

Summary
Aims
Cerebral small vessel disease (SVD) is the leading cause of vascular dementia. Although the most of cases are sporadic, familial monogenic causes have been identified in a growing minority of patients. CADASIL, due to mutations of NOTCH3 gene, is the most common genetic SVD, and CARASIL, linked to HTRA1 gene mutations, is a rare but well known autosomal recessive SVD. Recently, also heterozygous HTRA1 mutations have been described in patients with familial SVD. To detect a genetic cause of familial SVD, we performed mutational analysis of HTRA1 gene in a large cohort of Italian NOTCH3‐negative patients.
Methods
We recruited 142 NOTCH3‐negative patients and 160 healthy age‐matched controls. Additional control data were obtained from five pathogenicity prediction software.
Results
Five different HTRA1 heterozygous mutations were detected in nine patients from five unrelated families. Clinical phenotype was typical of SVD, and the onset was presenile. Brain magnetic resonance imaging (MRI) showed a subcortical leukoencephalopathy, with involvement of the external and internal capsule, corpus callosum, and multiple lacunar infarcts. Cerebral microbleeds were also seen, while anterior temporal lobes involvement was not present.
Conclusion
Our observation further supports the pathogenic role of the heterozygous HTRA1 mutations in familial SVD.
Keywords: CADASIL, CARASIL, HTRA1, NOTCH3, small vessel disease
1. INTRODUCTION
Cerebral small vessel disease (SVD) is a heterogeneous group of disorders affecting small vessels of the brain and is commonly recognized to be the leading cause of vascular cognitive impairment and dementia. While in the great majority of cases cerebral SVD is an age‐related sporadic disorder with multifactorial origin, familial monogenic causes have been identified in the last decades in a minority of patients.1 The monogenic SVDs, which share overlapping clinical and radiological features among them and with the sporadic form, are generally early onset and transmitted with a dominant inheritance pattern. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL),2 due to NOTCH3 gene mutations, is the most common dominantly inherited SVD, hundreds of subjects having been diagnosed worldwide with a prevalence of about 4/100 000 in European countries.3, 4, 5, 6 Therefore, NOTCH3 gene analysis is currently performed in familial cases with evidence of dominant transmission. However, it is common experience the finding of a considerable number of patients with typical CADASIL phenotype who do not carry NOTCH3 mutations.
Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL)7, 8, 9 is a very rare autosomal recessive familial SVD reported, with the exception of few cases,10, 11, 12, 13, 14, 15 only in a small number of Japanese and Chinese families. CARASIL distinctive features include the onset in early adulthood (20‐30 years) of a nonhypertensive cerebral arteriopathy with rapidly progressive cognitive and motor disability in association with extraneurological manifestations, namely alopecia and spondylosis. Brain magnetic resonance imaging (MRI) shows diffuse leukoencephalopathy and multiple subcortical infarcts. HtrA serine protease 1 gene (HTRA1) which encodes a serine protease that represses signaling by TGF‐ β family members has been recognized16 as the only causative gene of CARASIL.17
Recently, heterozygous HTRA1 mutations have been described in patients with late‐onset familial SVD.18, 19
To better investigate our undiagnosed cases of familial SVD, we performed mutational analysis of HTRA1 gene in a large cohort (142 cases) of NOTCH3‐negative patients. Our study resulted in the detection of heterozygous HTRA1 mutations in nine patients from five families, accounting for about 3.5% of cases. These findings further support the role of heterozygous HTRA1 mutations in the development of SVD.
2. METHODS
2.1. Standard protocol approvals, registration, and patient consent
We recruited 142 Italian patients (mean age 58.2±12,7, 69 females) with cerebral ischemic SVD and 160 healthy age‐matched Caucasian controls among three Italian reference centers for cerebrovascular disorders (Siena, Florence, Modena). The inclusion criteria were as follows: age 18‐75 years, moderate/severe leukoaraiosis (grade 2‐3 of the modified Fazekas scale),20 a family history of stroke/transient ischemic attack (TIA) or dementia, a Mendelian pattern of inheritance, and negative screening for NOTCH3‐gene (exons 2‐24). Demographic, clinical, and MRI data were collected for each patient. In our cohort of 142 patients, 80 patients had a history of stroke/TIA, 60 patients a history of cognitive decline, and 30 patients both stroke/TIA and dementia. The rest of patients were asymptomatic and had leukoaraiosis and a familial history of cerebrovascular disease. The occurrence of typical disease features in the family and the presence of vascular risk factors were also recorded. Family history was defined, if available, with direct examination, otherwise with interview of the probands. Vascular risk factors, if present, were well controlled: 7/142 presented diabetes mellitus, 20/142 hypertension, 30/142 dyslipidemia, and 35/142 smoking. No other significant comorbidities were present.
The study was approved by the local Ethic Committee. All patients and healthy controls provided written informed consent before participation in the study.
2.2. Genetic analysis
Total genomic DNA from patients and control subjects was extracted from whole blood. Then, the entire coding regions and adjacent intronic regions of HTRA1 (current RefSeq: NM_002775) were amplified by PCR and directly sequenced in patients; the presence of genetic variants was screened in sex‐ and age‐matched healthy control subjects. When available, relatives of HTRA1 mutation carriers were also sequenced. Additional control data were obtained from five pathogenicity prediction software (Polyphen‐2, SIFT, Mutation Tester, Panther, and Mutation Assessor) and four freely available databases of genetic variants (dbSNP, 1000Genomes, ExAc, and EVS).
3. RESULTS
3.1. Genetic results
Among 142 NOTCH3‐negative patients, five different HTRA1 heterozygous mutations were detected in nine patients from five unrelated families.
These mutations included two new genomic variants, p.Ser136Gly and p.Gly206Glu, found in two patients, and three previously reported variants found in seven patients: p.Gln151Lys, p.Val175Met, and p.Gly295Arg.
All variants affect highly conserved amino acids (from human to fish) and are absent in 320 chromosomes from matched healthy controls.
In details, p.Gly206Glu and p.Gly295Arg are predicted to be deleterious by all in silico tools and are located in the serine protease domain. The p.Gly206Glu variant is absent in 1000 Genomes, Exac, and EVS databases. The p.Gly295Arg variant is absent in 1000 Genomes and EVS databases, but it is found in ExAc database in one nonFinnish European out of 60 546 subjects. Furthermore, this variant has been reported in homozygousity in a Caucasian man with CARASIL.11
p.Ser136Gly and p.Gln151Lys, predicted to be deleterious by four out of five in silico tools, are located in the Kazal‐like domain.
p.Gln151Lys has been found in Exac database in two South Asians out of 5352 subjects, whereas p.Ser136Gly is absent in all databases.
The p.Val175Met variant is predicted to be deleterious by four out of five in silico tools and it affects the interdomain region, a linker region from aminoacid position 157‐204. The variant is absent in 1000 Genomes and EVS databases, but it is found in ExAc database in one nonFinnish European out of 60706 subjects.
The localization of the five variants in the protein structure, the ID SNP, and the frequency in public variant databases (where available) and in silico analysis with pathogenicity prediction are reported in Table 1.
Table 1.
Heterozygous HTRA1 variants identified in the familial SVD probands
| Mutation | Protein domain | ID SNP | 1000Genomes Frequency (MAF) | Allele frequency ExAC | EVS frequency | Polyphen‐2 | SIFT | Mutation taster | nsSNPanalyzer | Mutation Assessor |
|---|---|---|---|---|---|---|---|---|---|---|
| p.Ser136Gly (F3) | Kazal‐like | New | ‐ | ‐ | NA | Benign (score 0.118) | Damaging (score 0.04) | Disease causing | Disease | Medium functional impact |
| p.Gln151Lys (F5) | Kazal‐like | rs754645487 | ‐ | 0.0001868 | NA | Possibly damaging (score 0.567) | Tolerated (score 0.16) | Disease causing | Disease | Medium functional impact |
| p.Val175Met (F4) | None | rs768665565 | NA | 0.000008236 | Absence | Probably damaging (score 1.00) | Damaging (score 0) | Disease causing | Neutral | Medium functional impact |
| p.Gly206Glu (F2) | Serine protease | New | ‐ | ‐ | Absence | Probably damaging (score 1.00) | Damaging (score 0) | Disease causing | Disease | High functional impact |
| p.Gly295Arg (F1) | Serine protease | rs587776873 | NA | 0.000008258 | Absence | Probably damaging (score 1.00) | Damaging (score 0) | Disease causing | Disease | High functional impact |
3.2. Clinical results
Clinical characteristics of patients are detailed in Table 2. As a whole, the phenotype was quite homogeneous among them, the clinical picture being typical of the SVD (subcortical ischemic events and/or progressive cognitive impairment). The age at onset was presenile (mean 61.3 years±4.2 SD).
Table 2.
Main clinical features of the heterozygous HTRA1 mutation carriers
| Patients | Mutation | Sex | Age | Onset (y) | Vascular risk factors | Symptoms at onset | TIA/stroke | Cognitive decline | Gait disturbances | Affected relatives |
|---|---|---|---|---|---|---|---|---|---|---|
| F1 (II,3) | p.Gly295Arg | M | 73 | 65 | ‐ | Cognitive decline | ‐ | + | + | 5 |
| F1 (II,5) | p.Gly295Arg | M | 71 | 67 | Hypertension | Cognitive decline | + | + | ‐ | 5 |
| F1 (II,9) | p.Gly295Arg | M | 63 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 5 |
| F1 (III,3) | p.Gly295Arg | M | 49 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 5 |
| F2 (II,1) | p.Gly206Glu | M | 65 | 55 | Hypertension | Gait disturbances | ‐ | + | + | 1 |
| F3 (IV,1) | p.Ser136Gly | F | 77 | 63 | ‐ | Gait disturbances | + | + | + | 2 |
| F4 (II,1) | p.Val175Met | M | Died (77) | 59 | Hypertension Dyslipidemia | Gait disturbances | + | + | + | 1 |
| F4 (III,2) | p.Val175Met | F | 46 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 |
| F5 (II,1) | p.Gln151Lys | M | 61 | 59 | ‐ | Gait disturbances | ‐ | + | + | 2 |
None of our patients had early‐onset alopecia and/or spondylosis, explored with lumbar MRI, usually described in CARASIL. When they were present, they manifested over age 40. The frequency of vascular risk factors is reported in Table 2. Pedigrees of the families carrying heterozygous HTRA1 mutations are shown in Figure 1, and a short description of them is reported below.
Figure 1.
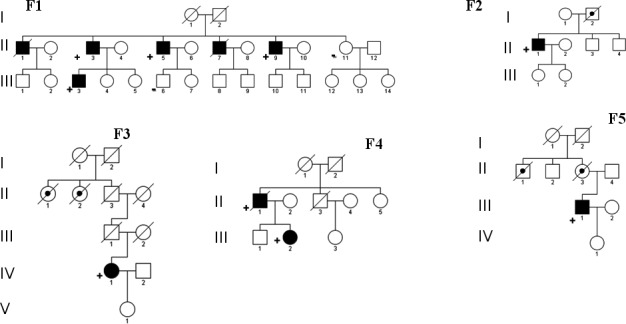
Pedigrees of the probands. Black filled symbol: patient with leukoencephalopathy and/or neurological signs/symptom of SVD. Black dot: patient with positive past history for SVD. +: patient with single HTRA1 mutation. ‐: individual without single HTRA1 deleterious variant
Family 1 (F1): The proband (II,3) was a 73‐year‐old man, with a progressive cognitive decline and mood depression since he was 65. To date, the patient is bedridden with pseudobulbar palsy. Brain MRI showed a severe subcortical leukoencephalopathy and mild diffuse atrophy.
His 71‐year‐old brother (II,5), at the age of 67, experienced two TIAs, and in the same period, he developed a mild reduction in cognitive performances and mood disorders. Clinical conditions have remained substantially stable. Brain MRI showed a severe leukoencephalopathy (Figure 2). Two brothers (II,1 and II,7) died of complications of a presenile dementia, respectively at age 71 and 52. For both of them, a severe leukoencephalopathy had been described. Two asymptomatic carriers (II,9 aged 63, III,3 aged 49) showed moderate leukoencephalopathy.
Figure 2.
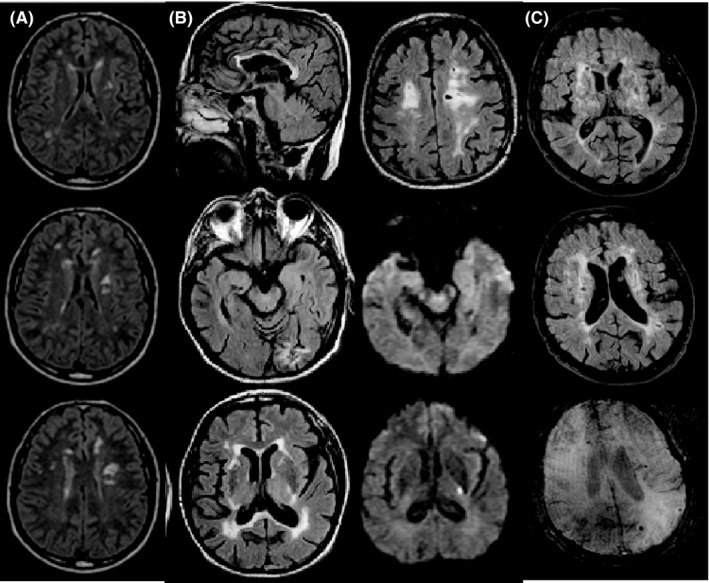
Brain MRI findings. (A) Periventricular white matter Fazekas grade 1 and deep white matter Fazekas grade 2 involvement including internal capsules (Patient F1‐III,3, age 46, serial consecutive axial FLAIR images); (B) Periventricular and deep white matter Fazekas grade 3 involvement including external and internal capsules and corpus callosum, lacunar infarcts, subacute ischemia in the left internal capsule, previous infarction in the left occipital lobe, and supratentorial subcortical atrophy, without involvement of anterior temporal lobes (Patient F1‐II,5, age 67, serial nonconsecutive axial FLAIR and DWI images); (C) Periventricular and deep white matter Fazekas grade 3 involvement including external and internal capsule, lacunar infarcts, microbleeds, and supratentorial subcortical atrophy (Patient F5‐III,1, age 60, sagittal nonconsecutive axial FLAIR and SWI images)
Family 2 (F2): The index case (II,1), since he was 55, had progressive gait disturbances, mood depression, and cognitive impairment. Brain MRI showed diffuse leukoencephalopathy with temporal involvement and mild cerebral atrophy. His father (I,2) died of stroke at 55.
Family 3 (F3): The proband (IV,1) was a 77‐year‐old woman with dementia, gait disturbances, urinary incontinence, and dysarthria. She had a history of mood depression and a stroke at the age of 63, and some TIAs during the years. History of stroke, cognitive decline, and psychiatric disorders from her father’ side was reported. Brain MRI showed diffuse subcortical leukoencephalopathy.
Family 4 (F4): The proband (II,1) died at 77 years. He had several TIAs and progressive cognitive decline since he was 59, and a major stroke at 61 years. Brain MRI showed a moderate leukoencephalopathy and ischemic lacunes. An asymptomatic daughter (III,2) performed MRI which showed white matter abnormalities.
Family 5 (F5): The proband (III,1) was a 61‐year‐old man who presented, since a couple of years, progressive ataxic gait, dysarthria and dysphagia, and a moderate cognitive impairment. The same clinical pattern was referred in his mother and his maternal uncle. In his past, history was unremarkable. Brain MRI showed confluent subcortical leukoencephalopathy, microbleeds, and mild diffuse atrophy.
3.3. MRI description
Brain MRI of HTRA1 heterozygous patients was centrally reviewed by an expert neuroradiologist (A.C.). Figure 2 shows the main observed findings. Typical MRI findings included foci to confluent areas of high signal intensity on T2‐weighted and FLAIR images in the periventricular and deep white matter,21 with involvement of the external and internal capsule and multiple lacunar infarcts in most of them. U fibers were always spared. Corpus callosum was involved in one patient (Figure 2B), while anterior temporal lobes were not involved. Dilated interstitial perivascular spaces or so‐called Virchow‐Robin spaces showing “status cribrosum” in the basal ganglia, were present. Microbleeds were observed peripherally in two patient (II,5‐F1,III,1‐F5), however, due to the absence of T2*‐ or susceptibility‐weighted images in most of patients, we were unable to estimate the MRI frequency of microbleeds. Gliotic‐malacic changes in the left occipital lobe from previous infarction were seen in one patient (II,5‐F1). Supra‐ and subtentorial atrophy was seen in one patient (III,1‐F5). In one patient (II,5‐F1), an acute‐to‐early subacute ischemic lesion was observed in the left posterior limb of the internal capsule.
4. DISCUSSION
Herein, we reported clinical, MRI, and genetic details of nine individuals from five unrelated families with familial cerebral SVD related to heterozygous mutations of HTRA1 gene. The pathogenic role in late‐onset cerebrovascular phenotype of heterozygous HTRA1 gene mutations, otherwise causative in homozygosity of CARASIL, was recently hypothesized by Verdura et al.18
Moreover, familial recurrence of SVD and/or leukoencephalopathy has been outlined in obligate carriers of CARASIL patients11, 13, 14 and in CARASIL pedigrees.22, 23
The pathogenicity of HTRA1 variants found in our patients is supported by the following observations: all variants were (i) located in highly conserved positions; (ii) absent in our control subjects; (iii) absent (p.Ser136Gly, p.Gly206Glu) or rare (MAF<0.01: p.Gln151Lys, p.Val175Met, p.Gly295Arg) in databases; (iv) predicted as deleterious at least by four out of five in silico tools (p.Gly206Glu and p.p.Gly295Arg are classified pathogenic by all five in silico tools); and e) located in regions of the protein where pathogenic mutations were already found (Kazal‐like domain, linker region, and serine protease domain) both in CARASIL and in HTRA1‐related SVD patients. Moreover, segregation analysis was positive in the two families (F1 and F4) in which it was possible to perform it.
We point out the limitation of our study consisting in the absence of functional studies, although functional studies on the HTRA1 mutant protein does not necessarily resolve the question regarding the pathogenicity: Verdura et al.18 show a complete loss of protease activity only in five of seven pathogenic variants described, and Nozaki et al.24 present HTRA1 mutant with no reduction in protease activity.
The role of single mutations of putative recessive genes, included HTRA1 gene, is still matter of debate. Mild phenotypic expression in heterozygotes for recessive diseases has largely described for an increasing number of diseases.25, 26, 27, 28 As regards to the pathogenic mechanism of heterozygous HTRA1 mutations, Nozaki et al.24 suggested a dominant‐negative effect.
The deleterious effect of HTRA1 mutations on vascular homeostasis is not yet fully understood but is probably related to the dysregulation of TGF‐β signaling. Recently, the TGF‐β binding protein (LTBP), an important component of the extracellular matrix (ECM), has been shown to be a substrate of proteolytic HTRA1 activity.29 Human HTRA1, like all eukaryotic HTRA proteins, is not completely structurally and biochemically characterized30 in particular in its N‐domain: The autolysis of HTRA1 N‐terminal domain is observed in vivo, without decreasing the protease activity.31 The detection of HTRA1 mutations even in the Kazal‐like domain, both in CARASIL and in HTRA1‐related small vessel disease patients, suggests for the N‐terminal domain a function not fully understood.
Clinical features of this novel familial condition,18 which include acute cerebrovascular events, cognitive impairment, progressive gait disturbances, and presenile onset, overlap with common sporadic SVD and other adult rare monogenic cerebrovascular disorders, specifically CADASIL and CARASIL.
Compared to CARASIL, in heterozygous HTRA1, the age of onset is older, the phenotype is milder, the history of illness is long‐lasting, and extraneurological signs (early‐onset spondylosis and alopecia) are usually lacking. No significant clinical differences are present with respect to CADASIL except for the age at onset, usually earlier in CADASIL. Migraine with aura, a common early clinical manifestation of CADASIL,32 is very rarely observed in heterozygous HTRA1.18 With respect to common sporadic SVD, the main differentiating features are usually younger age at onset with a presenile pattern, the familial recurrence, and the substantial lack of correlation of clinical and MRI phenotype with vascular risk factors which may be occasionally present.
Brain MRI in HTRA1‐related SVD presents the typical neuroimaging pattern of SVD, with confluent subcortical T2‐weighted white matter hyperintensities with relative sparing of U fibers, lacunar infarcts, status cribrosum, microbleeds and sparing of anterior temporal lobes.18 Of note, we found corpus callosum involvement in one patient and this is a finding rarely observed in CADASIL and generally absent in CARASIL and SVD.
In conclusion, we have confirmed single HTRA1 mutations as a cause of late‐onset familial SVD. Therefore, we suggest to perform the screening of HTRA1 gene in all patients with an undiagnosed SVD, also in the absence of an evident familial pattern, which otherwise could be overlooked for the late onset of clinical symptoms. Further studies could better define the genetic spectrum of mutations, the phenotypic picture of this novel condition, and the possible still unproved genotype‐phenotype correlation.
DISCLOSURE
All authors declare no conflict of interest.
ACKNOWLEDGMENT
This research was in part supported by a grant from MIUR to MTD (prot. 20095JPSNA_005).
Di Donato I, Bianchi S, Gallus GN, et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci Ther. 2017;23:759–765. 10.1111/cns.12722
REFERENCES
- 1. Tan RYY, Markus HS. Monogenic causes of stroke: now and the future. J Neurol. 2015;262:2601‐2606. [DOI] [PubMed] [Google Scholar]
- 2. Tournier‐Lasserve E, Joutel A, Melki J, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet. 1993;3:256‐259. [DOI] [PubMed] [Google Scholar]
- 3. Razvi SS, Davidson R, Bone I, Muir KW. The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) in the west of Scotland. J Neurol Neurosurg Psychiatry. 2005;76:739‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narayan SK, Gorman G, Kalaria RN, Ford GA, Chinnery PF. The minimum prevalence of CADASIL in northeast England. Neurology. 2012;78:1025‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moreton FC, Razvi SS, Davidson R, Miur KW. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol Scand. 2014;130:197‐203. [DOI] [PubMed] [Google Scholar]
- 6. Bianchi S, Zicari E, Carluccio A, et al. CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262:134‐141. [DOI] [PubMed] [Google Scholar]
- 7. Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis. 2011;20:85‐93. [DOI] [PubMed] [Google Scholar]
- 8. Yanagawa S, Ito N, Arima K, Ikeda S. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Neurology. 2002;58:817‐820. [DOI] [PubMed] [Google Scholar]
- 9. Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45:3447‐3453. [DOI] [PubMed] [Google Scholar]
- 10. Zheng DM, Xu FF, Gao Y, Zhang H, Han SC, Bi GR. A Chinese pedigree of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): clinical and radiological features. J Clin Neurosci. 2009;16:847‐849. [DOI] [PubMed] [Google Scholar]
- 11. Mendioroz M, Fernández‐Cadenas I, Del Río‐Espinola A, et al. A missense HTRA1 mutation expands CARASIL syndrome to the Caucasian population. Neurology. 2010;75:2033‐2035. [DOI] [PubMed] [Google Scholar]
- 12. Wang XL, Li CF, Guo HW, Cao BZ. A novel mutation in the HTRA1 gene identified in Chinese CARASIL pedigree. CNS Neurosci Ther. 2012;18:867‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, He Z, Meng S, Li L, Yang H, Zhang X. A novel mutation of the high‐temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy(CARASIL). J Int Med Res. 2013;41:1445‐1455. [DOI] [PubMed] [Google Scholar]
- 14. Bianchi S, Di Palma C, Gallus GN, et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology. 2014;82:898‐900. [DOI] [PubMed] [Google Scholar]
- 15. Cai B, Zeng J, Lin Y, et al. A frameshift mutation in HTRA1 expands CARASIL syndrome and peripheral small arterial disease to the Chinese population. Neurol Sci. 2015;36:1387‐1391. [DOI] [PubMed] [Google Scholar]
- 16. Singh N, Kuppili RR, Bose K. The structural basis of mode of activation and functional diversity: a case study with HtrA family of serine proteases. Arch Biochem Biophys. 2011;516:85‐96. [DOI] [PubMed] [Google Scholar]
- 17. Hara K, Shiga A, Fukutake T, et al. Association of HTRA1 mutations and familial ischemic cerebral small‐vessel disease. N Engl J Med. 2009;360:1729‐1739. [DOI] [PubMed] [Google Scholar]
- 18. Verdura E, Hervé D, Scharrer E, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain. 2015;138:2347‐2358. [DOI] [PubMed] [Google Scholar]
- 19. Bougea A, Velonakis G, Spantideas N, et al. The first Greek case of heterozygous cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy: an atypical clinico‐radiological presentation. Neuroradiol J. 2017. [Epub ahead of print]. 10.1177/1971400917700168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pantoni L, Basile AM, Pracucci G, et al. Impact of age‐related cerebral white matter changes on the transition of disability‐The LADIS study: rationale, design and methodology. Neuroepidemiology. 2005;24:51‐62. [DOI] [PubMed] [Google Scholar]
- 21. Nozaki H, Sekine Y, Fukutake T, et al. Characteristic features and progression of abnormalities on MRI for CARASIL. Neurology. 2015;85:459‐463. [DOI] [PubMed] [Google Scholar]
- 22. Fukutake T, Hirayama K. Familial young‐adult‐onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur Neurol. 1995;35:69‐79. [DOI] [PubMed] [Google Scholar]
- 23. Nishimoto Y, Shibata M, Nihonmatsu M, et al. A novel mutation in the HTRA1 gene causes CARASIL without alopecia. Neurology. 2011;76:1353‐1355. [DOI] [PubMed] [Google Scholar]
- 24. Nozaki H, Kato T, Nihonmatsu M, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology. 2016;86:1964‐1974. [DOI] [PubMed] [Google Scholar]
- 25. Plomp AS, Hu X, de Jong PT, Bergen AA. Does autosomal dominant pseudoxanthoma elasticum exist? Am J Med Genet A. 2004;126A:403‐412. [DOI] [PubMed] [Google Scholar]
- 26. Illa I, De Luna N, Domínguez‐Perles R, et al. Symptomatic dysferlin gene mutation carriers: characterization of two cases. Neurology. 2007;68:1284‐1289. [DOI] [PubMed] [Google Scholar]
- 27. Klein C, Lohmann‐Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652‐662. [DOI] [PubMed] [Google Scholar]
- 28. Schapira AH. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci. 2015;66:37‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beaufort N, Scharrer E, Kremmer E, et al. Cerebral small vessel disease‐related protease HtrA1 processes latent TGF‐β binding protein 1and facilitates TGF‐β signaling. Proc Natl Acad Sci USA. 2014;111:16496‐16501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eigenbrot C, Ultsch M, Lipari MT, et al. Structural and functional analysis of HtrA1 and its subdomains. Structure. 2012;20:1040‐1050. [DOI] [PubMed] [Google Scholar]
- 31. Risør MW, Poulsen ET, Thomsen LR, et al. The autolysis of human HtrA1 is governed by the redox state of its N‐terminal domain. Biochemistry. 2014;53:3851‐3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long‐term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. 2004;127:2533‐2539. [DOI] [PubMed] [Google Scholar]