3.4. Autophagy activation rescued HT‐22 cells from ox‐LDL‐induced decrease in cell viability
To further assess neuroprotective effects of autophagy against ox‐LDL‐induced cell injury, HT‐22 cells were treated with ox‐LDL (100 μg/mL) together with autophagy flux inhibitor CQ (30 μM) or autophagy inducer RAP (400 nM) for 24 hours, followed by CCK‐8 assay. Cell viability of HT‐22 cells in ox‐LDL‐treated group was profoundly declined as compared with that in untreated group (Figure 4). As expected, coadministration with CQ significantly reduced cell activity of HT‐22 cells induced by ox‐LDL (Figure 4), suggesting that blockage of autophagy flux in HT‐22 cells aggravated neurotoxic effects of ox‐LDL. However, compared with HT‐22 cells in ox‐LDL group, cell viability of HT‐22 cells in cotreatment of RAP and ox‐LDL group remarkably increased (Figure 4), implying that activation of autophagy ameliorated neurotoxic effects of ox‐LDL. These data demonstrate that autophagy plays a protective role against neurotoxicity of ox‐LDL.
Figure 4.
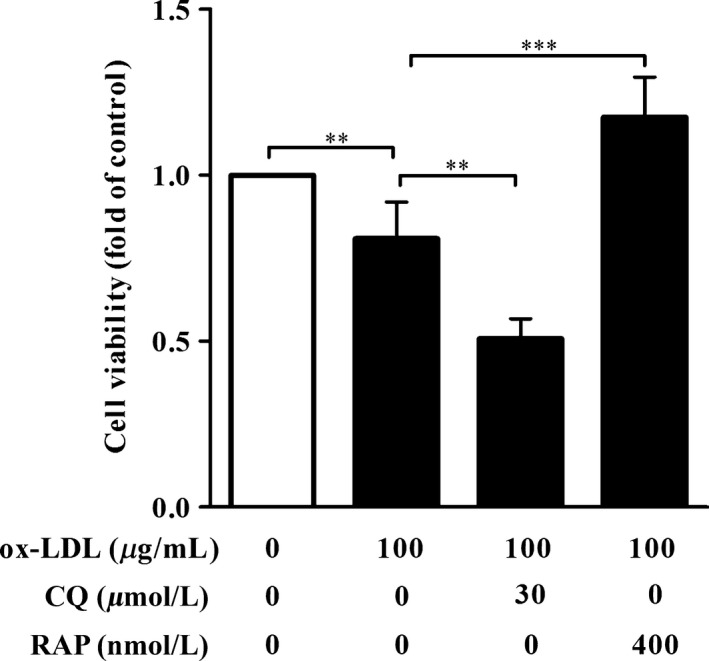
Autophagy protected HT‐22 cells against a decrease in cell viability induced by ox‐LDL. HT‐22 cells were treated with ox‐LDL (100 μg/mL) in the presence of chloroquine (30 μM) or rapamycin (400 nM) for 24 hours. Cell viability was detected by CCK‐8 assay. Data are expressed as mean ± SEM. (n=3). **P<.01, *** P<.001
3.5. Autophagy activation alleviated ox‐LDL‐induced cell apoptosis in HT‐22 cells
To further confirm the relationship between autophagy and cell apoptosis in HT‐22 cells exposed to ox‐LDL (100 μg/mL), apoptotic cells of each group were assessed by flow cytometry with annexin V/PI double‐staining method (Figure 5). Flow cytometry analysis demonstrated that untreated HT‐22 cells had very low apoptotic rates. As expected, HT‐22 cell apoptotic rates of ox‐LDL treatment group were significantly higher than those of the untreated group. Furthermore, HT‐22 cell apoptotic rates in cotreatment of CQ and ox‐LDL group significantly elevated as compared with those in ox‐LDL group, indicating that blockage of autophagy flux aggravated ox‐LDL‐caused HT‐22 cell apoptosis. In contrast, the cell apoptotic rates of HT‐22 cells in RAP treatment group obviously lowered as compared with those in ox‐LDL treatment group, implying that activation of autophagy could protect HT‐22 cells from cell apoptosis induced by ox‐LDL. Collectively, these results suggest that autophagy exerts a protective role against ox‐LDL‐induced cell apoptosis of HT‐22 cells.
Figure 5.
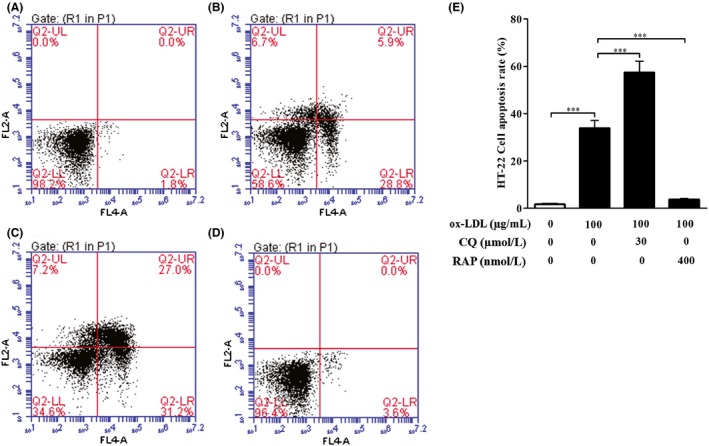
Effect of autophagy on HT‐22 cell apoptosis induced by ox‐LDL. Flow cytometry was used to measure the effect of autophagy on HT‐22 cell apoptosis induced by ox‐LDL. (A–D) HT‐22 cells were treated with ox‐LDL (100 μg/mL) in the presence of chloroquine (30 μM) or rapamycin (400 nM) for 24 hours. Apoptosis of HT‐22 cells was assessed by flow cytometry as described in 2 section. (A) Control (untreated) group; (B) ox‐LDL group; (C) ox‐LDL + CQ group; (D) ox‐LDL + RAP group. (E) HT‐22 cell apoptotic rates were quantified as described in 2 section. All the data were shown as mean ± SEM of three independent experiments. *** P<.001
3.6. ox‐LDL elevated Bax/Bcl‐2 ratio which could be aggravated by CQ and rescued by RAP
Because decreases in Bcl‐2 and increases in Bax levels can promote cell apoptosis, in this study, Western blot assay was carried out to evaluate changes in Bax and Bcl‐2 protein expression in HT‐22 cells. Bcl‐2 levels notably declined and Bax levels significantly elevated in ox‐LDL‐treated HT‐22 cells as compared with those in untreated ones, respectively (Figure 6). Furthermore, this trend was more significant in ox‐LDL combined with CQ group than that in ox‐LDL group, indicated by higher Bax and lower Bcl‐2 levels. However, this trend remarkably reversed in ox‐LDL combined with RAP group, demonstrated by increased Bcl‐2 and decreased Bax levels in HT‐22 cells. Taken together, these results demonstrate that autophagy plays a critical role in regulating the expressions of Bcl‐2 and Bax in ox‐LDL‐induced HT‐22 cells.
Figure 6.
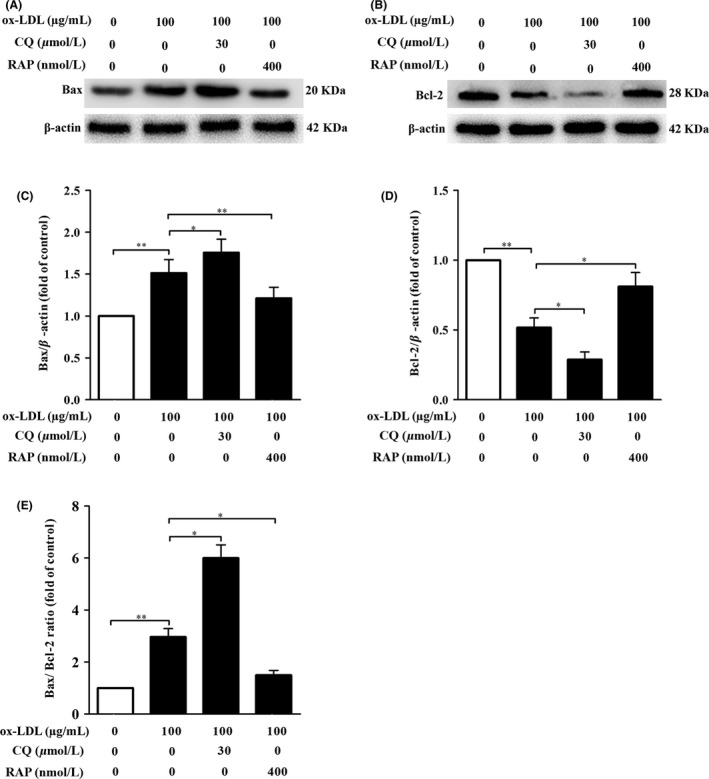
Effects of CQ and RAP on the expression of apoptosis‐related proteins Bcl‐2 and Bax in HT‐22 cells induced by ox‐LDL. (A) and (B) HT‐22 cells were treated with ox‐LDL (100 μg/mL) in the presence of chloroquine (30 μM) or rapamycin (400 nM) for 24 hours. After 24‐hour treatment, proteins were extracted from cell lysates and analyzed by Western blotting assay for Bax (20 kDa) and Bcl‐2 (28 kDa). Each lane was loaded with 30 μg of proteins for all experiments. (C–E) The relative optical density values of Bax and Bcl‐2 to β‐actin were quantified with Sigma Scan Pro5 software, respectively. All the data were shown as mean ± SEM of three independent experiments. * P<.05, ** P<.01
4. Discussion
ox‐LDL has been well known for its ability to induce neuronal injury and loss. However, the precise mechanism by which ox‐LDL contributes to neuronal death has not been elucidated. Recent studies have indicated that induction of autophagy can prevent the neural cell death and injury in brain and neurodegenerative diseases.12, 26, 27 Thus, we investigated the role of autophagy in regulating ox‐LDL‐induced neuronal cell viability and apoptosis. In this work, we found that ox‐LDL obviously declined the cell viability and elevated cell apoptotic rates of HT‐22 cells. Furthermore, ox‐LDL significantly impaired autophagy flux and decreased Bcl‐2 and increased Bax levels in HT‐22 cells. Cotreatment with autophagy flux inhibitor CQ further aggravated ox‐LDL‐induced HT‐22 cell injury and apoptosis. In contrast, cotreatment of autophagy inducer RAP led to activating autophagy, enhancing cell viability and decreasing apoptotic rate, and lowering Bax expression. Collectively, our findings revealed that disruption of autophagy may be a crucial mechanism of ox‐LDL‐induced HT‐22 cell damage and apoptosis.
Autophagy is essential for maintaining neuronal homeostasis and is disturbed by oxidative stress. In the present study, we first explored the effects of ox‐LDL on HT‐22 cell damage and autophagy. Our findings demonstrated that exposure of HT‐22 cells to ox‐LDL decreased cell viability and increased cell apoptotic rate, indicating the neurotoxicity of ox‐LDL. Recently, ox‐LDL has been proved to directly promote neuronal injury,28 negatively affect memory of animals,29 and be closely related to the major depression and neurodegenerative diseases.30 All these above provide strong evidences to support the neurotoxicity of ox‐LDL. The mechanism for ox‐LDL‐induced neuronal injury and death is still unclear. ox‐LDL‐induced increases in reactive oxygen species, calcium, and caspases might be associated with this neurotoxicity. Notably, our results indicated that 100 μg/mL of ox‐LDL treatment for 24 hours significantly impaired autophagy of HT‐22 cells, indicated by declined LC‐3II level and elevated p62 level, and decreased APs. Our findings were not consistent with Zhang's results that ox‐LDL enhanced autophagy of cultured endothelial cells.31 There are several reasons accounted for this phenomenon, including different cell lines, ox‐LDL concentrations, and treatment time. These data indicated that ox‐LDL promoted cell apoptosis and inhibited autophagy of HT‐22 cells.
Many studies have indicated that disrupted autophagy flux contributes to neuronal cell injury and death in brain of patients with neurodegenerative diseases, including amyotrophic lateral sclerosis, PD‐like dementia, and Huntington's disease.32, 33, 34 Thus, we next investigated whether autophagy implicated in ox‐LDL‐induced HT‐22 cell injury and apoptosis. Our data showed that CQ profoundly decreased cell viability and increased cell apoptosis rate of ox‐LDL‐induced HT‐22 cells through blocking autophagy flux. Furthermore, the ultrastructure of these cells exhibited more serious injury than that of cells treated with ox‐LDL alone. In contrast, RAP exerted protective effects against neurotoxicity of ox‐LDL via activating autophagy. These findings were in agreement with our previous study indicating that activation of autophagy attenuated neuronal apoptosis in hippocampal CA1 regions of rats exposed to chronic mild unpredictable stress.35 Furthermore, several studies confirmed that autophagy prevents intracellular lipid accumulation and foam cell formation in THP‐1 cells.21 As autophagy facilitates abnormal aggregated proteins such as ox‐LDL and Aβ degradation, we inferred that the protective function of autophagy against ox‐LDL‐induced apoptosis of HT‐22 cells at least partially contributed to its role in decreasing intracellular ox‐LDL accumulation. These results suggest that regulation of autophagy may serve as a novel avenue to protect neuronal cells from ox‐LDL‐induced injury and apoptosis.
Then, we further confirmed whether the shift from autophagy to apoptosis contributed to neurotoxicity of ox‐LDL. Autophagy and apoptosis pathways share several upstream signaling molecules, such as mTOR, Bcl‐2, and Bax, which serve as a bistable switch to mediate the interplay between autophagy and apoptosis.15, 16 Bcl‐2 is the antiapoptotic protein that can regulate the permeability of outer membrane of mitochondria. Bcl‐2/beclin‐1 complex is a critical switch that regulates the balance between autophagy and apoptosis.36 Growing evidence indicates that silencing or blocking beclin‐1 promotes cell apoptosis. Recent studies have demonstrated that proapoptotic protein Bax inhibits autophagy by enhancing caspase‐dependent degradation of beclin‐1.37 In the present study, we found that ox‐LDL elevated Bax expression and declined Bcl‐2 level. Furthermore, ox‐LDL significantly suppressed autophagy of HT‐22 cells, indicated by decreased LC‐3 II and increased p62 protein expression, and reduced APs. Bax can accelerate beclin‐1 degradation in a caspase‐dependent manner, and beclin‐1 is essential for LC‐3 II formation. Therefore, LC‐3 II level and AP number of ox‐LDL group decreased as compared with those of the untreated group, respectively. Cotreatment with CQ did not cause significant increases in LC‐3 II and p62 expression in ox‐LDL‐induced HT‐22 cells, suggesting autophagy flux impairment occurred. These data suggest that ox‐LDL results in a switch from autophagy to apoptosis by increasing Bax and decreasing Bcl‐2 expression. Interestingly, our results suggested that CQ facilitated ox‐LDL‐induced transition from autophagy to apoptosis maybe via increasing Bax and decreasing Bcl‐2 expression. However, autophagy inducer RAP obviously protected HT‐22 cells against ox‐LDL‐induced switch to apoptosis from autophagy, indicated by activation of autophagy and elevation of cell viability. The present results were in agreement with previous studies that dysfunction of autophagy could induce neuronal apoptosis.12, 38, 39 Taken together, these results suggest that ox‐LDL causes HT‐22 cell apoptosis and the mechanism for this role may depend on the balance between autophagy and apoptosis.
In conclusion, our findings indicate that ox‐LDL significantly induces HT‐22 cell injury and apoptosis and significantly disrupts autophagy flux. Our findings also suggest ox‐LDL‐induced switch from autophagy to apoptosis may be through upregulating the expression of Bax and decreasing the expression of Bcl‐2. Importantly, the present study implies that insufficient autophagy is a crucial mechanism for ox‐LDL‐induced cell damage to HT‐22 cells. Our results provide important insights into the association between autophagy and apoptosis induced by ox‐LDL in neuronal cells.
Conflict of interest
The authors declare no conflict of interest, and all the authors listed have approved the manuscript that is enclosed.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grant nos. 31371161, 81500349), the construct program of the key discipline in Hunan Province, China (Basic Medicine Sciences in University of South China), and Postdoctoral Science Foundation of China (2012M521525). The authors gratefully acknowledge Dr. Zhao‐Yang Luo for technical support with the transmission electron microscopy at Key Laboratory for Arteriosclerology of Hunan Province, Hunan, China.
Gu H‐F, Li H‐Z, Xie X‐J, et al. Oxidized low‐density lipoprotein induced mouse hippocampal HT‐22 cell damage via promoting the shift from autophagy to apoptosis. CNS Neurosci Ther. 2017;23:341–349. 10.1111/cns.12680
The first two authors contributed equally to this work.
Contributor Information
Ya‐Xiong Nie, Email: nieyx-usc@qq.com.
Duan‐Fang Liao, Email: dfliao66@aliyun.com.
References
- 1. Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386:896–912. [DOI] [PubMed] [Google Scholar]
- 2. Saul A, Sprenger F, Bayer TA, Wirths O. Accelerated tau pathology with synaptic and neuronal loss in a novel triple transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:2564–2573. [DOI] [PubMed] [Google Scholar]
- 3. Ettcheto M, Petrov D, Pedros I, et al. Hypercholesterolemia and neurodegeneration. Comparison of hippocampal phenotypes in LDLr knockout and APPswe/PS1dE9 mice. Exp Gerontol. 2015;65:69–78. [DOI] [PubMed] [Google Scholar]
- 4. Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. [DOI] [PubMed] [Google Scholar]
- 5. Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood‐brain barrier. J Cell Biol. 1997;138:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Esterbauer H, Gebicki J, Puhl H, Jurgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341–390. [DOI] [PubMed] [Google Scholar]
- 7. Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid β‐protein via a scavenger receptor. Neuron. 1996;17:553–565. [DOI] [PubMed] [Google Scholar]
- 8. Wang XJ, Cao Q, Zhang Y, Su XD. Activation and regulation of caspase‐6 and its role in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2015;55:553–572. [DOI] [PubMed] [Google Scholar]
- 9. Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Söllvander S, Nikitidou E, Brolin R, et al. Accumulation of amyloid‐β by astrocytes result in enlarged endosomes and microvesicle‐induced apoptosis of neurons. Mol Neurodegener. 2016;11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. [DOI] [PubMed] [Google Scholar]
- 13. Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- 15. Mariño G, Niso‐Santano M, Baehrecke EH, Kroemer G. Self‐consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu HJ, Pu JL, Krafft PR, Zhang JM, Chen S. The molecular mechanisms between autophagy and apoptosis: potential role in central nervous system disorders. Cell Mol Neurobiol. 2015;35:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li M, Gao P, Zhang J. Crosstalk between autophagy and apoptosis: potential and emerging therapeutic targets for cardiac diseases. Int J Mol Sci. 2016;17:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang J, Yao S. JNK‐Bcl‐2/Bcl‐xL‐Bax/Bak pathway mediates the crosstalk between matrine‐induced autophagy and apoptosis via interplay with Beclin 1. Int J Mol Sci. 2015;16:25744–25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muller C, Salvayre R, Nègresalvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011;18:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei DH, Jia XY, Liu YH, et al. Cathepsin L stimulates autophagy and inhibits apoptosis of ox‐LDL‐induced endothelial cells: potential role in atherosclerosis. Int J Mol Sci. 2013;31:400–406. [DOI] [PubMed] [Google Scholar]
- 21. Gu HF, Li HZ, Tang YL, Tang XQ, Zheng XL, Liao DF. Nicotinate‐curcumin impedes foam cell formation from THP‐1 cells through restoring autophagy flux. PLoS ONE. 2016;11:e0154820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J, Li L, Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267–271. [DOI] [PubMed] [Google Scholar]
- 23. Sugawa M, Ikeda S, Kushima Y, Takashima Y, Cynshi O. Oxidized low density lipoprotein caused CNS neuron cell death. Brain Res. 1997;761:165–172. [DOI] [PubMed] [Google Scholar]
- 24. Nowicki M, Serke H, Kosacka J, Muller K, Spanel‐Borowski K. Oxidized low‐density lipoprotein (oxLDL) induces cell death in neuroblastoma and survival autophagy in schwannoma cells. Exp Mol Pathol. 2010;89:276–283. [DOI] [PubMed] [Google Scholar]
- 25. Keller JN, Hanni KB, Markesbery WR. Oxidized low‐density lipoprotein induces neuronal death: implications for calcium, reactive oxygen species, and caspases. J Neurochem. 1999;72:2601–2609. [DOI] [PubMed] [Google Scholar]
- 26. Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. [DOI] [PubMed] [Google Scholar]
- 27. Lee S, Sato Y, Nixon RA. Primary lysosomal dysfunction causes cargo‐specific deficits of axonal transport leading to Alzheimer‐like neuritic dystrophy. Autophagy. 2011;7:1562–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Papassotiropoulos A, Ludwig M, Naib‐Majani W, Rao GS. Induction of apoptosis and secondary necrosis in rat dorsal root ganglion cell cultures by oxidized low density lipoprotein. Neurosci Lett. 1996;209:33–36. [DOI] [PubMed] [Google Scholar]
- 29. Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem Soc Trans. 2005;33:1033–1036. [DOI] [PubMed] [Google Scholar]
- 30. Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E. Increased plasma peroxides and serum oxidized low density lipoprotein antibodies in major depression: markers that further explain the higher incidence of neurodegeneration and coronary artery disease. J Affect Disord. 2010;125:287–294. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Han Q, You S, et al. Rapamycin promotes the autophagic degradation of oxidized low‐density lipoprotein in human umbilical vein endothelial cells. J Vasc Res. 2015;52:210–219. [DOI] [PubMed] [Google Scholar]
- 32. Walter C, Clemens LE, Müller AJ, et al. Activation of AMPK‐induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology. 2016;108:24–38. [DOI] [PubMed] [Google Scholar]
- 33. Wei ZB, Yuan YF, Jaouen F, et al. SLC35D3 increases autophagic activity in midbrain dopaminergic neurons by enhancing BECN1‐ATG14‐PIK3C3 complex formation. Autophagy. 2016;12:1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vidal RL, Matus S, Bargsted L, Hetz C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. 2014;35:583–591. [DOI] [PubMed] [Google Scholar]
- 35. Gu HF, Nie YX, Tong QZ, et al. Epigallocatechin‐3‐gallate attenuates impairment of learning and memory in chronic unpredictable mild stress‐treated rats by restoring hippocampal autophagic flux. PLoS ONE. 2014;9:e112683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X, Su J, Xia M, et al. Caspase‐mediated cleavage of Beclin1 inhibits autophagy and promotes apoptosis induced by S1 in human ovarian cancer SKOV3 cells. Apoptosis. 2016;21:225–238. [DOI] [PubMed] [Google Scholar]
- 38. Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994–2014. [DOI] [PubMed] [Google Scholar]
- 39. Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–153. [DOI] [PubMed] [Google Scholar]