

Summary
Aims
To explore the common effects of the clusterin (CLU) rs11136000 variant on the default mode network (DMN) in amnestic mild cognitive impairment (aMCI) subjects and remitted geriatric depression (RGD) subjects.
Methods
Fifty‐one aMCI subjects, 38 RGD subjects, and 64 cognitively normal elderly subjects underwent resting‐state fMRI scans and neuropsychological tests at both baseline and a 35‐month follow‐up. Posterior cingulate cortex seed‐based functional connectivity (FC) analysis was used to obtain the DMN patterns.
Results
A CLU gene×disease×time interaction for aMCI subjects was mainly detected in the core cortical midline structures of the DMN, and the interaction for RGD subjects was mainly detected in the limbic system. However, they overlapped in two frontal regions, where consistent effects of the CLU gene on FC alterations were found between aMCI and RGD groups. Furthermore, the alterations of FC with frontal, parietal, and limbic regions compensated for episodic memory impairments in CLU‐CT/TT carriers, while no such compensation was found in CLU‐CC carriers.
Conclusion
The CLU gene could consistently affect the DMN FC with frontal regions among individuals at risk for Alzheimer's disease, and the CLU‐T allele was associated with more compensatory neural processes in DMN changes.
Keywords: Alzheimer's disease, clusterin, default mode network, geriatric depression, mild cognitive impairment
1. Introduction
Clusterin (CLU), an extracellular chaperone protein, is richly expressed within the central nervous system and plays an important role in Alzheimer's disease (AD) pathogenesis.1 CLU can inhibit amyloid beta (Aβ) deposition and facilitate Aβ transport across the blood‐brain barrier.2 The C allele of the rs11136000 single nucleotide polymorphism in the CLU gene is the third strongest genetic risk factor for sporadic AD.3 The C allele confers greater cognitive impairments and a 1.16 greater odds of developing sporadic AD than the T allele.4 In a sense, the minor T allele has been considered as a protective factor.
Using resting‐state fMRI, a series of functionally relevant resting‐state networks were identified according to the temporal correlations between intrinsic fluctuations of blood‐oxygen‐level‐dependent signals in different brain areas, also known as functional connectivity (FC).5 Altered FC patterns of the default mode network (DMN) have been demonstrated across the AD spectrum and can predict the severity and progression of AD.6, 7 Studies exploring the association between Aβ deposition and DMN patterns have suggested that aberrant DMN changes may be induced by Aβ deposition before the onset of any clinical symptoms.8, 9 As mentioned above, the CLU gene could affect Aβ pathology, and thus, it would not be surprising for there to exist a regulation of the CLU gene on DMN patterns. However, our recent cross‐sectional study did not find any impact of the CLU rs11136000 variant on the DMN itself in either healthy elderly subjects or subjects with amnestic mild cognitive impairment (aMCI, an intermediate state between normal aging and early AD that is characterized by episodic memory loss).10 The reasons may include the limited sample size and the multiple regions‐of‐interest‐based methods used in DMN reconstruction, both of which may reduce the sensitivity of DMN alterations.
Both aMCI and geriatric depression are very common in the elderly population and confer an increased risk for developing AD.11 A high prevalence of depressive symptoms has been found among MCI patients,12 and cognitive impairments are also common in depressed patients.13 The coexistence of aMCI and depression significantly increase the risk for AD.14 Furthermore, increased Aβ deposition has been shown not only in aMCI patients but also in geriatric depression patients.15, 16 As described above, increased Aβ deposition may induce aberrant DMN changes, and thus, DMN patterns have been investigated in MCI patients and geriatric depression patients. For example, both decreased FC from the posterior to anterior portions of DMN and increased FC with multiple brain regions have been identified in MCI patients.17, 18, 19 Similarly, both decreased DMN FC with the frontal cortex and increased FC with the parietal cortex and the anterior cingulate cortex have been found in geriatric depression patients.20, 21 However, whether the DMN patterns in these subjects can be regulated by the CLU gene remains relatively unknown. This study hypothesized that the CLU gene could affect the DMN patterns in both aMCI subjects and geriatric depression subjects. In addition, in view of the distinct clinical link between MCI and geriatric depression, we also hypothesized that similar effects of the CLU gene on the DMN might exist between aMCI subjects and geriatric depression subjects.
This study recruited cognitively normal elderly subjects, aMCI subjects, and remitted geriatric depression (RGD) subjects. Each group was divided into CLU‐CC carriers and CLU‐CT/TT carriers. All subjects underwent resting‐state fMRI scans and neuropsychological tests at both baseline and a 35‐month follow‐up. A series of quantitative DMN FC analyses were performed. The aims of this study were (i) to explore the effects of the CLU rs11136000 variant on the longitudinal changing patterns of the DMN in aMCI, RGD, and cognitively normal elderly subjects; (ii) to determine whether aMCI and RGD share a common regulation of the DMN by the CLU; and (iii) to identify the behavioral significance of CLU gene‐related modulation of the DMN.
2. Methods
2.1. Participants
This study was approved by the Affiliated ZhongDa Hospital of Southeast University Research Ethics Committee. Chinese Han participants were recruited using community health screening and media advertisements. Written informed consent was provided by each participant. We initially recruited 87 aMCI subjects, 72 RGD subjects, and 135 cognitively normal elderly subjects. During the follow‐up period, 36 aMCI subjects and 34 RGD subjects were lost due to the development of neurological or other psychiatric diseases, relocation to other cities, passing by, nonresponders, and subjective unwillingness. The follow‐ups of cognitively normal elderly subjects were paused after comparative numbers of aMCI subjects, RGD subjects, and cognitively normal elderly subjects were obtained. A total of 51 aMCI subjects, 38 RGD subjects, and 64 cognitively normal elderly subjects underwent resting‐state fMRI scans and neuropsychological tests at both baseline and follow‐up. The mean follow‐up period was 35 months. Eight aMCI subjects, five RGD subjects, and four cognitively normal elderly subjects were excluded after the evaluation of head motion artifacts. During the follow‐up period, six aMCI subjects converted to cognitively normal elderly subjects and nine cognitively normal elderly subjects converted to aMCI subjects. These subjects were also excluded to decrease the heterogeneity in each group. Finally, there were 37 aMCI subjects, 33 RGD subjects, and 51 control subjects. In the remaining RGD subjects, the average age of onset of depression was 60±9.43 years, and 28 subjects developed depression after the age of 50 years, also known as late‐onset depression.22 These groups were matched for the duration of the follow‐up period.
2.2. Neuropsychological assessments
Each subject underwent a standardized diagnostic evaluation, including demographic information, medical history, and examination of neurological and mental status. See Supporting information.
2.3. Inclusion and exclusion criteria
All aMCI subjects were included according to the diagnostic criteria proposed by Petersen et al.23 and others.24 The inclusion criteria for RGD subjects and control subjects were described previously.25 See Supporting information.
2.4. CLU genotyping
See Supporting information. In consideration of a dominant model of minor T allele effects,26, 27 each group was divided into CLU‐CC carriers and CLU‐CT/TT carriers.
2.5. Magnetic resonance imaging procedures and image preprocessing
See Supporting information. Because the global signal has recently been thought to be related to major neuronal components, the global signal was not regressed in the present study.28, 29, 30
2.6. FC analysis
A 5‐mm‐radius sphere centered at posterior cingulate cortex (PCC) hub [Montreal Neurological Institute (MNI) space: −2, −45, 34] served as a seed region. For each subject, a mean time series of the seed region was computed as the reference time course. Pearson cross‐correlation analysis was carried out between the seed time course and the time course of the whole‐brain voxels. A Fisher's z‐transformation was applied to improve the normality of the correlation coefficients . Finally, individual maps of the DMN were obtained.
2.7. Statistical analysis
2.7.1. Demographic and neuropsychological data
Analysis of variance (ANOVA) and chi‐square tests (applied for the comparisons of gender and ApoE status) were used to compare the demographic data. Mixed analysis of covariance (ANCOVA), with disease, gene, and time as fixed factors, was used to analyze the neuropsychological performances among the six groups with statistically significant differences (P<.05), controlling for age, gender, years of education, and ApoE status. Two‐sample t tests and chi‐square tests (applied in the comparisons of gender and ApoE status) were used for the comparison of the demographic data between included subjects and excluded subjects with significance of P<.05. As described in our previous study,29 the individual raw scores of each cognitive test (except for MMSE) were transformed to Z scores. See Supporting information. All statistical procedures utilized SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA).
2.7.2. Group‐level intrinsic connectivity analysis
To analyze the interaction between the CLU genotype, disease, and time on the DMN, a whole‐brain voxel‐wise mixed ANCOVA with CLU genotype (CLU‐CC and CLU‐CT/TT), disease status (control and aMCI or RGD), and time point (baseline and follow‐up) as fixed factors was performed, controlling for age, gender, and years of education (using the full factorial option in SPM8). ApoE status was also treated as a covariate. The thresholds were set at a corrected P<.05 determined by Monte Carlo simulation for multiple comparisons in the whole brain (voxel‐wise P<.01, FWHM=6 mm, cluster size>1080 mm3). Then, the average FC strength of each significant region associated with the CLU genotype×disease×time point interaction was extracted in each subgroup at baseline and follow‐up using the Resting‐State fMRI Data Analysis Toolkit (REST) 1.7. Post hoc tests were performed to explore the difference in FC between groups and longitudinal changes (baseline vs follow‐up) of FC within each group using SPSS 19.0 software. The significance level for post hoc tests was set at P<.05. It should be noted that ANCOVA was performed in aMCI subjects (aMCI vs control) and RGD subjects (RGD vs control) respectively. Then, the overlaps and nonoverlaps between the interactive regions in aMCI and RGD subjects were further analyzed.
2.7.3. Correlative analysis between FC alterations and episodic memory impairments
Correlation analyses were performed between the longitudinal changes of FC in each interactive region and the longitudinal changes in episodic memory scores in each group. It should be noted that the correlative analysis was focused on episodic memory for the following reasons. First, in the present study, episodic memory was the most impaired cognitive domain in both aMCI and RGD subjects. Second, many studies have suggested that episodic memory is subserved by the DMN.31, 32, 33 All statistical procedures for correlation analysis utilized SPSS 19.0 software. The significance level for correlation analysis was set at P<.05.
3. Results
3.1. Demographic and neuropsychological data
As shown in Table 1, compared with control subjects, RGD subjects had less years of education. No other significant difference was observed in the demographic characteristics or the ApoE status between all groups. A significant main effect of disease was shown in the neuropsychological data. Compared with control subjects, both aMCI subjects and RGD subjects displayed impairments in all cognitive domains, except the information processing speed for RGD subjects. Compared with aMCI subjects, RGD subjects displayed poorer performance in all cognitive domains except for visuospatial function. A main effect of the CLU gene was also shown. CLU‐CC carriers exhibited poorer performance in information processing speed than CLU‐CT/TT carriers. Furthermore, a main effect of time revealed longitudinal decreases in MMSE and executive function scores in the whole sample during the follow‐up period. There was no significant interaction of disease, CLU gene, and time on neuropsychological data (data on interactions of any two factors were not shown). Finally, it should be noted that although some subjects were excluded during analyses, there was no significant demographic difference between included subjects and excluded subjects (Table S1).
Table 1.
Demographic and neuropsychological data
| Items | Control | aMCI | RGD | P‐value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CLU‐CC (n=26) | CLU‐CT/TT (n=25) | CLU‐CC (n=24) | CLU‐CT/TT (n=13) | CLU‐CC (n=21) | CLU‐CT/TT (n=12) | Disease | Gene | Time | Disease×Gene×Time interaction or chi‐square | |
| Age (y) | 69.3±5.29 | 68.4±6.27 | 67.7±7.37 | 68.7±5.92 | 69.1±6.32 | 66.3±6.17 | .518 | .293 | — | — |
| Education (y) | 12.0±2.75 | 13.2±3.00 | 12.0±3.18 | 12.2±3.90 | 12.1±3.37 | 10.3±2.14 | .019b | .715 | — | — |
| Gender (male:female) | 10:16 | 12:13 | 14:10 | 8:5 | 8:13 | 2:10 | — | — | — | .156 |
| ApoE status (ε4+:−) | 6:20 | 3:22 | 9:15 | 3:10 | 2:19 | 1:11 | — | — | — | .139 |
| MMSE | ||||||||||
| Baseline | 28.4±1.06 | 28.2±1.55 | 27.2±2.74 | 26.8±1.80 | 27.6±1.12 | 27.7±1.44 | <.001a , b , c | .299 | .024 | .757 |
| Follow‐up | 28.0±1.54 | 28.4±1.58 | 26.1±3.53 | 25.4±4.54 | 27.2±1.78 | 27.1±1.62 | ||||
| Episodic memory | ||||||||||
| Baseline | 0.58±0.50 | 0.52±0.57 | −0.64±0.56 | −0.73±0.65 | −0.06±0.64 | −0.26±0.71 | <.001a , b , c | .301 | .901 | .698 |
| Follow‐up | 0.54±0.57 | 0.62±0.61 | −0.72±0.70 | −0.89±0.80 | −0.15±0.78 | −0.06±0.65 | ||||
| Executive function | ||||||||||
| Baseline | 0.25±0.43 | 0.51±0.71 | −0.23±0.48 | −0.46±0.58 | 0.03±0.58 | 0.04±0.42 | <.001a , b , c | .806 | .021 | .800 |
| Follow‐up | 0.14±0.52 | 0.30±0.74 | −0.53±0.46 | −0.67±0.94 | −0.12±0.75 | −0.01±0.59 | ||||
| Visuospatial function | ||||||||||
| Baseline | 0.28±0.51 | 0.32±0.80 | 0.04±0.68 | −0.33±0.84 | −0.13±0.75 | −0.26±0.73 | .001a , b | .373 | .960 | .980 |
| Follow‐up | 0.19±0.88 | 0.29±0.75 | 0.04±0.51 | −0.21±1.19 | −0.14±0.85 | −0.28±1.30 | ||||
| Information processing speed | ||||||||||
| Baseline | 0.22±0.70 | 0.44±0.84 | −0.38±0.59 | −0.37±0.49 | 0.20±0.75 | 0.26±0.82 | <.001a , c | .046 | .105 | .606 |
| Follow‐up | −0.01±0.77 | 0.23±0.86 | −0.65±0.66 | −0.59±0.50 | 0.04±0.76 | 0.50±0.96 | ||||
ANOVA, analysis of variance; ANCOVA, analysis of covariance; MMSE, Mini‐Mental State Examination.
P<.05, aMCI subjects differs from control subjects.
P<.05, RGD subjects differs from control subjects.
P<.05, RGD subjects differs from aMCI subjects.
Values are presented as the mean±standard deviation (SD). Chi‐square test was applied in the comparisons of gender and ApoE status. ANOVA was applied in the comparisons of age and years of education. ANCOVA (with disease, gene, and time as fixed factors) was applied in the comparisons of neuropsychological data, controlling for age, gender, years of education, and ApoE status.
3.2. DMN FC data
The main effects of CLU gene, disease, and time are shown in Figures S1 and S2. While comparing aMCI subjects and control subjects, the CLU gene×disease×time interaction was detected in the frontal cortex, parietal cortex, and cingulate cortex, most of which belong to the core cortical midline structures of the DMN (Figure 1A). While comparing RGD subjects and control subjects, the CLU gene×disease×time interaction was primarily observed in the limbic regions and small parts of cerebral cortex (Figure 1B). Specifically, the overlaps between the interactive regions in aMCI subjects and those in RGD subjects were found in the left middle frontal gyrus (LMFG) and the left medial orbital frontal gyrus (LMOFG; Figure 1C and Table 2).
Figure 1.
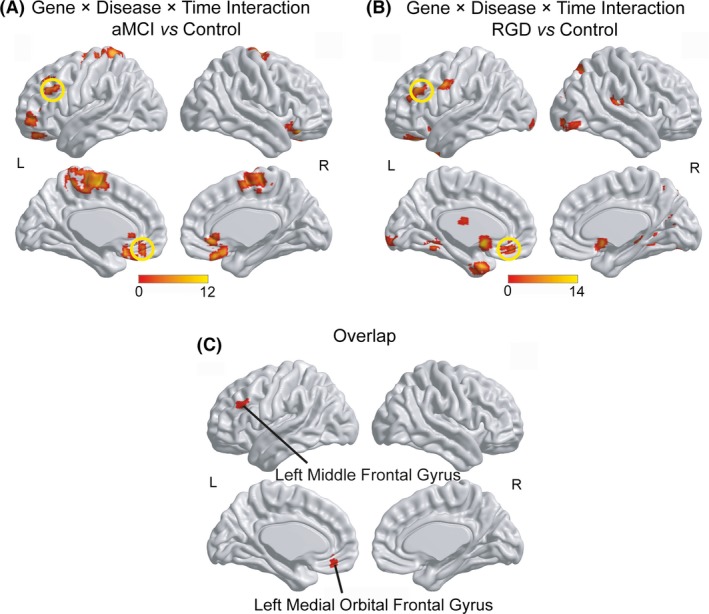
CLU gene×disease×time interaction on the DMN. (A) The CLU gene×disease×time interaction in aMCI subjects and control subjects. (B) The CLU gene×disease×time interaction in RGD subjects and control subjects. (C) The overlaps between the interactive regions in aMCI subjects and in RGD subjects. The thresholds were set at a corrected P<.05 determined by Monte Carlo simulation for multiple comparisons. L, left; R, right
Table 2.
Gene×disease×time interaction on DMN
| Brain region | BA | Peak MNI coordinates x, y, z (mm) | Peak F‐value | Cluster size (mm3) |
|---|---|---|---|---|
| (1) aMCI vs control | ||||
| Bilateral medial frontal gyri | 6 | −6, −9, 60 | 11.1 | 15 255 |
| Left medial orbital frontal gyrus | 11 | 3, 30, −24 | 9.98 | 2349 |
| Left middle frontal gyrus | 45 | −33, 45, −6 | 11.3 | 2727 |
| Left postcentral gyrus | 1 | −19, −43, 71 | 8.58 | 3213 |
| Right anterior cingulate gyrus | 32 | 6, 33, −6 | 7.61 | 1161 |
| Right inferior frontal gyrus | 47 | 36, 30, −18 | 9.56 | 1755 |
| (2) RGD vs control | ||||
| Left calcarine gyrus | 18 | −9, −99, −9 | 7.00 | 1107 |
| Left caudate | 25 | −3, 6, −9 | 13.5 | 1350 |
| Left lingual gyrus | 19 | −21, −48, −9 | 10.5 | 1269 |
| Left medial orbital frontal gyrus | 11 | −6, 39, −21 | 8.52 | 2106 |
| Left middle frontal gyrus | 45 | −48, 33, 36 | 11.8 | 1512 |
| Left precentral gyrus | 6 | −60, 3, 36 | 11.3 | 1080 |
| Left temporal pole | 38 | −18, 3, −33 | 13.1 | 2295 |
| Left thalamus | ‐ | −6, −15, 18 | 13.2 | 1620 |
| Right cuneus | 18 | 21, −81, 15 | 9.50 | 1080 |
| Right inferior occipital gyrus | 18 | 30, −81, −6 | 7.33 | 1215 |
| Right insula | 13 | 42, −27, 18 | 13.0 | 1296 |
| Right parahippocampal gyrus | 19 | 24, −33, −9 | 10.0 | 1377 |
| Right superior parietal lobule | 7 | 21, −66, 54 | 6.03 | 1107 |
| (3) Overlap | ||||
| Left middle frontal gyrus | 45 | 1323 | ||
| Left medial orbital frontal gyrus | 11 | 1863 | ||
DMN, default mode network; BA, Brodmann's area; MNI, Montreal Neurological Institute.
The thresholds were set at a corrected P<.05, determined by Monte Carlo simulation for multiple comparisons. The overlaps between the interactive regions in aMCI subjects and that in RGD subjects appear in bold.
Post hoc test: For each overlapping region, no significant difference in FC at baseline or FC changes during follow‐up were shown between control subjects, aMCI subjects, and RGD subjects (Table S2). Then, these three groups were divided into six subgroups according to the CLU genotype. The six subgroups displayed no significant difference in FC at baseline but did exhibit a significant difference of FC changes during follow‐up (Table 3). First, significant regulation by the CLU gene on the DMN was shown in each group. In the control group, CLU‐CT/TT carriers displayed greater increases of FC with the LMFG than CLU‐CC carriers (P=.003). In the aMCI group, CLU‐CT/TT carriers displayed greater decreases in FC with the LMOFG than CLU‐CC carriers (P=.030). In the RGD group, CLU‐CT/TT carriers also displayed greater decreases of FC with both the LMFG (P=.033) and the LMOFG (P=.041) than CLU‐CC carriers. Second, the trends of longitudinal changes of FC were consistently affected by the CLU gene in the aMCI group and the RGD group. In the LMFG, while the control CLU‐CC subgroup showed longitudinally decreased FC (P=.024), both the aMCI CLU‐CC subgroup (P=.047) and the RGD CLU‐CC subgroup (P=.012) displayed longitudinally increased FC; However, while longitudinally increased FC was found in the control CLU‐CT/TT subgroup (P=.038), no significant change was found in the aMCI CLU‐CT/TT subgroup or the RGD CLU‐CT/TT subgroup (Figure 2A). In the LMOFG, longitudinal decrease trends of FC were observed in both the aMCI CLU‐CT/TT subgroup (P=.118) and the RGD CLU‐CT/TT subgroup (P=.025; Figure 2B). Third, the extents of longitudinal changes of FC were also consistently affected by the CLU gene in the aMCI group and the RGD group (Table 3). In the LMFG, compared with the control CLU‐CC subgroup, both the aMCI CLU‐CC subgroup (P=.002) and the RGD CLU‐CC subgroup (P=.001) showed greater increases of FC. In the LMOFG, compared with the control CLU‐CT/TT subgroup, both the aMCI CLU‐CT/TT subgroup (P=.024) and the RGD CLU‐CT/TT subgroup (P=.008) showed greater decreases of FC. On the other hand, in most of the nonoverlapping regions associated with the interaction, CLU‐CT/TT carriers in both the aMCI group and the RGD group displayed significant longitudinally decreased FC (Figures S3 and S4).
Table 3.
Comparisons of FC according to both diagnosis and gene
| Brain region | Control | aMCI | RGD | F‐value | P‐value | |||
|---|---|---|---|---|---|---|---|---|
| CLU‐CC | CLU‐CT/TT | CLU‐CC | CLU‐CT/TT | CLU‐CC | CLU‐CT/TT | |||
| FC at baseline | ||||||||
| Left middle frontal gyrus | 0.18±0.25 | 0.07±0.19 | 0.12±0.23 | 0.09±0.20 | 0.01±0.24 | 0.09±0.19 | 1.47 | .204 |
| Left medial orbital frontal gyrus | 0.51±0.21 | 0.40±0.24 | 0.44±0.24 | 0.52±0.28 | 0.42±0.22 | 0.48±0.26 | 0.89 | .487 |
| Changes of FC (follow‐up−baseline) | ||||||||
| Left middle frontal gyrus | −0.09±0.19 | 0.07±0.17a | 0.09±0.21a | −0.01±0.21 | 0.10±0.16a | −0.05±0.22c , d | 3.77 | .003 |
| Left medial orbital frontal gyrus | −0.06±0.22 | 0.03±0.23 | 0.03±0.27 | −0.16±0.34b , c | −0.01±0.20 | −0.20±0.27b , c , d | 2.52 | .033 |
FC, functional connectivity.
P<.05 compared to the control CLU‐CC subgroup.
P<.05 compared to the control CLU‐CT/TT subgroup.
P<.05 compared to the aMCI CLU‐CC subgroup.
P<.05 compared to the RGD CLU‐CC subgroup.
Values are presented as the mean±standard deviation (SD). ANOVA was applied in each comparison.
Figure 2.
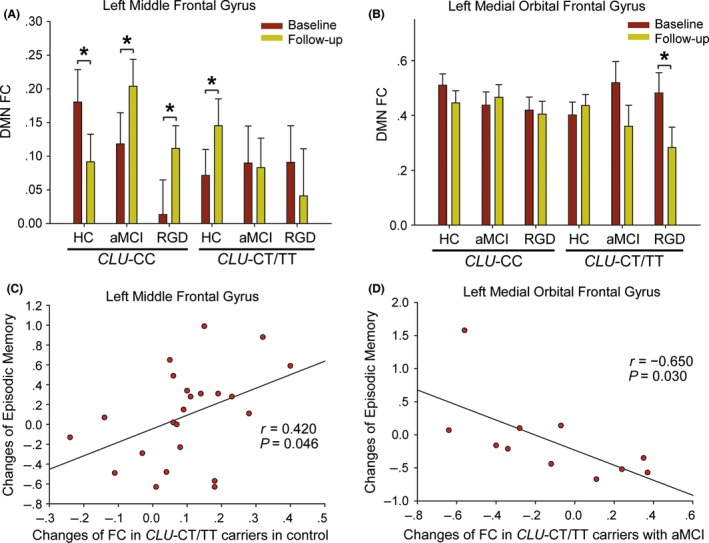
Post hoc tests and correlative analyses performed in overlapping regions. (A) In the left middle frontal gyrus, CLU‐CC carriers in both the aMCI group and the RGD group displayed longitudinally increased FC. (B) In the left medial orbital frontal gyrus, CLU‐CT/TT carriers in both the aMCI group and the RGD group displayed longitudinal decrease trends of FC. (C) For control CLU‐CT/TT subgroup, greater increase of FC with the left middle frontal gyrus was associated with less decline in episodic memory scores (r=.420, P=.046, two‐tailed). (D) For the aMCI CLU‐CT/TT subgroup, greater decrease of FC with the left medial orbital frontal gyrus was associated with less decline in episodic memory scores (r=−.650, P=.030, two‐tailed). * P<.05. DMN, default mode network; FC, functional connectivity
3.3. Longitudinal changes of DMN FC and behavioral significance
As shown in Table 4, for CLU‐CT/TT carriers, five longitudinal changes of FC compensated for the declines in episodic memory. For example, in the control CLU‐CT/TT subgroup, the longitudinal increase of FC with the LMFG was positively correlated with the changes in episodic memory scores (r=.420, P=.046, two‐tailed; Figure 2C); greater increases of FC corresponded to lower episodic memory scores. In the aMCI CLU‐CT/TT subgroup, greater decreases of FC in the LMOFG were associated with less decline of episodic memory scores (r=−.650, P=.030, two‐tailed; Figure 2D). Only one significant correlation in CLU‐CT/TT carriers showed harmful effects; decreased FC in the right parahippocampal gyrus was associated with greater decline of episodic memory scores in the RGD CLU‐CT/TT subgroup (Figure S5). The only significant correlation in CLU‐CC carriers was shown in the right cuneus in the control group, where the decreased FC was harmful for the changes of episodic memory.
Table 4.
Correlative analysis between DMN FC changes and impairments of episodic memory
| Group | Brain regions | Longitudinal changes of FC | r value | P‐value | Effects of FC changes on episodic memory |
|---|---|---|---|---|---|
| CLU‐CC | |||||
| Control | Right cuneus | Decreased | .457 | .022 | Harmful |
| CLU‐CT/TT | |||||
| Control | Left middle frontal gyrus | Increased | .420 | .046 | Compensatory |
| aMCI | Left medial orbital frontal gyrus | Decreased | −.650 | .030 | Compensatory |
| RGD | Right cuneus | Decreased | −.657 | .039 | Compensatory |
| RGD | Right parahippocampal gyrus | Decreased | .666 | .018 | Harmful |
| RGD | Right superior parietal lobule | Decreased | −.654 | .040 | Compensatory |
| aMCI | Right anterior cingulate | Decreased | −.592 | .045 | Compensatory |
FC, functional connectivity; DMN, default mode network.
The overlaps between the interactive regions in aMCI subjects and in RGD subjects appear in bold.
4. Discussion
The present study is the first to show the effects of the CLU gene on the DMN in subjects at high risk of AD. Some adjustments were made relative to our previous study. First, PCC seed‐based FC analysis was used to obtain DMN patterns. The PCC is a key region of the DMN, and the PCC seed‐based FC analysis has been widely used in DMN construction.31 Second, the present study employed longitudinal data from a larger sample size. One of the key strengths of the present study was that this study employed two different groups of subjects at high risk of AD. Despite the different core symptoms of aMCI subjects and RGD subjects, both displayed impaired performance in multiple cognitive domains. Differences and similarities were also found in the DMN patterns. The CLU gene×disease×time interaction for aMCI subjects was mainly detected in the core cortical midline structures of the DMN, and the interaction in RGD subjects was mainly detected in the limbic system. However, the interactive regions of the two groups overlapped in frontal regions, suggesting that the CLU gene could consistently affect the DMN changing patterns in the frontal cortex among subjects at high risk of AD. Interestingly, using DTI, common deficits of structural connectivity patterns between aMCI and RGD patients were also shown in frontal regions in our previous study.25 These data indicate that the frontal lobe plays an important role in the neural underpinnings of aMCI and RGD.
The other strength of the present work was that the participants were followed for an average of nearly 3 years. As the most important risk factor for AD,34 aging could affect the DMN patterns at different stages of the disease. For example, aging‐related decreases of the DMN FC have been widely reported in cognitively normal elderly subjects.35, 36 At the stage of MCI, both aging‐related decreases of DMN FC in the frontal cortex and increases in the prefrontal cortex and the anterior cingulate cortex have been shown.37 Moreover, the aging‐related changes of DMN FC, including increases in the frontal cortex and decreases in major parts of the network, were accelerated in subjects with AD.36 The present study also found both longitudinally increased DMN FC primarily in frontal regions and decreased FC in extensive regions. Notably, within the two disease groups, all of the significantly increased FC was shown in CLU‐CC carriers, and all of the decreased FC with significances was shown in CLU‐CT/TT carriers. The CLU gene‐related distribution suggested that the increased FC in frontal regions tends to occur in subjects at a much higher risk of AD, that is, with both CLU‐CC genotype and aMCI or RGD. On the other hand, disease groups with a protective factor, that is, the CLU‐T allele, tended to display extensively decreased DMN FC.
Increased fMRI activity and FC in the frontal cortex have been generally considered as compensatory recruitment or reallocation of cognitive resources because they have been associated with relatively better performance in cognitive tasks.38 However, several studies found that increased activity and FC were associated with relatively worse performance.39, 40 One study demonstrated that the reduction of brain activities could even improve cognitive function in aMCI patients.39 Another study found that reduced resting‐state FC was associated with increased task‐related activity, which could compensate to maintain cognitive performance.41 In the present study, for CLU‐CT/TT carriers, both the increased FC with frontal regions in the control group and the decreased FC with parietal and limbic regions in the aMCI and RGD groups compensated for episodic memory declines. We concluded that the effects of FC changes in episodic memory depended not only on the FC changing patterns, that is, increases or decreases, but also on the risk factors associated with each subject, including the disease status and the CLU rs11136000 variant; CLU‐CT/TT carriers tended to display increased DMN FC, especially in the frontal cortex, as compensatory neural processes during normal aging, but decreased FC in parietal and limbic regions as compensation after the onset of cognitive impairments. Increased FC compensation for cognitive decline may occur in normal aging. However, persistent hypersynchrony or hyperactivity could be harmful for cognitive development after the onset of cognitive impairments.39, 41 Thus, CLU‐CT/TT carriers displayed decreased FC to compensate for cognitive impairments. This might explain the compensatory effects of both increased and decreased FC in these subjects. However, the significance of the correlative analyses should be validated in a larger sample, and we treat this interpretation with caution. Nevertheless, under similar conditions, that is, similar sample sizes and the same analytic procedure, CLU‐CT/TT carriers displayed more compensatory FC alterations in the DMN relative to CLU‐CC carriers.
Thus far, no study has explored the interaction of geriatric depression and the CLU gene on the incidence of dementia. However, the present study found that geriatric depression interacted with the CLU gene to affect the changing patterns in the DMN. Importantly, the interaction between geriatric depression and the CLU gene was highly similar to that between aMCI and the CLU gene in the frontal cortex. Even in the nonoverlapping regions, CLU gene‐related changing trends of DMN FC were also similar between aMCI and RGD subjects. In summary, geriatric depression could interact with the CLU gene to result in aMCI‐like alterations of the DMN. The interactive effects might be associated with Aβ pathology. Increased brain Aβ deposition has been shown in geriatric depression patients compared with healthy elderly.16 Healthy elderly with depressive episodes also display high Aβ deposition.42 Subjects with both geriatric depression and MCI even had Aβ deposition comparable to that in AD.43 On the other hand, CLU gene could also modulate the brain Aβ loads.44 Therefore, the coexistence of geriatric depression and CLU risk variants might considerably influence Aβ pathology, which could be reflected by the DMN patterns. Future studies investigating the interaction of geriatric depression and the CLU gene on the incidence of AD will help to confirm these synergistic effects.
The present study has several limitations. First, the sample size was small, especially for testing three‐way interactions. During the follow‐up period, nine subjects in the control group converted to aMCI subjects and nine subjects in the aMCI group converted to dementia subjects. However, the small number of converters limited further analyses comparing alterations of the DMN between converters and nonconverters. Second, although the interaction of geriatric depression and the CLU gene was interpreted based on Aβ pathology, the present study did not utilize any techniques to measure Aβ pathology. Finally, in the post hoc tests, comparisons between subgroups or time points were not corrected for multiple comparisons. Thus, these interpretations should be treated with caution.
5. Conclusion
The CLU rs11136000 variant could consistently affect the changing of DMN patterns of aMCI and RGD subjects in the frontal cortex. The CLU‐T allele was associated with more compensatory neural processes in DMN changes. The findings may help to focus attention to the frontal cortex when investigating the effects of the CLU gene on the brain. The interaction of the CLU gene and the diagnosis should be considered in future studies. Furthermore, the present findings also indicate that the CLU gene might interact with geriatric depression and be related to risk of developing AD, which should be investigated in the future.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
We would like to thank Wenxiang Liao, Haifeng Chen, and Qin Xu for helping on the MRI scanning. This research was partly supported by the National Natural Science Foundation of China (No. 91332104; 81671665); Natural Science Foundation of Jiangsu Province (No. BK20160071); and National High‐tech R.D Program (863 Program) (No.2015AA020508).
Ye Q, Su F, Shu H, et al. Shared effects of the clusterin gene on the default mode network among individuals at risk for Alzheimer's disease. CNS Neurosci Ther. 2017;23:395–404. 10.1111/cns.12682
References
- 1. Yerbury JJ, Stewart EM, Wyatt AR, Wilson MR. Quality control of protein folding in extracellular space. EMBO Rep. 2005;6:1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oda T, Wals P, Osterburg HH, et al. Clusterin (apoJ) alters the aggregation of amyloid beta‐peptide (A beta 1‐42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp Neurol. 1995;136:22–31. [DOI] [PubMed] [Google Scholar]
- 3. Bertram L, Tanzi RE. Alzheimer disease: new light on an old CLU. Nat Rev Neurol. 2010;6:11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrasquillo MM, Crook JE, Pedraza O, et al. Late‐onset Alzheimer's risk variants in memory decline, incident mild cognitive impairment, and Alzheimer's disease. Neurobiol Aging. 2015;36:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damoiseaux JS, Rombouts SA, Barkhof F, et al. Consistent resting‐state networks across healthy subjects. Proc Natl Acad Sci USA. 2006;103:13848–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang HY, Wang SJ, Liu B, et al. Resting brain connectivity: changes during the progress of Alzheimer disease. Radiology. 2010;256:598–606. [DOI] [PubMed] [Google Scholar]
- 7. Wu X, Li R, Fleisher AS, et al. Altered default mode network connectivity in Alzheimer's disease–a resting functional MRI and Bayesian network study. Hum Brain Mapp. 2011;32:1868–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheline YI, Raichle ME, Snyder AZ, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hedden T, Van Dijk KR, Becker JA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci. 2009;29:12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai F, Shi Y, Yuan Y, Xie C, Zhang Z. Immunity factor contributes to altered brain functional networks in individuals at risk for Alzheimer's disease: neuroimaging‐genetic evidence. Brain Behav Immun. 2016;56:84–95. [DOI] [PubMed] [Google Scholar]
- 11. Panza F, Frisardi V, Capurso C, et al. Late‐life depression, mild cognitive impairment, and dementia: possible continuum? Am J Geriatr Psychiatry. 2010;18:98–116. [DOI] [PubMed] [Google Scholar]
- 12. Solfrizzi V, D'Introno A, Colacicco AM, et al. Incident occurrence of depressive symptoms among patients with mild cognitive impairment – the Italian longitudinal study on aging. Dement Geriatr Cogn Disord. 2007;24:55–64. [DOI] [PubMed] [Google Scholar]
- 13. Butters MA, Young JB, Lopez O, et al. Pathways linking late‐life depression to persistent cognitive impairment and dementia. Dialogues Clin Neurosci. 2008;10:345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Modrego PJ, Ferrandez J. Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Arch Neurol. 2004;61:1290–1293. [DOI] [PubMed] [Google Scholar]
- 15. Wolk DA, Price JC, Saxton JA, et al. Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol. 2009;65:557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar A, Kepe V, Barrio JR, et al. Protein binding in patients with late‐life depression. Arch Gen Psychiatry. 2011;68:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wee CY, Yap PT, Zhang D, et al. Identification of MCI individuals using structural and functional connectivity networks. NeuroImage. 2012;59:2045–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sperling RA, Dickerson BC, Pihlajamaki M, et al. Functional alterations in memory networks in early Alzheimer's disease. Neuromolecular Med. 2010;12:27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gardini S, Venneri A, Sambataro F, et al. Increased functional connectivity in the default mode network in mild cognitive impairment: a maladaptive compensatory mechanism associated with poor semantic memory performance. J Alzheimers Dis. 2015;45:457–470. [DOI] [PubMed] [Google Scholar]
- 20. Andreescu C, Tudorascu DL, Butters MA, et al. Resting state functional connectivity and treatment response in late‐life depression. Psychiatry Res. 2013;214:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexopoulos GS, Hoptman MJ, Kanellopoulos D, Murphy CF, Lim KO, Gunning FM. Functional connectivity in the cognitive control network and the default mode network in late‐life depression. J Affect Disord. 2012;139:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hickie IB, Naismith SL, Norrie LM, Scott EM. Managing depression across the life cycle: new strategies for clinicians and their patients. Intern Med J. 2009;39:720–727. [DOI] [PubMed] [Google Scholar]
- 23. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. [DOI] [PubMed] [Google Scholar]
- 24. Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256:240–246. [DOI] [PubMed] [Google Scholar]
- 25. Bai F, Shu N, Yuan Y, et al. Topologically convergent and divergent structural connectivity patterns between patients with remitted geriatric depression and amnestic mild cognitive impairment. J Neurosci. 2012;32:4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roussotte FF, Gutman BA, Madsen SK, Colby JB, Thompson PM. Combined effects of Alzheimer risk variants in the CLU and ApoE genes on ventricular expansion patterns in the elderly. J Neurosci. 2014;34:6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mengel‐From J, Christensen K, McGue M, Christiansen L. Genetic variations in the CLU and PICALM genes are associated with cognitive function in the oldest old. Neurobiol Aging. 2011;32:554 e557‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo CC, Kurth F, Zhou J, et al. One‐year test‐retest reliability of intrinsic connectivity network fMRI in older adults. NeuroImage. 2012;61:1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shu H, Shi Y, Chen G, et al. Opposite neural trajectories of apolipoprotein E 4 and 2 alleles with aging associated with different risks of Alzheimer's disease. Cereb Cortex. 2014;26:1421–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hahamy A, Calhoun V, Pearlson G, et al. Save the global: global signal connectivity as a tool for studying clinical populations with functional magnetic resonance imaging. Brain Connect. 2014;4:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buckner RL, Andrews‐Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. [DOI] [PubMed] [Google Scholar]
- 32. Jeong W, Chung CK, Kim JS. Episodic memory in aspects of large‐scale brain networks. Front Hum Neurosci. 2015;9:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sestieri C, Corbetta M, Romani GL, Shulman GL. Episodic memory retrieval, parietal cortex, and the default mode network: functional and topographic analyses. J Neurosci. 2011;31:4407–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Riedel BC, Thompson PM, Brinton RD. Age, APOE and sex: triad of risk of Alzheimer's disease. J Steroid Biochem Mol Biol. 2016;160:134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang CC, Hsieh WJ, Lee PL, et al. Age‐related changes in resting‐state networks of a large sample size of healthy elderly. CNS Neurosci Ther. 2015;21:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones DT, Machulda MM, Vemuri P, et al. Age‐related changes in the default mode network are more advanced in Alzheimer disease. Neurology. 2011;77:1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z, Liang P, Jia X, et al. The baseline and longitudinal changes of PCC connectivity in mild cognitive impairment: a combined structure and resting‐state fMRI study. PLoS One. 2012;7:e36838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Obler LK, Rykhlevskaia E, Schnyer D, et al. Bilateral brain regions associated with naming in older adults. Brain Lang. 2010;113:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bakker A, Krauss GL, Albert MS, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duverne S, Motamedinia S, Rugg MD. The relationship between aging, performance, and the neural correlates of successful memory encoding. Cereb Cortex. 2009;19:733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Trujillo JP, Gerrits NJ, Veltman DJ, Berendse HW, van der Werf YD, van den Heuvel OA. Reduced neural connectivity but increased task‐related activity during working memory in de novo Parkinson patients. Hum Brain Mapp. 2015;36:1554–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yasuno F, Kazui H, Morita N, et al. High amyloid‐beta deposition related to depressive symptoms in older individuals with normal cognition: a pilot study. Int J Geriatr Psychiatry. 2016;31:920–928. [DOI] [PubMed] [Google Scholar]
- 43. Butters MA, Klunk WE, Mathis CA, et al. Imaging Alzheimer pathology in late‐life depression with PET and Pittsburgh Compound‐B. Alzheimer Dis Assoc Disord. 2008;22:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan L, Wang HF, Tan MS, et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer's disease cohorts. Sci Rep. 2016;6:26027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials